

Abstract
Previously we found that α3β1 integrin–deficient neonatal mice develop micro-blisters at the epidermal–dermal junction. These micro-blisters were associated with poor basement membrane organization. In the present study we have investigated the effect of α3β1-deficiency on other keratinocyte integrins, actin-associated proteins and F-actin organization. We show that the absence of α3β1 results in an increase in stress fiber formation in keratinocytes grown in culture and at the basal face of the basal keratinocytes of α3-null epidermis. Moreover, we see a higher concentration of actin-associated proteins such as vinculin, talin, and α-actinin at focal contact sites in the α3-deficient keratinocytes. These changes in focal contact composition were not due to a change in steady-state levels of these proteins, but rather to reorganization due to α3β1 deficiency. Apart from the loss of α3β1 there is no change in expression of the other integrins expressed by the α3-null keratinocytes. However, in functional assays, α3β1 deficiency allows an increase in fibronectin and collagen type IV receptor activities. Thus, our findings provide evidence for a role of α3β1 in regulating stress fiber formation and as a trans-dominant inhibitor of the functions of the other integrins in mouse keratinocytes. These results have potential implications for the regulation of keratinocyte adhesion and migration during wound healing.
Keywords: α3β1 integrin, trans-dominant inhibition, keratinocyte, wound healing
The epidermis is composed of multiple layers of cells known as keratinocytes. In normal skin, keratinocyte proliferation is restricted to the basal layer of the epidermis and cells undergo differentiation as they migrate up through the suprabasal layers. Basal keratinocytes adhere to the underlying basement membrane via the extracellular matrix receptors known as integrins, a family of heterodimeric glycoproteins each composed of an α- and a β-subunit. Heterodimer composition confers extracellular matrix ligand specificity (Hynes, 1992). Keratinocytes express a subset of integrins. These include: α2β1, a receptor for collagen; α3β1, a receptor for laminin 5; α5β1, a fibronectin receptor; αvβ5, known to be a receptor for various substrates including vitronectin and fibronectin; α6β4, another laminin-5 receptor known to be important in hemidesmosome formation (for review see Watt and Hertle, 1994); and α9β1, a tenascin receptor (Palmer et al., 1993).
Cultured keratinocytes adhere to substrates via at least two structures: focal contacts and stable anchoring contacts (SACs),1 the in vitro “equivalent” of hemidesmosomes (Carter et al., 1990b ; Stepp et al., 1990). Focal contacts are defined as clusters of integrins whose extracellular domains adhere to the extracellular matrix and whose cytoplasmic tails are linked to a concentration of actin-associated proteins that in turn link to actin filament bundles (Burridge et al., 1988). The β1 and αv integrins link the extracellular matrix outside the cell with the actin cytoskeleton inside the cell via a network of focal contact-associated proteins including talin, vinculin, and α-actinin (Otey et al., 1993; Clark and Brugge, 1995; Johnson and Craig, 1995). On the other hand, hemidesmosomes can be defined as electron-dense units containing a high concentration of α6β4 integrins whose cytoplasmic tails associate with cytoplasmic plaque proteins that in turn link to keratin filaments (Carter et al., 1990b ; Borradori and Sonnenberg, 1996). So, although focal contacts and SACs are distinct structures on the keratinocyte surface both are essential for effective adhesion to a common ligand, laminin 5.
In addition to being a receptor for laminin 5, α3β1 has been implicated as a weak receptor for fibronectin (FN), collagen type IV (Coll IV), and laminin 1 (Gelson et al., 1988; Elices et al., 1991) and is recruited to focal contacts in cells cultured on these proteins (Grenz et al., 1993; DiPersio et al., 1995). Adhesion-blocking assays using antibodies specific for α3β1 have suggested a variety of functions for α3β1 (Wayner and Carter, 1987; Wayner et al., 1988; Carter et al., 1990a ). For example, it has been reported that function-blocking antibodies to α3β1 can inhibit long-term keratinocyte–substrate adhesion and cell– cell contact (Carter et al., 1990a ), and that blocking α3β1 function induces keratinocyte differentiation (Symington and Carter, 1995). It has also been shown that antibodies to α3β1 may increase migration on FN and Coll IV (Kim et al., 1992). Studies in vivo have indicated changes in α3β1 integrin expression patterns during wound healing (Hertle et al., 1992; Cavani et al., 1993), psoriasis (Hertle et al., 1992; Pellegrini et al., 1992), and tumorigenesis (Bartolazzi et al., 1994). Taken together, these data suggest multiple roles for α3β1.
In this report we have used α3 integrin–deficient mice to address directly the roles of α3β1 in keratinocytes. α3-deficient mice suffer kidney and lung abnormalities with basement defects in the kidney (Kreidberg et al., 1996). We have shown that the α3-deficient mice form epidermal–dermal blisters that are associated with basement membrane defects (DiPersio et al., 1997). Mice deficient in α6 (Georges-Labouesse et al., 1996) or β4 integrin subunits (Dowling et al., 1996; van der Neut et al., 1996) also display blistering at the epidermal–dermal junction and are models for the human blistering disease Junctional Epidermolysis Bullosa with pyloric atresia (Niessen et al., 1996; Ruzzi et al., 1997). These mice have normal basement membranes, but hemidesmosomes are absent with alterations in associated keratin cytoskeletal morphology. Since α3β1 links to the actin cytoskeleton, we hypothesized that there might be alterations in the actin filament morphology in the α3-deficient mice.
We have analyzed the skin and isolated keratinocytes from the α3-deficient mice to explore further the roles of α3β1 in keratinocytes. Here we show that the absence of α3β1 results in a change in filamentous actin (F-actin) bundling both in keratinocytes grown in culture and in basal keratinocytes of α3-null epidermis. Moreover, we see a higher concentration of actin-associated proteins at focal contact sites in the α3-deficient keratinocytes. In addition, there is an increase in FN and Coll IV receptor activity in the α3-null keratinocytes. Our results suggest a role for α3β1 as a regulator of the actin cytoskeleton, focal contact composition, and as a trans-dominant inhibitor of fibronectin and collagen type IV integrin receptors in keratinocytes.
Materials and Methods
Antibodies
Rabbit sera against the cytoplasmic domains of human α5, β1, and chicken α3A integrin subunits were prepared as described (Marcantonio and Hynes, 1988; Hynes et al., 1989; DiPersio et al., 1995). Rabbit antiserum against α9 was a donation from Dr. D. Sheppard (University of California, San Francisco, CA) and the rabbit polyclonal antibodies to the cytoplasmic domain of α2 and α6 were gifts from Drs. M. Hemler (Dana-Farber, Boston, MA) and V. Quaranta (Scripps Research Institute, La Jolla, CA), respectively. Rabbit antiserum against the αv integrin subunit cytoplasmic domain was purchased from Chemicon International (Temecula, CA). Rat mAbs against mouse α5 and β1 extracellular domains were kind donations from Dr. B. Chan (University of Western Ontario, Canada), and rat mAb against mouse β4 was donated by S. Kennel (Oak Ridge National Laboratory, Oak Ridge, Tennessee). The rat anti–mouse α6 extracellular domain-specific mAb GoH3 was purchased from Immunotech (Westbrook, ME), and the mouse anti–human α3 mAb P1B5 was purchased from Gibco Laboratories, (Grand Island, NY).
The anti–mouse keratin 14 antibody was a donation from Dr. E. Fuchs (University of Chicago, Chicago, IL). The anti-actin rabbit antibody (751.1) was a generous donation from P. Matsudaira (Whitehead Institute, Massachusetts Institute of Technology, Cambridge, MA). Anti-vinculin and talin antibodies were obtained from Sigma Chemical Co. (St. Louis, MO) and the anti–α-actinin antibody was obtained from ICN Biochemicals (Costa Mesa, CA). All fluorescently conjugated secondary antibodies were obtained from TAGO BioSource (Camarillo, CA). The FITC-conjugated phalloidin was obtained from Sigma Chemical Co. (St. Louis, MO).
Extracellular Matrix Components
Purified human laminin 5 was a generous donation from Dr. R. Burgeson (Harvard/Massachusetts General Hospital, Charlestown, MA). Rat fibronectin was obtained from Gibco Laboratories and human collagen type IV and mouse laminin 1 were obtained from Collaborative Biomedical Products (Bedford, MA).
Preparation of Tissue for Histology
Animals were killed by CO2 narcosis and limbs removed. Tissue for paraffin embedding was fixed in 4% formaldehyde solution for 24 h at room temperature. Tissue for cryosectioning was snap frozen in OCT. Microscopy was carried out on a Zeiss Axiophot microscope (Carl Zeiss, Inc., Thornwood, NY).
Isolation and Culture of Primary Mouse Keratinocytes
Epidermal keratinocytes were prepared from neonatal mice essentially as described previously (Dlugosz et al., 1995). In brief, newborn mice were killed by CO2 narcosis, washed in 0.01 N iodine in PBS for 10 min, rinsed with PBS, washed in 70% ethanol for 10 min, and then rinsed in PBS. Tails were used for genotyping by PCR (as described in DiPersio et al., 1997), and limbs were prepared for frozen skin sections (see above). Skins were removed from the torso and head, and then floated on 0.25% trypsin solution (Gibco Laboratories) overnight at 4°C, with the epidermis facing upward. Skins were then transferred to a dry, sterile surface with the epidermis facing down, and the dermis was separated from the epidermis. The epidermis was minced, suspended in growth medium (see below), and then agitated to release keratinocytes. Suspensions were passed through a sterile, 70-μm nylon filter (Becton Dickinson, Franklin Lakes, NJ) to remove cornified sheets. Keratinocytes were seeded onto tissue culture plates coated with 30 μg/ml denatured rat tail collagen (Collagen Corp., Palo Alto, CA) at a density of ∼2–4 × 105 cells/cm2. To prevent differentiation of keratinocytes, cultures were grown in low calcium medium consisting of MEM (BioWhittaker, Walkersville, MD) supplemented with 4% FBS (Intergen Co., Purchase, NY) from which Ca2+ had been removed by chelation (Brennen et al., 1982), 0.05 mM CaCl2, “HICE” mix (5 μg/ml insulin [Sigma Chemical Co.], 0.5 μg/ml hydrocortisone [Calbiochem-Novabiochem Corp., La Jolla, CA], 10−10 M cholera toxin [ICN Biomedicals, Costa Mesa, CA], 10 ng/ml EGF [Upstate Biotechnology Inc., Lake Placid, NY], 2 × 10−9 M T3 [Sigma Chemical Co.]), 100 units/ ml penicillin, and 100 μg/ml streptomycin (Gibco Laboratories). Mouse keratinocytes were cultured at 33°C, 7.5% CO2.
