

Abstract
γ-Sarcoglycan is a transmembrane, dystrophin-associated protein expressed in skeletal and cardiac muscle. The murine γ-sarcoglycan gene was disrupted using homologous recombination. Mice lacking γ-sarcoglycan showed pronounced dystrophic muscle changes in early life. By 20 wk of age, these mice developed cardiomyopathy and died prematurely. The loss of γ-sarcoglycan produced secondary reduction of β- and δ-sarcoglycan with partial retention of α- and ε-sarcoglycan, suggesting that β-, γ-, and δ-sarcoglycan function as a unit. Importantly, mice lacking γ-sarco- glycan showed normal dystrophin content and local- ization, demonstrating that myofiber degeneration occurred independently of dystrophin alteration. Furthermore, β-dystroglycan and laminin were left intact, implying that the dystrophin–dystroglycan–laminin mechanical link was unaffected by sarcoglycan deficiency. Apoptotic myonuclei were abundant in skeletal muscle lacking γ-sarcoglycan, suggesting that programmed cell death contributes to myofiber degeneration. Vital staining with Evans blue dye revealed that muscle lacking γ-sarcoglycan developed membrane disruptions like those seen in dystrophin-deficient muscle. Our data demonstrate that sarcoglycan loss was sufficient, and that dystrophin loss was not necessary to cause membrane defects and apoptosis. As a common molecular feature in a variety of muscular dystrophies, sarcoglycan loss is a likely mediator of pathology.
Keywords: muscular dystrophy, sarcolemma, dystrophin, extracellular matrix, apoptosis
Progress in human genetics has identified a number of proteins at the muscle membrane that are associated with muscular dystrophy. Dystrophin, the protein product of the Duchenne muscular dystrophy (DMD)1 gene, is an elongated cytoskeletal protein that binds actin and a complex of dystrophin-associated proteins, forming the dystrophin-glycoprotein complex (DGC; Burghes et al., 1987; Campbell and Kahl, 1989; Ervasti and Campbell, 1991; Koenig et al., 1987; Monaco et al., 1986; Rybakova et al., 1996; Yoshida and Ozawa, 1990). More recently, a subcomplex of the DGC, sarcoglycan, was associated with mutations that result in human autosomal recessive muscular dystrophy (Bönnemann et al., 1996 a; Campbell, 1995; Duggan et al., 1997; Straub and Campbell, 1997). There are at least five subunits of sarcoglycan: α, β, γ, δ, and ε (Ettinger et al., 1997; Lim et al., 1995; McNally et al., 1998; Nigro et al., 1996; Noguchi et al., 1995; Roberds et al., 1993). Three of these sarcoglycans— β, γ, and δ—share homology as type II transmembrane proteins with cysteine-rich EGF-like repeats in their extracellular domains. Mutations in any single sarcoglycan gene result in varying degrees of secondary reduction of the other sarcoglycan units, suggesting interaction between the sarcoglycan subunits. α-sarcoglycan is a type I transmembrane protein expressed exclusively in skeletal and cardiac muscle. A widely expressed fifth sarcoglycan, ε-sarcoglycan, was recently identified with significant homology to α-sarcoglycan (Ettinger et al., 1997; McNally et al., 1998). ε-sarcoglycan is present in skeletal and cardiac muscle, but is also expressed in most tissues studied, demonstrating that sarcoglycan-like proteins are present in nonmuscle tissues.
In the presence of β-d-octyl-glycoside, the sarcoglycan subunits can be separated from the remainder of the DGC, but the exact interaction between sarcoglycan and the remainder of the DGC, particularly dystrophin, is ill-defined (Ozawa et al., 1995; Yoshida et al., 1994). Studies of human muscle biopsies have provided limited information regarding the nature of the sarcoglycan–dystrophin interaction. Quantitative assessment of dystrophin content showed a secondary decrease of dystrophin content in patients with mutations in β-, γ-, and δ-sarcoglycan; the reduction in dystrophin content ranged from 40 to 90% of normal (Vainzof et al., 1996). This finding suggests that a secondary decrease in dystrophin may be responsible for the pathology of muscular dystrophy in patients with primary sarcoglycan mutations.
