

Abstract
Mitochondria have a well-established capacity to detect cytoplasmic Ca2+ signals resulting from the discharge of ER Ca2+ stores. Conversely, both the buffering of released Ca2+ and ATP production by mitochondria are predicted to influence ER Ca2+ handling, but this complex exchange has been difficult to assess in situ using conventional measurement techniques. Here we have examined this interaction in single intact BHK-21 cells by monitoring intraluminal ER [Ca2+] directly using trapped fluorescent low-affinity Ca2+ indicators. Treatment with mitochondrial inhibitors (FCCP, antimycin A, oligomycin, and rotenone) dramatically prolonged the refilling of stores after release with bradykinin. This effect was largely due to inhibition of Ca2+ entry pathways at the plasma membrane, but a significant component appears to arise from reduction of SERCA-mediated Ca2+ uptake, possibly as a consequence of ATP depletions in a localized subcellular domain. The rate of bradykinin-induced Ca2+ release was reduced to 51% of control by FCCP. This effect was largely overcome by loading cells with BAPTA-AM, highlighting the importance of mitochondrial Ca2+ buffering in shaping the release kinetics. However, mitochondria-specific ATP production was also a significant determinant of the release dynamic. Our data emphasize the localized nature of the interaction between these organelles, and show that competent mitochondria are essential for generating explosive Ca2+ signals.
Keywords: metabolic microdomains, calcium homeostasis, organelle interactions, mitochondrial uncouplers, signal transduction
Cellular Ca2+ homeostasis reflects an intricate balance of many factors (pumps, leaks, release channels, and buffer systems; Berridge, 1993; Clapham, 1995) that are expected to directly or indirectly influence Ca2+ handling by internal stores. Among these are interactions with other organelles, including mitochondria. By now it has been firmly established using a variety of techniques (for example, using targeted recombinant aequorin; Rizzuto et al., 1992) that mitochondria sense Ca2+ released into the cytoplasm during Ca2+ signaling events (Rizzuto et al., 1993; Sparagna et al., 1995; Babcock et al., 1997), and that this uptake of Ca2+ constitutes a physiologically important mechanism for activating mitochondrial metabolism (Denton and McCormack, 1990; Rutter et al., 1996; Hajnóczky et al., 1995).
Thus far this problem has been analyzed from the point of view of the mitochondria or of the cytoplasm. But how do mitochondrial metabolism and Ca2+ uptake influence Ca2+ release from the ER? In fact, interactions between these two organelles may be expected to occur on multiple levels.
First, with respect to the refilling of internal stores with Ca2+, it is obvious that ATP (derived from mitochondrial and glycolytic sources) is required to fuel accumulation of Ca2+ by SERCAs, the Ca2+-ATPases resident on the ER membrane. Second, previous studies have demonstrated that store-operated Ca2+ entry pathways (those that are activated by store depletion; Putney, 1990) are influenced by mitochondrial metabolism (Gamberucci et al., 1994; Hoth et al., 1997; Marriot and Mason, 1995). Since the refilling of internal stores after agonist-induced Ca2+ release relies principally on this pathway (see for example Hofer et al., 1998), mitochondrial inhibition will also be expected to impede the recharging of internal stores.
Regarding the mobilization of Ca2+ from stores, ATP is known to modulate Ca2+ release channels in the ER, including inositol 1,4,5-trisphosphate (InsP3) receptors (Bezprozvanny and Ehrlich, 1995; Ferris et al., 1990; Smith et al., 1985), as well as Ca2+ leaks (Hofer et al., 1996). The allosteric modulation of InsP3 receptors by ATP shows a bell-shaped dependence on [ATP], with progressive reduction in open probability of the channel at ATP concentrations below 500 μM and above 4 mM (Bezprozvanny and Ehrlich, 1993). It has been questioned in the past whether local [ATP] can vary sufficiently under physiological conditions to effect any sort of regulation on the release channels in situ.
The ability of mitochondria to buffer cytoplasmic Ca2+ transients is expected to influence InsP3 receptor opening further, since both this release channel and the ryanodine receptor (that mediates Ca2+-induced Ca2+ release) have also been shown to exhibit a bell-shaped dependence of open probability on cytosolic [Ca2+] (Bezprozvanny et al., 1991; Finch et al., 1991). In support of this view is the recent demonstration by Jouaville et al. (1995) that mitochondrial substrates alter the propagation of Ca2+ waves in oocytes, a finding confirmed by another study using blockers of mitochondrial respiration in cultured oligodendrocytes (Simpson and Russell, 1996). These phenomena have been attributed to local alterations in Ca2+ buffering as a consequence of Ca2+ uptake into energized mitochondria, resulting in modulation of InsP3 receptor opening.
Here we have addressed the integrated actions of the above factors from the perspective of the ER, using a fluorescence technique that permits selective measurement of free [Ca2+] changes in the agonist-sensitive store of single intact cells. Our work demonstrates that not only is the ability of mitochondria to buffer Ca2+ transients important, but also that mitochondrial ATP production alters the dynamics of Ca2+ release, the two factors acting in concert to permit explosive discharge of Ca2+ from the store under conditions where mitochondria are actively respiring. Our results also indicate that the effects of mitochondrial inhibitors on the Ca2+ entry pathways at the plasma membrane result in considerable inhibition of store refilling. At the same time, SERCAs in BHK-21 cells appear to depend on mitochondrial ATP production for Ca2+ sequestration to a surprising extent, a result best explained by the existence of subcellular metabolic microdomains. Our findings suggest that there is an intimate functional (and possibly physical) relationship between these two organelles.
Materials and Methods
Cell Culture and Dye Loading
BHK-21 cells (purchased from Consorzio. Gest. Biotec. Avanzate, Genova, Italy) were grown in Earle's MEM medium containing 10% FBS, and were maintained in a humidified incubator at 37°C in the presence of 5% CO2/95% air. Cells were seeded at low density on glass coverslips and used the following day for microspectrofluorimetric or ratio imaging measurements of cytoplasmic or intraorganellar free [Ca2+] with fura-2-AM (Grynkiewicz et al., 1985), or mag-fura-2-AM (Raju et al., 1989), respectively. Cells were loaded in tissue culture medium for 20–30 min at room temperature with fura-2-AM (5 μM) or at 37°C for 45–60 min with mag-fura-2-AM (5–10 μM). Subcultures were prepared by trypsinization, and the cells used for not more than 10 passages after receipt from the distributor.
