

Abstract
Actin with a Val 159 to Asn mutation (V159N) forms actin filaments that depolymerize slowly because of a failure to undergo a conformational change after inorganic phosphate release. Here we demonstrate that expression of this actin results in reduced actin dynamics in vivo, and we make use of this property to study the roles of rapid actin filament turnover. Yeast strains expressing the V159N mutant (act1-159) as their only source of actin have larger cortical actin patches and more actin cables than wild-type yeast. Rapid actin dynamics are not essential for cortical actin patch motility or establishment of cell polarity. However, fluid phase endocytosis is defective in act1-159 strains. act1-159 is synthetically lethal with cofilin and profilin mutants, supporting the conclusion that mutations in all of these genes impair the polymerization/ depolymerization cycle. In contrast, act1-159 partially suppresses the temperature sensitivity of a tropomyosin mutant, and the loss of cytoplasmic cables seen in fimbrin, Mdm20p, and tropomyosin null mutants, suggesting filament stabilizing functions for these actin-binding proteins. Analysis of the cables in these double-mutant cells supports a role for fimbrin in organizing cytoplasmic cables and for Mdm20p and tropomyosin in excluding cofilin from the cables.
Keywords: actin, dynamics, ATP hydrolysis, yeast, cytoskeleton
In most eukaryotic cells, actin filaments undergo dynamic cycles of assembly and disassembly. Control of actin filament dynamics is thought to be essential for a wide range of cellular processes, including cytokinesis, cell motility, response to external signals, and establishment and maintenance cell shape and polarity. There are numerous regulatory proteins that modulate actin dynamics. However, fundamental to the regulation of actin dynamics is the ATP cycle of actin itself. Actin polymerizes from ATP-bound actin, and the ATP is hydrolyzed subsequent to assembly. The release of inorganic phosphate (Pi) after ATP hydrolysis destabilizes the filament and promotes actin filament disassembly (Carlier and Pantaloni, 1986; Carlier, 1990). Yeast actin with a single amino acid change of the highly conserved Val 159 to Asn (V159N) forms filaments that depolymerize approximately three times more slowly than wild-type filaments. The V159N mutant filaments also nucleate assembly slightly faster than wild-type, but then assemble at a slower rate. The critical concentration of V159N actin is more than fivefold lower than that of wild-type actin, suggesting that slow depolymerization and rapid nucleation dominate the overall effects on filament dynamics in this mutant. The slow depolymerization of V159N actin appears to be the result of a failure to make the conformational change that normally accompanies Pi release (Belmont, L.D., D.G. Drubin, and E. Egelman, submitted for publication). Previous studies of how actin filament dynamics are regulated by Pi release have all made use of phosphate analogues, such as BeF4 or free phosphate, methods that are too non-specific to be used in living cells. Therefore, the act1-159 mutant provides an excellent tool with which to probe the role of rapid actin filament dynamics in vivo.
The actin cytoskeleton of Saccharomyces cerevisiae is simple but dynamic (Ayscough et al., 1997; Lappalainen and Drubin, 1997). Filamentous actin (F-actin)1 in this yeast is found in cortical patches and cytoplasmic cables. Assembly of both types of structures is regulated spatially and temporally during bud emergence. The cortical patches are concentrated in the bud, and the cables run between the mother cell and the bud. When the bud is large, the actin patches become depolarized for isotropic growth, and late in the cell cycle they become polarized at the neck immediately before cytokinesis (for review see Welch et al., 1994). The cortical actin patches move quite rapidly within the plane of the cell cortex at rates of ≤1.5 μm/s (Waddle et al., 1996; Doyle and Botstein, 1996). The relative contributions of actin dynamics and actin patch motility to the establishment and maintenance of polarity in yeast have not been established.
Genetic and biochemical approaches have identified numerous proteins that regulate actin dynamics and organization in yeast. Proteins that increase actin dynamics include cofilin and profilin. Yeast cofilin is an essential protein that is localized to actin cortical patches (Moon et al., 1993) where it increases the rate of actin filament depolymerization (Lappalainen and Drubin, 1997). Profilin binds to actin monomers and has been reported to accelerate nucleotide exchange and the addition of actin to the barbed ends of actin filaments (Goldschmidt-Clermont et al., 1991; Pantaloni and Carlier, 1993). Deletion of the profilin gene in yeast (PFY1) results in a severe phenotype of extremely slow growth, depolarized actin patches, loss of cables, and very large cells that lyse easily (Haarer et al., 1990). Proteins that are thought to stabilize actin filaments include fimbrin and tropomyosin. Yeast fimbrin (SAC6) is localized to actin patches and cables and tropomyosin (TPM1) is localized exclusively to cables (Drubin et al., 1988; Liu and Bretscher, 1989). Deletion of the genes that encode either of these proteins results in a loss of actin cables and partial depolarization of patches (Liu and Bretscher, 1989; Adams et al., 1991). Deletion of MDM20, a gene required for mitochondrial inheritance, also causes loss of actin cables (Hermann et al., 1997). It still remains unclear how these proteins interact with each other and with actin to form a highly polarized, yet dynamic, actin cytoskeleton.
Here we use the act1-159 mutation to study the role of rapid actin dynamics in yeast and to elucidate the in vivo functions of several of these actin regulatory proteins.
Materials and Methods
Strains and Cell Culture
Standard media and cell culture conditions were used (Guthrie and Fink, 1991). The strains used in this study are listed in Table I.
Table I.
Strains Used in This Study
| Strain number | Genotype | |
|---|---|---|
| DDY216 | α sac6Δ::URA3 lys2 trp1 his3 lue2 ura3 | |
| DDY479 | α/a ACT1::HIS3/+ his3Δ200/his3Δ200 lue2-3, 112/lue2-3, | |
| 112 ura3-52/ura3-52 ade2-101/+ ade4/+ cry1/+ can1-1/+ tub2-101/+ | ||
| DDY487 | α tpm1::LUE2 ura3 lys2 | |
| DDY1277 | a cof1-5::LUE2 ura3-52 his3Δ200 lys2-801 ade2-101 | |
| DDY1279 | a cof1-22::LUE2 ura3-52 his3Δ200 lys2-801 ade2-101 | |
| DDY1415 | α mdm20::LEU2 his3Δ200 leu2Δ1 ura3-52 | |
| DDY1418 | a pfy1-111::LUE2 ura3 his3 lue2 ade2 ade3 | |
| DDY1420 | a pfy1Δ4C::LUE2 ura3 his3 lue2 ade2 ade3 lys2 | |
| DDY1490 | α ACT1::HIS3 his3Δ200 tub2-101 ura3-52 lue2-3, 112 ade2-101 ade4 | |
| DDY1491 | α/a ACT1/act1-159::HIS3 his3Δ200/ his3Δ200 lue2-3,112/lue2-3,112 ura3-52/ura3-52 | |
| ade2-101/+ cry1/+ can1-1/+ tub2-101/+ ade4/+ | ||
| DDY1492 | α act1-159::HIS3 his3Δ200 tub2-101 ade4 ura3-52 lue2-3, 112 | |
| DDY1493 | a act1-159::HIS3 his3Δ200 tub2-101 ura3-52 lue2-3, 112 | |
| DDY1495 | a ACT1::HIS3 his3Δ200 tub2-101 ura3-52 lue2-3, 112 | |
| DDY1507 | a pca1::HIS3 his3Δ200 leu2-3,112 lys2-801 ura3-52 | |
DDY1418 (BHY46) and DDY1420 (BHY70) were provided by Brian Haarar, DDY1415 (JSY999) was provided by Greg Hermann and Janet Shaw and DDY1507 (RLY148) was provided by Rong Li. The other strains are from this lab.
