

Abstract
α-Dystrobrevin is both a dystrophin homologue and a component of the dystrophin protein complex. Alternative splicing yields five forms, of which two predominate in skeletal muscle: full-length α-dystrobrevin-1 (84 kD), and COOH-terminal truncated α-dystrobrevin-2 (65 kD). Using isoform-specific antibodies, we find that α-dystrobrevin-2 is localized on the sarcolemma and at the neuromuscular synapse, where, like dystrophin, it is most concentrated in the depths of the postjunctional folds. α-Dystrobrevin-2 preferentially copurifies with dystrophin from muscle extracts. In contrast, α-dystrobrevin-1 is more highly restricted to the synapse, like the dystrophin homologue utrophin, and preferentially copurifies with utrophin. In yeast two-hybrid experiments and coimmunoprecipitation of in vitro–translated proteins, α-dystrobrevin-2 binds dystrophin, whereas α-dystrobrevin-1 binds both dystrophin and utrophin. α-Dystrobrevin-2 was lost from the nonsynaptic sarcolemma of dystrophin-deficient mdx mice, but was retained on the perisynaptic sarcolemma even in mice lacking both utrophin and dystrophin. In contrast, α-dystrobrevin-1 remained synaptically localized in mdx and utrophin-negative muscle, but was absent in double mutants. Thus, the distinct distributions of α-dystrobrevin-1 and -2 can be partly explained by specific associations with utrophin and dystrophin, but other factors are also involved. These results show that alternative splicing confers distinct properties of association on the α-dystrobrevins.
Keywords: dystrophin complex, neuromuscular junction, postsynaptic folds, high revolution immunofluorescence, isoform-specific antibodies
Mutations that disrupt the expression or function of dystrophin cause Duchenne muscular dystrophy, a myopathy that leads to muscle cell degeneration (3, 34). Dystrophin is a large, 427-kD protein composed of an NH2-terminal actin-binding domain (33), a series of spectrin-like repeats, and a cysteine-rich (CR)1 region, and a COOH-terminal (CT) domain, both of which contain sites for interaction with other proteins (3). Association between dystrophin and β-dystroglycan, a transmembrane protein, provides a link to the extracellular α-dystroglycan, which in turn binds laminin and agrin (for review see reference 33). Thus, one function of the dystrophin complex is to link the extracellular matrix to cortical actin. A group of four proteins, the sarcoglycans, are also part of the dystrophin complex. Although the function of the sarcoglycans are unknown, they are altered in several of the limb girdle muscular dystrophies in which dystrophin is found to be normal (40, 42). Other proteins in the complex include dystrobrevin and the syntrophins (25, 28, 29, 36).
Dystrophin is expressed at highest levels in skeletal muscle, where it may serve to stabilize the sarcolemma during cycles of muscle contraction and relaxation. In addition to this structural role, however, the dystrophin complex may also be involved in membrane signaling. Two lines of evidence support this idea. First, Grb2, a modular protein that mediates signals generated by receptor tyrosine kinases, can bind via its SH3 domain to β-dystroglycan (44). A second group of modular adapter proteins, the syntrophins, are associated directly with dystrophin via the CT domain and are major components of the dystrophin complex (1, 4, 5, 25, 28, 45). Three syntrophins, each encoded by separate genes, have a common domain structure. In addition to the COOH-terminal syntrophin-unique domain, the remainder of the protein is composed of two pleckstrin homology domains and one PDZ domain (a domain originally identified in postsynaptic density-95, discs large, ZO-1) (2, 5). In skeletal muscle, the syntrophin PDZ domain binds neuronal nitric oxide synthase (12) or voltage-activated sodium channels (21, 37).
Four other proteins related to dystrophin but encoded by separate genes have been identified. Utrophin is most closely similar to dystrophin and is widely expressed, although at only low levels in skeletal muscle (41). Dystrophin-related protein (DRP)2 lacks the actin-binding domain and most of the spectrin-like repeats, and is expressed at high levels in brain (32). Together, dystrophin, utrophin, and DRP2 comprise a subfamily, based on sequence comparison. A second subfamily is composed of the dystrobrevins (29). α-Dystrobrevin, first discovered as an 87-kD tyrosine-phosphorylated protein in Torpedo electric organ (14), has no spectrin-like repeats, but contains a CRCT domain that binds syntrophins (11, 35, 43, 46). In addition, α-dystrobrevin has a unique COOH-terminal extension of ∼180 amino acids, the dystrobrevin unique region (DUR) that contains multiple sites for phosphorylation on tyrosine. Several forms of α-dystrobrevin are generated by alternative splicing, including two that lack the DUR (10, 35). A second gene encoding a closely related protein, β-dystrobrevin, has recently been described (11, 29, 30). β-Dystrobrevin is quite similar to α-dystrobrevin, except that the DUR is significantly shorter.
α- and β-dystrobrevins are unique among the dystrophin family proteins in that they are both related to and associated with dystrophin (28, 29, 36). Coiled-coils in the CT domains of each protein mediate heterodimerization between the dystrobrevins and dystrophin (36). Thus, a refined model for the dystrophin and utrophin complexes contains two syntrophins, one bound to dystrophin (or utrophin) and one to dystrobrevin (28, 36). The presence of two syntrophins may provide a platform on which multicomponent signaling complexes can be assembled.
A membrane site that is particularly rich in dystrophin family proteins is the neuromuscular junction (NMJ) (for review see reference 38). Although utrophin is expressed only at very low levels in skeletal muscle as a whole, it is concentrated at and localized to the crests of the junctional folds in a distribution that closely matches that of nicotinic acetylcholine receptors (AChRs) and rapsyn (8, 27). Dystrophin is also concentrated at neuromuscular synapses but is restricted to the depths of the folds along with voltage-activated sodium channels (8, 13, 18, 39). Here, we have studied the distribution in skeletal muscle of two isoforms of α-dystrobrevin generated by alternative splicing (10, 35). α-Dystrobrevin-1 contains the COOH-terminal extension, while α-dystrobrevin-2 lacks this sequence. α-Dystrobrevin-1 is highly localized to the NMJ, while α-dystrobrevin-2 is concentrated at the synapse but also is present on the sarcolemma. In part, these unique distributions probably result from inherent specificities of the association of the two dystrobrevin isoforms with utrophin and dystrophin. However, our studies with skeletal muscle from mice lacking dystrophin, utrophin, or both proteins (24) suggest that additional regulatory factors, possibly including interactions with other proteins, are necessary for targeting of isoforms of dystrobrevin to different locations within the postsynaptic membrane.
Materials and Methods
Antibodies
Anti-dystrobrevin.
mAb 13H1 was prepared against Torpedo dystrobrevin (14) and was a gift from J.B. Cohen (Harvard Medical School, Cambridge, MA). Rabbit polyclonal antibodies (Abs) were prepared against synthetic dystrobrevin peptides with a terminal cysteine coupled to keyhole limpet hemocyanin according to standard methods (Covance Inc., Denver, PA). Isoform-specific synthetic peptides C-DMVPEDGDPYTQPEDGNYENE and C-EEYLKQKLQDEAYQVSLQG, which correspond to amino acids 638–658 and 670–688 of the mouse α-dystrobrevin-1 sequence originally published by Blake et al. (see reference 10), were used to produce Ab αDB638 and Ab αDB670, respectively. The preparation of Ab αDB2 against the peptide C-GVSYVPYCRS-COOH corresponding to the COOH-terminal 10 amino acids unique to α-dystrobrevin-2 has been described (29). All polyclonal antibodies were affinity purified from serum with relevant peptide coupled to Affi-Gel-10 or -15 (Bio-Rad Laboratories, Hercules, CA). The binding activity of each Ab was blocked by preincubation with the appropriate peptide (100 μM) for 30 min before use.
Anti-syntrophin.
mAb SYN1351 was raised against Torpedo syntrophin (19) and has been shown to recognize all three mammalian syntrophin isoforms (28).
Anti-dystrophin.
Ab DYS3669 was described previously (28). Mandra-1 was purchased from Sigma Chemical Co. (St. Louis, MO). DYS1 and DYS2 were purchased from Novacastra (Newcastle upon Tyne, UK).
Anti-utrophin.