Immunofluorescence Microscopy
Unfixed neonatal mouse limbs were embedded in OCT and snap-frozen in chilled isopentane in liquid nitrogen. The following procedure was used for the immunostaining of 8-μm cryosections. Sections were fixed either in 4% formaldehyde in PBS for 20 min at room temperature when staining for F-actin, α3, α6, or β4 integrin subunits or in 100% acetone for 10 min at −20°C when staining for β1, α5, or α9 integrin subunits. When staining for αv integrin subunits, sections were treated with 0.1% NP-40 in PBS for 10 min at room temperature, and then fixed in 4% paraformaldehyde (PFA) in PBS for 20 min at room temperature. All sections were blocked with 0.1% BSA, 0.2% Triton X-100 in PBS for 1 h before 1-h incubations with primary antibodies. Sections were washed in PBS and incubated with secondary antibodies at a 1:200 dilution in block solution. Finally, sections were washed in PBS, and then distilled water and mounted in Gelvatol containing the anti-bleaching agent, DABCO.
When staining for integrins at focal contacts, keratinocyte cultures were first cross-linked in 0.4 mM BS3 (Pierce Chemical Co., Rockford, IL) 10 min at room temperature, washed in 10 mM Tris-HCl, pH 7.4, in PBS, extracted in 0.5% NP-40 in PBS for 10 min and fixed in 4% PFA in PBS for 10 min before blocking and antibody incubations as described above (Enomoto-Iwamoto et al., 1993; DiPersio et al., 1995). To stain for filamentous actin, keratinocyte cultures were first permeabilized with 0.5% Triton X-100 in PBS for 5 min followed by 20 min in 4% formaldehyde in PBS before incubating in a 1:1,000 dilution of rhodamine-conjugated phalloidin. To stain keratinocyte cultures for cytoplasmic focal contact proteins, cells were fixed in 4% PFA for 20 min, permeabilized with 0.5% NP-40 for 10 min, and then blocked for 1 h before incubation with the appropriate antibodies.
Interference Reflection Microscopy
Keratinocytes were cultured on glass coverslips coated with denatured rat tail collagen (Vitrogen, Palo Alto, CA) and were photographed for interference reflection microscopy on a Zeiss Photo Microscope III (Carl Zeiss, Inc.).
Transfection of Primary Keratinocytes
An α3 expression plasmid was constructed by inserting an XbaI restriction fragment encompassing the cDNA for the human α3 integrin subunit (provided by M. Hemler, Dana Farber Cancer Institute, Boston, MA) into the XbaI site of the plasmid pcDNA3.1/Zeo(+) (Invitrogen Corporation, Carlsbad, CA). Primary keratinocytes were transfected using LipofectACE reagent according to the manufacturer's directions (Gibco Laboratories). In brief, 2 μg of the α3 expression plasmid was mixed with 12 μl of LipofectACE reagent (Gibco Laboratories) in 1 ml of Optimem and added to subconfluent cultures of keratinocytes on 35-mm plates. After a 6-h incubation at 33°C, the cells were fed with normal culture medium and grown for an additional 48 h, and then prepared for immunofluorescence as described above.
Adhesion Assays
96-well bacteriological plates (Nunc Inc., Naperville, IL) were coated with 10 μg/ml of fibronectin, collagen type IV, EHS laminin (laminin 1), or laminin 5 in PBS overnight at 4°C. The plates were washed briefly and then blocked in 10 mg/ml BSA in PBS for 2 h at 37°C. Cells were trypsinized, washed in MEM medium containing 10% serum, and then resuspended at 3 × 104 cells/100 μl of serum-free medium containing 25 μM cycloheximide. 3 × 104 cells were plated per well for 1 h at 37°C. Non-adherent cells were washed off in PBS and the remaining adherent cells were fixed and stained in 0.1% methylene blue in H2O and counted.
Phagokinetic Assays
Phagokinetic assays were carried out as described by Albrecht-Buehler (1977). In brief, 8-chamber Nunc slides were coated in BSA and colloidal gold (prepared from gold chloride from Sigma Chemical Co.). The colloidal gold was then coated in 50 μg/ml of either fibronectin, collagen type IV, EHS laminin, or laminin 5 overnight at 4°C. 103 cells were plated in serum-free medium overnight at 33°C (the temperature of mouse keratinocyte culture), fixed in 4% formaldehyde, and then viewed under dark field on an inverted Zeiss IM35 microscope. Using a Hamamatsu CCD camera linked to a Hamamatsu Argus 10 image processor (Hamamatsu Phototonics, Bridgewater, NJ) the area translocated per cell per hour was calculated.
FACS® Analysis of Surface Integrins
Keratinocytes were trypsinized, washed in medium containing serum, and then resuspended in PBS containing a 1:100 dilution of antibody for 30 min at 4°C. Cells were then washed three times in PBS and incubated with a suitable FITC-conjugated secondary antibody for 30 min at 4°C, washed three times in PBS and resuspended in PBS-containing propidium iodide, and then analyzed on a FACScan® from Becton Dickinson.
Surface Iodination and Immunoprecipitation of Integrins
Monolayers of mouse keratinocytes that had been passaged once were surface labeled with 1 mCi/10 cm plate of Na-[125I] (New England Nuclear, Boston, MA) using the lactoperoxidase-glucose oxidase method (Hynes, 1973). Cells were washed four times with 50 mM NaI in PBS/ Ca2+/Mg2+ and lysed for 15 min on ice in 1 ml of a detergent buffer containing 200 mM octyl-β-d-glucopyranoside (Calbiochem-Novabiochem Corp.), 50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 2 mM PMSF (Sigma Chemical Co.), 0.02 mg/ml aprotinin (Sigma Chemical Co.), and 0.0125 mg/ml leupeptin (Calbiochem-Novabiochem Corp.). Lysates were sedimented for 10 min at 10,000 g. Supernatants were preincubated with 100 μl of protein A–Sepharose (1:1 slurry; Pharmacia Biotech, Inc., Piscataway, NJ) for 1 h and the beads sedimented for 2 min at 10,000 g. Protein concentrations of supernatants were determined using a Bio-Rad kit. To compare the relative amounts of each integrin between wild-type and α3-null cells an equal amount of lysate was immunoprecipitated with each anti-integrin antibody as described (Marcantonio and Hynes, 1988). In brief, BSA was added to lysates (60–100 μg total protein) to a final concentration of ∼5 mg/ml, followed by 5–10 μl of antiserum. After incubation at 4°C for 1 h, 50 μl of protein A–Sepharose (1:1 slurry preabsorbed with 10 mg/ml BSA in lysis buffer) were added, and reactions were incubated overnight at 4°C. Samples were washed extensively with cold lysis buffer plus protease inhibitors, and then suspended in sample buffer (5% SDS, 80 mM Tris-HCl, pH 6.8, 2 mM EDTA, 10% glycerol and bromophenol blue) and boiled for 5 min. Non-reducing SDS-PAGE was performed by the method of Laemmli (1970) using 5% acrylamide and a 3% stacking gel.
Analysis of the Levels of F-Actin in Keratinocyte Cultures
Keratinocytes from wild-type or α3-null mice were grown to 75% confluency in 60-mm dishes as described above. Cells were stained for F-actin as described in “Immunofluorescence Microscopy.” The adherent cells were then washed in PBS and extracted in 600 μl of 100% methanol for 2 h at room temperature in a humidified box. The extract was then read on a fluorescent plate reader (Titertek Fluoroskan II). Unstained, equivalent plates were fixed in 4% formaldehyde, stained in 0.1% crystal violet, extracted in 10% acetic acid, and then read at 600 nm on a Titertek Multiscan Plus plate reader. These readings were used to determine protein concentrations of the samples and indicate relative cell numbers. Results were calculated as fluorescent units for equal cell numbers from wild-type or α3-null cells.
Western Blotting
Keratinocytes were grown in 10-cm dishes, extracted in 1 ml of radioimmunoprecipitation assay buffer (2% Triton X-100, 2% NaDOC, 0.2% SDS, 316 mM NaCl, 20 mM Tris-HCl, pH 7.3, 2 mM EDTA with protease inhibitors (as for surface iodinations) on ice for 10 min, extracts were centrifuged and protein concentration assessed by Bio-Rad “Dc” Protein Assay. 5 μg of each sample mixed with non-reducing sample buffer were boiled and run on a 7.5 or 5% acrylamide gel (Laemmli, 1970). Samples were then transferred onto nitrocellulose (Schleicher & Schuell, Keene, NH), blocked (5% “Marvel,” 0.1% Tween-20 in PBS) overnight at 4°C, incubated with primary antibody in block solution for 2 h at room temperature, washed in 0.1% Tween-20 in PBS, incubated with appropriate HRP-conjugated secondary antibody, washed again, and then bands detected by chemiluminescence (Renaissance NEN, Life Science Products).