Construction of Mutations
The mutation was constructed as described previously (Wertman et al., 1992), and was confirmed in the final haploid yeast strain by sequencing a PCR product from the region containing the mutation.
Actin Purification
Actin was purified from strains DDY1492 and DDY1495 by DNaseI affinity chromatography as described (Ayscough et al., 1997) and frozen as G-actin in liquid N2. Before experiments measuring ATP exchange, the actin was subjected to an assembly and disassembly cycle as follows. Actin was thawed and immediately polymerized by addition of 0.1 M KCl, 0.5 mM ATP, and 2 mM MgCl2. After 45 min, additional KCl was added to a final concentration of 0.3 M and the F-actin was sedimented in a Beckman TLA100 rotor (Beckman Instruments, Inc., Fullerton, CA) at 90,000 rpm for 20 min at 25°C. The F-actin was resuspended at ∼30 μM in G buffer (10 mM Tris, pH 7.5, 0.2 mM CaCl2, 0.5 mM ATP, 1 mM DTT), and dialyzed for at least 16 h at 4°C against two changes of G buffer. The G-actin was then clarified by centrifugation at 90,000 rpm for 30 min at 4°C in a TLA100 rotor. The actin concentration was measured first by absorbance at 290 nm (e = 26,600 M−1), and then by densitometry of SDS-PAGE gel bands. The cycled actin was used within 8 h.
ATP Exchange
ATP exchange was measured by diluting the actin into ε-ATP (1,N6-ethenoadenosine-5′ triphosphate; Molecular Probes, Inc., Eugene, OR), with a final concentration of 20 μM ATP, 200 μM ε-ATP, 1.6 μM actin, 10 mM Tris, pH 7.5, 0.2 mM CaCl2, and the appropriate concentration of latrunculin A (LAT-A). The sample was then immediately placed into a thermostatted cuvette at 18°C, and the change in fluorescence was monitored (excitation = 360 nm; emission = 410 nm). There was a dead time of ∼4 s between addition of ε-ATP and recording fluorescence.
Fluorescence Staining and Microscopy
Phalloidin staining and immunofluorescence were performed as described (Ayscough et al., 1997). Images were acquired with a Zeiss Axioskop microscope (Carl Zeiss Inc., Thornwood, NY) equipped with a Sony CCD (Sony Corp., Park Ridge, NJ) camera controlled by Northern Exposure Software (Phase 3 Imaging). Patch motility (see Fig. 6) was recorded at 4-s intervals with shuttering of light between images. Fluorescence intensity of phalloidin stained structures in yeast, and actin filament lengths were measured with Image Pro Plus software (Phase 3 Imaging). Fluorescence intensities were compared by measuring the percent area of a cell that contained phalloidin-stained structures above a defined threshold. Immunofluorescence images of double mutants and patch motility in the presence of LAT-A were collected with a Nikon TE300 microscope equipped with a Hamamatsu Orca-100 cooled CCD camera (Hamamatsu Photonics, Bridgewater, NJ). Images of patch motility in LAT-A were collected continuously without shuttering. Exposure time was 500 ms per image.
Figure 6.

Rates of actin patch motility are unaffected by the act1-159 mutation. Actin cortical patches were visualized using GFP-ABP1, and images were collected at 4-s intervals. The speed of each moving patch was measured at every interval. n = 114 (wild-type actin); n = 132 (V159N actin). There was no significant difference in the speed of patch movement.
Latrunculin A Assays
Halo assays to measure sensitivity to LAT-A and actin turnover assays were performed as described (Ayscough et al., 1997; Lappalainen and Drubin, 1997). For pelleting assays, 4 μM actin was polymerized at room temperature for 1 h. LAT-A (or control buffer) was added, the samples incubated for an additional hour, and then spun at 90,000 rpm for 15 min in a TLA100 rotor. The pellet and supernatant fractions were dissolved in equivalent volumes of sample buffer, analyzed by SDS-PAGE and densitometry to determine the amount of G-actin.
Western Blots
4 OD units at 600 nm (8 × 107 cells) of log phase cells were pelleted in a clinical centrifuge and washed with distilled water. 100 μl of glass beads and 50 μl of 2× sample buffer were added to the cell pellet and this was boiled immediately for 3 min. The sample was then vortexed for 1 min, an additional 75 μl of sample buffer was added, and then the sample was boiled for an additional 2 min. The sample was spun for 1 s in a microfuge and the supernatant was loaded onto a 10% acrylamide SDS gel. Proteins were transferred to nitrocellulose and probed for actin and tubulin using standard techniques (Harlow and Lane, 1988). Band intensity was quantified by densitometry using an IS-2000 Digital Imaging System.
Endocytosis Assay
Yeast cells were grown to an OD at 600 nm of 0.25 (5 × 106 cells/ml). 2 ml of cells were sedimented at low speed in a clinical centrifuge and resuspended in 40 μl of media. 15 μl of resuspended cells were added to 7.5 μl of 40 mg/ml Lucifer yellow (Molecular Probes Inc.) and incubated at 25°C for 2 h. Cells were then washed three times in ice-cold succinate/azide buffer (50 mM succinic acid, 20 mM NaN3, pH adjusted to 5 with NaOH) and then resuspended in 6 μl succinate/azide buffer. 2 μl were placed on a slide for immediate observation.
Results
The V159N Mutation in Actin Results in Yeast with More Pronounced Cables and Larger Cortical Actin Patches, but Has Only Modest Effects on Actin Cytoskeleton Organization
The actin gene with Val 159 mutated to Asn (V159N) was integrated by homologous recombination into the genome of diploid yeast cells, and the resulting transformants were sporulated. The phenotypes of haploid progeny expressing only the mutated copy of the actin gene were then characterized. Yeast expressing V159N actin (act1-159) have a dramatically altered actin cytoskeleton. In wild-type yeast cortical actin patches are concentrated in the bud, and actin cables are aligned along the mother–bud axis (Fig. 1 a). In yeast expressing act1-159, the cortical actin patches are much brighter and many cells appear to have more pronounced cables (Fig. 1 b). The images in Fig. 1, a and b were recorded using identical exposure and gain settings to allow direct comparison of fluorescence intensity of rhodamine phalloidin, which binds specifically to F-actin. In fluorescence intensity measurements of 40 rhodamine phalloidin–stained wild-type and act1-159 cells, the act1-159 cells had ∼25% higher fluorescence intensity. F-actin structures observed by antibody staining also appear to be brighter in act1-159 strains compared with wild-type (see Fig. 8, a and b), suggesting that the increased brightness is not due to enhanced phalloidin binding to V159N actin filaments. The actin patches in a heterozygous strain carrying one wild-type and one V159N mutant copy of the actin gene (Fig. 1 d) also appear brighter compared with a wild-type diploid (Fig. 1 c). However, there appears to be little or no increase in the number or brightness of cables.