In addition to the sarcoglycans, the DGC includes dystroglycan, a highly glycosylated protein whose α subunit directly binds the extracellular matrix (ECM) protein laminin (Ibraghimov-Beskrovnaya et al., 1992). Dystroglycan is widely expressed, and mice homozygously lacking dystroglycan have an early embryonic lethality (day 6.5 after conception) arising from the role of dystroglycan in nonmuscle tissue, particularly the extraembryonic structure Reichert's membrane (Williamson et al., 1997). Dystroglycan binds laminin-α2, an extracellular matrix protein expressed in peripheral nerve, skeletal, and cardiac muscle (Ervasti and Campbell, 1993; Yamada et al., 1994). In humans, laminin-α2 (merosin) mutations have been associated with a severe phenotype, congenital muscular dystrophy (Helbling-Leclerc et al., 1995; Nissinen et al., 1996). A murine model for laminin-α2 deficiency, the dy mouse, demonstrates a severe phenotype resulting in marked skeletal muscle degeneration and early death (Sunada et al., 1994; Xu et al., 1994). In contrast, the murine model for dystrophin deficiency, the mdx mouse (Sicinski et al., 1989), has a milder phenotype than its human counterpart. mdx mice have a normal lifespan, but demonstrate many of the histologic features seen in human muscular dystrophy, including fiber size variation and centrally placed nuclei. Human DMD patients exhibit replacement of myofibers with fatty and fibrous infiltration within the first decade, yet mdx mice show progressive fibrosis relatively later in life. Although the mdx mouse has a milder phenotype than human DMD patients, it has proved useful for studies identifying the nature of the membrane defect in muscular dystrophy. Vital staining studies of mdx and dy mice suggested different mechanisms of membrane degeneration since dystrophin deficiency produces membrane defects, while laminin deficiency did not (Matsuda et al., 1995; Straub et al., 1997). This observation and its cytoskeletal localization has lead to the hypothesis that dystrophin participates in a mechanical link that stabilizes muscle membrane. Contraction-induced force exerted on a membrane defective in dystrophin is thought to lead directly to membrane disruptions and myofiber degeneration.
To investigate the role of sarcoglycan in muscular dystrophy, we targeted the murine γ-sarcoglycan gene. In early life, mice lacking γ-sarcoglycan developed muscular dystrophy that preferentially affected the proximal musculature. Mice lacking γ-sarcoglycan developed cardiomyopathy that affected both the right and left ventricle. Vital staining of mice lacking γ-sarcoglycan revealed membrane disruptions demonstrating that sarcoglycan deficiency has a similar mechanism to dystrophin-deficiency and is unlike laminin deficiency. Membrane disruptions in γ-sarcoglycan-deficient muscle were primarily observed in proximal skeletal muscles similar to humans with limb-girdle muscular dystrophy (LGMD). Moreover, apoptotic myonuclei were abundant in γ-sarcoglycan–deficient muscle. Importantly, dystrophin, dystroglycan, and laminin were intact and normally present at the sarcolemma in γ-sarcoglycan– deficient muscle. Therefore, γ-sarcoglycan deficiency was sufficient to cause muscle membrane instability and the dystrophic process independently of dystrophin, placing sarcoglycan genetically downstream of dystrophin.
Materials and Methods
Gene Targeting
Genomic phage encoding exon 2 of γ-sarcoglycan was isolated from a murine 129SVJ library (Stratagene, La Jolla, CA) and characterized by restriction mapping, nucleotide sequencing, long-range PCR, and Southern hybridization. 5 kb of the first intron and 1.7 kb of the second intron were cloned into pPNT (Tybulewicz et al., 1991). Targeted RW4 ES cells were selected as described previously (Scott et al., 1994). Recombinants were screened by PCR and confirmed by Southern blotting of EcoRI-digested genomic DNA using a probe shown in Fig. 1 a. Recombinant ES cell clones were injected into C57BL/6 blastocysts and implanted into pseudopregnant foster mothers. Chimeric males were identified by coat color, and were mated to C57BL/6 females. Germline transmission was identified by coat color, and was confirmed by PCR and Southern blotting. PCR genotyping of progeny was performed on genomic DNA isolated from tail clippings using the primers shown in Fig. 1 a. Animals were housed and treated according to standards set by the University of Chicago Institutional Animal Care and Use Committee.