For Western blotting of integrin immunoprecipitations, unlabeled lysates from wild-type or α3-null keratinocyte cultures were prepared in 200 mM octyl-β-d-glucopyranoside buffer, preabsorbed, and then immunoprecipitated as described above. For each immunoprecipitation, 565 μg of lysate were incubated with 5–10 μl of rat mAb against the β1 (PharMingen, San Diego, CA), α6 (GoH3), or β4 integrin subunit, followed by 100 μl of goat anti–rat IgG conjugated to Sepharose-4B (ICN Biomedicals, Costa Mesa, CA); 1:1 slurry preabsorbed with 20 mg/ml BSA in lysis buffer; control reactions contained Sepharose beads only. Immunoprecipitates were resolved on non-reducing, 7% polyacrylamide gels, and assayed by Western blotting, as described above, with an antiserum against the cytoplasmic domain of the α6 integrin subunit.
Results
α3 Integrin Deficiency Is Associated with Changes in F-Actin Localization in Basal Keratinocytes
Previous work showed that deficiency in α3β1 integrin resulted in areas of disorganized basement membrane and microblister development at the epidermal–dermal junction (DiPersio et al., 1997). Here, we examine the nature of the skin defects in further detail with a focus on the expression and function of integrins, cytoskeleton, and focal contact proteins in the α3-null keratinocytes.
Newborn mice from α3+/− crosses were examined immediately after birth for skin defects. At the microscopic level micro-blisters were observed in 80% of α3-null neonate feet and ∼15% of these were blood-filled blisters (Fig. 1, A–F). From previous work it was not clear whether the blisters in the α3-null mice were caused by handling the pups during examination. The presence of blood-filled blisters on live newborn mice demonstrates that the blisters observed in the α3-null mice are not due to post-mortem trauma (Fig. 1, A and B). Wild-type and heterozygous α3-knockout mice had indistinguishable and normal skin morphology (Fig. 1, C and D). However, in the α3-knockout mice, microblisters were evident at the epidermal–dermal interface (Fig. 1, E and F). Note the presence of blood in the blister shown in Fig. 1 F. We did not observe any abnormalities in epidermal stratification or proliferation in the α3-deficient mice.
Figure 1.

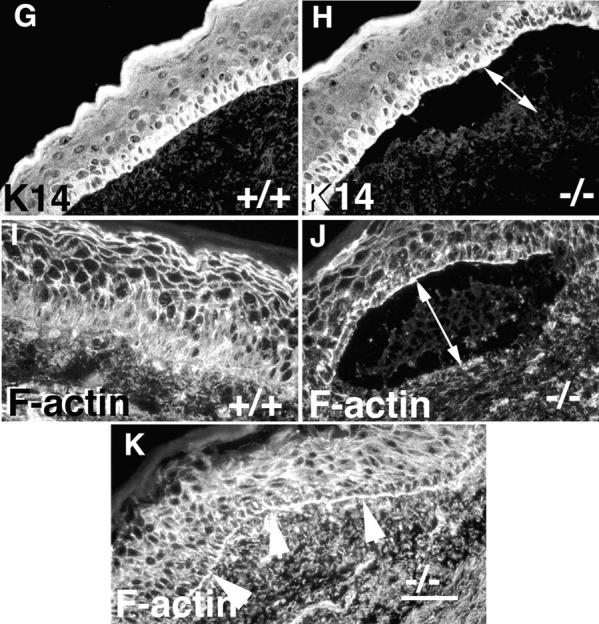
α3 integrin deficiency leads to blister formation at the epidermal–dermal interface and reorganization of the actin cytoskeleton. (A and B) Low power photographs of foot pads from newborn wild-type (A) and α3-null (B) mice. 18% of the α3-null mice display blood-filled blisters (arrowheads in B). (C–F) Hematoxylin and eosin-stained sections of neonatal skin from wild-type (C), heterozygous (D), and α3-null mice (E and F). Blisters were not observed in either wild-type or heterozygous animals. However, they were seen in 80% of α3-null feet (E) and some of these were filled with blood (arrow in F). (G–K) Immunofluorescence staining using antibodies to keratin 14 (G and H) or F-actin using phalloidin (I–K) of skin cryosections; wild-type (G and I) and α3-null skin (H, J, and K). Keratin 14 expression was normal in the α3-deficient skin (H). However, F-actin showed increased concentration at the basal face of basal keratinocytes in both blistering (J) and non-blistering (K) areas of α3-deficient skin. Double-headed arrows, blisters; arrowheads, dermal–epidermal junction. Bar, 50 μm.
The cytoplasmic tails of α6β4 integrins are linked via hemidesmosomal proteins to the keratin cytoskeleton. Mice deficient in α6 and β4 integrins develop blisters that are associated with altered keratin expression (Dowling et al., 1996; Georges-Labouesse et al., 1996; van der Neut et al., 1996). Since the cytoplasmic tail of α3β1 is associated with focal contact-associated proteins such as talin, vinculin, and α-actinin, which link integrins with the actin cytoskeleton, we explored the possibility that α3 deficiency would be associated with changes in actin cytoskeletal morphology. In Fig. 1, G–K we show that there are no changes in keratin 14 localization in the basal keratinocytes, but that there are changes in the actin cytoskeleton.
Immunostaining of cryosections from skin showed that expression of keratin 14 (Fig. 1, G and H) and keratin 5 (data not shown) were restricted to the basal keratinocytes and appeared normal in both non-blistering and blistering regions of the skin of α3-null mice. Since keratin 14 or 5 were not observed in suprabasal layers, or split between the roof and the floor of the blisters, the results show that neither hyperproliferation nor basal keratinocyte lysis occurred in the α3-null blisters and that cleft formation was truly at the epidermal–dermal interface.
Using rhodamine-conjugated phalloidin, F-actin had a pericellular distribution in all layers of the epidermis of both wild-type (Fig. 1 I) and α3-deficient skin (Fig. 1, J and K). In addition, F-actin staining was also concentrated at the basal face of the basal keratinocytes in the α3-null skin but not in the wild type. Increased concentration of F-actin in the basal keratinocytes was observed not only over blistering regions of the α3-nulls (Fig. 1 J), but also in intact areas of skin (Fig. 1 K, arrowheads). These results suggested that α3β1 integrin regulates the expression pattern of F-actin in the epidermis.
α3 Integrin Deficiency Results in Enhanced F-actin Stress Fibers and Focal Contacts
Since F-actin localization was altered in α3-null skins, we investigated the effect of α3 integrin deficiency on the distribution of F-actin and focal contact proteins in keratinocytes grown in culture (Fig. 2). In wild-type cells, phalloidin-stained actin bundles appeared thin and cobweb-like (Fig. 2 A). In contrast, F-actin in the α3-null keratinocytes was arranged into thick stress fibers especially at the basal face of the cells (Fig. 2 B). This was reminiscent of the altered F-actin staining seen in skin sections, where it also appeared to be concentrated at the basal face of the basal keratinocytes in the α3-null mice (see Fig. 1, J and K).
Figure 2.
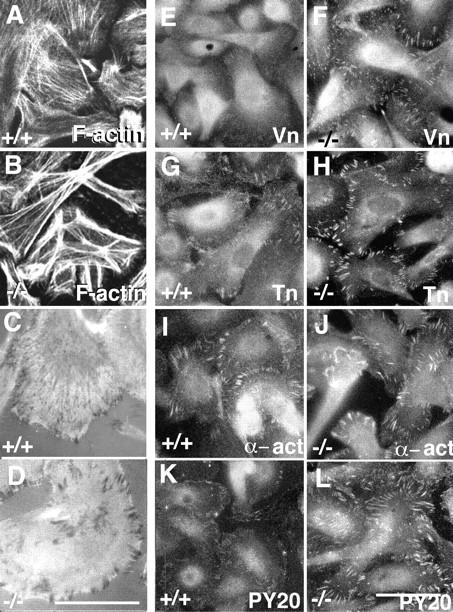
Loss of α3 integrin enhances formation of stress fibers and focal contacts. Wild-type (A, C, E, G, I, and K) and α3-deficient (B, D, F, H, J, and L) keratinocytes were grown under normal culture conditions and examined for F-actin (A and B); by interference reflection microscopy for focal contacts (C and D) and by immunofluorescence for vinculin (E and F), talin (G and H), α-actinin (I and J) and phosphotyrosine (K and L). Staining of F-actin with phalloidin showed larger stress fibers in the α3-null keratinocytes (B) than in the wild-type (A) cells (basal-most confocal images). Interference reflection microscopy shows darker and larger focal contacts in the α3-null cells (D) than in the wild-type cells (C). The levels of vinculin, talin, α-actinin, and phosphotyrosine were all increased in the focal contacts of the α3-null keratinocytes (F, H, J, and L) when compared with the wild-type cells (E, G, I, and K). Bars, 10 μm.
F-actin stress fibers in cultured cells are known to terminate at focal contact sites. Therefore, we asked whether the increase in stress fiber thickness in the α3-null keratinocytes was associated with any changes in focal contacts. Interference reflection microscopy revealed that focal contact sites in the α3-null keratinocytes (Fig. 2 D) were darker and larger than those observed in the wild-type cells (Fig. 2 C). This result implies that α3β1 deficiency leads to closer apposition of the ventral cell surface to the substrate at focal contact sites and also suggests that the focal contacts in the α3-null keratinocytes may be structurally different from wild-type focal contacts.
Consistent with the increased stress fiber formation and focal contact apposition to substrate in the α3-null keratinocytes, all the focal contact–associated proteins tested, including vinculin (Fig. 2, E and F), talin (Fig. 2, G and H), and α-actinin (Fig. 2, I and J), were more concentrated in the focal contacts of the α3-null keratinocytes than in the wild-type cells. In addition, PY20 staining for phosphotyrosine in the focal contacts was stronger in the α3-null keratinocytes than in wild-type cells (Fig. 2, K and L). Thus, in the α3-null keratinocytes there is a change in focal contact composition. The increase in phosphotyrosine expression in the focal contacts of the α3-null keratinocytes likely reflects a change in focal contact signaling activities.