Figure 1.

Rhodamine-phalloidin staining of yeast expressing V159N actin. Yeast cells were grown in rich media at 25°C. The F-actin was stained with rhodamine-phalloidin. (a) Wild-type haploids (DDY1495), (b) act1-159 haploids (DDY1493), (c) wild-type diploids (DDY479), and (d) act1-159/ACT1 heterozygotes (DDY1491). Images for haploids (a and b) and diploids (c and d) were captured using identical exposure times and gain settings to allow comparison of the intensity of rhodamine-phalloidin staining. Bar, 5 μm.
Figure 8.
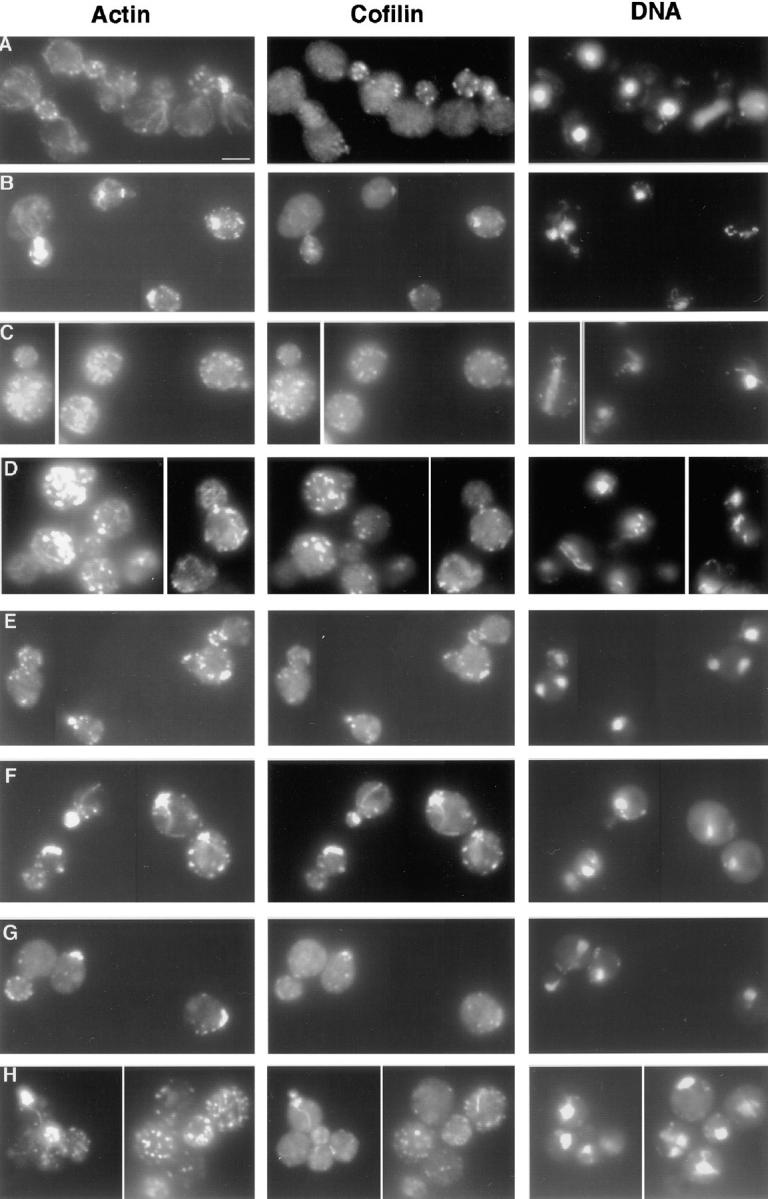
Immunofluorescence of double mutants. Log phase cells grown at 25°C were fixed with formaldehyde and stained with anti-actin and anti-cofilin antibodies. (A) Wild-type yeast (DDY1495), (B) act1-159 (DDY1493), (C) sac6Δ (haploid progeny of DDY216 × DDY1493), (D) sac6Δ act1-159 (from DDY216 × DDY1493), (E) tpm1Δ (from DDY487 × DDY1493), (F) tpm1Δ act1-159 (from DDY487 × DDY1493), (G) mdm20Δ (from DDY1415 × DDY1493), (H) act1-159 mdm20Δ (from DDY1415 × DDY1493). Cofilin is localized exclusively to cortical actin patches in wild-type, act1-159, and act1-159 sac6Δ cells, but in the tpm1Δ act1-159 and act1-159 mdm20Δ double mutants, cofilin is seen on structures that look like cables. Bar, 5 μm.
Despite these effects on the appearance of the actin cytoskeleton, and those described below, most act1-159 yeast were able to develop a polarized organization of their actin cytoskeleton with cortical patches concentrated on the surface of the bud (see Table II). Approximately one-third of the cells had additional patches in both the mother and bud, making the polarity less pronounced. In addition, approximately one-quarter of small-budded cells have depolarized actin or actin structures that are not clearly identifiable as cortical patches or cables. Many of the act1-159 cells with depolarized actin are larger and rounder than wild-type cells, a phenotype commonly associated with the failure to properly polarize the yeast actin cytoskeleton. Approximately 13% of act1-159 cells appear to have excess cables and these are often not properly organized. These cables frequently extend at right angles to the mother–bud axis, rather than running along the mother– bud axis as they do in wild-type strains.
Table II.
Characterization of Actin Polarity Defects in act1-159 Strains
| Polarized | Partially polarized | Depolarized | ||||
|---|---|---|---|---|---|---|
| % | % | % | ||||
| ACT1 | 93 | 7 | 0 | |||
| act1-159 | 40 | 36 | 24 |
Cells were grown at 25°C. Actin structures were observed by rhodamine-phalloidin staining. To assess actin polarity, only small-budded cells were counted. Terms are defined as follows. Polarized, the majority of actin patches were in the bud; Partially polarized, more than half of the patches were in the bud, but there were also many additional patches in the mother, or the patches polarized to the neck instead of the bud; Depolarized, actin patch distribution was nearly random or the phalloidin staining structures were not easily identified as patches or cables.