Ab UTR3165 was prepared previously (25). mAb MANCHO-3 was a gift of G.E. Morris (26). mAb DRP1 was purchased from Novacastra (Newcastle upon Tyne, UK).
Animals
Control (C57BL/10ScSn) and mdx mice (C57BL/10ScSn mdx) were obtained from Jackson Laboratories (Bar Harbor, ME). utrn−/− and mdx/ utrn−/− mice have been described (23, 24).
Immunohistochemistry
Unfixed hind limb skeletal muscles were flash frozen in liquid nitrogen-cooled isopentane. Cryosections (7 μm) were labeled with primary antibodies at 30 nM as described previously (23, 28). α-Bungarotoxin (α-BgTx) conjugated to BODIPY was used at 1:300 (Molecular Probes Inc., Eugene, OR).
High resolution localization of the dystrobrevins by confocal microscopy was done on mouse sternomastoid muscle. α-Dystrobrevin-1 and α-dystrobrevin-2 were localized at NMJs using the respective rabbit antibodies, followed by Texas red–conjugated secondary and tertiary antibodies (Jackson ImmunoResearch). AChR were labeled with biotin-conjugated α-Bgtx followed by CY2-conjugated streptavidin (Molecular Probes Inc.). Appropriate control experiments confirmed the specificity of all secondary and tertiary antibodies.
Mouse sternomastoid muscles were dissected into physiological saline containing 0.5% PFA, fixed for 30 min, and then washed in saline. Blocks containing the innervation zone were flash frozen for microtomy. Cryostat sections (15 μm) were labeled with antibodies as described above, fixed in 4% PFA, and embedded in Araldite. Sections (100-nm thick) of small junction-containing areas were prepared with an ultramicrotome equipped with a diamond knife and mounted on No. 1.5 coverslips without mounting medium. Two-channel images were recorded with a Leica TCS-NT confocal microscope. To verify that bleed-through of signal from α-BgTx labeling (green) was not significantly contaminating the signal from antibody labeling (red), selected images were taken in the identical way except that either the 568-nm or the 488-nm excitation beam was turned off at the acoustically tunable filter.
Immunoaffinity Purifications and Immunoblotting
Immunoaffinity purifications and immunoblotting were performed as previously described (28).
Yeast Two-Hybrid Assay
cDNAs encoding full-length α-dystrobrevin-1 and the CT region of utrophin (CTDRP) (amino acids 2,753–3,432) were specifically amplified by PCR as previously described (35) and cloned into both the pGBT9 and the pGAD vectors for the two-hybrid assay (CLONTECH, Palo Alto, CA). The α-dystrobrevin-2 construct and the COOH-terminal dystrophin construct corresponding to exons 61–79 (DYS e61–79), were previously described (35). The yeast two-hybrid analysis was performed according to published methods (17). The HF7c yeast strain, containing two Gal4-inducible reporter genes, HIS3 and LacZ, was transformed simultaneously with both a DNA-binding domain plasmid (trp−) and a transactivating domain plasmid (leu−). Double transformants were plated onto selection plates lacking tryptophan, leucine, and histidine. After 5 d at 30°C, the colonies that grew on this selective medium (His+ colonies) were lifted onto 3-mm Whatman paper, immersed in liquid nitrogen for a few seconds, and then layered onto a second piece of Whatman paper, humidified with 3 ml of Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 30 mM β-mercaptoethanol, 5-bromo-4-chloroindolyl-β-D-galactoside [XGal]; final concentration of 2 mg/ml) for 8 h at room temperature.
In Vitro Translation and Immunoprecipitation Assay
Full-length dystrobrevin (α-dystrobrevin-1) was amplified by PCR as previously described (36) and cloned into the expression vector pMGT. The α-dystrobrevin-2 construct, the COOH-terminal region of utrophin construct (TDRII), and the COOH-terminal region of dystrophin construct (C2979) have been described (4, 36). Proteins encoded by the dystrobrevin, dystrophin, and utrophin constructs were produced by in vitro transcription/ translation of the expression vector pMGT (4) using the TNT T7–coupled reticulocyte lysate system (Promega Corp., Madison, Wl) in a reaction volume of 50 μl as per the manufacturer's protocol. Some reactions were carried out in a reaction buffer containing 0.2 μCi (4 μl) of l-[U-14C]leucine (>300 mCi/mmol; Amersham Corp., Arlington Heights, IL).
Coprecipitation assays were performed with protein taken directly from the translation reactions. Aliquots (5 μl) of each protein of interest were incubated together in 20 μl of TBST buffer (10 mM Tris, pH 8.0, 0.1% Tween 20, 150 mM NaCl). After 1.5–2 h of incubation on ice, 20 μl of Ab (1:10 dilution in TBST) directed against one of the proteins (the cognate protein) were added. After 1-h incubation on ice, interacting proteins were precipitated by the addition of 50 μl of a 50% suspension of protein G–Sepharose (Sigma Chemical Co.) and incubated for 30 min on ice. The protein–antibody bead complexes were pelleted at 10,000 g for 2 min at room temperature, and the supernatant was removed. The beads were then washed three times with 1 ml of TBST buffer, and the pellet was resuspended in 10 μl of 2× SDS sample buffer, and stored at −20°C. All samples were separated by electrophoresis on SDS-PAGE gels, and protein sizes were compared with [14C]methylated high molecular weight standards (10–50 μCi/mg) (Amersham Corp.). Gels were dried onto 3-mm Whatman paper, and exposed to a storage phosphor plate for 1–7 d, which was then scanned by a PhosphorImager (Molecular Dynamics, Sunnyvale, CA) and analyzed with ImageQuant software (Molecular Dynamics).
Results
Isoform Specificity of Dystrobrevin Antibodies
Analysis of α-dystrobrevin cDNA products predicts five proteins. Four of these contain the coiled-coils and syntrophin-binding regions needed for assembly into dystrophin–dystrobrevin–syntrophin complexes (Fig. 1 A) (6, 10, 35). To investigate the distribution and protein associations of dystrobrevin isoforms, we prepared polyclonal antibodies against isoform-specific peptides. Antibody αDB2 was generated against a peptide corresponding to the COOH-terminal 10 amino acids unique to α-dystrobrevin-2 and -5 (Fig. 1 A). Antibodies αDB638 and αDB670 were generated against peptides corresponding to regions of the COOH-terminal tail unique to α-dystrobrevin-1 and -4 (Fig. 1 A). Since none of these peptides are conserved in the recently identified β-dystrobrevin (11, 29, 30), the antibodies should be specific for α-dystrobrevin splice forms.
Figure 1.

Antibodies to α-dystrobrevin isoforms in skeletal muscle. (A) Schematic of the known α-dystrobrevin cDNAs identified in mouse (10) and human tissues (35). The coding region includes a syntrophin-binding region (for review see reference 20) and a pair of coiled-coils (9). Abs were generated against peptides (shown as bars) corresponding to regions specific for isoforms. Predicted molecular weights for mouse α-dystrobrevin-1 and -2 were calculated from the mouse sequences while α-dystrobrevin-3, -4, and -5 were estimated based on the corresponding sequences in human. (B) The specificities of dystrobrevin antibodies were tested by immunoblot analysis. Dystrobrevin–dystrophin–syntrophin complexes were partially purified with mAb SYN1351 from Triton X-100–solubilized skeletal muscle extracts and immunoblotted with affinity-purified antibodies prepared against sequences specific to α-dystrobrevin-2 (αDB2) or α-dystrobrevin-1 (αDB638, αDB670). Labeling with antibodies (−) was eliminated by preincubation of the antibody with antigenic peptide (+). Molecular weight markers are shown in kD.
These antibodies were characterized by immunoblotting of syntrophin–dystrophin–dystrobrevin preparations isolated from skeletal muscle using a syntrophin antibody (mAb SYN1351). A pan-specific dystrobrevin antibody (14), DB13H1, recognized polypeptides of apparent molecular weights ∼ 84 kD and ∼ 65 kD, similar to the predicted values for α-dystrobrevins-1 and -2 (Fig. 1 B). Ab αDB2 recognized only the ∼ 65-kD polypeptide, whereas Abs αDB638 and αDB670 labeled only the ∼ 84-kD polypeptide (Fig. 1 B). In each case, labeling was judged to be specific since preincubating the antibody with the appropriate peptide eliminated all labeling (Fig. 1 B).