Microspectrofluorimetric Measurements
Coverslips with dye-loaded cells were mounted into a heated metal flow-through perfusion chamber described previously (Negulescu and Machen, 1990) placed on the stage of an inverted Zeiss IM 35 microscope and perfused by gravity feed at a rate of 1.5–2 ml/min. Change of solutions was made by a remote-controlled electronic manifold. Emitted fluorescence from single cells was measured in response to alternate pulses of excitation light (5-msec duration) at 340 nm and 380 nm using a computer-controlled four-place sliding filter holder manufactured in-house. The emitted fluorescence (510 nm) was focused on a photomultiplier tube, amplified, digitally converted, and sampled on an IBM-compatible computer. The filter exchange system and data sampling software were designed by Giuseppe and Antonio Troccoli (Bari, Italy). All measurements were automatically corrected for background. The ratio of emitted light from the two excitation wavelengths (340/380) of fura-2 or mag-fura-2 provide a measure of ionized cytoplasmic [Ca2+] (Grynkiewicz et al., 1985) or intrastore [Ca2+] (Hofer and Machen, 1993; Hofer et al., 1998), respectively. Mag-fura-2 data are presented as uncalibrated ratio changes and not free [Ca2+] due to uncertainties in the calibration procedure as described previously by Hofer and Schulz (1996).
Ratio Imaging Experiments
Some measurements in this study were made using a commercial imaging system (Georgia Instruments, Roswell, Georgia) described previously in more detail (Gamberucci et al., 1994). The 345/375 nm excitation ratio (emission 450 nm) was acquired from individual cells within the microscope field every 4 s. Cells were superfused continuously on the heated microscope stage in an open Leiden chamber equipped with gravity feed inlets and vacuum outlets for solution changes.
Solutions and Materials
Unless otherwise stated, all chemicals were purchased from Farmitalia Carlo Erba (Milano, Italy), Fluka AG (Buchs, Switzerland) or Sigma Chemical Co. (St. Louis, MO). Experiments were performed with a Ringer's solution containing (in mM) 121 NaCl, 2.4 K2HPO4, 0.4 KH2PO4, 1.2 CaCl2, 1.2 MgCl2, 5.5 glucose, 10 Hepes/NaOH, pH 7.20. Bradykinin and ionomycin were from Calbiochem-Novabiochem Corp. (La Jolla, CA); fura-2-AM and mag-fura-2-AM were obtained from Molecular Probes (Eugene, OR); InsP3 was from L.C. Services (Woburn, MA). When DMSO or ethanol were used as a solvent, their final concentration never exceeded 0.01 or 0.1%, respectively. Where applicable, data are expressed as means ± SEM, with n equal to the number of experimental runs.
Cell Permeabilization
As described previously (Scheenen et al., 1998; Hofer et al., 1995), dye-loaded cells were rinsed briefly in a high K+ solution (in mM: 125 KCl, 25 NaCl, 10 Hepes, pH 7.25, 0.1 MgCl2), and then exposed for 2–3 min to an intracellular buffer at 37°C (the same solution supplemented with 0.5 mM MgATP, pH 7.25, and Ca2+/EGTA buffers, 0.1 total [EGTA], 200 nM free Ca2+, calculated according to the computer program described in Bers et al., 1994) also containing 5 mg/ml digitonin. After plasma membrane permeabilization, cells were continuously superfused with intracellular buffer (without digitonin). Imaging measurements were performed as above for intact cells.
Results
Mitochondria are known to take up Ca2+ released into the cytoplasm during agonist stimulation via an electrogenic Ca2+ uniporter, a process dependent on the membrane potential across the inner membrane (Rizzuto et al. 1992, 1993; Pozzan et al., 1994). Carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (FCCP)1 is a protonophore that uncouples mitochondrial respiration and ATP production by dissipating the proton gradient across the inner mitochondrial membrane. As it abolishes the membrane potential normally maintained by oxidative phosphorylation, FCCP also completely prevents mitochondrial Ca2+ uptake (Gunter and Pfeiffer, 1990; Gunter et al., 1994; Rizzuto et al., 1994).
Mitochondrial Inhibitors Impede Ca2+ Uptake into Internal Stores by at Least Two Mechanisms
We tested the effects of FCCP and other mitochondrial inhibitors on Ca2+ handling by the ER of BHK-21 cells loaded with the AM-ester of the fluorescent Ca2+ dye mag-fura-2 (Fig. 1). We have shown previously in this cell type that prolonged loading with this indicator results in preferential accumulation of dye into organelles (Hofer et al., 1998; see also Golovina and Blaustein, 1997). The low affinity of the dye for Ca2+ allows for sensitive measurements almost exclusively in organelles where Ca2+ is high, i.e., the ER (Hofer and Schulz, 1996; Hofer et al., 1995). Adding 1 μM of FCCP to resting cells (in the presence of glucose) frequently caused a slow, persistent, and reversible decrease in the ratio (Fig. 1 A; n = 8 out of 13 cells) comparable to that elicited by treatments that block SERCA-mediated Ca2+ uptake (Hofer et al., 1995; Hofer et al., 1996). A similar, although poorly reversible decrease in the ratio was observed upon acute treatment with 1 μM oligomycin, which blocks the ATP synthetase responsible for coupling the proton gradient to ATP production (n = 3; not shown).
Figure 1.