V159N Actin Forms Exceptionally Stable Filaments In Vivo
We tested the sensitivity of act1-159 strains to the actin binding drug LAT-A using a halo assay in which various concentrations of the drug are pipetted onto a sterile filter disc and placed on a nascent lawn of yeast cells (Fig. 2 A). The size of the halo within which no cell growth occurred was measured to determine the relative resistance to the drug. Haploid yeast expressing V159N actin are fourfold more resistant to LAT-A than wild-type cells (Fig. 2 B). In addition to the halo of complete growth inhibition, a halo of reduced growth was observed for the act1-159 strain reflecting slower growth in lower concentrations of LAT-A. This suggests that while the act1-159 mutant is able to tolerate high concentrations of LAT-A, its growth rate is reduced by LAT-A, even at concentrations that do not affect wild-type cells. One possible explanation for this effect is that the act1-159 strain already has a reduced monomer pool (see Discussion), and the LAT-A reduces it further.
Figure 2.

LAT-A halo assay and Western blot of actin. (A) LAT-A was placed on sterile disks at concentrations of 0, 1 mM, 2 mM, and 4 mM. These disks were placed on nascent lawns of wild-type (left) or act1-159 (right) yeast and allowed to grow at 25°C for 3 d. (B) The concentration of LAT-A required to produce a halo of complete growth inhibition of a given size is fourfold higher in the act1-159 mutant compared with wild-type yeast. (C) Western blot of an SDS polyacrylamide gel loaded with 3, 6, and 9 μl of yeast extracts from wild-type and act1-159 strains. Actin bands from three separate blots were measured by densitometry. Tubulin was measured as a loading control for normalization (not shown). The expression levels of wild-type and V159N actin were the same within an experimental error of 15%.
One direct explanation for the increased LAT-A resistance and the presence of more F-actin would be if act1-159 strains contained more actin. However, quantitative immunoblotting revealed that actin levels are indistinguishable between mutant and wild-type cells (Fig. 2 C). Therefore it seems likely that the biochemical properties of the mutant actin itself are responsible for the increased LAT-A resistance and the increased levels of F-actin observed in vivo.
Because LAT-A binds and sequesters actin monomers, but does not increase the off rate of actin from filaments (Coue et al., 1987), this drug can be used to measure the rate of filament turnover in vivo (Ayscough et al., 1997). To measure filament turnover, 400 μM LAT-A was added to the media and yeast were fixed with formaldehyde at various time points. F-actin was visualized with rhodamine-phalloidin. Consistent with previous reports, there are almost no visible actin structures in wild-type yeast after 2 min in 400 μM LAT-A, suggesting very rapid filament turnover. However, in the V159N mutant yeast, actin structures disappear much more slowly, and are still visible in 30% of cells for >40 min after LAT-A addition (Fig. 3). Therefore, this mutation appears to result in slow actin dynamics in vivo.
Figure 3.

Actin patch persistence in 400 μM LAT-A. LAT-A, a drug that sequesters actin monomers, was added to the growth medium. Cells were fixed in formaldehyde at various time points, and stained with rhodamine phalloidin. (A) Wild-type (DDY1495) and act1-159 mutant (DDY1493) cells are shown at 0, 2, and 10 min after drug addition. (B) The percentage of cells with detectable actin structures at various time points. More than 200 cells were counted for each condition. Bar, 5 μm.
For this conclusion to be valid, the slow kinetics of disassembly must not be due to the fact that the mutant starts out with higher levels of assembled actin, and must not be due to reduced affinity of the mutant actin for LAT-A. First, the difference is unlikely to be due simply to the fact that act1-159 actin patches contain more F-actin because rhodamine-phalloidin–stained patches are only 25% brighter than wild-type, yet they take 10–20 times longer to fall below the detection limit upon treatment with LAT-A.
Two experiments demonstrate that LAT-A binds to wild-type and V159N actin with similar affinity. First, we looked at effects of LAT-A on nucleotide exchange. The ATP bound to monomeric actin exchanges spontaneously with ATP in solution. We compared the rate of ATP exchange in wild-type and V159N mutant actin by monitoring the increase of ε-ATP fluorescence at 410 nm upon binding to actin. As shown in Fig. 4 A, the rate of ATP exchange is increased in V159N actin. The t 1/2 of ATP exchange in V159N actin is 15 s, as compared with 100 s for wild-type. 5–10 μM LAT-A completely inhibits nucleotide exchange on purified wild-type yeast actin. 5 μM LAT-A inhibits nearly all nucleotide exchange for both wild-type and V159N actin, and 2.5 μM LAT-A increases the t 1/2 of ATP exchange by approximately twofold for both wild-type and V159N actin (Fig. 4 B), demonstrating that LAT-A binds to wild-type and the V159N mutant actin with similar affinities.
Figure 4.
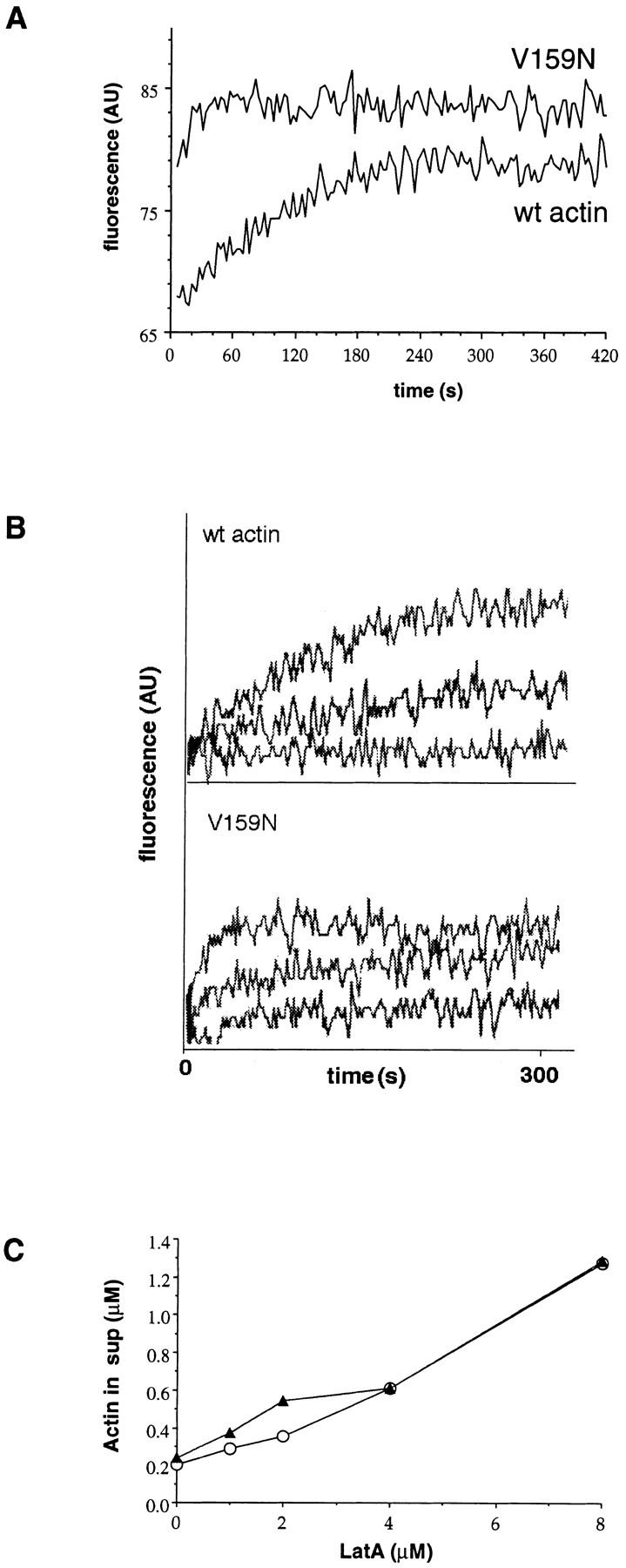
LAT-A binding and ATP exchange. (A) ATP exchange was measured by monitoring the increase in fluorescence of ε-ATP upon binding to monomeric actin. Each measurement was repeated three times, and the t 1/2 of V159N actin was 15 s (15 s, 15 s, 14 s), and the t 1/2 of wild-type actin was ∼100 s (100 s, 102 s, 100 s). Typical plots of fluorescence are shown. The beginning and ending fluorescence levels for wild-type actin are 68 and 80. The beginning and ending fluorescence levels for the V159N actin are 80 and 85. There is a dead time of ∼4 s before the first measurement can be recorded, so it is possible that the starting point for the V159N actin is similar to that of the wild-type actin. This would make the value of 15 s an overestimate of the t 1/2 of ATP exchange, because most of the exchange would have occurred in the dead time. (B) LAT-A was added to the ATP exchange reactions to a final concentration of 0, 2.5, and 5 μM. The t 1/2 of ATP exchange in 2.5 μM LAT-A is approximately doubled for both V159N and wild-type actin, suggesting similar affinities for LAT-A. (C) Increasing concentrations of LAT-A were added to 4 μM polymerized actin. After 1 h, the polymerized actin was pelleted and the amount of actin left in the supernatant was quantified (○, wild-type actin; ▴, V159N actin).