Two other isoforms predicted to bind syntrophin and dystrophin are α-dystrobrevin-4 and -5. We did not detect α-dystrobrevins of the size expected for these isoforms in our syntrophin preparations from skeletal muscle. The possibility remains that these proteins are present at low levels, or in a form not associated with syntrophin. However, our results are consistent with the fact that α-dystrobrevin-5 was not found in immunoblots of normal muscle tissue (10, 11) and with mRNA analysis showing that transcripts corresponding to α-dystrobrevins-1 and -2 are expressed at high levels in skeletal muscle, while α-dystrobrevins-4 and -5 as well as β-dystrobrevin mRNAs are expressed at much lower levels (10, 29, 35).
Localization of α-Dystrobrevins in Skeletal Muscle
Previous immunofluorescence studies on skeletal muscle found dystrobrevins on the sarcolemma, but with particular enrichment at the NMJ (10, 14). We now know that these studies used antibodies that recognize both α-dystrobrevin-1 and -2, as well as β-dystrobrevin (29). Using the new antibodies described above, we examined the distribution of α-dystrobrevin isoforms in skeletal muscle. Regions rich in NMJs were identified with α-BgTx (Fig. 2, A, C, E, G, I, and K). The anti–α-dystrobrevin-2 antibody strongly labeled the sarcolemma with particular enrichment at the NMJ (Fig. 2 B). In contrast, labeling with both α-dystrobrevin-1 Abs (αDB638 and αDB670) was highly restricted to the NMJ (Fig. 2, F and J). Occasionally, faint but specific extrasynaptic labeling could be detected with Abs αDB638 and αDB670 (not apparent in Fig. 2, F and J, but can be seen in Fig. 5 B, panel b; and Fig. 6 A). Labeling with each antibody was blocked by preincubation with the antigenic peptide (Fig. 2, D, H, and L). Thus, two α-dystrobrevin isoforms generated by alternative splicing are differentially targeted in skeletal muscle.
Figure 2.

Localization of α-dystrobrevin isoforms in skeletal muscle. The distribution of α-dystrobrevins in gastrocnemius skeletal muscle was compared with α-bungarotoxin (α-BgTx) staining of AChRs (A, C, E, G, I, and K). α-Dystrobrevin-2 antibodies (αDB2) stained the entire sarcolemma with particular concentration at the NMJ (B). Abs αDB638 and αDB670 directed against COOH-terminal tail of α-dystrobrevin-1 labeled NMJs with little or no extrasynaptic sarcolemmal labeling (F and J). Labeling for each antibody was dramatically reduced by preincubation of antibody with the appropriate antigenic peptide (D, H, and L). Bar, 50 μm.
Figure 5.

α-Dystrobrevins in skeletal muscle from wild-type, mdx, utrn−/−, and mdx:utrn−/− mice. (A) Sections of gastrocnemius muscle lacking NMJs from wild-type and genetically altered mice were stained with antibody to α-dystrobrevin-2 (αDB2). Staining in wild-type (a) and utrn−/− muscle (b) were indistinguishable. In mdx (c) and mdx:utrn−/− (d) muscles, the sarcolemmal staining of most fibers was greatly diminished. (B) Sections of gastrocnemius muscle containing NMJs were double labeled with α-bungarotoxin (α-BgTx) (primed letters) and antibodies to α-dystrobrevin-2 (a, c, e, g) or α-dystrobrevin-1 (b, d, f, h). Staining for α-dystrobrevin-2 in the synaptic and perisynaptic membrane was essentially unaffected by the absence of utrophin (utrn−/−), dystrophin (mdx), or both proteins (mdx:utrn−/−). Stain-ing for α-dystrobrevin-1 was strong at synapses in wild-type (wt), utrn−/−, and mdx muscle, but was lost in the double mutant. Bars: (c and h′) 40 μm.
Figure 6.
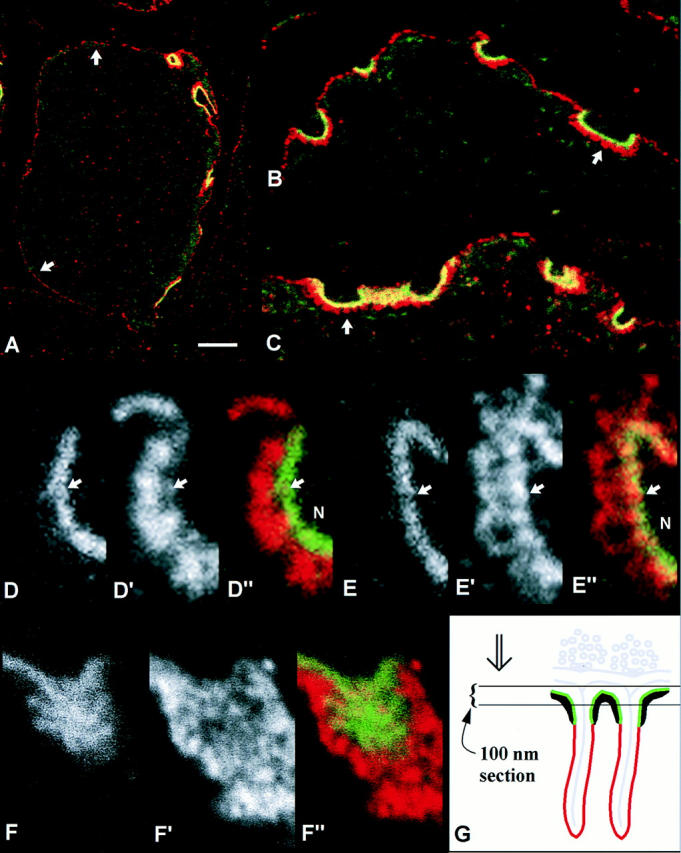
High resolution localization of α-dystrobrevin-1 and α-dystrobrevin-2 at the NMJ. Cryostat sections of mouse sternomastoid muscle were double-labeled on AChRs (with α-bungarotoxin; green) and on α-dystrobrevins (antibodies; red), then prepared for confocal microscopy. Yellow, overlap of strong red and green signals. (A) Muscle fiber labeled with Ab αDb638, against α-dystrobrevin-1. The four nerve– muscle contacts on the fiber are strongly labeled (right-hand side of the fiber). The extrajunctional sarcolemma is weakly labeled (arrows). (B and C) Higher magnification views of junctions labeled with Ab αDb2 against α-dystrobrevin-2 (B) and Ab αDb638 (C). The folds are strongly labeled (arrows). Extensive overlap of the red and green labels on the crests is suggested by the yellow. (D–D″ and E–E″) Partial views of single nerve–muscle contacts labeled with Ab αDb2 (D–D″) and Ab αDb638 (E–E″). N, nerve terminal. (D and E) The separated green channels showing receptor labeling. (D′ and E′) Red channels showing antibody labeling. (D″ and E″) the combined red and green channels. Arrows pointing to the extracellular side of the α-BgTx images in D″ and E″ are reproduced at equivalent positions in D and D′, and in E and E′, respectively. Ab αDb638 (α-dystrobrevin-1) gave stronger labeling of the crests than the deep portions (E′), while Ab αDb2 (α-dystrobrevin-2) labeled the deep portions more strongly (D′). (F–F″) En face view of a receptor-rich area in a cell labeled with Ab αDb2. Extensive overlap of α-BgTx and antibody labeling is apparent. The deep portions of the folds, surrounding the toxin-labeled area, are more strongly labeled by the antibody than the crest area. (G) A schematic representation of the sectioning that gives rise to en face views involving two adjacent folds. Green, receptor-rich membrane. Red, antigen-rich membrane in the deep folds. Black, submembrane cytoskeletal specialization the antigen content of which is to be evaluated. The parallel lines indicate, to scale, a plane of section that projects from the page; in the region shown, the section includes receptor-rich regions, but no membrane from the deep folds. Double arrow, direction of viewing, parallel to the page. Gray, synaptic basal lamina, presynaptic membrane, synaptic vesicles. Bars: (A) 8 μm; (B) 3.3 μm; (C) 2.8 μm; (D) 0.7 μm; (E), 0.6 μm; (F) 1.0 μm.