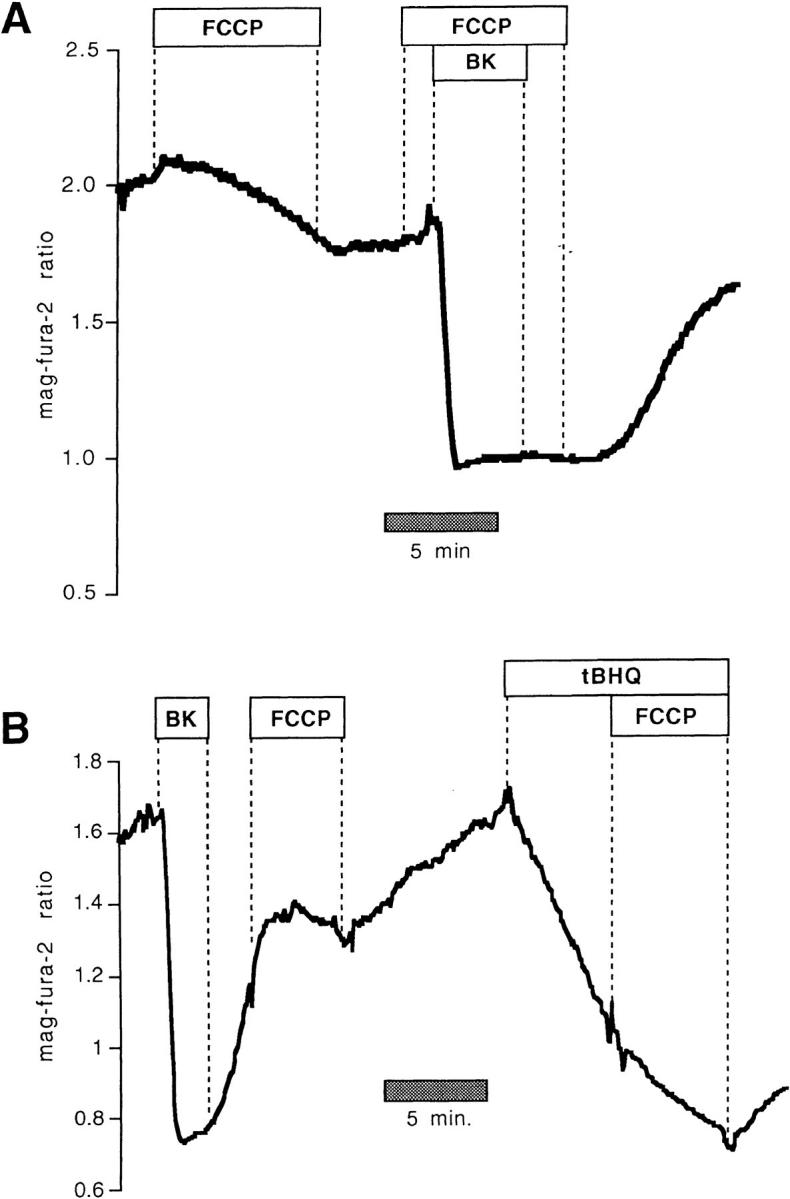
FCCP inhibits ER Ca2+ uptake. Mag-fura-2 measurements of [Ca2+] in agonist-sensitive internal stores in intact cells. (A) Resting intact cell treated with FCCP (1 μM) followed by FCCP 1 BK (100 nM). (B) FCCP administered during store refilling subsequent to BK-induced release inhibited Ca2+ sequestration, but had little effect on the mag-fura-2 ratio when given after treatment with a SERCA blocker, tBHQ (10 μM).
As further illustrated by Fig. 1 A, when intact cells were stimulated in the presence of FCCP with 100 nM bradykinin (BK) to release stored Ca2+, the rapid recovery of Ca2+ that was normally seen when the agonist was removed (see for example Fig. 3) was not observed until FCCP was washed out. Adding FCCP during the recovery phase in BK-stimulated cells prevented store refilling (Fig. 1 B). When SERCA activity was abolished with 2, 5-Di(tert-butyl)hydroquinone (tBHQ), however, (resulting in a relatively slow leak of Ca2+ out of the store) FCCP did not further reduce the mag-fura-2 ratio, showing that tBHQ and FCCP act on the same Ca2+ pool (Fig. 1 B; n = 6). In fact, FCCP caused a slight reversal of the downward trend in the signal, a result we attribute to effects of ATP depletion on the passive Ca2+ leak of internal Ca2+ stores (Hofer et al. 1996).
Figure 3.




FCCP and oligomycin impede store refilling after BK-induced Ca2+ release in intact mag-fura-2-loaded cells. (A) Control recovery and response in the same cell to hormone after brief pretreatment with FCCP. (Inset) Entire experimental record. (Bottom) Enlarged overlay emphasizing the recovery phases with and without FCCP. (B) As above, profile of store reloading before and after treatment with 1 μM oligomycin. (C) Comparison of store refilling after three separate stimulations with 100 nM BK: first in the presence of 1 μM FCCP in Ca2+-containing medium; second after a 15-min incubation with 40 μM BAPTA-AM (added at arrow); cells were washed with normal Ringer's for 5 min, and were then stimulated with BK in Ca2+-free medium containing 1 μM FCCP. Finally, cells were stimulated in the absence of FCCP, still in Ca2+-free external medium. (D) Cells were loaded with 40 μM BAPTA-AM for 15 min before the start of experiment. (Inset) Responses to 100 nM BK in Ca2+-free medium before and after treatment with 1 μM oligomycin. (Bottom) Enlarged overlay of intraluminal [Ca2+] changes, oligomycin vs. control.
Collectively, the data of Fig. 1 strongly suggest that uncoupling mitochondria grossly affects Ca2+ uptake into the ER. However, mitochondrial inhibitors, particularly protonophores such as FCCP, are subject to several nonspecific effects (e.g., membrane potential and pH changes) that could influence our measurements. It should be stressed that in previous experiments on permeabilized BHK-21 cells where [ATP] in the superfusion was kept constant at 3 mM, there was no effect of 1 μM FCCP on the mag-fura-2 ratio (Hofer et al., 1995), nor was there any consequence of adding ruthenium red, valinomycin, or oligomycin + azide (Hofer et al., 1995), agents that either prevent mitochondrial Ca2+ uptake and/or induce Ca2+ release from that organelle. Thus, these drugs have no effect per se on the fluorescence of mag-fura-2, and do not cause release of Ca2+ from mitochondria that can be detected by this low-affinity fluorophore in permeabilized BHK-21 cells. Furthermore, as shown in Fig. 2, InsP3-induced release and reloading of internal stores of permeabilized cells was unaffected by FCCP. The response to a supramaximal dose of InsP3 (6 μM) was similar before and after treatment with 1 μM FCCP under conditions where [Ca2+] and [ATP] of the intracellular-like buffer were clamped to 170 nM and 0.5 mM, respectively. When FCCP was given to cells during the recovery phase after InsP3 washout, there was no consequence on the mag-fura-2 ratio (Fig. 2).
Figure 2.