To verify that the change in actin cytoskeletal organization observed in α3-null keratinocytes was due to a deficiency in α3β1 integrin, α3-null primary keratinocytes were transiently transfected with a recombinant plasmid expressing the α3 subunit. Double-label immunofluorescence revealed that ∼80% of cells expressing the exogenous α3 (Fig. 3, A and C), no longer displayed the thick stress fibers typical of α3-null keratinocytes (Fig. 3, B and D). These α3-transfected keratinocytes also showed reduced vinculin staining in focal contacts (Fig. 3, E and F). These results support a role for α3β1 integrin in regulating the organization of the actin cytoskeleton.
Figure 3.
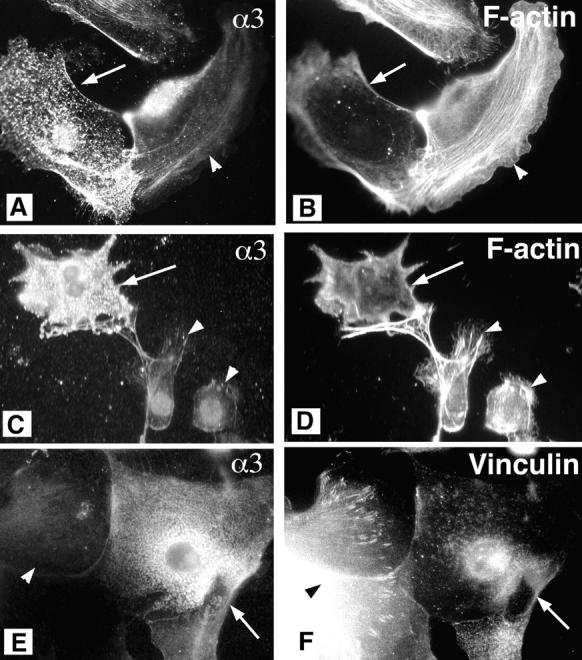
Reconstitution of α3β1-expression in α3-null keratinocytes reduces the thick actin bundles. α3-null primary keratinocytes were transiently transfected with a recombinant plasmid expressing the α3-subunit. Two examples of cells transfected with α3 are given. Cells were double labeled for α3 integrin (mAb P1B5) and F-actin (A and C, and B and D, respectively), or for α3 integrin (polyclonal antiserum) and vinculin (E and F, respectively). Arrows, transfected cells. Untransfected cells (arrowheads) served as controls within the same fields. Transfection with α3 reduced both the thick actin bundles and vinculin localization to focal contacts.
The Enhancement in Stress Fibers and Focal Contacts in the α3-null Keratinocytes Is Due to Redistribution and Not Elevated Levels of Proteins
We wished to determine whether the altered F-actin and focal contact protein staining patterns observed in the α3-null keratinocytes were due to changes in protein levels. To compare the levels of F-actin in the α3-deficient and wild-type keratinocytes, cultures were stained with fluorescently conjugated phalloidin, washed, and then the bound phalloidin extracted and quantified. The results showed that for equal protein concentrations of wild-type and α3-null keratinocytes the levels of F-actin were the same (Fig. 4 A). Combined with the immunofluorescence staining of F-actin (Fig. 2), this result indicated that the increased stress fiber formation in α3-null keratinocytes is not due to an increase in F-actin assembly, but rather to a redistribution of existing F-actin.
Figure 4.

The expression levels of focal contact-associated proteins in the α3-null keratinocytes are normal. (A) The levels of fluorescent-phalloidin–labeled F-actin in the α3-null and wild-type keratinocytes were the same. Student's t test showed no statistical difference in three experiments for a total of 11 samples. (B) Western blots of α3 integrin, actin, vinculin, talin, and α-actinin. The α3-subunit (α3, 150 kD) was absent in the α3-null keratinocytes. The levels of total actin (actin, 42 kD), vinculin (Vn, 110 kD), talin (Tn, 220 kD), and α-actinin (α-act, 100 kD) were all the same in the wild-type (+) and α3-null keratinocytes (−). Molecular weight markers represented in kD (kD). All cells were grown on vitrogen-coated plates.
Western blot analyses of protein extracts from α3-deficient (−) and wild-type (+) keratinocyte cultures showed that, although the α3-null cells were indeed α3 integrin deficient, the levels of total actin, vinculin (Vn), talin (Tn), and α-actinin (α-act) were each the same in the α3-null and wild-type keratinocytes (Fig. 4 B). Once again, these results, in combination with the immunofluorescence studies of focal contact-associated proteins (Fig. 2), demonstrate that α3 integrin deficiency results in a redistribution of existing proteins rather than increased steady state levels of these proteins.
Expression of Other Integrins Is Normal in α3-deficient Skin and Keratinocytes in Culture
Since integrins are known to be critical in regulating focal contact functions, one might predict that α3-deficiency could result in changes in other integrins that may compensate for the absence of α3β1. The effect of α3 integrin deficiency on the expression patterns of other integrins in the skin was investigated by immunofluorescence microscopy. Cryosections from wild-type and α3-null mice were stained with antibodies to various integrin subunits including α3, αv, β1, and β4 (Fig. 5, A–H). As expected, α3 was present on all surfaces of the basal keratinocytes in wild-type epidermis (Fig. 5 A) and was completely absent from α3-null skin (Fig. 5 E). There were no detectable differences in distributions of the other integrins tested in the skins of the wild-type or α3-null mice. αv and β1 subunits were confined to the basal keratinocytes in both wild-type (Fig. 5, B and C, respectively) and α3-null skin (Fig. 5, F and G). β4 integrins were concentrated at the basal face of the basal keratinocytes in both wild-type (Fig. 5 D) and α3-null skin (Fig. 5 H). It should be noted that, although α6β4 expression was strongest on the basal face of the basal keratinocytes, faint α6β4 staining was occasionally also observed on the lateral faces of basal keratinocytes in the α3-deficient mice.
Figure 5.
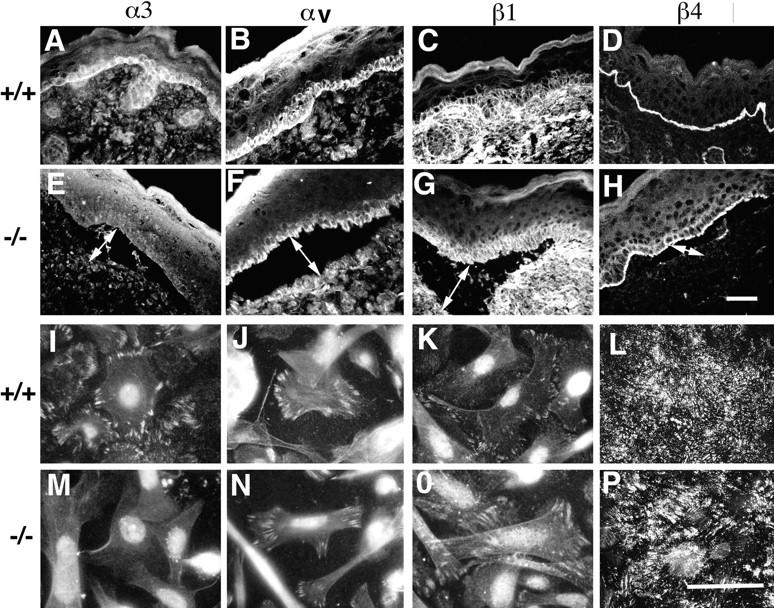
Immunofluorescence patterns of integrins in vivo and in vitro. Integrin expression patterns were examined by immunofluorescence microscopy in wild-type (A–D and I–L) and α3-deficient (E–H and M–P) skin sections (A–H) and in keratinocytes grown in culture (I–P). Immunofluorescence staining of α3 (A, E, I, and M), αv (B, F, J, and N), β1 (C, G, K, and O), and β4 (D, H, L, and P). Apart from the loss of α3, the staining patterns for other integrins in the α3-deficient skin and cells were normal. Bars: (H) 100 μm; (P) 10 μm.
The distributions of integrin subunits were also tested on keratinocytes grown in culture (Fig. 5, I–P). α3 integrin was present in the focal contacts of wild-type (Fig. 5 I), but not α3-null cells (Fig. 5 M, nuclei showed background staining). The expression of all the other integrins tested was normal in the α3-null cells. αv and β1 subunits both localized to focal contacts (Fig. 5, J and N, and K and O, respectively). α6β4 distribution was also normal in the α3-null keratinocytes, localizing to structures known as SACs.
Cells were also stained for α2, α5, and α9. α2 and α5 showed very low or undetectable levels of expression with no differences between wild-type and α3-null cells in vivo and in vitro (data not shown). α9 staining was confined to the basal layer of keratinocytes and was normal in wild-type and α3-null skin (data not shown). However, α9 was not detectable in wild-type or α3-null cells in culture. The distribution of the α6-subunits was identical with the β4 staining patterns both in vivo and in vitro.
α3 Integrin Deficiency Does Not Result in Changes in the Surface Expression of Other Integrins
To investigate whether there were any changes in processing and surface expression levels of other integrins, primary and secondary passage mouse keratinocyte cultures from wild-type and α3-null mice were either stained with anti-integrin antibodies and used in flow cytometric assays (Fig. 6 A) or were surface iodinated and immunoprecipitated with anti-integrin antibodies (Fig. 6, B and C).
Figure 6.