Immunoaffinity Purification of Dystrobrevin Complexes
Dystrophin and utrophin complexes were partially purified from detergent extracts of skeletal muscle by immunoaffinity purification and analyzed by immunoblotting. It should be noted that the bulk of utrophin in skeletal muscle tissue is derived from non-muscle cells and not from the postsynaptic membrane (31). Gel sample loadings were adjusted to contain similar amounts of total syntrophin (Fig. 3, Anti-Syn). As previously found (28), dystrophin preparations were highly enriched in dystrophin but were free of detectable utrophin (Fig. 3). Likewise, utrophin preparations were highly enriched in utrophin but lacked detectable dystrophin. Dystrophin and utrophin preparations contain approximately equal amounts of total dystrobrevin immunoreactivity (detected with the pan-specific antibody DB13H1). However, the molecular mass of these dystrobrevins was clearly distinct (Fig. 3). Dystrophin preparations contained predominantly an ∼65-kD dystrobrevin (α-dystrobrevin-2), whereas the utrophin preparations were highly enriched in an ∼84-kD dystrobrevin (α-dystrobrevin-1). In each case, however, the specific copurification was not absolute. Low levels of α-dystrobrevin-2 were detected in utrophin preparations, whereas dystrophin preparations contained very small amounts of α-dystrobrevin-1. These results suggest that α-dystrobrevins-1 and -2 are preferentially associated with utrophin and dystrophin, respectively.
Figure 3.
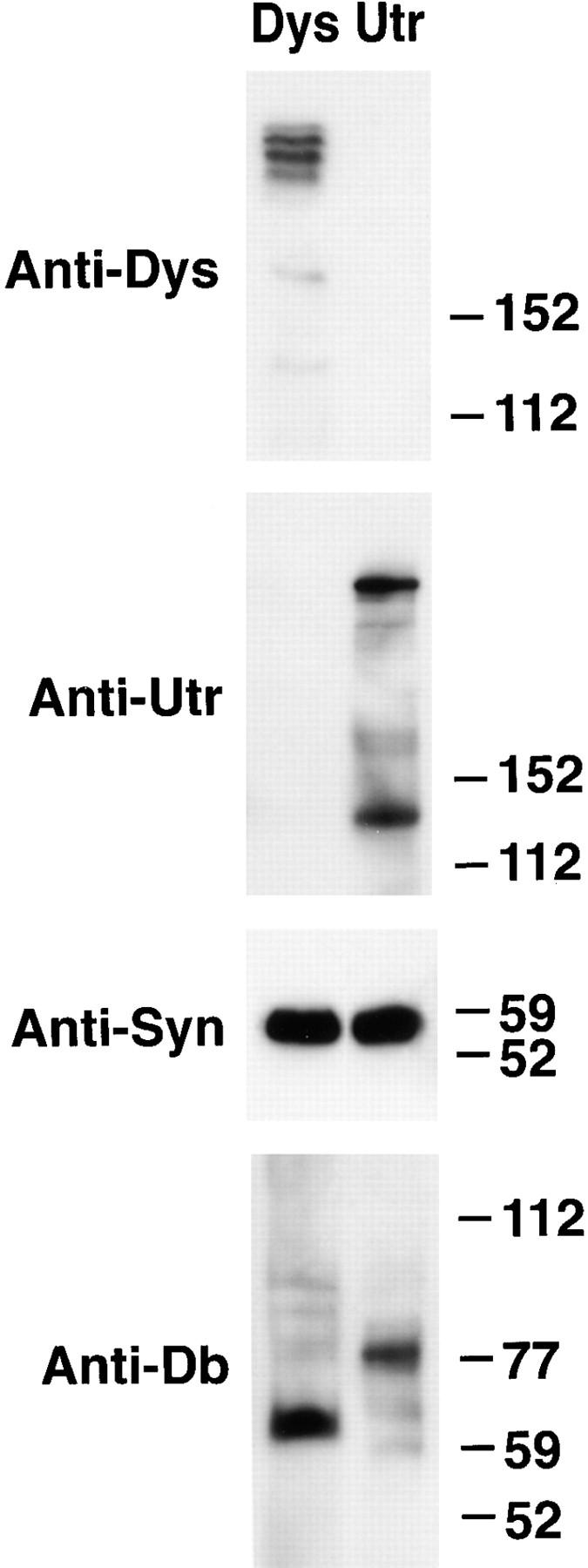
Preferential copurification of α-dystrobrevin-1 with utrophin and α-dystrobrevin-2 with dystrophin. Dystrophin (Dys) and utrophin (Utr) complexes were isolated from Triton X-100–solubilized extracts of mouse skeletal muscle with Ab DYS3669 and with Ab UTR 3165, respectively. For immunoblotting, sample loads were adjusted to contain approximately equal amounts of syntrophin, detected by mAb SYN1351 (Anti-Syn). Immunoblotting these preparations with MANDRA-1 (Anti-Dys) and MANCHO-3 (Anti-Utr) demonstrated specific enrichment for dystrophin and utrophin. Dystrobrevin in these preparations was identified with DB13H1, a pan-specific mAb (Anti-Db). Dystrophin and utrophin complexes purified with the mAbs MANDRA-1 and MANCHO-3, respectively, gave very similar results (data not shown). Molecular weight markers are shown in kD.
Regulation of Dystrobrevin Isoform Pairing with Dystrophin and Utrophin
The association of α-dystrobrevin and dystrophin is mediated by the coiled-coil regions found in both proteins (36). Similar mechanisms probably drive the association of utrophin and α-dystrobrevin. Thus, alternative splicing of α-dystrobrevin sequences immediately adjacent to these coiled-coils could alter the specificity of their interactions with dystrophin and utrophin (10, 35). A related possibility is that other proteins that associate preferentially with utrophin or dystrophin, or with the isoforms of α-dystrobrevin, regulate the interactions. We have used several approaches, including association of in vitro–translated proteins, yeast two-hybrid, and analysis of the distributions in mice genetically altered in expression of utrophin and dystrophin to investigate these possibilities.
Association of In Vitro–translated Proteins
To test for specific associations of dystrobrevin isoforms with dystrophin and utrophin, we studied the interaction of in vitro–translated proteins (4). Recombinant proteins corresponding to the COOH-terminal domains of dystrophin and utrophin (which contain the dystrobrevin binding site) were incubated in pairwise combinations with recombinant full-length α-dystrobrevin-1 and -2, and then analyzed for the formation of complexes by immunoprecipitation with Abs to one of the proteins. The dystrophin construct (C2979) corresponds to amino acids 2,980–3,685 and gives rise to an 84-kD protein (4). The utrophin construct (TDRII) corresponds to amino acids 2,753–3,432 and produces an 81-kD protein (4). The full-length α-dystrobrevin-l construct encodes exons 1–21 (amino acids 1–666) and produces a 79-kD protein, whereas the α-dystrobrevin-2 construct contains exons 1–17B (amino acids 1–567) and encodes a 68-kD product. The dystrobrevin proteins were radiolabeled by incorporation of [14C]leucine and the conditions of coprecipitation were optimized by using the documented interactions of β1-syntrophin and dystrophin or dystrobrevin (25, 28, 29, 36) (data not shown). Both dystrobrevin isoforms tend to aggregate and to be immunoprecipitated non-specifically. Therefore, in each case, a control immunoprecipitation reaction was included in which either anti-dystrophin or anti-utrophin antibody was omitted.
In vitro interactions between dystrobrevins and either dystrophin or utrophin were found with this assay (Fig. 4). An antibody directed against the COOH-terminal region of dystrophin (Dys2) coprecipitated radiolabeled α-dystrobrevin-1 and α-dystrobrevin-2 in the presence of dystrophin (Fig. 4 A, lanes 3 and 5, respectively). By withholding from the reaction either cognate protein (data not shown) or antibodies (Fig. 4 A, lanes 2 and 4), or by using an antibody directed against the NH2 terminus of dystrophin (data not shown), precipitation of the complex was significantly reduced. Thus, dystrophin appears to interact with either α-dystrobrevin-1 or α-dystrobrevin-2. In a similar experiment, utrophin coprecipitated with α-dystrobrevin-l, but not α-dystrobrevin-2 (Fig. 4 B, compare lanes 3 and 5). By withholding from the reaction either respective cognate protein (data not shown) or antibodies (Fig. 4 B, lanes 2 and 4), little or no dystrobrevin was coprecipitated.