Figure 1.

Homologous recombination in the murine γ-sarcoglycan locus. (a) The strategy for replacing exon 2 of murine γ-sarcoglycan with neomycin phosphotransferase (neo) is shown. The probe used for Southern blotting and the primers (arrows) used for genotyping progeny from gsg +/− crosses are indicated. E, EcoRI; H, HindIII; N, NotI; Xb, XbaI; Xh, XhoI. (b) Southern blot of EcoRI-digested genomic DNA from ES cells. Homologous recombination in the γ-sarcoglycan locus produces the loss of a single EcoRI site. (c) PCR genotyping of progeny from gsg +/− interbreeding. The PCR product generated by the mutant allele is 410 bp, and that generated by the wild-type allele is 376 bp. (d) Immunoblotting of total protein from wild-type (lanes 1 and 5) and mutant gsg− /− (lanes 2 and 6) muscle with a monoclonal antibody directed at γ-sarcoglycan amino acids encoded by exon 6 (NCL-35DAG, lanes 1 and 2), or with a polyclonal anti-dystrophin antibody (AB6-10, lanes 5 and 6). No γ-sarcoglycan is detectable in gsg −/− muscle. Dystrophin is normal in gsg −/− muscle. Polyclonal antisera raised to the complete γ-sarcoglycan protein also shows the absence of γ-sarcoglycan protein in gsg −/− mice (data not shown). A Coomassie-stained gel, gsg +/+ (lane 3) and gsg −/−(lane 4), demonstrates equivalent loading. γ-sarcoglycan and dystrophin were resolved on 12.5% and 5% SDS-PAGE, respectively.
Immunoblotting
Immunoblots to detect γ-sarcoglycan protein were performed as described previously (Nicholson et al., 1989), using either affinity-purified polyclonal (McNally et al., 1996) or monoclonal (NCL-35DAG; NovoCastra Laboratories, Newcastle upon Tyne, UK) anti-γ-sarcoglycan.
General Characterization, Vital Staining, and Terminal Deoxynucleotidyl Transferase-mediated dUTP Nick End-labeling (TUNEL) Assay
Animals were observed for gait abnormalities weekly through 24 wk of age. Serum creatine kinase (CK) levels were measured on blood drawn from tail veins at 4 (n = 24) and 8 (n = 24) wk of age. Blood was collected in a Microstainer Serum Separator tube (Becton Dickinson, Franklin Lakes, NJ) and analyzed using a Vitros DT60II discrete chemistry analyzer (Eastman Kodak, Rochester, NY). Vital staining was performed as described previously using filter-sterilized Evans blue dye (Sigma-Aldrich, St. Louis, MO) resuspended in PBS at 1 mg/0.1 ml/10 g body weight (Matsuda et al., 1995; Straub et al., 1997). Intraperitoneal injections were performed on mice at 2 (n = 5), 4 (n = 4) and 8 (n = 4) wk of age; mice were killed 12 h after injection. TUNEL was performed on fixed, paraffin-embedded muscle using the ApopTag™-fluorescein kit (Oncor Inc., Gaithersburg, MD). Double-labeling with polyclonal anti-dystrophin antibodies was performed after TUNEL-labeling, as described below.