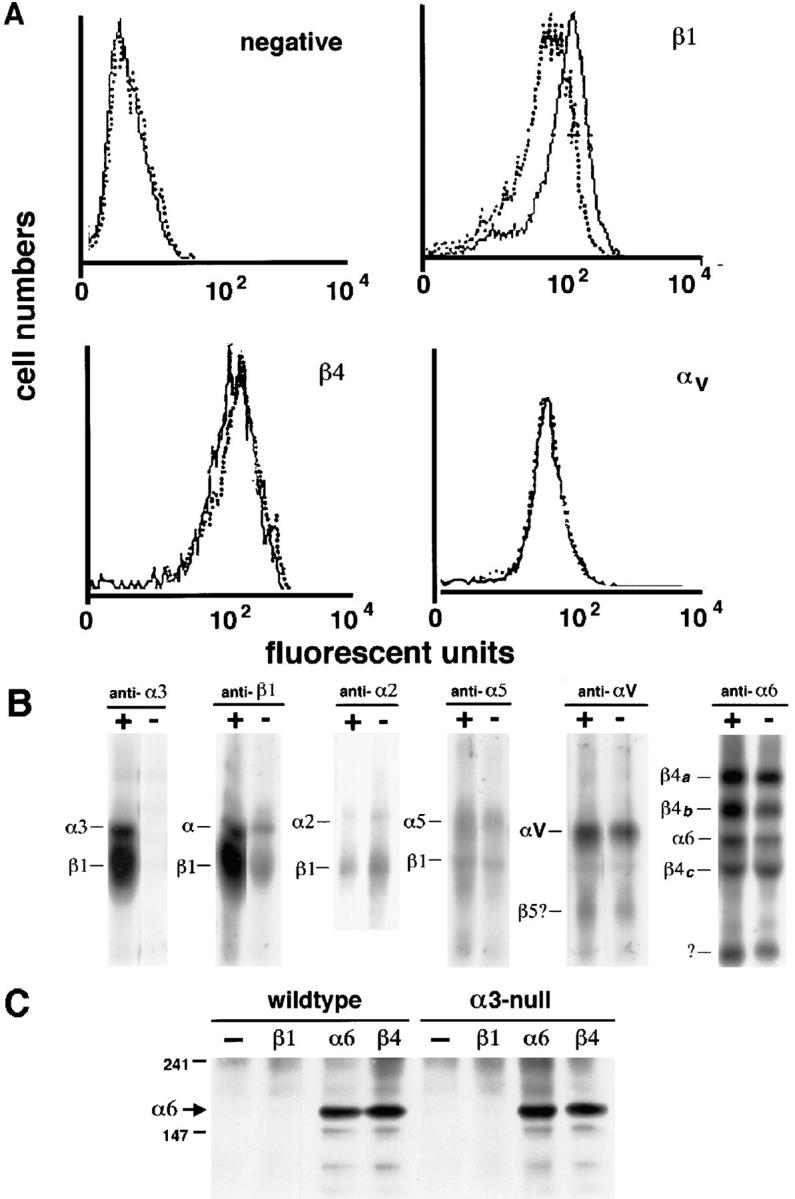
Expression levels of integrin subunits in wild-type and α3-null keratinocytes. (A) FACS® profiles of wild-type (solid line) and α3-null (dotted line) keratinocytes stained with antibodies to various integrins. The negative control shows background levels of staining. β1 levels were reduced in the α3-null keratinocytes, probably corresponding to deficiency of α3-subunits in the mutant cells. β4 and αv levels were normal. (B) Surface iodination and immunoprecipitation of integrins from wild-type (+) and α3-null (−) keratinocytes. There was no α3β1 detectable on the α3-null keratinocytes. The β1 immunoprecipitation showed decreased levels of surface β1 and coprecipitating α-subunits corresponding to the loss of surface α3. The surface expression of α2, α5, αv, and α6β4 remained the same in the α3-null keratinocytes. (C) Immunoprecipitation and Western blots illustrate the absence of detectable α6β1 in wild-type and α3-null keratinocytes. Immunoprecipitation and Western blotting of α6 integrins from wild-type and α3-null keratinocytes. Whole cell lysates were immunoprecipitated either with mAbs specific for the β1, α6, or β4 integrin subunits as indicated, or without primary antibody (−), separated on a non-reducing, 7% polyacrylamide gel, and then immunoblotted with a rabbit antiserum against the cytoplasmic domain of the α6 subunit. The α6 subunit (∼150 kD, arrow) was detected in immunoprecipitations for the α6 and β4 subunits, but not in that for β1 or in the control reaction, indicating the absence of α6β1 from these cells. Positions for 147-kD and 241-kD MW markers are indicated at left.
In FACS® analysis (Fig. 6 A) pools of either wild-type or α3-null cultured keratinocytes were labeled with anti-integrin extracellular domain–specific antibodies to β1, β4, or αv, or with IgG2a as a negative control. The levels of β1 expression were considerably reduced in the α3-null cells. This reflects the fact that α3β1 constitutes a large proportion of the β1 integrins on the keratinocyte cell surface. β4 and αv showed no consistent changes in levels of surface expression (Fig. 6 A).
Surface iodination and immunoprecipitation with antibodies to integrin subunits α3, β1, α2, α5, αv, or α6 was carried out on wild-type (+) and α3-null (−) keratinocytes (Fig. 6 B). Immunoprecipitations with anti-α3 and anti-β1 antibodies confirmed the absence of α3β1 in the α3-null keratinocytes and the expected reduction of β1. There were no detectable differences in surface expression of α2, α5, αv, or α6β4 between the wild-type and α3-null cells.
Thus, the surface iodination and immunoprecipitation data confirm the absence of detectable differences between wild-type and α3-null cells in the surface expression of the different integrin subunits. The α6 integrin subunit can also pair with the β1 subunit in some cell types, and α6β1 can function as a weak receptor for laminin 5. Western blot analysis demonstrated that the loss of α3β1 from the surfaces of α3-null keratinocytes resulted in an increase in the intracellular pool of β1 (data not shown); therefore, it seemed possible that some of the excess β1 might dimerize with endogenous α6 (normally dimerized with β4) and cause an upregulation of α6β1 on the cell surface. Although the immunoprecipitation data in Fig. 6 B do not support this idea, we addressed this possibility directly by immunoprecipitation of β1 integrins followed by immunoblotting with antiserum against the α6 subunit. For both wild-type and α3-null keratinocytes α6 was present in immunoprecipitates of either α6 or β4 integrins, but was not detected in immunoprecipitates of β1 integrins or in control immunoprecipitations lacking antibody (Fig. 6 C). Similar immunoblots with antiserum against the β1 subunit confirmed the presence of β1 in immunoprecipitates of β1 integrins, and the absence of β1 from immunoprecipitates of α6 integrins (data not shown). These results confirm the absence of detectable levels of α6β1 in both wild-type and α3-null keratinocytes. Thus, although α3 deficiency affects F-actin and focal contact protein recruitment to focal contact sites, it has no effect on the expression patterns or levels of the other integrins in keratinocytes.
The Presence of α3β1 Inhibits Fibronectin and Collagen Type IV Receptor Functions
We have demonstrated that α3 integrin deficiency enhances accumulation of actin-associated proteins to focal contacts, but does not change other integrin expression patterns. To examine the effect of α3 integrin deficiency on keratinocyte behavior, secondary passage wild-type and α3-null keratinocytes were tested in adhesion and migration assays. The numbers of α3-null or wild-type keratinocytes able to adhere to laminin 1 (LM-1) or laminin 5 (LM-5) were the same. This result implied that, in the absence of α3 integrins, α6β4 (another receptor for LM-5) was sufficient to allow cell attachment to LM-5. Saturating concentrations of GoH3 (an anti–α6 integrin function-blocking antibody; Sonnenberg et al., 1987) had no effect on the number of wild-type cells adherent to LM-5 since α3β1 is sufficient for adhesion (Delwel et al., 1994; DiPersio et al., 1997). In the presence of GoH3, α3-null keratinocytes were completely inhibited from adhering to LM-5 (Fig. 7 A) demonstrating that either α3β1 or α6β4 is sufficient for adhesion to LM-5. Surprisingly, the numbers of α3-null cells able to adhere to FN and Coll IV were significantly higher than the numbers of wild-type cells able to adhere (P = 0.01 and 0.05, respectively) (Fig. 7 A). That is, the fibronectin and collagen receptors are more active in the α3-null cells.
Figure 7.