The ATP exchange experiments address the interaction of LAT-A with actin monomers in depolymerizing conditions. We addressed the binding of LAT-A to V159N actin under more physiological conditions by adding LAT-A to polymerized actin in F-buffer and measuring the amount of actin that was sequestered as monomer. Fig. 4 C shows that the amount of actin sequestered by increasing amounts LAT-A is similar between wild-type and V159N actin. Therefore, the increased resistance of act1-159 strains to LAT-A is most likely not due to a defect in LAT-A binding to the mutant actin.
The V159N Mutation Results in Slow Growth, Salt Sensitivity, a Defect in Endocytosis and Random Budding
We next determined the effects of the V159N mutation on various growth properties. At 25°C, the haploid act1-159 mutant strain (DDY1493) has a doubling time of 252 min, as compared with 126 min in an isogenic ACT1 wild-type strain (DDY1495). The act1-159 strain fails to grow on plates containing 0.9 M NaCl and grows poorly at 37°C (data not shown), conditions that support growth of wild-type strains. Defects in nuclear segregation and budding patterns are common phenotypes associated mutations that disrupt the actin cytoskeleton. act1-159 strains show an increase in the number of binucleate plus multinucleate cells (7%) compared with wild-type cells (<0.5%). Wild-type diploid yeast exhibit a pattern of bipolar budding, resulting in bud scars that are localized to opposite poles of the cell, but act1-159 homozygous diploids exhibit a random budding pattern (Fig. 5 A). We measured fluid phase endocytosis by the uptake of Lucifer yellow. There is a pronounced defect in endocytosis in act1-159 strains, demonstrated by weaker vacuolar staining after 2 h in Lucifer yellow (Fig. 5 B). A similar endocytosis defect was seen in cofilin mutants, suggesting that rapid actin filament turnover is required for fluid phase endocytosis (Lappalainen and Drubin, 1997).
Figure 5.

act1-159 mutants exhibit random budding and a defect in endocytosis. (A) Calcofluor staining showing bud scars in wild-type and act1-159 diploids. (B) Endocytosis was evaluated by observing the uptake of Lucifer yellow into the vacuole using fluorescence microscopy. The weak fluorescence from the act1-159 mutant strain indicates a defect in fluid phase endocytosis. Bar, 5 μm.
Cortical Actin Patch Motility Does Not Require Rapid Actin Dynamics
Cortical actin patches move rapidly within the plane of the cell cortex. To test whether rapid dynamics are required for actin patch motility we constructed an act1-159 strain expressing a fusion between Abp1p, an actin-binding protein that is localized to the patches (Drubin et al., 1988), and green fluorescent protein (GFP-Abp1p) (Doyle and Botstein, 1996). As shown in Fig. 6, there is no significant difference in the rates of patch motility between act1-159 strains and isogenic wild-type strains. Thus, rapid actin filament turnover is not required for patch motility. Patch motility was recorded at 4-s intervals, which does not allow the observation of the fastest patch movements (1–2 μm/s). Since it is possible that the fastest patch movements represent a mechanistically distinct type of motility, we also wanted to determine if fast patch movement was inhibited in the mutant. We observed 10 wild-type and 10 act1-159 cells for 1 min each by direct real-time observation under fluorescence microscopy and counted the number of fast patch movements. In both the wild-type and the act1-159 strain we observed a total of 12 fast patch movements during 10 min of observation, suggesting that fast patch movements are also uninhibited in act1-159 strains.
It has been observed that patches continue to move as they are depolymerizing in the presence of LAT-A. However, it was difficult to document this observation because most patches are not visible by fluorescence microscopy of GFP-labeled patch proteins 1 min after treatment with LAT-A. Because cortical actin patches persist in LAT-A–treated act1-159 strains for much longer periods of time than in wild-type cells, we were able to use an act1-159 mutant to test whether actin polymerization is required for actin patch motility. When 400 μM LAT-A is added to act1-159 strains to sequester free actin monomers, actin patches continue to move for >90 min after the addition of the drug. Fig. 7 shows actin patch motility in act1-159 yeast cells 86 min after LAT-A addition. The low signal to noise ratio of the fluorescence signal is a result of the LAT-A addition.
Figure 7.

Cortical actin patches in act1-159 continue to move in the presence of LAT-A. LAT-A was added to 400 μM, and samples were taken during a 2-h time period and images were recorded to assess patch motility using GFP-ABP1. The cells pictured here had been growing in LAT-A–containing media for 86 min. The top white arrow points to a patch that moves away from the arrow towards the center of the cell. In the cell on the bottom right, the white arrow marks the starting position of an actin patch, and the black arrow points to its final destination. The low signal to noise ratio is due to decreased actin patch fluorescence intensity resulting from sequestration of actin monomers by LAT-A. Frames are 2-s apart. Bar, 5 μm.