Histology and Immunocytochemistry
Muscle from 4-, 8-, and 20-wk-old animals was fixed for 24–48 h in 10% neutral buffered formalin and embedded in paraffin. 10-μM sections were cut and stained with Masson trichrome. Immunostaining was performed as described (McNally et al., 1996). The following primary antibodies were used: polyclonal anti-dystrophin AB6-10 (Lidov et al., 1990), polyclonal anti-γ-sarcoglycan (McNally et al., 1996), polyclonal anti-β-sarcoglycan (Bönnemann et al., 1996 b), monoclonal anti-α and γ-sarcoglycans (NCL-50DAG and NCL-35DAG, respectively), monoclonal anti-β-dystroglycan (NCL-β-DG), monoconal anti-utrophin (NCL-DRP2), and monoclonal anti-merosin (NCL-merosin; NovoCastra Laboratories). Polyclonal anti-δ-sarcoglycan was raised to a glutathione-S-transferase fusion protein containing amino acids 85–291 of the human protein (Genbank number X95191), as previously described (McNally et al., 1996). Polyclonal anti-ε-sarcoglycan was raised similarly using amino acid residues 31–413 of the human ε-sarcoglycan sequence (Genbank no. AF036364). Cy3-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) were used. Integrin-β1 antibody (MAB1997) was from Chemicon International, Inc. (Temecula, CA). Counterstaining with 4′,6-diamidino-2-phenylindole (DAPI) was performed at 20 μg/ml for 1 min. Sections were photographed on a Zeiss Axiophot epifluorescence microscope.
Electron Microscopy
Mice were perfusion-fixed with 2% paraformaldehyde and 2% gluteraldehyde. Skeletal muscle was processed for EM by postfixation with 1% osmium tetrozide in water for 1 h, washed in water, and dehydrated with ethanol and propylene oxide. Dehydrated tissues were embedded in plastic, sectioned at 60 nm, and stained with 5% uranyl acetate and 1% lead. Sections were viewed and photographed on a Hitachi 600 EM.
Results
Targeted Disruption of γ-Sarcoglycan and Generation of Null Mice
Given the large size of the γ-sarcoglycan gene, exon 2 was chosen for targeting. Exon 2 specifies the initiator methionine, the cytoplasmic tail, and the transmembrane domain. Homologous recombination replaces exon 2 with the phosphoglycerate kinase promoter/neomycin phosphotransferase cDNA (Fig. 1 a). A representative Southern blot of a targeted ES cell clone is shown in Fig. 1 b. Heterozygous gsg +/− mice were viable, fertile, and appeared healthy. gsg +/− mice were bred to obtain gsg −/− mice (Fig. 1 c). Immunoblot analysis showed no detectable γ-sarcoglycan protein in homozygous mutant gsg −/− muscle (Fig. 1 d), indicating that this mutation created a null allele. Notably, immunoblotting for dystrophin revealed that normal amounts of dystrophin remain in γ-sarcoglycan–deficient muscle (Fig. 1 d).
γ-Sarcoglycan-deficient Mice Develop Muscular Dystrophy
Most mutant gsg −/−mice were of normal size and appearance, but displayed a variably abnormal gait that was characterized by a stiff walking posture and a widened hind-leg spacing (Fig. 2 a). Less than 10% (4 of 50) of mutant gsg −/− mice had stunted growth or died as early as 3 wk of age. By 5 mo of age, 50% of gsg −/− animals died (Fig. 2 b). The average weight of mutant, heterozygous, and wild-type mice did not differ significantly at 8 wk of age. However, by 20 wk of age mutant mice weighed less than wild-type (28.3 ± 1.9 vs. 34.1 ± 0.3 g). Mutant mice were less active than wild-type littermates, exhibited less climbing and burrowing activity, and were slow to initiate walking or running from a sitting position. Elevation of serum CK, indicating disruption of muscle membrane integrity, was detected at 2 wk of age (1.7× wild-type controls). By 4 wk of age, serum CK levels were elevated in gsg −/− mice to an average of ten times that seen in normal mice (Fig. 2 c). This elevation continued in 8-wk-old gsg −/− mice and ranged from 3–240× the serum CK levels of normal controls (Fig. 2 c).
Figure 2.