Figure 4.
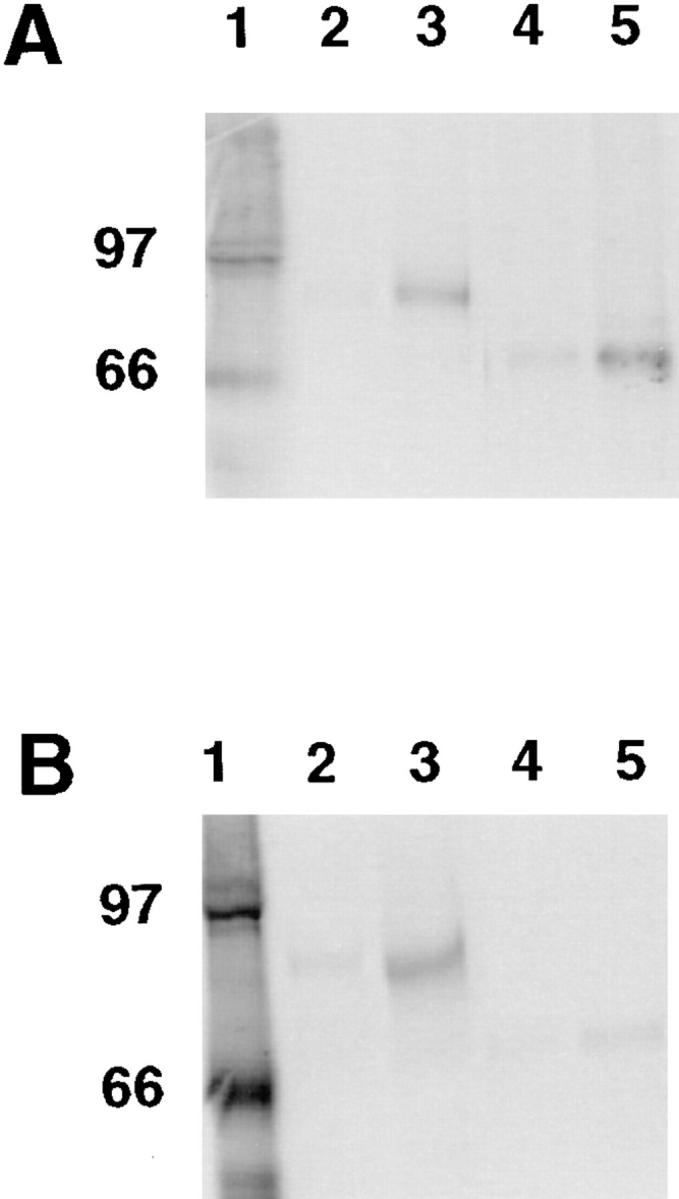
Analysis of dystrobrevin– dystrophin and dystrobrevin–utrophin interactions by coimmunoprecipitation of in vitro–translated proteins. α-Dystrobrevin-1 or -2 was synthesized in the presence of 14C-leucine; utrophin and dystrophin were synthesized as non-radioactive forms. In vitro–translated proteins were incubated together, and then precipitated with a specific antibody in the presence of protein G–Sepharose. After centrifugation and washing, bound proteins were eluted in sample buffer and analyzed by SDS-PAGE and autoradiography. (A) Lane 1, [14C]methylated molecular weight standards. Lane 2, control in which radiolabeled α-dystrobrevin-1 was incubated with dystrophin, but anti-dystrophin was excluded from the immunoprecipitation. Lane 3, radiolabeled α-dystrobrevin-1 was incubated with dystrophin and complexes were immunoprecipitated with anti-dystrophin. Lane 4, control in which radiolabeled α-dystrobrevin-2 was incubated with dystrophin, but anti-dystrophin was excluded from the immunoprecipitation. Lane 5, radiolabeled α-dystrobrevin-2 was incubated with dystrophin and complexes were immunoprecipitated with anti-dystrophin. (B) Lane 1, [14C]methylated molecular weight standards. Lane 2, control in which radiolabeled α-dystrobrevin-1 was incubated with utrophin, but anti-utrophin was excluded from the immunoprecipitation. Lane 3, radiolabeled α-dystrobrevin-1 was incubated with utrophin and complexes were immunoprecipitated with anti-utrophin. Lane 4, control in which radiolabeled α-dystrobrevin-2 was incubated with utrophin, but anti-utrophin was excluded from the immunoprecipitation. Lane 5, radiolabeled α-dystrobrevin-2 was incubated with utrophin and complexes were immunoprecipitated with anti-utrophin. A background of nonspecific aggregation and precipitation of α-dystrobrevin-1 and -2 was seen, regardless of whether a specific antibody was used.
To provide a more quantitative assessment of these interactions, the coprecipitation experiments were replicated five times and the amounts of dystrobrevins associated with either dystrophin or utrophin were determined by band densitometry (Table I). These results confirmed the findings shown in Fig. 4. α-Dystrobrevin-1 interacts specifically with both dystrophin and utrophin. α-Dystrobrevin-2 also interacts with dystrophin, but not with utrophin.
Table I.
Quantification of α-Dystrobrevin-Dystrophin and α-Dystrobrevin-Utrophin Coprecipitation
| Expt. No. | Dys/αDB-1 | Dys/αDB-2 | Utr/αDB-1 | Utr/αDB-2 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −Ab | +Ab | fold** | −Ab | +Ab | fold | −Ab | +Ab | fold | −Ab | +Ab | fold | |||||||||||||
| 1 | 14,270* | 38,134 | 2.7 | 17,995 | 66,851 | 3.7 | 19,449 | 60,339 | 3.1 | 14,009 | 18,355 | 1.3 | ||||||||||||
| 2 | 884 | 1,918 | 2.2 | 1,057 | 4,411 | 4.2 | 33,097 | 134,637 | 4.0 | 47,288 | 44,523 | 0.9 | ||||||||||||
| 3 | 47,436 | 170,415 | 3.6 | 487 | 1,865 | 3.8 | 2,966 | 8,904 | 3.0 | 2,638 | 1,974 | 0.8 | ||||||||||||
| 4 | 9,493 | 28,209 | 3.0 | 20,182 | 67,344 | 3.3 | 3,840 | 13,792 | 3.6 | 1,343 | 1,472 | 1.1 | ||||||||||||
| 5 | 19,009 | 61,822 | 3.3 | 1,124 | 3,565 | 3.2 | 3,451 | 9,940 | 2.9 | 1,244 | 1,277 | 1.0 | ||||||||||||
| 3.0 | 3.6 | 3.3 | 1.0 | |||||||||||||||||||||
| Avg. | (σ ± 0.5) | (σ ± 0.4) | (σ ± 0.4) | (σ ± 0.2) | ||||||||||||||||||||
The band activities were quantified with ImageQuant software and are in units pixel. The positive band activities of at least twofold over the background pixel values were easily discernable.
fold, Fold difference between the band activities of the reaction containing the specific antibody and the control experiment (no antibody).
Yeast Two-Hybrid Analysis of Interactions
The preferential interaction of α-dystrobrevin-2 with dystrophin was also studied using the yeast two-hybrid system (Table II). α-Dystrobrevin-1 and -2 were each tested for interactions with dystrophin, utrophin, and each other by co-transforming into the Hf7c yeast strain a DNA-binding domain plasmid (pGBT9) encoding one of the proteins and an activation domain plasmid (pGAD) encoding the other protein. In this assay, α-dystrobrevin-1 interacted with both utrophin and dystrophin, while α-dystrobrevin-2 interacted only with dystrophin (Table II). These results confirm those obtained with in vitro–translated proteins.
Table II.