Double Mutant Analysis Provides Insights into Actin-binding Protein Functions In Vivo
Because act1-159 cells have decreased actin turnover rates and in some cases have more cables than are found in wild-type cells, we tested for genetic interactions between act1-159 and mutations or deletions in the genes that encode cofilin, profilin, fimbrin, tropomyosin, and Mdm20p (Table III). These proteins are thought to drive actin dynamics or regulate actin cable stability in vivo. We found that act1-159 is synthetically lethal with two temperature-sensitive (ts) alleles of cofilin that cause a decrease in the rate of actin filament turnover (Lappalainen et al., 1997; Lappalainen and Drubin, 1997). act1-159 is also synthetically lethal with two mutant alleles of profilin, pfy1-111 and pfy1-Δ4C. pfy1-111 has a defect in binding to PIP2, and pfy1-Δ4C has a defect in binding to polyproline, as well as a slight defect in binding to actin (Haarer et al., 1993). In addition, act1-159 shows negative growth synergism with a fimbrin null mutant (sac6Δ). In contrast, the temperature sensitivity of a tropomyosin (tpm1Δ) deletion mutant is partially suppressed by act1-159. This is not true for an mdm20 deletion mutant. We also crossed act1-159 to a strain with a deletion of the gene encoding Pca1p, a protein proposed to be involved in nucleating actin filament assembly (Lechler and Li, 1997), and observed no synergistic effects.
Table III.
Genetic Interactions of act1-159 with Mutants of Other Actin Cytoskeleton Genes
| Mutations | Growth | |
|---|---|---|
| act1-159 cof1-5 | lethal | |
| act1-159 cof1-22 | lethal | |
| act1-159 pfy1-111 | lethal | |
| act1-159 pfy1-Δ4C | lethal | |
| act1-159 sac6Δ | increased temperature sensitivity | |
| act1-159 tpm1Δ | partial suppression of tpm1 temperature sensitivity | |
| act1-159 mdm20Δ | no change in mdm20 temperature sensitivity | |
| act1-159 pca1Δ | same as act1-159 alone |
For each mutant, 8–12 tetrads were dissected and the genotypes of each segregant was determined by marker segregation. None of the haploid progeny predicted to have inherited act1-159 and either a profilin or a cofilin mutation were viable. Each viable double mutant was grown at a variety of temperatures in parallel with the single mutants to determine whether the double mutants had an altered permissive temperature range. All of the act1-159 sac6 double mutants were viable at 25°C, but dead at 34°C, whereas all of the resultant single mutants were viable at 34°C. The tpm1Δ act1-159 double mutants were viable at 34°C (4 out of 5) while the tpm1Δ alone failed to grow 34°C. Both mdm20Δ and act1-159 mdm20Δ strains were inviable at 34°C. act1-159 pca1Δ double mutants were indistinguishable from act-159 mutants.
We next examined actin staining in the double mutants (when viable) and single mutant progeny of the same crosses. The sac6 deletion results in depolarized actin patches and loss of cables (Fig. 8 C). The double sac6Δ act1-159 mutant, however, has actin cables of normal number and staining intensity, but these cables are frequently mislocalized (in the bud or running across the width of the mother). This double mutant also has depolarized actin patches (Fig. 8 D). TPM1 deletion results in loss of cables and a slight increase in depolarized actin patches (Liu and Bretscher, 1989) (Fig. 8 E). act1-159 tpm1Δ double mutants have a few structures that resemble cables (Fig. 8 F). However, these structures are recognized by antibodies raised against cofilin, a protein normally localized exclusively to actin cortical patches. Therefore, these structures either are not cables, or in the absence of tropomyosin cofilin is localized to both actin cables and patches. act1-159 tpm1Δ double mutants display a distribution of polarized, partially polarized, and depolarized patches that is similar to that of the act1-159 mutant alone. mdm20 deletion strains also lack cables (Hermann et al., 1997). act1-159 mdm20Δ double mutants have a few cable-like structures that are recognized by cofilin antibodies, similar to those seen in act1-159 tpm1Δ strains. The act1-159 mdm20Δ double mutant shows slight depolarization defects similar to the act1-159 mutant alone. This analysis demonstrates different roles for fimbrin compared with tropomyosin and Mdm20p, even though their deletion phenotypes are similar.
Discussion
Characterization of act1-159 Strains
We have demonstrated by two criteria that act1-159 mutants have more stable actin filaments than wild-type cells. First, yeast strains expressing act1-159 as their sole source of actin appear to have more F-actin than wild-type strains, despite having normal levels of total actin as measured by Western blotting. Second, actin structures in act1-159 strains persist for much longer in the presence of LAT-A. Because LAT-A does not accelerate depolymerization, but sequesters free actin monomers and prevents reassembly, these observations suggest that monomers are released from actin filaments at a much slower rate in act1-159 strains. This could be the result of slower depolymerization, longer filament length, or altered interactions with actin regulatory proteins. Biochemical data (Belmont, L.D., D.G. Drubin, and E. Egelman, submitted for publication) suggest that the slow turnover is at least partially due to a slow depolymerization of V159N actin. The observation of increased levels of F-actin that result from slow filament dynamics suggests the actin monomer pool in wild-type yeast is higher than the previously reported value of only 0.3% of the total actin (Karpova et al., 1995).
The rate of ATP exchange is faster in the purified V159N actin. This is consistent with the predictions one can make from the crystal structure of rabbit muscle actin. The amide group of V159 makes a hydrogen bond with the γ-phosphate of the bound ATP (Kabsch et al., 1990). The V to N change might be expected to weaken this interaction because the pKa of the α-amino group of Asn is significantly lower than that of Val. This is consistent with our observation of an increased off-rate of ATP from the monomer. It is possible that rapid nucleotide exchange contributes to the apparent increase in F-actin in act1-159 strains, but the reduced dynamics are likely the result of the slow dissociation of monomers from filaments.
Endocytosis, Actin Cytoskeleton Organization, and Patch Motility
Because act1-159 strains have slower actin filament turnover, this mutant is a useful tool with which to probe the role of rapid actin filament dynamics in vivo. The act1-159 strains grow at half the rate of wild-type yeast in liquid media, and are sensitive to high salt, demonstrating the importance of actin dynamics for normal growth properties. A severe defect in fluid-phase endocytosis is a pronounced phenotype of act1-159 mutants. This observation is consistent with previous results with cofilin mutants, and provides further evidence that rapid actin filament turnover is essential for endocytosis (Lappalainen and Drubin, 1997).
More than 75% of act1-159 cells maintain at least partially polarized actin cortical patches. This suggests either that selective stabilization of polarized cortical actin filaments, and/or destabilization of depolarized actin filaments, is not the only mechanism by which actin cytoskeleton polarity is maintained, or that the residual dynamics of V159N actin are sufficient to establish and maintain partially polarized actin patches.