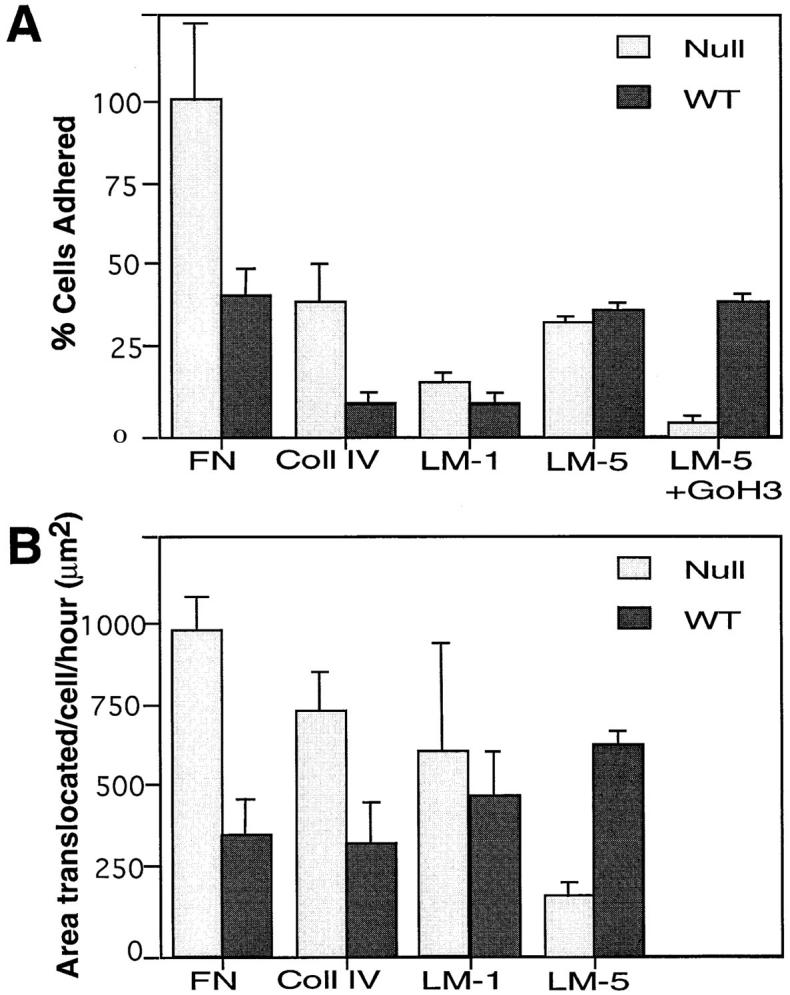
Adhesive and migratory behavior of α3-null and wild-type keratinocytes on various substrates. (A) Adhesion assays. Cell adhesion was measured as the percentage of cells adherent compared with the number of α3-null cells adherent to fibronectin (FN). A significantly higher number of α3-null keratinocytes adhered to fibronectin (FN), and collagen type IV (Coll IV) when compared with wild-type keratinocytes (P = 0.01 and P = 0.05, respectively). The numbers of wild-type and α3-null keratinocytes that adhered to either laminin 1 or laminin 5 were indistinguishable. When adhesion to laminin-5 was carried out in the presence of an α6β4 blocking antibody (GoH3), adhesion of the α3-null keratinocytes was significantly reduced whereas adhesion of the wild-type cells was not (P < 0.005). Results are an average of four experiments each done in triplicate. (B) Migration assays. Cell migration was measured as the area cleared by a single cell per hour. The α3-null keratinocytes migrated significantly further than wild-type cells on FN (P < 0.005) and Coll IV (P < 0.005). The migration of α3-null keratinocytes and wild-type cells on laminin 1 was not significantly different. In contrast, wild-type keratinocytes migrated significantly better than α3-null keratinocytes on laminin 5 (P < 0.005).
To compare the migration of wild-type and α3-null keratinocytes, phagokinetic assays were performed (Fig. 7 B). The phagokinetic assays involved plating cells on an even field of matrix-coated, colloidal gold particles and allowing the cells to attach and migrate over the substrate. As the cells migrate they phagocytose the gold particles; the area of the particle-free traces was measured and quantitated as described in Materials and Methods. The α3-null keratinocytes migrated significantly further on FN and Coll IV than did the wild-type cells (P = 0.005 and 0.005, respectively). In contrast to the migration on FN the average area cleared by the α3-null keratinocytes on LM-1 was not significantly different between the α3-deficient and wild-type keratinocytes. The α3-null keratinocytes showed a significantly reduced level of migration on LM-5 when compared with wild-type cells (P = 0.005). This was consistent with our previous observation that α3-null keratinocytes do not spread well on LM-5, but the wild-type cells do (DiPersio et al., 1997). Thus, although the α3-null cells can adhere as well as the wild-type cells to LM-5, they do not migrate as well.
Taken together, these data suggest that the changes in focal contact protein composition observed in the α3-null keratinocytes may be associated with the functional upregulation of fibronectin and collagen type IV receptors in these cells.
Discussion
α3β1 is an abundant integrin on the surfaces of keratinocytes. However, to date, the precise roles of α3β1 in skin biology have been unclear. The generation of α3 integrin knockout mice has permitted us to isolate α3-deficient keratinocytes, which we have used to investigate the roles of α3β1. Comparison of the characteristics and functional behaviors of these cells with those of wild-type keratinocytes has shed light on novel functions of α3β1 as a regulator of cytoskeletal organization and as a trans-dominant inhibitor of other integrins. Our major findings are as follows: (a) α3β1-deficient mice show an enhanced concentration of F-actin at the basal face of the basal keratinocytes; (b) in α3-null keratinocytes there is an increase in stress fiber formation and actin-associated protein localization to focal contacts, and this is associated with increased levels of phosphotyrosine in focal contacts, and (c) in functional assays the absence of α3β1 in the α3-null keratinocytes permits increased function of fibronectin and collagen receptors when compared with wild-type cells, implying a novel role of α3β1 as a trans-dominant inhibitor of other integrins.
The blisters observed in the α3-null mice are small compared with the extensive blistering found in the α6- (Georges-Labouesse et al., 1996) or β4-knockout mice (van der Neut et al., 1996; Dowling et al., 1996) and are particularly evident on the foot pads where they are sometimes filled with blood (Fig. 1). Mutations in α6β4 and laminin 5 have been shown to be causes of severe human blistering disorders such as Junctional Epidermolysis Bullosa with pyloric atresia (for review see Burgeson and Christiano, 1997). However, the etiologies of many other human blistering diseases are still unknown. It will be of interest to determine whether mutations in α3β1 may also contribute to human blistering disorders.
α3β1 Deficiency Results in F-Actin Reorganization That Is Reminiscent of Wound Healing
Although α6β4 and α3β1 integrins are both receptors for laminin 5, they differ in that the cytoplasmic tails of α6β4 interact with the keratin cytoskeleton, whereas the α3β1 cytoplasmic tails interact with the F-actin cytoskeleton. At the ultrastructural level, the α6- and β4-knockout skins have altered keratin filament organization (Dowling et al., 1996; Georges-Labouesse et al., 1996; van der Neut et al., 1996). Our immunofluorescence studies show that, although keratin 14 expression patterns are similar in α3-null and wild-type skin, the pattern of F-actin changes. In the α3-null epidermis F-actin was concentrated at the basal face of the basal keratinocytes in both blistering and non-blistering areas (Fig. 1, I–J). These observations were reflected in cultured keratinocytes where thin fibers of F-actin became concentrated into thick bundles in the absence of α3β1 integrins (Fig. 2, A and B). Reconstitution of α3β1-expression in α3-null keratinocytes reduced the thick actin bundles typically presented in these cells (Fig. 3).
Our findings both in vivo and in vitro show a striking resemblance to the patterns of reorganized F-actin seen during epithelial and corneal endothelial wound healing (Gabbiani et al., 1978; Brock et al., 1996; Gordon and Buxar, 1997) and suggest that the basal keratinocytes in the α3-null skin may be mimicking some kind of wound healing process. We postulate that deficiency for α3β1 results in a wound healing–like response, which in turn is manifested by the repeated deposition of basement membrane proteins during epidermal development, thus giving rise to the “layered” appearance of the basement membrane in the α3-null skin (DiPersio et al., 1997).
α3β1 Is a Trans-dominant Inhibitor of Fibronectin and Collagen Type IV Receptor Function
In the α3-null keratinocytes the changes seen in F-actin organization are also associated with an increased recruitment of actin-associated proteins such as vinculin, talin and α-actinin to focal contacts (Fig. 2). Immunochemical data show that this increased recruitment is not due to an increase in production of these proteins, but rather to changes in localization (Fig. 4). A similar change in actin organization was also observed in corneal epithelial cells during wound healing with no change in actin synthesis (Gordon and Buxar, 1997). The phenomenon of enhanced focal contacts in the α3-null keratinocytes implies that, when α3β1 is present, it prevents these events from occurring (Fig. 2, C–L). This is a novel function for α3β1, but the mechanism is unclear.
Since α3β1 is such an abundant integrin on the surface of keratinocytes and most of it is not concentrated in focal contact sites (DiPersio et al., 1995), it is possible that α3β1 competes for certain proteins thereby regulating focal contact composition. Hence, when α3β1 is absent, actin-associated proteins may be “free” to localize more readily to focal contact sites. The increase in PY20 staining at the focal contacts in the α3-null keratinocytes demonstrates that there is an increase in tyrosine phosphorylation at these sites. Elevated tyrosine phosphorylation is associated with activation of focal adhesion proteins and this in turn with integrin-mediated cell binding to the extracellular matrix (Hynes, 1992; Juliano and Haskill, 1993). Thus, in the absence of α3β1 integrin, such an increase in phosphotyrosine at focal contacts may reflect a change in the signaling of other integrins.
Another way in which α3β1 might regulate stress fiber and focal contact assembly is via effects on cell polarization and cadherin function. Antibodies to α3β1 inhibit cell–cell adhesion (Carter et al., 1990a ), so loss of α3β1 could similarly affect cadherin function including recruitment of F-actin and certain focal contact proteins to cell– cell adherens junctions. This in turn would release these components for assembly at the basal surface in response to other integrins. We have observed that E-cadherin is poorly organized in α3-null epidermis that overlies “diffuse” areas of basement membrane (data not shown). This preliminary evidence supports the idea that α3 deficiency may affect keratinocyte polarization in skin and is in accordance with the disorganized cadherins observed in immortalized kidney epithelial cells (Wang, Z., J.M. Symons, and J.A. Kreidberg, personal communication). Whatever the precise mechanism, it is clear that the absence of α3β1 allows greater recruitment of cytoskeletal components by other integrins.
Functional assays revealed that α3-null keratinocytes adhere and migrate better on fibronectin and collagen type IV than do wild-type controls (Fig. 7). Using P1B5, a function-blocking anti-α3 antibody, Kim et al. (1992) showed evidence that blocking α3 integrin function enhanced the migration of keratinocytes on FN. However, they and others (Carter et al., 1990a ) have suggested that P1B5 blocked keratinocyte adhesion to FN. Our data instead support a role for α3β1 as a trans-dominant inhibitor of FN and Coll IV receptor function.