Effects of FCCP on Ca2+ homeostasis in internal stores of digitonin-permeabilized mag-fura-2-loaded BHK-21 cells. Cells were continuously superfused with intracellular buffer. [Ca2+] was clamped with EGTA buffers to 170 nM, and a constant [ATP] of 0.5 mM was maintained. FCCP had no effect on the mag-fura-2 ratio or InsP3-induced release and reloading of internal stores under these conditions.
In contrast to the data of Fig. 2, measurements of store [Ca2+] in intact cells revealed that metabolic poisons dramatically prolonged the time course of store refilling (Fig. 3). Direct comparison of the rates of recovery after BK stimulation between control and FCCP-treated cells (Fig. 3 A) showed that refilling rates were on average only 18.3 ± 2.5% of control values in the same cell (n = 23), with some variability at the single-cell level. Similar experiments using oligomycin to block ATP synthesis revealed that the inhibitor reduced the rate of recovery to 25 ± 4% of control (Fig. 3 B). Comparable results were obtained using 4 μM rotenone (not shown; blocks oxidative phosphorylation at complex I; n = 3), oligomycin plus rotenone (n = 3), or combinations of rotenone, oligomycin, and 1 μM atractiloside (inhibits ATP/ADP exchange across the inner mitochondrial membrane; n = 4). Taken together, these data indicate that a specific effect on mitochondrial function accounts for the inhibition of store refilling in the intact cell.
How do mitochondrial inhibitors impede internal store refilling? While the simplest explanation for the effects of such pharmacologically diverse compounds as FCCP, oligomycin, rotenone, and atractiloside in the intact cell (Figs. 1 and 3) is that the energy supply for Ca2+ uptake had been rapidly compromised upon mitochondrial poisoning, another important consideration is the effect of these agents on capacitative Ca2+ entry at the plasma membrane. Since store refilling depends strictly on Ca2+ supplied from the extracellular milieu in many cell types, including BHK-21 cells (Hofer et al., 1998), inhibition of this pathway will have important consequences on Ca2+ reuptake. To bypass the requirement for Ca2+ entry, we took advantage of a phenomenon that we have described recently, permitting internal store refilling in Ca2+-free external medium, provided that the cells have been preloaded with BAPTA-AM (Hofer et al., 1998). As seen in Fig. 3 C (typical of n = 14), the rate of recovery in the presence of FCCP after agonist washout was considerably faster in the Ca2+-free solution/BAPTA-AM pretreated condition, where Ca2+ entry pathways had been bypassed (FCCP alone was 71 ± 7% of the BAPTA/FCCP recovery rate). However, the rate of refilling was even faster in zero Ca2+/BAPTA conditions in the absence of FCCP (147 ± 6% of the FCCP + BAPTA recovery rate).
A similar protocol using BAPTA preloading to circumvent the plasma membrane Ca2+ entry pathway showed that oligomycin reduced the rate of recovery in BAPTA/0 Ca2+ to 47 ± 7% of that of the control (n = 19; Fig. 3 D). It should be noted that somewhat higher concentrations of oligomycin have been reported to inhibit store-operated channels by a means independent of cellular ATP depletion (Cho et al., 1997).
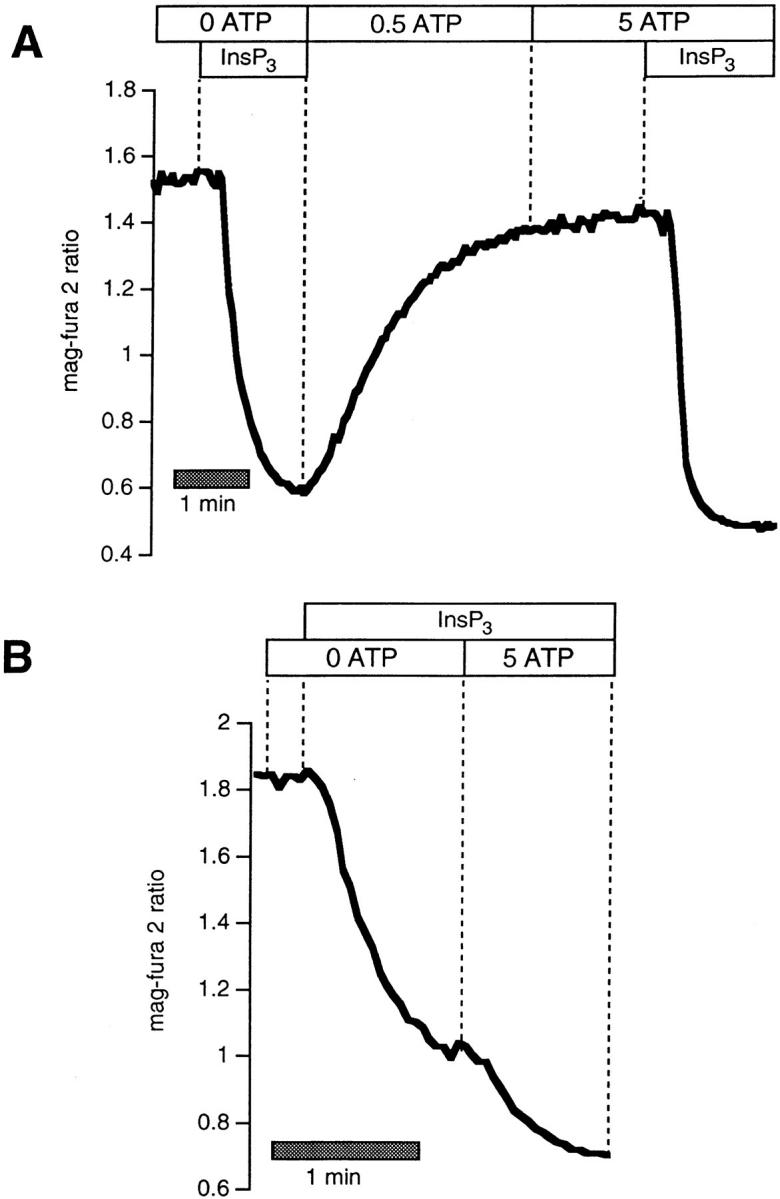
These data imply that a major action of mitochondrial inhibitors on store refilling operates via an indirect mechanism as a consequence of inhibition of capacitative Ca2+ entry pathways at the plasma membrane. However, Fig. 3, C and D also indicates that mitochondrial ATP production (or alterations in the ATP/ADP ratio) likely influence Ca2+ reaccumulation by the ER to a significant degree. This result was quite surprising, as the available literature suggests that rather dramatic reductions in [ATP] would be required to block SERCA-mediated Ca2+ uptake (see Discussion). We therefore checked this point in permeabilized BHK-21 cells by comparing the rates of Ca2+ reaccumulation after InsP3-induced release while clamping [ATP] (and [ADP]) in the perfusate to various levels. As shown in Fig. 4 A, rapid recovery was observed even when [ATP] in the bath was reduced to 50 μM. Stores reaccumulated Ca2+ at near maximal speed with [ATP] fixed at 200 μM (not shown). Thus, profound cellular [ATP] depletions are necessary to impede Ca2+ reuptake.