The mechanism of actin cortical patch motility remains a mystery. Movement of actin patches by myosin motors is an attractive hypothesis, however, none of the myosin mutants tested showed a defect in patch motility (Waddle et al., 1996). In contrast, a temperature sensitive mutation in the ARP3 gene was reported to disrupt patch motility. The authors proposed that patch motility is driven by actin polymerization, much like the actin based motility of Listeria monocytogenes (Winter et al., 1997). This possibility is tenable because the Arp2/3 complex was recently shown to nucleate actin filament assembly (Mullins et al., 1998; Welch et al., 1998). However, we find that the rate of patch motility is normal in the act1-159 mutant, suggesting that rapid filament depolymerization is not required for motility. In addition, when observing actin patch motility in this strain, we do not see any tail-like structures akin to Listeria tails. The presence of such tails is predicted if the patches move by actin filament turnover (Theriot and Mitchison, 1992) especially if actin depolymerization is inhibited (Carlier et al., 1997; Rosenblatt et al., 1997) as it is in the act1-159 mutant. Because actin patches in the act1-159 mutant persist for much longer in the presence of LAT-A than they do in wild-type cells, we were able to record patch motility in the presence of this drug. Patches continued to move for up to 2 h in the presence of 400 μM LAT-A before depolymerizing. This observation strongly suggests that patch motility does not require new actin assembly. Therefore, neither actin assembly nor rapid filament turnover appears to be required for actin patch motility in yeast.
Evidence for Actin-binding Protein Functions In Vivo
We used the act1-159 mutant to obtain novel insights into the in vivo roles of proteins that regulate actin dynamics. The insights from these studies are valuable because many actin-binding proteins have similar in vitro activities, making it difficult to differentiate between their in vivo functions. A powerful approach that can provide insights into the functional relationships between proteins is to make strains with mutations in two genes and observe any synergistic effects and compare phenotypes of different double mutant combinations. The synthetic lethality of act1-159 with cofilin mutants is consistent with the previously demonstrated role of cofilin in increasing filament turnover by promoting disassembly (Carlier et al., 1997; Lappalainen and Drubin, 1997). It is interesting that act1-159 mutants have a slightly different phenotype than temperature-sensitive cofilin mutants. Mutations in the yeast cofilin gene result in cells that have larger cortical actin patches (Lappalainen and Drubin, 1997), but cytoplasmic cables appear unaffected. In contrast, while the act1-159 mutation also results in larger cortical actin patches, there is an additional phenotype of an increase in the number and apparent size of actin cables in ∼13% of cells. This observation supports the conclusion that actin cables normally turn over dynamically (Ayscough et al., 1997), and suggests that cable stability is determined by different factors from those that regulate actin patch stability.
The in vivo role of profilin is less clear and we have used the act1-159 mutant to gain new insights into its role in yeast. Profilin is a small protein capable of sequestering actin monomers, increasing the actin monomer pool, and accelerating the addition of actin monomers to filament barbed ends (Pantaloni and Carlier, 1993). The role of profilin appears to differ between cell types, acting either to sequester monomers and inhibit assembly, or to promote assembly at specific sites within a cell (Sun et al., 1995). act1-159 mutant strains are predicted to have a smaller actin monomer pool, based on the observation of an increase in rhodamine-phalloidin fluorescence intensity in situ. Therefore, the simplest interpretation of the observed synthetic lethality between act1-159 and pfy1 mutants is that yeast need to maintain a monomer pool of a certain size to allow rapid assembly of new actin filaments, and are unable to do so with the combination of a hyper-stable actin and an impaired monomer binding protein. However, the pfy1-111 mutant has no demonstrated defect in actin monomer binding, making this interpretation unlikely. pfy1-111 has a defect in binding to PIP2, and pfy1-Δ4C has a defect in binding to polyproline, as well as a slight defect in binding to actin (Haarer et al., 1993). PIP2 binding displaces actin from profilin (Lassing and Lindberg, 1985), and polyproline binding is thought to localize profilin to sites of filament assembly. Therefore, both of these mutants are predicted have defects in promoting polarized actin assembly. The defects in pfy1-111 and pfy1-Δ4C single-mutant strains are subtle. There is a slight depolarization of actin at higher temperatures as well as a decrease in cable staining (data not shown). Despite the subtlety of the single-mutant phenotypes, both profilin mutants are lethal in combination with act1-159. This is consistent with a model in which organization of the actin cytoskeleton involves both polarized assembly as well as polarized disassembly. Individually the mutations have only slight defects in actin organization, but if polarized assembly as well as disassembly are inhibited the effects are more severe.
To investigate the regulation of actin cables, we crossed act1-159 to tropomyosin (TPM1), MDM20 and fimbrin (SAC6) deletion strains. Because absence of these proteins results in partial or complete loss of cables, we thought that deletions of TPM1, MDM20, or SAC6 might suppress the act1-159 phenotype (or vice versa). In the sac6Δ act1-159 double mutant the intensity of cable staining is returned to near wild-type levels. However, these yeast are more temperature sensitive than either of the parents. The cables are often mislocalized, either running across the width of the mother, or residing entirely within the bud. These mislocalized cables usually terminated at or near depolarized patches, suggesting a functional link between patch and cable organization. One model to explain these data is that having mislocalized cables is more deleterious than having no cables at all, and that fimbrin might have a roles in both stabilizing cables and directing their orientation. The tpm1Δ act1-159 double mutant also has some cable-like structures, and these cables are properly oriented. In this case, the temperature sensitivity of the tpm1Δ mutant is partially suppressed by act1-159, suggesting that the temperature sensitivity of the tpm1Δ is caused, at least in part, by the destabilization of actin filaments. Interestingly, the actin cables in the tpm1Δ act1-159 double mutants are recognized by antibodies raised against cofilin, a protein normally localized exclusively to patches. The mdm20Δ act1-159 double mutant also has cables that can be stained by cofilin antibodies. The data for the mdm20Δ and tpm1Δ double mutants with act1-159 are consistent with the model that cofilin is normally excluded from the cables by Tpm1p (and perhaps Mdm20p, although Mdm20p localization is not known). Tpm1p could stabilize actin cables both by direct filament binding and by excluding cofilin, an actin filament depolymerizing protein, from actin cables. This is consistent with biochemical studies that showed tropomyosin can partially protect actin from the depolymerizing activity of ADF (Berstein and Bamburg, 1982).
The act1-159 mutation has a semi-dominant effect in heterozygotes carrying one wild-type (ACT1) and one mutant (act1-159) copy of the actin gene. The actin patches in these heterozygotes are bigger and more numerous than in ACT1/ACT1 homozygotes, although overall actin organization is fairly normal. The two most likely explanations for the semi-dominant effect are (a) there are exceptionally stable filaments composed only of V159N actin, or (b) this actin co-assembles with the wild-type actin and increases the stability of the mixed polymer. Regardless of the mechanism, the fact that the V159N mutation has a semi-dominant phenotype promises to make this actin mutation useful for the study of the role of actin filament dynamics in more complex organisms that have multiple actin genes, or for which gene replacement experiments are not possible.
Acknowledgments
We wish to thank B. Haarar, G. Hermann, J. Shaw, and R. Li for providing yeast strains. We also thank K. Kozminski for critically reading the manuscript, and P. Lappalainen and B. Goode for helpful discussions.