α-Dystrobrevin-Dystrophin and α-Dystrobrevin-Utrophin Interactions in the Yeast Two-Hybrid Assay
| Expressed proteins | Dystrophin | Utrophin | α-Dystrobrevin-1 | α-Dystrobrevin-2 | ||||
|---|---|---|---|---|---|---|---|---|
| Dystrophin | −− | −− | ++ | ++ | ||||
| Utrophin | −− | −− | ++ | −− | ||||
| α-Dystrobrevin-1 | ++ | ++ | −− | −− | ||||
| α-Dystrobrevin-2 | ++ | −− | −− | −− |
−− = no growth on selective media, and no positive β-galactosidase activity. ++ = growth on selective media, and positive β-galactosidase activity.
Distribution of Dystrobrevin Isoforms in Skeletal Muscle Lacking Dystrophin and Utrophin
The distribution and levels of dystrophin-associated proteins in skeletal muscle are known to be altered by the absence of dystrophin (16, 28). Prominent examples of these changes occur in skeletal muscle from humans with Duchenne muscular dystrophy and in the mdx mouse. At the NMJ, however, dystrophin-associated proteins are retained, probably because they are associated with utrophin at this site. We have examined the distribution of α-dystrobrevin isoforms in skeletal muscle from mdx mice, from mice in which utrophin is absent because of targeted disruption of the gene (utrn−/−) (23), and from mice obtained by crossing mdx and utrn−/− mice, which thus lack both dystrophin and utrophin (24).
For α-dystrobrevin-2, different results were obtained depending on the region of muscle examined. In regions away from the neuromuscular synapses, staining for α-dystrobrevin-2 appears normal on the sarcolemma of skeletal muscle from utrn−/− mice, but is dramatically reduced on the sarcolemma of mdx and mdx:utrn−/− mice in regions of muscle fibers away from the central zone where NMJs are located (Fig. 5 A). This result suggests that association with dystrophin is probably the major factor in the sarcolemmal association of α-dystrobrevin-2. In contrast, labeling for α-dystrobrevin-2 is retained at NMJs and on the perijunctional sarcolemma, even in mice lacking both dystrophin and utrophin (Fig. 5 B, left column). This labeling presumably reflects retention of α-dystrobrevin-2. The alternative possibility is that the labeling is due to α-dystrobrevin-5, an NH2-terminal–truncated form that shares the αDB2 epitope with α-dystrobrevin-2 (35). The only available evidence on this possibility is that α-dystrobrevin-5 is not expressed at significant levels in normal skeletal muscle. In either case, however, our result suggests that factors in addition to dystrophin and utrophin are required in the association of α-dystrobrevins with the perijunctional sarcolemma (10, 35).
The synaptic localization of α-dystrobrevin-1 is retained in mdx muscle, as expected if it is associated with utrophin. However, synaptic staining of α-dystrobrevin-1 is also present in muscles from utrn−/− mice. Only when both dystrophin and utrophin were eliminated from the postsynaptic membrane was α-dystrobrevin-1 lost from the synapse. These results suggest that α-dystrobrevin-1 is associated with both dystrophin and utrophin at the synapse, an interaction consistent with the biochemical and yeast two hybrid findings described above.
High Resolution Analysis of α-Dystrobrevins in the Postsynaptic Membrane
Within the NMJ, utrophin is sharply confined to the AChR- and rapsyn-rich superficial zones, or crests, of the junctional folds (8; see Fig. 6 G for a schematic representation). Dystrophin occurs in the deep portions of the folds (13, 39), which are also the site of sodium channel concentration (18). To determine whether this may also be true of α-dystrobrevin-1 and α-dystrobrevin-2, we applied a high resolution immunofluorescence method in which labeled cryostat sections of mouse sternomastoid muscle are embedded in Araldite, sectioned at 100-nm thickness and examined by confocal microscopy (Kramarcy, N.R., and R. Sealock, manuscript in preparation).
A low magnification view of a muscle fiber stained with Ab αDB638 against α-dystrobrevin-1 is shown in Fig. 6 A (antibody staining in red, α-BgTx staining of AChRs in green; overlap in yellow). As previously shown in Fig. 2, NMJs were strongly labeled by Ab αDB638. Weak but specific extrajunctional labeling was also consistently detected by the thin-section method. At intermediate magnifications, the deep portions of the junctional folds were labeled for both α-dystrobrevins-1 and -2 (Fig. 6, B and C, respectively). Labeling for α-dystrobrevin-1, and to a lesser extent for α-dystrobrevin-2, was also apparent in the receptor- and utrophin-rich crests, as suggested by the yellow in Figs. 6, B and C. Viewing of the separated red and green channels at high magnification showed that the crests were strongly labeled by Ab αDB638, while the deep portions were also labeled, but more weakly (Fig. 6, E–E″). Ab αDB2, against α-dystrobrevin-2, gave the opposite result: the deep portions were strongly labeled, while the crests were more weakly labeled, and labeled to various extents (Fig. 6, D–D″). On the basis of these results, α-dystrobrevin-1 and α-dystrobrevin-2 appear to have distinct but overlapping distributions within the NMJ. To test whether the appearance of labeling on the crests reflected genuine presence of antibody, rather than arising from the low resolution of light microscopy, we concentrated on sections that just grazed individual nerve–muscle contacts. Fig. 6 G shows schematically why such views contain patches of receptor-rich membrane only. An example from a junction labeled with Ab αDB2 is shown in Fig. 6, F–F″. Overlapping labels for receptor (Fig. 6 F) and α-dystrobrevin-2 (Fig. 6 F′) are clearly apparent. As in the transverse image (Figs. 6, D–D″), the deep regions are more strongly labeled than the crests. Greater than 90% of such images show distinct labeling of the receptor-rich zone by Ab αDB2. In contrast, labeling for ankyrin G, a protein that has been shown quantitatively to be confined to the deep portions (18), is very weak or absent from most en face views of the crests (Kramarcy, N.R., and R. Sealock, unpublished observations). This suggests that the labeling of the crest region by Ab αDB2 is accurate. Thus, α-dystrobrevin-2 is present on the receptor-rich membranes but is most concentrated on the receptor-poor membranes of the junction.
The presence of α-dystrobrevin-2 on the crests, albeit at lower amounts than in the deep portions of the folds, could be due to association of this isoform with utrophin. This interaction may be of too low an affinity to be detected by in vitro association or yeast two-hybrid analyses, but could be driven by the high local concentrations of the two proteins at the junction. Alternatively, occurrence of α-dystrobrevin-2 on the crests could reflect its association with other proteins, as in its persistence at the junction in mdx:utrn−/− mice.
Discussion
Originally identified as an 87-kD protein associated with AChR-enriched membranes in Torpedo electric organ (14), dystrobrevin was subsequently found to be related to dystrophin and utrophin (43). Dystrophin and dystrobrevin are homologous throughout their CT and CR regions (10, 36, 43). Although the extent of homology is rather low, defined sites for interaction with other proteins, including the syntrophins, are highly similar. Outside the CRCT domain, dystrophin and dystrobrevin are largely unrelated. The actin-binding domain and spectrin-like repeats of dystrophin (and utrophin) are absent from dystrobrevin, while the COOH-terminal extension of ≈180 amino acids of α-dystrobrevin-1 is not found in dystrophin and utrophin. Thus, dystrobrevin and dystrophin may serve similar functions through the CRCT domains, while at the same time having different functions mediated by their unique domains.
The demonstration that dystrobrevins are, in fact, dystrophin-associated proteins, an interaction mediated by two coiled-coil heptad repeats in the COOH-terminal regions of both proteins (36), formed the basis for a new model of the dystrophin complex (28, 36). A key feature of this model is the presence of two syntrophins, one bound to dystrophin and one to dystrobrevin. Both syntrophins are presumably available to bind via their PDZ domains to signaling proteins such as neuronal nitric oxide synthase and sodium channels (12, 21, 37). Consequently, the existence of multiple forms of α-dystrobrevin generated by alternative splicing and the use of different promoters (10, 35), as well as a second, closely related protein, β-dystrobrevin, encoded by a separate gene (11, 29, 30), implies that the composition of proteins in the dystrophin complex, and therefore the function of the complex, can be quite diverse. In results reported here, we show that alternative splicing of α-dystrobrevin regulates its interaction with dystrophin and utrophin, and that the unique COOH-terminal extension of α-dystrobrevin-1 may have a special function at the neuromuscular synapse.