Figure 4.

(A) Effects of changing [ATP] on rates of refilling in digitonin-permeabilized BHK-21 cells loaded with mag-fura-2. Rates of refilling after stimulation with 6 μM InsP3 in the presence of 50 μM ATP is compared with that in 0.5 mM. Maximal recovery velocity was obtained when [ATP] was 200 μM (not shown). (B) Rates of recovery after release with 6 μM InsP3 were barely affected by adding 0.5 mM ADP during the recovery phase. (C) 4 mM ADP (in the presence of 0.5 mM ATP) blocked refilling.
A further consideration, however, is that metabolic inhibition may alter not only [ATP], but also the ATP/ADP ratio (which is the true index of the driving force for Ca2+ reaccumulation). There was very little effect on the recovery when the ATP/ADP ratio was set to one (Fig. 4 B; 0.5 mM ATP, and 0.5 mM ADP or 0.05 ATP and 0.05 ADP; n = 6). In contrast, the recovery after InsP3-induced Ca2+ release was almost completely blocked when [ATP] was 0.5 mM and [ADP] was 4 mM (n = 4; Fig. 4 C).
Mitochondrial Inhibitors Impede Ca2+ Release from Internal Stores by at Least Two Mechanisms
In addition to interfering with the refilling process, mitochondrial inhibitors also affect the kinetics of Ca2+ release in intact cells. These results can be better appreciated when the time scale of the data presented in the inset of Fig. 3 is expanded, as shown in Fig. 5.
Figure 5.

Effects of mitochondrial inhibitors on rates of release in intact mag-fura-2–loaded cells. Data are taken from the records shown in Fig. 3, and are represented on an expanded time scale to allow better appreciation of the release kinetics. (A) FCCP vs. control, (B) oligomycin vs. control, (C) as in Fig. 3 C, comparison in the same cell between rates of release: (a) in the presence of 1 μM FCCP (FCCP); (b) after a 15-min incubation with 40 μM BAPTA-AM, after which cells were washed with normal Ringer's for 5 min and then stimulated with BK in Ca2+-free medium containing 1 μM FCCP (BAPTA/ FCCP); (c) cells stimulated in the absence of FCCP, still in Ca2+-free external medium (BAPTA control). (D) Data from Fig. 3 D: rates of release after BAPTA-AM pretreatment in the presence and absence of oligomycin.
As seen in Fig. 5 A after a 1-min pretreatment with FCCP, cells were stimulated briefly (to minimize receptor desensitization), consistently resulting in a marked slowing of the rate and magnitude of Ca2+ release compared with the control BK response in the same cell (rate 51.6 + 4.5% of control; n = 23). This result was observed irrespective of whether the control stimulation was performed before or after the stimulation plus FCCP. On the other hand, the rates of release for two successive control challenges with BK were not significantly different from one another (not shown; n = 16; see also additional data in Hofer et al., 1998).
Using the same experimental protocol, oligomycin was also shown to alter the kinetics of Ca2+ release (Fig. 5 B), although to a lesser extent (73 ± 4% of control, n = 23) than FCCP. Direct comparison in the same cells further confirmed that the uncoupler was more effective in slowing the release of Ca2+ than was oligomycin (the rate in FCCP was 68 ± 6% of that in oligomycin; n = 22).
The question then arises as to whether the effects of FCCP or oligomycin in intact cells are due to impairment of ATP production, inhibition of Ca2+ uptake by mitochondria, or both. It should be stressed that oligomycin does not block Ca2+ uptake by mitochondria, but only inhibits ATP synthesis. Thus, the most plausible explanation for inhibition of Ca2+ release by oligomycin alone is that in the absence of mitochondrial ATP production, the release channels in the store become susceptible to reductions in [ATP] (possibly in a localized domain), and to an extent that can compromise the Ca2+ release process. The fact that FCCP was more effective in slowing the discharge from internal stores (Fig. 3, A and C) could therefore be explained by the fact that FCCP produces a more profound depletion of cytoplasmic [ATP] than does oligomycin (see for example Budd and Nicholls, 1996). Alternatively, there may be an additional effect of FCCP because of the failure of mitochondria to buffer cytoplasmic Ca2+ changes near the InsP3 receptor, resulting in a reduced rate of Ca2+ release from the stores. To distinguish between these possibilities, we compared the rates of release after FCCP alone with those after treatment with a combination of FCCP and oligomycin (not shown). The rationale for these experiments is as follows: with oligomycin present, the ATP consumption by the mitochondrial F0-F1 ATPase that normally occurs upon FCCP addition will be prevented so that the degree of ATP depletion with the combination of FCCP and oligomycin will be the same as with oligomycin alone. Therefore, if the effect that we observe on Ca2+ release is due only to ATP depletion, then FCCP alone should slow down the response to BK much more than the combination of FCCP and oligomycin. However, we observed that the rate of Ca2+ release under these two conditions was essentially identical in paired experiments conducted on the same cells (n = 44 cells). This result indirectly suggests that Ca2+ uptake by mitochondria (abolished by FCCP) also plays a role in modulating Ca2+ release from the stores.
We next undertook a series of experiments to more directly assess the role of mitochondrial Ca2+ buffering in shaping the profile of Ca2+ discharge by comparing the rates of release in the presence of FCCP before and after loading of the cytoplasm with the rapid Ca2+ buffer BAPTA-AM. The exogenous Ca2+ buffer should prevent local increases in [Ca2+]cyt near the InsP3 receptor, in effect rescuing the physiological Ca2+-buffering activity that we predict to be furnished by the mitochondria. Fig. 5 C shows that the inhibitory effect of FCCP on BK-induced Ca2+ release was largely overcome when the same cells were loaded on the microscope stage for 15 min with 40 μM BAPTA-AM. The rates of release for the first stimulation in the presence of FCCP alone were 59 ± 7% (n = 51) of those with FCCP and BAPTA. This rate was in turn generally slightly slower than the BK-induced Ca2+ release measured in the BAPTA-loaded cells in the absence of FCCP, and as illustrated by Fig. 5 C, with an altered kinetic profile.