This work was supported by an National Institutes of Health (NIH) fellowship (5 F32 CA69804-02) awarded to L.D. Belmont, and grants from the NIH (GM42759), and American Cancer Society (FRA-442) to D.G. Drubin.
Abbreviations used in this paper
- F-actin
filamentous actin
- LAT-A
latrunculin-A
Footnotes
Address all correspondence to David G. Drubin, Department of Molecular and Cell Biology, 401 Barker Hall, University of California, Berkeley, CA 94720-3202. Tel.: (510) 642-3692. Fax: (510) 642-6420. E-mail: drubin@uclink4.berkeley.edu
References
- Adams AE, Botstein D, Drubin DG. Requirement of yeast fimbrin for actin organization and morphogenesis in vivo. Nature. 1991;354:404–408. doi: 10.1038/354404a0. [DOI] [PubMed] [Google Scholar]
- Ayscough KR, Stryker J, Pokala N, Sanders M, Crews P, Drubin DG. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J Cell Biol. 1997;137:399–416. doi: 10.1083/jcb.137.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berstein BW, Bamburg JR. Tropomyosin binding to F-actin protects the F-actin from disassembly by brain actin-depolymerizing-factor (ADF) Cell Motil. 1982;2:1–8. doi: 10.1002/cm.970020102. [DOI] [PubMed] [Google Scholar]
- Carlier MF. Actin polymerization and ATP hydrolysis. Adv Biophys. 1990;26:51–73. doi: 10.1016/0065-227x(90)90007-g. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Pantaloni D. Direct evidence for ADP-Pi-F-actin as the major intermediate in ATP-actin polymerization. Rate of dissociation of Pi from actin filaments. Biochemistry. 1986;25:7789–7792. doi: 10.1021/bi00372a001. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: Implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coue M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS (Fed Eur Biochem Soc) Lett. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- Doyle T, Botstein D. Movement of yeast cortical actin cytoskeleton visualized in vivo. Proc Nat Acad Sci USA. 1996;93:3886–3891. doi: 10.1073/pnas.93.9.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Miller KG, Botstein D. Yeast actin-binding proteins: Evidence for a role in morphogenesis. J Cell Biol. 1988;107:2551–2561. doi: 10.1083/jcb.107.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt-Clermont PJ, Machesky LM, Doberstein SK, Pollard TD. Mechanism of the interaction of human platelet profilin with actin. J Cell Biol. 1981;113:1081–1089. doi: 10.1083/jcb.113.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie, C., and G. Fink. 1991. Guide to yeast genetics and molecular biology. In Methods Enzymology. Vol. 194. Academic Press, San Diego, CA. 3–20. [PubMed]
- Haarer BK, Lillie SH, Adams AE, Magdolen V, Bandlow W, Brown SS. Purification of profilin from Saccharomyces cerevisiaeand analysis of profilin-deficient cells. J Cell Biol. 1990;110:105–114. doi: 10.1083/jcb.110.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarer BK, Petzold AS, Brown SS. Mutational analysis of yeast profilin. Mol Cell Biol. 1993;13:7864–7873. doi: 10.1128/mcb.13.12.7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 471–510.
- Hermann GJ, King EJ, Shaw JM. The yeast gene, MDM20 is necessary for Mitochondrial Inheritance and organization of the actin cytoskeleton. J Cell Biol. 1997;137:141–153. doi: 10.1083/jcb.137.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Atomic structure of the actin: DNase I complex. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- Karpova TS, Tatchell K, Cooper JA. Actin filaments in yeast are unstable in the absence capping protein or fimbrin. J Cell Biol. 1995;131:1483–1493. doi: 10.1083/jcb.131.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen P, Drubin DG. Cofilin promotes rapid actin filament turnover in vivo. Nature. 1997;388:78–82. doi: 10.1038/40418. [DOI] [PubMed] [Google Scholar]
- Lappalainen P, Fedorov EV, Fedorov AA, Almo SC, Drubin DG. Essential functions and actin-binding surfaces of yeast cofilin revealed by systematic mutagenesis. EMBO (Eur Mol Biol Organ) J. 1997;16:5520–5530. doi: 10.1093/emboj/16.18.5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassing I, Lindberg U. Specific interaction between phosphatidylinositol 4,5-bisphosphate and profilactin. Nature. 1985;314:472–474. doi: 10.1038/314472a0. [DOI] [PubMed] [Google Scholar]
- Lechler T, Li R. In vitro reconstitution of cortical actin assembly sites in budding yeast. J Cell Biol. 1997;138:95–103. doi: 10.1083/jcb.138.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HP, Bretscher A. Disruption of the single tropomyosin gene in yeast results in the disappearance of actin cables from the cytoskeleton. Cell. 1989;57:233–242. doi: 10.1016/0092-8674(89)90961-6. [DOI] [PubMed] [Google Scholar]
- Moon AL, Janmey PA, Louie KA, Drubin DG. Cofilin is an essential component of the yeast cortical cytoskeleton. J Cell Biol. 1993;120:421–435. doi: 10.1083/jcb.120.2.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: Nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni D, Carlier MF. How profilin promotes actin filament assembly in the presence of thymosin β4. Cell. 1993;75:1007–1014. doi: 10.1016/0092-8674(93)90544-z. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Agnew BJ, Abe H, Bamburg JR, Mitchison TJ. Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenestails. J Cell Biol. 1997;136:1323–1332. doi: 10.1083/jcb.136.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HQ, Kwiatkowska K, Yin HL. Actin monomer binding proteins. Curr Opin Cell Biol. 1995;7:102–110. doi: 10.1016/0955-0674(95)80051-4. [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ. The rate of actin-based motility of intracellular Listeria monocytogenesequals the rate of actin polymerization. Nature. 1992;357:257–260. doi: 10.1038/357257a0. [DOI] [PubMed] [Google Scholar]
- Waddle JA, Karpova TS, Waterston RH, Cooper JA. Movement of cortical actin patches in yeast. J Cell Biol. 1996;132:861–870. doi: 10.1083/jcb.132.5.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MD, Holtzman DA, Drubin DG. The yeast actin cytoskeleton. Curr Opin Cell Biol. 1994;6:110–119. doi: 10.1016/0955-0674(94)90124-4. [DOI] [PubMed] [Google Scholar]
- Welch MD, Rosenblatt J, Skoble J, Portnoy DA, Mitchison TJ. Interaction of human Arp2/3 complex and the Listeria monocytogenesActA protein in actin filament nucleation. Science. 1998;281:105–108. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- Wertman KF, Drubin DG, Botstein D. Systematic mutational analysis of the yeast ACT1 gene. Genetics. 1992;132:337–350. doi: 10.1093/genetics/132.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter D, Podtelejnikov A, Mann M, Li R. The complex containing actin-related proteins Arp2 and Arp3 is required for the motility and integrity of yeast actin patches. Curr Biol. 1997;7:519–529. doi: 10.1016/s0960-9822(06)00223-5. [DOI] [PubMed] [Google Scholar]