Previous work suggests that occupancy of one integrin can suppress the functions of other integrins in the same cell. For example, anti-αvβ3 antibodies suppress α5β1-dependent phagocytosis (Blystone et al., 1994) and ligation of α4β1 inhibits α5β1-dependent expression of metalloproteinases (Huhtala et al., 1995). Diàz-Gonzàlez et al. (1996) showed that ligand-specific ligation of a “suppressor” integrin such as αIIbβ3 is associated with a blockade in functioning of “target” integrins such as α5β1. Diàz-Gonzàlez's work also indicates that αllbβ3 is dependent on ligand interaction to inhibit α5β1 function. Our in vitro results show that the trans-dominant role of α3β1 can be independent of ligation with LM5 and relies on the presence of α3β1. It should be noted that unlike αllbβ3, α3β1 appears to be constitutively active in cultured cells in that focal contact localization may be ligand-independent in some cases (DiPersio et al., 1995). It is quite possible that ligation of α3β1 with laminin 5 may enhance α3β1 activity to send an inhibitory signal to the FN and Coll IV receptors on the surface of the cell and thus inhibit their functions further.
Potential Implications for Trans-dominant Inhibition During Wound Healing
Given the results reported here, one can make some hypotheses about the role of α3β1 in regulating keratinocyte behavior in resting skin and during wound healing. In resting skin α3β1 is prevalent and is engaged with LM5 in the basement membrane and the cells are sessile. This situation would be predicted to lead to α3β1-dependent suppression of the functions of other migratory integrins. At the time of wounding, α5β1 and αv integrins are upregulated and are believed to promote keratinocyte migration on the provisional matrix, which is rich in ligands for these integrins (fibronectin, fibrin, and vitronectin). α2β1-collagen interactions could also play a role (Larjava et al., 1993). Our results suggest that migration would be enhanced if trans-dominant inhibition by α3β1 were relieved (Fig. 8). There are several ways in which this could arise. Metalloproteinases, which are upregulated on wounding (for review see Clark, 1996) could cleave constituents in the basement membrane such as LM5 or α3β1 itself. This might cause uncoupling of the α3β1-LM5 adhesion system, allowing efficient functioning of migratory integrins (including α5β1, α2β1, and αv) and epithelial closure of the wound. Relief of α3β1 trans-dominant inhibition could also occur by some form of inactivation of α3β1 from inside the cell (i.e., inside-out signaling). Then, once the wound is closed, reestablishment of a newly formed basement membrane and of functional α3β1-LM5 interactions, could be hypothesized to restore trans-dominant inhibition of the other integrins, bringing migration to an end and returning the keratinocytes to their sessile state.
Figure 8.

A model for α3β1 integrin as a trans-dominant inhibitor during wound healing. In resting skin α3β1 engages LM5 and this results in trans-dominant inhibition of α5β1 and α2β1 functions. This confers an adherent non-migratory state on the keratinocytes. When skin is wounded, the α3β1– LM5 interactions are altered, due to changes in α3β1 and/or LM5, relieving trans-dominant inhibition of the functions of other integrins. α3β1 and/or LM5 may show changes in conformation switching α3β1 form an adherent, inhibitory mode to a migratory, non-inhibitory mode. Once the wound is closed, a normal basement membrane is reestablished, α3β1 binds resting LM5 and inhibits α5β1 and α2β1 functions again.
In addition to the above hypothesis, α3β1–LM5 interactions may occur in two different modes. The first would be as a trans-dominant inhibitor just as in the model above and we would suggest that this is the role of this interaction in resting skin and after completion of healing. However, during migration, α3β1–LM5 interactions could also play a role in migration. This change from a resting, adhesive, and trans-dominant inhibitory state of α3β1 to a migratory and non-inhibitory state could occur by alterations in LM5 and/or in intracellular regulation of α3β1 function. Such a model would accommodate a role for the high levels of LM5 during re-epithelialization (Larjava et al., 1993; Zhang and Kramer, 1996; Lotz et al., 1997).
Either model invokes α3β1 as a suppressor of keratinocyte migration in non-wounded skin and could explain the apparent formation of multiple layers of basement membrane in the α3-null skin (DiPersio et al., 1997). The models also lead to testable predictions about the behavior of α3-null skin before and after wounding; such predictions could be tested by transplanting neonatal skin (Medalie et al., 1996) from α3-null pups to immuno-deficient recipient mice. These hypotheses also raise questions about α3β1 signaling processes which will be readily investigated using keratinocytes established from wild-type and α3-null mice and transfected with α3 cDNAs.
In conclusion, using α3-deficient mice and keratinocytes, we have uncovered a novel role for α3β1 in the regulation of focal contact composition, F-actin organization and as a trans-dominant inhibitor of FN and Coll IV receptor functions. This new role for α3β1 may be important in understanding more about the regulation of keratinocyte migration in wound healing physiology.
Acknowledgments
The authors are grateful to K. Mercer for her excellent help with histology. Thanks also to all those who kindly provided antibodies and reagents. And finally, thanks to B. Burgeson, E. Clark, and M. Borowsky for their valuable comments while preparing this paper.
Abbreviations used in this paper
- F-actin
filamentous actin
- FN
fibronectin
- Coll IV
collagen type IV
- LM1 laminin 1
LM5 laminin 5
- SAC
stable achoring contact
Footnotes
This work was sponsored in part by The Human Frontiers Science Programme and The Dystrophic Epidermolysis Bullosa Research Association, by a grant from the National Cancer Institute (RO1 CA7007), and by the Howard Hughes Medical Institute (HHMI). R.O. Hynes is an Investigator of the HHMI.
The first two authors contributed equally to the work presented.
Please address all correspondence to R.O. Hynes, Howard Hughes Medical Institute, Center for Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139. Tel.: (617) 253-6422. Fax: (617) 253-8357.
References
- Albrecht-Buehler G. The phagokinetic tracks of 3T3 cells. Cell. 1977;11:395–404. doi: 10.1016/0092-8674(77)90057-5. [DOI] [PubMed] [Google Scholar]
- Bartolazzi A, Cerboni C, Nicotra MR, Mottolese M, Bigotti A, Natali P. Transformation and tumor progression are frequently associated with expression of the α3/β1 heterodimer in solid tumors. Int J Cancer. 1994;58:488–491. doi: 10.1002/ijc.2910580405. [DOI] [PubMed] [Google Scholar]
- Blystone SD, Graham IL, Lindberg FP, Brown EJ. Integrin αvβ3 differentially regulates adhesive and phagocytic functions of the fibronectin receptor α5β1. J Cell Biol. 1994;127:1129–1137. doi: 10.1083/jcb.127.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland S, Boisvieux-Ulrich E, Houcine O, Baeza-Sqiban A, Pouchelet M, Schoëvaërt D, Marano F. TGFβ1 promotes actin cytoskeleton reorganisation and migratory phenotype in epithelial tracheal cells in primary culture. J Cell Sci. 1996;109:2207–2219. doi: 10.1242/jcs.109.9.2207. [DOI] [PubMed] [Google Scholar]
- Borradori L, Sonnenberg A. Hemidesmosomes: roles in adhesion, signalling and human diseases. Curr Opin Cell Biol. 1996;8:647–656. doi: 10.1016/s0955-0674(96)80106-2. [DOI] [PubMed] [Google Scholar]
- Brennen JK, Mansky J, Roberts G, Litchman MA. Inproved methods for reducing calcium and magnesium concentrations in tissue culture medium. In Vitro. 1982;11:693–703. doi: 10.1007/BF02616371. [DOI] [PubMed] [Google Scholar]
- Brock J, Midwinter K, Lewis J, Martin P. Healing of incisional wounds in the embryonic chick wing bud: Characterization of the actin purse-string and demonstration of a requirement for Rho activation. J Cell Biol. 1996;135:1097–1107. doi: 10.1083/jcb.135.4.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgeson RE, Christiano AM. The dermal-epidermal junction. Curr Biol. 1997;9:651–658. doi: 10.1016/s0955-0674(97)80118-4. [DOI] [PubMed] [Google Scholar]
- Burridge K, Fath K, Kelly T, Nuckolls G, Turner C. Focal adhesions: transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu Rev Cell Biol. 1988;4:487–525. doi: 10.1146/annurev.cb.04.110188.002415. [DOI] [PubMed] [Google Scholar]
- Carter WG, Wayner EA, Bouchard TS, Kaur P. The role of integrins α2β1 and α3β1 in cell–cell and cell–substrate adhesion of human epidermal cells. J Cell Biol. 1990a;110:1387–1404. doi: 10.1083/jcb.110.4.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter WG, Kaur P, Gil SG, Gahr PJ, Wayner EA. Distinct functions for integrins α3β1 in focal adhesions and α6β4/bullous antigen in a new stable anchoring contact (SAC) of keratinocytes: Relation to hemidesmosomes. J Cell Biol. 1990b;111:3141–3154. doi: 10.1083/jcb.111.6.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavani A, Zambruno G, Marconi A, Manca V, Marchetti M, Giannetti A. Distinctive integrin expression in the newly forming epidermis during wound healing in humans. J Invest Dermatol. 1993;101:600–604. doi: 10.1111/1523-1747.ep12366057. [DOI] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Clark, R.A.F. 1996. The Molecular and Cellular Biology of Wound Repair. 2nd Ed. Plenum Press, New York and London. 442–447.