The coiled-coil region of α-dystrobrevin that mediates binding to dystrophin (36) and utrophin is identical in both α-dystrobrevin-1 and -2. Thus, the selective interaction of α-dystrobrevin-2 with dystrophin in in vitro association and yeast two-hybrid tests was not anticipated. In part, the specificity of the interactions between α-dystrobrevins and dystrophin/utrophin is inherent in the structures of the proteins themselves. The failure of α-dystrobrevin-2 to bind recombinant utrophin suggests that sequences present in this form prevent the coiled-coil–mediated interaction that occurs between dystrophin or utrophin and α-dystrobrevin-1. A plausible explanation for this result is that sequences adjacent to the coiled-coils in α-dystrobrevin-1 are responsible for this regulation. Regions immediately after the coiled-coils are subject to alternative splicing and, depending on the exons used, α-dystrobrevin-1 and α-dystrobrevin-2 could differ significantly at this site (6, 10, 35). In the same manner, the DUR or two additional splice sites upstream of the coiled-coils could influence specificity.
The selective interaction of α-dystrobrevin-2 with dystrophin is entirely consistent with the copurification and colocalization of these two proteins, and with the loss of α-dystrobrevin-2 from the sarcolemma in mdx muscle. Similarly, the ability of α-dystrobrevin-1 to bind both dystrophin and utrophin could explain the presence of α-dystrobrevin-1 throughout the junctional folds, its retention at the synapse in mdx and utrn−/−, and its absence from the junction in the double mutant. However, the retention of α-dystrobrevin-2 at synaptic and perisynaptic sites in muscle lacking both dystrophin and utrophin is not easily explained by these interactions. Rather, this finding implies that α-dystrobrevin-2 interacts with another protein at these sites whose presence is independent of dystrophin or utrophin. This protein could be another member of the dystrophin family, a protein with a compatible coiled-coil domain, or a protein that interacts with other regions of α-dystrobrevin-2. This protein would not interact with α-dystrobrevin-1, however, since this form is not present at synapses in the double mutant.
Proteins at the neuromuscular synapse are generally confined to a specific region of the postsynaptic folds (for review see reference 38). The segregation of nicotinic receptors at the crests and sodium channels in the bottoms of the folds are classic examples. Submembrane proteins, such as rapsyn and utrophin at the crests and dystrophin in the bottoms of the folds, are similarly distributed. In this sense, α-dystrobrevin-1 is unusual in that it is found throughout the folds. Thus, its function, while likely to be synaptically related, probably is not unique to either sodium channels or nicotinic receptors. The synaptic function of α-dystrobrevin-1 most likely involves the DUR, which contains numerous potential sites for tyrosine phosphorylation. Tyrosine phosphorylation stimulated by agrin activation of a muscle-specific kinase has been linked to synapse formation (7, 15, 22). Although Torpedo dystrobrevin is endogenously phosphorylated on tyrosine (43), neither the identity of the kinase(s) responsible for its phosphorylation nor the downstream consequences are known.
The association of particular α-dystrobrevin isoforms with dystrophin or utrophin is not random, but rather appears to be highly regulated. Furthermore, this regulatory event may be important for targeting of dystrophin or utrophin complexes to different membrane domains. Although much less is known about the existence of alternatively spliced forms of β-dystrobrevin, similar regulation of its interaction with dystrophin and utrophin may occur. The wide expression of α-dystrobrevin-1 and -2 (and other isoforms) and β-dystrobrevin in a variety of tissues suggests that targeting to specialized membrane domains may occur in many cell types. Thus, the assembly of multi-component complexes containing dystrophin, utrophin, and dystrobrevin, and their associated signaling proteins, may be a common feature of specialized membrane domains in many cell types.
Acknowledgments
We thank our colleagues for helpful discussions and comments on the manuscript. We are indebted to K. McNaughton and C. Conner for extensive assistance with cryosectioning; to G. Morris for donating MANCHO-3; to J. Cohen for providing mAb 13H1; and to K. Davies and D. Blake for providing mouse α-dystrobrevin sequence before publication.
This work was supported by National Institutes of Health Grants NS33145 (to S.C. Froehner and R. Sealock), NS14871 (to S.C. Froehner), NS23740 (to L.M. Kunkel), and NS29172 (to J.R. Sanes); Muscular Dystrophy Association Grants (to S.C. Froehner, R. Sealock and J.R. Sanes); and a grant from the Council for Tobacco Research (to R. Sealock). L.M. Kunkel is an Investigator of the Howard Hughes Medical Institute.
Abbreviations used in this paper
- Ab
polyclonal antibody
- AChR
acetylcholine receptor
- CR
cysteine rich
- CT
COOH-terminal
- DRP
dystrophin-related protein
- DUR
dystrobrevin-unique region
- NMJ
neuromuscular junction
- PDZ
protein domain originally identified in postsynaptic density-95, discs large, ZO-1
- α-BgTx
α-bungarotoxin
Footnotes
Address all correspondence to S.C. Froehner, Department of Cell and Molecular Physiology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7545. Tel.: (919) 966-1239. Fax: (919) 966-6413. E-mail: froehner@med.unc.edu
References
- 1.Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC. Two forms of mouse syntrophin, a 58 kD dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron. 1993;11:531–540. doi: 10.1016/0896-6273(93)90157-m. [DOI] [PubMed] [Google Scholar]
- 2.Adams ME, Dwyer TM, Dowler LL, White RA, Froehner SC. Mouse α1- and β2-syntrophin gene structure, chromosome localization, and homology with a discs large domain. J Biol Chem. 1995;270:25859–25865. doi: 10.1074/jbc.270.43.25859. [DOI] [PubMed] [Google Scholar]
- 3.Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet. 1993;3:283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- 4.Ahn AH, Kunkel LM. Syntrophin binds to an alternatively spliced exon of dystrophin. J Cell Biol. 1995;128:363–371. doi: 10.1083/jcb.128.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahn AH, Freener CA, Gussoni E, Yoshida M, Ozawa E, Kunkel LM. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J Biol Chem. 1996;271:2724–2730. doi: 10.1074/jbc.271.5.2724. [DOI] [PubMed] [Google Scholar]
- 6.Ambrose HE, Blake DJ, Nawrotzki RA, Davies KE. Genomic organization of the mouse dystrobrevin gene: comparative analysis of the dystrophin gene. Genomics. 1997;39:359–369. doi: 10.1006/geno.1996.4515. [DOI] [PubMed] [Google Scholar]
- 7.Apel ED, Glass GJ, Moscoso LM, Yancopoulos GD, Sanes JR. Rapsyn is required for MuSK signaling and recruits synaptic components to a MuSK-containing scaffold. Neuron. 1997;18:623–635. doi: 10.1016/s0896-6273(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 8.Bewick GS, Nicholson LVB, Young C, O'Donnell E, Slater CR. Different distributions of dystrophin and related proteins at nerve-muscle junctions. Neuroreport. 1992;3:857–860. doi: 10.1097/00001756-199210000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Blake DJ, Tinsley JM, Davies KE, Knight AE, Winder SJ, Kendrick-Jones J. Coiled-coil regions in the carboxy-terminal domains of dystrophin and related proteins: potentials for protein-protein interactions. Trends Biochem Sci. 1995;20:133–135. doi: 10.1016/s0968-0004(00)88986-0. [DOI] [PubMed] [Google Scholar]
- 10.Blake DJ, Nawrotzki R, Peters MF, Froehner SC, Davies KE. Isoform diversity of dystrobrevin, the murine 87-kDa postsynaptic protein. J Biol Chem. 1996;271:7802–7810. doi: 10.1074/jbc.271.13.7802. [DOI] [PubMed] [Google Scholar]