Fig. 5 D shows that the rates of release were also attenuated by oligomycin under these conditions; noteworthy were the kinetic differences between the two conditions.
The results of Fig. 5 C attest to the importance of mitochondrial Ca2+ buffering in modulating the kinetics of ER Ca2+ discharge. However, the data of Fig. 5, B and D also strongly suggest that [ATP] depletion alters the dynamics of Ca2+ mobilization, possibly through a direct interaction with the InsP3 receptor. We again checked this point in permeabilized cells. Alterations in [ATP] over a wide range of concentrations had clear effects on the kinetics of Ca2+ release. As shown in Figs. 4 and 6 A, while the initial rates of release were somewhat slower when [ATP] was reduced from 5 to 0.5 to 0 mM, the most striking action was on the later phases of the response (n = 18). The drop in luminal [Ca2+] was consistently less abrupt (less quantal; Meyer and Stryer, 1990) as [ATP] was lowered, as further illustrated by Fig. 6 B. Our results in BHK-21 cells differ in part from those of Bezprozvanny and Ehrlich (1993) regarding the type I InsP3 receptor, which was found to have a reduced open probability at [ATP] above 4 mM. No effect of ADP (used at concentrations of 0.5 to 4 mM) was seen (not shown; n = 6). These results in permeabilized cells confirm that alterations in intracellular [ATP] can potentially influence the profile of Ca2+ release after agonist activation.
Figure 6.
ATP modulates InsP3-induced Ca2+ release in digitonin-permeablized cells. (A) Comparison of two sequential stimulations with 6 μM InsP3 in the absence of ATP and in the presence of 5 mM ATP. (B) Increasing [ATP] to 5 mM during the release phase enhances the rate of InsP3-induced Ca2+ discharge.
Discussion
A large literature exists concerning the effects of mitochondrial inhibitors on Ca2+ homeostasis in intact cells (see for example Mohr and Fewtrell, 1990; Babcock et al., 1997; Drummond and Fay, 1996; Friel and Tsien, 1994). These studies relied principally on measurements of Ca2+ changes in the cytoplasm (for example, using fluorescent indicators), or used 45Ca2+ to make population measurements of the total cellular Ca2+ content. While some investigators concluded that poisoning of mitochondrial metabolism could compromise Ca2+ pumping by the SERCAs (resulting in loss of Ca2+ from the ER; Fasolato et al., 1991; al-Baldawi et al., 1993), many other studies have promoted the idea that the mitochondria themselves serve as a large reservoir of Ca2+ that can be released into the cytoplasm after treatment with inhibitors (Babcock et al., 1979; Fulceri et al., 1991).
Here we have reexamined this complex issue by measuring Ca2+ in the ER directly using a low-affinity fluorescent indicator in intact cells. Our results show that brief pretreatment with a wide range of mitochondrial inhibitors (atractiloside, rotenone, oligomycin, and FCCP) all resulted in a dramatic inhibition in store refilling after release of stored Ca2+ with agonists (Fig. 2). In addition, FCCP or oligomycin added acutely to resting cells frequently caused a slow but measurable loss of stored Ca2+ that cannot be attributed to release of the cation from the mitochondria themselves, as the effect was not additive to that obtained by blocking SERCAs with tBHQ. Furthermore, the lack of effect of FCCP in the permeabilized cell preparation, where the [Ca2+] (buffered by EGTA) and the [ATP] of the extraorganellar milieu are fixed (Fig. 2), are consistent with the idea that physiological interactions between mitochondria and other cellular components (the ER and plasma membrane) account for the actions of these drugs in the intact cell.
The data of Fig. 3 C, in which stores were refilled in the intact BAPTA-loaded cells in the absence of external Ca2+, indicate that a significant mechanism by which mitochondrial inhibitors block store refilling is through an indirect effect on plasma membrane Ca2+ entry pathways. Consistent with this idea is the finding that FCCP significantly inhibited the plateau phase of Ca2+ entry as measured by fura-2 (data not shown; see also Hoth et al., 1997; Marriot and Mason, 1995; Gamberucci et al., 1994). These data illustrate that the entry pathway was potently inhibited in spite of the fact that stores remained relatively empty after treatment with the uncoupler (Fig. 3 A).
The results of Fig. 3, C and D also show, however, that mitochondrial inhibitors influence Ca2+ reuptake by an additional mechanism besides that at the plasma membrane, as the rate of refilling in BAPTA-pretreated cells was significantly slower in the presence of oligomycin or FCCP compared with the control condition. We attribute this difference to effects on mitochondrial ATP production. Interpreted in this way, however, these results are somewhat surprising. In the presence of glucose in the bathing medium (such as was the case here), mitochondrial inhibitors are known to cause only a relatively slow and partial (50%) depletion of intracellular ATP in BHK-21 cells (Burkhardt and Argon, 1989), and in a variety of other cell types as well (see for example Mohr and Fewtrell, 1990; Fulceri et al., 1991; Luo et al., 1997). Meanwhile, SERCA pumps, like other ion-motive ATPases, are known to function even when [ATP] falls to quite low levels. In fact, our data from permeabilized BHK-21 cells (Fig. 6) show that stores are able to recover Ca2+ at maximal speed when the [ATP] in the bath is clamped to just 200 μM. Reducing the [ATP] in the bath to 50 μM only slowed the rate of refilling to ∼65% (n = 37 cells), and still allowed complete recovery. Our data may therefore suggest that local ATP depletions (and/or extreme local alterations in ATP/ADP) near the SERCAs that result from the increased pumping activity after agonist activation occur before global changes in cellular [ATP] (or the ATP/ ADP ratio) are evident, making the Ca2+ uptake process more susceptible to mitochondrial inhibition than might be expected from measurements of total cellular adenine nucleotide content. A wide variety of morphological evidence showing close physical apposition between mitochondria and ER lends support to this idea (McGraw et al., 1980; Nixon et al., 1994; Takei et al., 1992; Rizeuto et al., 1998).