- Delwel GO, de Melker AA, Hogervorst F, Jaspars LH, Fles DLA, Kuikman I, Lindblom A, Paulsson M, Timpl R, Sonnenberg A. Distinct and overlapping ligand specificities of the α3Aβ1 and α6Aβ1 integrins: recognition of laminin isoforms. Mol Biol Cell. 1994;5:203–215. doi: 10.1091/mbc.5.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dìaz-Gonzàlez F, Forsyth J, Steiner B, Ginsberg MH. Trans-dominant inhibition of integrin function. Mol Biol Cell. 1996;7:1939–1951. doi: 10.1091/mbc.7.12.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPersio CM, Shah S, Hynes RO. α3β1 integrin localizes to focal contacts in response to diverse extracellular matrix proteins. J Cell Sci. 1995;108:2321–2336. doi: 10.1242/jcs.108.6.2321. [DOI] [PubMed] [Google Scholar]
- DiPersio CM, Hodivala-Dilke KM, Jaenisch R, Kreidberg JA, Hynes RO. α3β1 integrin is required for normal development of the epidermal basement membrane. J Cell Biol. 1997;137:729–742. doi: 10.1083/jcb.137.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling J, Yu Q-C, Fuchs E. β4 integrin is required for hemidesmosomal formation, cell adhesion and cell survival. J Cell Biol. 1996;134:559–572. doi: 10.1083/jcb.134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugosz AA, Glick AB, Tennenbaum T. Isolation and utilization of epidermal keratinocytes for oncogene research. Methods Enzymol. 1995;254:3–20. doi: 10.1016/0076-6879(95)54003-2. [DOI] [PubMed] [Google Scholar]
- Elices MJ, Urry LA, Hemler ME. Receptor functions for the integrin VLA-3: Fibronectin, collagen, and laminin binding are differentially influenced by ARG-GLY-ASP peptide and by divalent cations. J Cell Biol. 1991;112:169–181. doi: 10.1083/jcb.112.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto-Iwamoto M, Menko AS, Philip N, Boettiger D. Evaluation of integrin molecules involved in substrate adhesion. Cell Adhes Commun. 1993;1:191–202. doi: 10.3109/15419069309097253. [DOI] [PubMed] [Google Scholar]
- Gabbiani G, Chaponnier C, Hüttner I. Cytoplasmic filaments and gap junctions in epithelial cells and myofibroblasts during wound healing. J Cell Biol. 1978;76:561–568. doi: 10.1083/jcb.76.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehlsen KR, Dillner L, Engvall E, Ruoslahti E. The human laminin receptor is a member of the integrin family of cell adhesion receptors. Science. 1988;241:1228–1229. doi: 10.1126/science.2970671. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Messaddeq N, Yehia G, Cadalbert L, Dierich A, Le Meur M. Absence of integrin α6 leads to epidermolysis bullosa and neonatal death in mice. Nat Genet. 1996;13:370–373. doi: 10.1038/ng0796-370. [DOI] [PubMed] [Google Scholar]
- Gordon SR, Buxar RM. Inhibition of cytoskeletal reorganisation stimulates actin and tubulin synthesies during injury-indued cell migration in the corneal endothelium. J Cell Biochem. 1997;67:409–421. [PubMed] [Google Scholar]
- Grenz H, Carbonetto S, Goodman SL. α3β1 integrin is moved into focal contacts in kidney mesangial cells. J Cell Sci. 1993;105:739–751. doi: 10.1242/jcs.105.3.739. [DOI] [PubMed] [Google Scholar]
- Hertle MD, Kubler M-D, Leigh IM, Watt FM. Aberrrant integrin expression during epidermal wound healing and psoriatic epidermis. J Clin Invest. 1992;89:1892–1901. doi: 10.1172/JCI115794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huhtala P, Humphries MJ, McCarthy JB, Tremble P, Werb Z, Damsky CH. Cooperative signalling by α5β1 and α4β1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J Cell Biol. 1995;129:867–879. doi: 10.1083/jcb.129.3.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Alterations of cell surface proteins by viral transformation and by proteolysis. Proc Natl Acad Sci USA. 1973;70:3170–3174. doi: 10.1073/pnas.70.11.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signalling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Hynes RO, Marcantonio EE, Stepp MA, Urry LA, Yee GH. Integrin heterodimer and receptor complexity in avian and mammalian cells. J Cell Biol. 1989;109:409–420. doi: 10.1083/jcb.109.1.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RP, Craig SW. F-actin binding site masked by the intramolecular association of vinculin head and tail domains. Nature. 1995;373:261–264. doi: 10.1038/373261a0. [DOI] [PubMed] [Google Scholar]
- Juliano RL, Haskill S. Signal transduction from the extracellular matrix. J Cell Biol. 1993;120:577–585. doi: 10.1083/jcb.120.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JP, Zhang K, Kramer RH, Schall TJ, Woodley DT. Integrin receptors and RGD sequences in human keratinocyte migration: unique anti-migratory function of α3β1 epiligrin receptor. J Invest Dermatol. 1992;98:764–770. doi: 10.1111/1523-1747.ep12499947. [DOI] [PubMed] [Google Scholar]
- Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K, Jones RC, Jaenisch R. α3β1 integrin has a crucial role in kidney and lung organogenesis. Development (Camb) 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Larjava H, Salo T, Haapasalmi K, Kramer RH, Heino J. Expression of integrins and basement membrane components by wound keratinocytes. J Clin Invest. 1993;92:1425–1435. doi: 10.1172/JCI116719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz MM, Nusrat A, Madara JL, Ezell R, Wewer UM, Mercurio AM. Involvement of specific laminin isoforms and integrin laminin receptors in wound closure of a transformed model epithelium. Am J Pathol. 1997;150:747–760. [PMC free article] [PubMed] [Google Scholar]
- Marcantonio EE, Hynes RO. Antibodies to the conserved cytoplasmic domain of the integrin β1 cytoplasmic domain by site directed mutagenesis. J Cell Biol. 1988;106:1765–1772. doi: 10.1083/jcb.106.5.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medalie DA, Eming SA, Tompkins RG, Yarmush ML, Krueger GG, Morgan JR. Evaluation of human skin reconstituted from composite grafts of cultured keratinocytes and human dermis transplanted to athymic mice. J Invest Dermol. 1996;107:121–127. doi: 10.1111/1523-1747.ep12298363. [DOI] [PubMed] [Google Scholar]
- Niessen CM, van der Raaij-Helmer LMH, Hulsman EHM, van der Neut R, Jonkman MF, Sonnenberg A. Deficiency of the integrin β4 subunit in junctional epidermolysis bullosa with pyloric atresia: consequences for hemidesmosome formation and adhesion properties. J Cell Sci. 1996;109:1695–1706. doi: 10.1242/jcs.109.7.1695. [DOI] [PubMed] [Google Scholar]
- Otey CA, Vasquez GB, Burridge K, Erickson BW. Mapping of the α-actinin binding site within the β1 integrin cytoplasmic domain. J Biol Chem. 1993;268:21193–21197. [PubMed] [Google Scholar]
- Palmer EL, Rüegg C, Ferrando R, Pytela R, Sheppard D. Sequence and tissue distribution of the integrin α9 subunit, a novel partner of β1 that is widely distributed in epithelia and muscle. J Cell Biol. 1993;123:1289–1297. doi: 10.1083/jcb.123.5.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini G, DeLuca M, Orecchia G, Balzac F, Cremona O, Savoia P, Cancedda R, Marchisio PC. Expression, topography, and function of integrin receptors are severely altered in keratinocytes from involved and uninvolved psoriatic skin. J Clin Invest. 1992;89:1783–1795. doi: 10.1172/JCI115782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzzi L, Gagnoux-Palacious L, Pinola M, Belli S, Meneguzzi G, D'Alessio M, Zambruno G. A homozygous mutation in the integrin α6 gene in junctional epidermolysis bullosa with pyloric atresia. J Clin Invest. 1997;99:2826–2831. doi: 10.1172/JCI119474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg A, Janssen H, Hogervorst F, Calafat J, Hilgers J. A complex of platelet glycoprotein Ic and Ia identified by a rat monoclonal antibody. J Biol Chem. 1987;262:10376–10383. [PubMed] [Google Scholar]
- Stepp MA, Spurr-Michaud S, Tisdale A, Elwell J, Gipson IK. α6β4 integrin heterodimer is a component of hemidesmosomes. Proc Natl Acad Sci USA. 1990;87:8970–8974. doi: 10.1073/pnas.87.22.8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington BE, Carter WE. Modulation of epidermal differentiation by epiligrin and integrin α3β1. J Cell Sci. 1995;108:831–838. doi: 10.1242/jcs.108.2.831. [DOI] [PubMed] [Google Scholar]
- van der Neut R, Krimpenfort P, Calafat J, Niessen CM, Sonnenberg A. Epithelial detachment due to absence of hemidesmosomes in integrin β4 null mice. Nat Genet. 1996;13:366–369. doi: 10.1038/ng0796-366. [DOI] [PubMed] [Google Scholar]
- Watt, F.M., and M.D. Hertle. 1994. Keratinocyte integrins. In The Keratinocyte Handbook. I.M. Leigh, E.B. Lane, and F.M. Watt, editors. Cambridge University Press, Cambridge. 153–164.
- Wayner EA, Carter WG. Identification of multiple cell adhesion receptors for collagen and fibronectin in human fibrosarcoma cells possessing unique a and common b subunits. J Cell Biol. 1987;105:1873–1884. doi: 10.1083/jcb.105.4.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayner EA, Carter WG, Piotrowicz RS, Kunicki TJ. The function of multiple extracellular matrix receptors in mediating cell adhesion to extracellular matrix: Preparation of monoclonal antibodies to the fibronectin receptor that specifically inhibit cell adhesion to fibronectin and react with platelet glycoproteins IIc-IIa. J Cell Biol. 1988;107:1881–1891. doi: 10.1083/jcb.107.5.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kramer RH. Laminin 5 deposition promotes keratinocyte motility. Exp Cell Res. 1996;227:309–322. doi: 10.1006/excr.1996.0280. [DOI] [PubMed] [Google Scholar]