- 11.Blake DJ, Nawrotzki R, Loh NY, Gorecki DC, Davies KE. β-dystrobrevin, a member of the dystrophin-related protein family. Proc Natl Acad Sci USA. 1998;95:241–246. doi: 10.1073/pnas.95.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 13.Byers TJ, Kunkel LM, Watkins SC. The subcellular distribution of dystrophin in mouse skeletal, cardiac, and smooth-muscle. J Cell Biol. 1991;115:411–421. doi: 10.1083/jcb.115.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carr C, Fischbach GD, Cohen JB. A novel Mr87,000 protein associated with acetylcholine receptors in Torpedo electric organ and vertebrate skeletal muscle. J Cell Biol. 1989;109:1753–1764. doi: 10.1083/jcb.109.4.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, et al. The receptor tyrosine kinase MuSk is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- 16.Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 17.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–256. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 18.Flucher BE, Daniels MP. Distribution of sodium channels and ankyrin in the neuromuscular junction is complementary to that of acetylcholine receptors and the 43kD protein. Neuron. 1989;3:163–175. doi: 10.1016/0896-6273(89)90029-9. [DOI] [PubMed] [Google Scholar]
- 19.Froehner SC, Murnane AA, Tobler M, Peng HB, Sealock R. A postsynaptic Mr58,000 (58K) protein concentrated at acetylcholine receptor-rich sites in Torpedo electroplaques and skeletal muscle. J Cell Biol. 1987;104:1633–1646. doi: 10.1083/jcb.104.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Froehner, S.C., M.E. Adams, M.F. Peters, and S.H. Gee. 1997. Syntrophins: modular adapter proteins at the neuromuscular junction and the sarcolemma. In Cytoskeletal Regulation of Membrane Function. S.C. Froehner and G.V. Bennett, editors. Rockefeller University Press, New York. 197–207. [PubMed]
- 21.Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998;18:128–137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattson K, Burden SJ, DiStefano PS, et al. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- 23.Grady RM, Merlie JP, Sanes JR. Subtle neuromuscular defects in utrophin-deficient mice. J Cell Biol. 1997;136:871–882. doi: 10.1083/jcb.136.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 25.Kramarcy NR, Vidal A, Froehner SC, Sealock R. Association of utrophin and multiple dystrophin short forms with the mammalian Mr58,000 dystrophin-associated protein (syntrophin) J Biol Chem. 1994;269:2870–2876. [PubMed] [Google Scholar]
- 26.Nguyen TM, Ellis JM, Love DR, Davies KE, Gatter KC, Dickson G, Morris GE. Localization of the DMDL gene-encoded dystrophin-related protein using a panel of nineteen monoclonal antibodies: Presence at neuromuscular junctions, in the sarcolemma of dystrophic skeletal muscle, in vascular and other smooth muscles, and in proliferating brain cell lines. J Cell Biol. 1991;115:1695–1700. doi: 10.1083/jcb.115.6.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohlendieck K, Ervasti JM, Matsumura K, Kahl SD, Leveille CJ, Campbell KP. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991;7:499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- 28.Peters MF, Adams ME, Froehner SC. Differential association of syntrophin pairs with the dystrophin complex. J Cell Biol. 1997;138:81–93. doi: 10.1083/jcb.138.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters MF, O'Brien KF, Sadoulet-Puccio HM, Kunkel LM, Adams ME, Froehner SC. β-dystrobrevin, a new member of the dystrophin family: identification, cloning, and protein associations. J Biol Chem. 1997;272:31561–31569. doi: 10.1074/jbc.272.50.31561. [DOI] [PubMed] [Google Scholar]
- 30.Puca AA, Nigro V, Piluso V, Belsito A, Sampaolo S, Quaderi N, Rossi E, Di Iorio G, Ballabio A, Franco B. Identification and characterization of a novel member of the dystrobrevin gene family. FEBS (Fed Eur Biochem Soc) Lett. 1998;425:7–13. doi: 10.1016/s0014-5793(98)00097-0. [DOI] [PubMed] [Google Scholar]
- 31.Rivier F, Robert A, Hugon G, Mornet D. Different utrophin and dystrophin properties related to their vascular smooth muscle distributions. FEBS (Fed Eur Biochem Soc) Lett. 1997;408:94–98. doi: 10.1016/s0014-5793(97)00398-0. [DOI] [PubMed] [Google Scholar]
- 32.Roberts RG, Freeman TC, Kendall E, Vetrie DL, Dixon AK, Shaw-Smith C, Bone Q, Bobrow M. Characterization of DRP2, a novel human dystrophin homologue. Nat Genet. 1996;13:223–226. doi: 10.1038/ng0696-223. [DOI] [PubMed] [Google Scholar]
- 33.Rybakaova IN, Amann KJ, Ervasti JM. A new model for the interaction of dystrophin with F-actin. J Cell Biol. 1996;135:661–671. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sadoulet-Puccio HM, Kunkel LM. Dystrophin and its isoforms. Brain Pathol. 1996;6:25–35. doi: 10.1111/j.1750-3639.1996.tb00780.x. [DOI] [PubMed] [Google Scholar]
- 35.Sadoulet-Puccio HM, Khurana TS, Cohen JB, Kunkel LM. Cloning and characterization of the human homologue of a dystrophin related phosphoprotein found at the Torpedo electric organ post-synaptic membrane. Hum Mol Gen. 1996;5:489–496. doi: 10.1093/hmg/5.4.489. [DOI] [PubMed] [Google Scholar]
- 36.Sadoulet-Puccio HM, Rajala M, Kunkel LM. Dystrobrevin and dystrophin: an interaction through coiled-coil motifs. Proc Natl Acad Sci USA. 1997;94:12413–12418. doi: 10.1073/pnas.94.23.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schultz J, Hoffmuller U, Krause G, Ashurst J, Macias MJ, Schmieder P, Schneider-Mergener J, Oschkinat H. Specific interactions between the syntrophin PDZ domain and voltage-gated sodium channels. Nat Struct Biol. 1998;5:19–24. doi: 10.1038/nsb0198-19. [DOI] [PubMed] [Google Scholar]
- 38.Sealock, R., and S.C. Froehner. 1997. Formation of the Neuromuscular Junction. In Dystrophin: Gene, Protein, and Cell Biology. S. Brown and J.A. Lucy, editors. Cambridge University Press, Cambridge, UK. 139–162.
- 39.Sealock R, Butler MH, Kramarcy NR, Gao KX, Murnane AA, Douville K, Froehner SC. Localization of dystrophin relative to acetylcholine receptor domains in electric tissue and adult and cultured skeletal muscle. J Cell Biol. 1991;113:1133–1144. doi: 10.1083/jcb.113.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol. 1997;10:168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- 41.Tinsley JM, Blake DJ, Roche A, Fairbrother U, Riss J, Byth BC, Knight AE, Kendrick-Jones J, Suthers GK, Love DR, Edwards YH, Davies KE. Primary structure of dystrophin-related protein. Nature. 1992;360:591–593. doi: 10.1038/360591a0. [DOI] [PubMed] [Google Scholar]
- 42.Vainzof M, Passos-Bueno MR, Canovas M, Moreira ES, Pavanello RCM, Marie SK, Anderson LVB, Bonnemann CG, McNally EM, Nigro V, et al. The sarcoglycan complex in the six autosomal recessive limb-girdle muscular dystrophies. Hum Mol Genet. 1996;5:1963–1969. doi: 10.1093/hmg/5.12.1963. [DOI] [PubMed] [Google Scholar]
- 43.Wagner KR, Cohen JB, Huganir RL. The 87K postsynaptic membrane protein from Torpedo is a protein-tyrosine kinase substrate homologous to dystrophin. Neuron. 1993;10:511–522. doi: 10.1016/0896-6273(93)90338-r. [DOI] [PubMed] [Google Scholar]
- 44.Yang B, Jung D, Motto D, Meyer J, Koretzy G, Campbell K. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270:11711–11714. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]
- 45.Yang B, Jung D, Rafael JA, Chamberlain JS, Campbell KP. Identification of α-syntrophin binding to syntrophin triplet, dystrophin, and utrophin. J Biol Chem. 1995;270:4975–4978. doi: 10.1074/jbc.270.10.4975. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida M, Yamamoto H, Noguchi S, Mizuno Y, Hagiwara Y, Ozawa E. Dystrophin-associated protein A0 is a homologue of the Torpedo 87K protein. FEBS (Fed Eur Biochem Soc) Lett. 1995;367:311–314. doi: 10.1016/0014-5793(95)00574-s. [DOI] [PubMed] [Google Scholar]