Ca2+ buffers have well-established effects on the dynamics of Ca2+ release from internal stores (Hajnóczky and Thomas, 1997; Montero et al., 1997). A number of other studies have stressed the importance of mitochondrial Ca2+ uptake and release in modulating cytosolic Ca2+ signals (Mohr and Fewtrell, 1990; Babcock et al., 1997; Drummond and Fay, 1996; Loew et al., 1994; Jouaville et al., 1995; Simpson and Russel, 1996; Hoth et al., 1997), and our direct measurements of intraluminal ER Ca2+ dynamics (Fig. 5, A and C) also support this view. In addition, however, our data using oligomycin alone to inhibit ATP synthesis also indicates that local ATP availability can subtly alter the responsiveness of stores to agonists. This effect is most readily explained by the ATP dependence of the InsP3 receptor (Smith et al., 1985; Ferris et al., 1990; Bezprozvanny and Ehrlich, 1993), although the possibility that ATP depletion can impede InsP3 production must also be seriously considered. The data of Figs. 4 and 6 show that altering [ATP] in the intracellular buffer in permeabilized BHK-21 cells had clear effects on the kinetics of InsP3-induced release, which were largely compatible with the observed effects of mitochondrial inhibitors on ER [Ca2+] in intact cells. Again, it is possible that local [ATP] changes may be responsible for the observed differences.
In any event, our results clearly demonstrate that active mitochondrial respiration modulates the dynamics of Ca2+ release, allowing for explosive efflux of Ca2+ from internal stores. How these kinetic differences influence cytosolic or intraluminal Ca2+-binding effectors, thereby influencing the signal transduction potential of intracellular [Ca2+] changes, remains to be established. These findings prompt the interesting possibility that subtle alterations in the release dynamics may somehow provide a signal to the rest of the cell, conveying information about the cellular metabolic status.
Acknowledgments
The authors thank Dr. Paolo Pinton for help with some preliminary measurements, and Dr. Rosario Rizzuto for helpful comments on the manuscript. B. Landolfi was a recipient of a fellowship from the E.E.C.
This work was supported by a grant from Ministero Universitá Ricerche Scientifica e Tecnologica (40% fund) to S. Curci and by grants to T. Pozzan from Telethon (N845), Consiglio Nazionale delle Ricerche Biotechnology, Human Science Frontiers, and the Armenise-Harvard Foundation. A.M. Hofer was supported by Human Science Frontiers.
Abbreviations used in this paper
- BK
bradykinin
- FCCP
carbonyl cyanide p-trifluoromethoxy-phenylhydrazone
- InsP3
inositol 1,4,5-trisphosphate
- tBHQ
2, 5-Di(tert-butyl)hydroquinone
Footnotes
Address all correspondence to Aldebaran M. Hofer, University of Padova, CNR Center for Biomembranes, Viale G. Colombo 3, I-35121 Padova, Italy. Tel.: +39-49-827-6065. Fax: +39-80-5443388. E-mail: wim@civ.bio.unipd.it
References
- al-Baldawi NF, Moore JE, Abercrombie RF. Calcium accumulation by organelles within Myxicola axoplasm. J Physiol. 1993;461:633–646. doi: 10.1113/jphysiol.1993.sp019533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+network. J Cell Biol. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DF, Chen JL, Yip BP, Lardy HA. Evidence for mitochondrial localization of the hormone-responsive pool of Ca2+in isolated hepatocytes. J Biol Chem. 1979;254:8117–8120. [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signaling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca2+buffers. Methods Cell Biol. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Ehrlich BE. The inositol 1,4,5-trisphosphate (InsP3) receptor. J Membr Biol. 1995;145:205–216. doi: 10.1007/BF00232713. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Ehrlich BE. ATP modulates the function of inositol 1,4,5-trisphosphate-gated channels at two sites. Neuron. 1993;10:1175–1184. doi: 10.1016/0896-6273(93)90065-y. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+homoestasis. J Neurochem. 1996;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, Argon Y. Intracellular transport of VSV is inhibited by CCCP at a late stage of post-translational processing. J Cell Sci. 1989;92:633–642. doi: 10.1242/jcs.92.4.633. [DOI] [PubMed] [Google Scholar]
- Cho JH, Balasubramanyam M, Chernaya G, Gardner JP, Aviv A, Reeves JP, Dargis PG, Christian EP. Oligomycin inhibits store-operated channels by a mechanism independent of its effects on mitochondrial ATP. Biochem J. 1997;324:971–980. doi: 10.1042/bj3240971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Denton RM, McCormack JG. Ca2+as second messenger within the mitochondria of the heart and other tissues. Annu Rev Physiol. 1990;52:451–466. doi: 10.1146/annurev.ph.52.030190.002315. [DOI] [PubMed] [Google Scholar]
- Drummond RM, Fay FS. Mitochondria contribute to Ca2+removal in smooth muscle cells. Pflügers Arch Eur J Physiol. 1996;431:473–482. doi: 10.1007/BF02191893. [DOI] [PubMed] [Google Scholar]
- Fasolato C, Zottini M, Clementi E, Zacchetti D, Meldolesi J, Pozzan T. Intracellular Ca2+pools in PC12 cells. J Biol Chem. 1991;266:20159–20167. [PubMed] [Google Scholar]
- Ferris CD, Huganir RL, Snyder SH. Calcium flux mediated by purified inositol 1,4,5-trisphosphate receptor in reconstituted lipid vesicles is allosterically regulated by adenine nucleotides. Proc Natl Acad Sci USA. 1990;87:2147–2151. doi: 10.1073/pnas.87.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch EA, Turner TJ, Goldin SM. Calcium as a co-agonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i . J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulceri R, Bellomo G, Mirabelli F, Gamberucci A, Benedetti A. Measurement of mitochondrial and non-mitochondrial Ca2+in isolated intact hepatocytes: a critical re-evaluation of the use of mitochondrial inhibitors. Cell Calcium. 1991;12:431–439. doi: 10.1016/0143-4160(91)90069-q. [DOI] [PubMed] [Google Scholar]
- Gamberucci A, Innocenti B, Fulceri R, Banhegyi G, Giunti R, Pozzan T, Benedetti A. Modulation of Ca2+influx dependent on store depletion by intracellular adenine-guanine nucleotide levels. J Biol Chem. 1994;269:23597–23602. [PubMed] [Google Scholar]
- Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–1648. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Thomas AP. Minimal requirements for calcium oscillations driven by the IP3 receptor. EMBO (Eur Mol Biol Organ) J. 1997;16:3533–3543. doi: 10.1093/emboj/16.12.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer AM, Machen TE. Technique for in situ measurement of calcium in intracellular InsP3-sensitive stores using the fluorescent indicator mag-fura-2. Proc Natl Acad Sci USA. 1993;90:2598–2602. doi: 10.1073/pnas.90.7.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer AM, Schlue W-R, Curci S, Machen TE. Spatial distribution and quantitation of free luminal [Ca2+] within the InsP3-sensitive internal store of individual BHK-21 cells. Ion dependence of InsP3-induced Ca2+release and reloading. FASEB J. 1995;9:788–798. doi: 10.1096/fasebj.9.9.7601343. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Curci S, Machen TE, Schulz I. ATP regulates the passive leak from agonist-sensitive internal calcium stores. FASEB J. 1996;10:302–303. doi: 10.1096/fasebj.10.2.8641563. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Schulz I. Quantification of intraluminal free [Ca2+] in the agonist-sensitive internal calcium store using compartmentalized fluorescent indicators: some considerations. Cell Calcium. 1996;20:235–242. doi: 10.1016/s0143-4160(96)90029-9. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Landolfi B, Debellis L, Pozzan T, Curci S. Free [Ca2+] dynamics measured in agonist-sensitive stores of single living intact cells: a new look at the refilling process. EMBO (Eur Mol Biol Organ) J. 1998;17:1986–1996. doi: 10.1093/emboj/17.7.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature. 1995;377:438–441. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Loew LM, Carrington W, Tuft RA, Fay FS. Physiological cytosolic Ca2+transients evoke concurrent mitochondrial depolarizations. Proc Natl Acad Sci USA. 1994;91:12579–12583. doi: 10.1073/pnas.91.26.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Bond JD, Ingram VM. Compromised mitochondrial function leads to increased cytosolic calcium and to activation of MAP kinases. Proc Natl Acad Sci USA. 1997;94:9705–9710. doi: 10.1073/pnas.94.18.9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriot I, Mason MJ. ATP depletion inhibits capacitative Ca2+entry in rat thymic lymphocytes. Am J Physiol. 1995;269:C766–C774. doi: 10.1152/ajpcell.1995.269.3.C766. [DOI] [PubMed] [Google Scholar]
- McGraw CF, Somlyo AV, Blaustein MP. Localization of calcium in presynaptic nerve terminals: an ultrastructure and electron microprobe analysis. J Cell Biol. 1980;85:228–241. doi: 10.1083/jcb.85.2.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Stryer L. Transient calcium release induced by successive increments of inositol 1,4,5-trisphosphate. Proc Natl Acad Sci USA. 1990;87:3841–3845. doi: 10.1073/pnas.87.10.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr CF, Fewtrell C. The effect of mitochondrial inhibitors on calcium homeostasis in tumor mast cells. Am J Physiol. 1990;258:C217–C226. doi: 10.1152/ajpcell.1990.258.2.C217. [DOI] [PubMed] [Google Scholar]
- Montero M, Barrero MJ, Alvarez J. [Ca2+] microdomains control agonist-induced Ca2+release in intact HeLa cells. FASEB J. 1997;11:881–885. doi: 10.1096/fasebj.11.11.9285486. [DOI] [PubMed] [Google Scholar]
- Negulescu PA, Machen TE. Intracellular ion activities and membrane transport in parietal cells measured with fluorescent dyes. Methods Enzymol. 1990;192:38–81. doi: 10.1016/0076-6879(90)92062-i. [DOI] [PubMed] [Google Scholar]
- Nixon GF, Mignery GA, Somlyo AV. Immunogold localization of inositol 1,4,5-trisphosphate receptors and characterization of ultrastructural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. J Muscle Res Cell Motil. 1994;15:682–700. doi: 10.1007/BF00121075. [DOI] [PubMed] [Google Scholar]
- Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- Raju B, Murphy E, Levy LA, Hall RD, London RE. A fluorescent indicator for measuring cytosolic free magnesium. Am J Physiol. 1989;256:C540–C548. doi: 10.1152/ajpcell.1989.256.3.C540. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Simpson AVM, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+revealed by specifically targeted recombinant aequorin. Nature. 1992;358:325–327. doi: 10.1038/358325a0. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains of high Ca2+close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+homeostasis in intact cells. J Cell Biol. 1994;126:1183–1194. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Rutter GA, Burnett P, Rizzuto R, Brini M, Murgia M, Pozzan T, Tavaré JM, Denton RM. Subcellular imaging of intramitochondrial Ca2+with recombinant targeted aequorin: significance for the regulation of pyruvate dehydrogenase activity. Proc Natl Acad Sci USA. 1996;93:5489–5494. doi: 10.1073/pnas.93.11.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheenen, W.J.J.M., A.M. Hofer, and T. Pozzan, 1998. Intracellular measurement of calcium using fluorescent probes. In Cell Biology: A Laboratory Handbook, J.E. Celis, editor. Academic Press, Inc., New York. 363–374.
- Simpson PB, Russel JT. Mitochondria support inositol 1,4,5-trisphosphate-mediated waves in cultured oligodendrocytes. J Biol Chem. 1996;271:33493–33501. doi: 10.1074/jbc.271.52.33493. [DOI] [PubMed] [Google Scholar]
- Smith JB, Smith L, Higgins BL. Temperature and nucleotide dependence of calcium release by myo-inositol 1,4,5-trisphosphate in cultured vascular smooth muscle cells. J Biol Chem. 1985;260:14413–14416. [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu SS, Gunter TE. Mitochondrial calcium uptake from physiological pulses of calcium. A description of the rapid uptake mode. J Biol Chem. 1995;270:27510–27515. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- Takei K, Stukenbrok H, Metcalf A, Mignery GA, Sudhof TC, Volpe P, DeCamilli P. Ca2+ stores in Purkinje neurons: endoplasmic reticulum subcompartments demonstrated by the heterogeneous distribution of the InsP3 receptor, Ca2+-ATPase, and calsequestrin. J Neurosci. 1992;12:489–505. doi: 10.1523/JNEUROSCI.12-02-00489.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]