Characterization of mice lacking γ-sarcoglycan. (a) Left, wild-type gsg +/+ mouse; right, gsg −/− mouse displaying stunted growth and abnormal stance. (b) Survival curve for gsg −/− vs. wild-type gsg +/+ littermates. By 4–5 mo of age, nearly 50% of gsg −/− animals die. (c) Serum creatine kinase (CK) levels of wild-type gsg +/+ (white), heterozygous gsg +/− (gray), and mutant gsg −/− (black) animals at 4 and 8 wk of age. At 4 wk, the mean serum CK was elevated ∼10× that seen in wild-type mice, and at 8 wk the mean CK was elevated more than 40× that seen in wild-type mice. The standard deviation (*) of serum CK values in 8-wk-old mice was ±22,830; 4/13 mice in this group had serum CK values of 17,750, 28,400, 33,400, and 80,500 U/liter (normal range 68– 1070 U/liter). (d) The percentage of myofibers with centrally placed nuclei is greatly increased in gsg −/−mice.
Diaphragm muscle from gsg−/− mice appeared hypertrophied and opaque when compared with gsg +/+ and gsg +/− littermates. The thickness of gsg −/− diaphragm muscle was greater than that of wild-type controls without an increase in myofiber size or content (Fig. 3 a). Such pseudohypertrophy is a hallmark of progressive proximal muscular dystrophy, and commonly affects the gastrocnemius muscles of patients with LGMD and DMD (Dubowitz, 1995). Histologic analyses of muscle from gsg −/− mice revealed severe dystrophic changes, including wide variation in fiber size, inflammatory infiltrate, and abnormal calcification with fatty and fibrous replacement. Dystrophic changes were evident throughout the entire diaphragm muscle of gsg− /− mice (Fig. 3 a). Quadriceps and gastrocnemius muscle also showed pronounced dystrophic changes (Fig. 3, b and c). In these muscles, unlike the diaphragm, regions of marked degeneration were juxtaposed with segments of intact myofibers. Most of these intact myofibers, however, had centrally placed nuclei consistent with regeneration having occurred. The number of centrally placed nuclei in 8-wk-old gsg− /−quadriceps muscle was greatly increased (45×) above wild-type littermate controls (Figs. 2 d and 3 d).
Figure 3.

Histologic analysis of skeletal muscle lacking γ-sarcoglycan. a–c show Masson Trichrome staining of muscle. (a) Low-power view of diaphragm muscle from normal (gsg +/+) and mutant (gsg −/−) mice. Of note, the magnification is equal in the two photos. Diaphragm tissue from gsg −/− mice was hypertrophied. Marked replacement by connective and adipose tissue is seen throughout the diaphragm of gsg −/− muscle. Areas of calcification are visible. (b) Low-power magnification of gsg −/− quadriceps muscle (right) shows regions of inflammatory infiltrate adjacent to normal appearing muscle. (a and b) Bar, 100 μm. (c) High-power view of quadriceps muscle showing an area of infiltrate and regeneration. (d) Immunostaining of muscle plasma membrane with an anti-dystrophin antibody counterstained with DAPI to reveal centrally placed nuclei seen in gsg −/− muscle. (c and d) Bar, 25 μm.
Cardiomyopathy in gsg− /− Mice
The hearts of 20-wk-old gsg −/− mice were examined. Both the right and left ventricular walls appeared thickened, resulting in near obliteration of the ventricular cavities. gsg −/− hearts weighed less than wild-type hearts (167 ± 17 vs. 190 ± 0 mg, but total body mass was decreased in gsg −/− mice). The heart-to-body mass index was increased (5.91 ± 0.30 mg/g for gsg −/− vs. 5.57 ± 0.05 mg/g for wild-type animals). Mice examined at earlier time points, including 4 and 8 wk of age, did not show the same degree of cardiomyopathy (data not shown). Fig. 4 a shows representative hearts from 20-wk-old gsg −/− animals that were killed for the study. Mutant hearts showed marked thickening of the ventricular walls; the heart in Fig. 4 a also showed significant fibrosis. Fibrosis was evident throughout the right and left ventricle, but a prominent fibrotic area was present in the posterior segment of the left ventricle (Fig. 4 c). Perivascular regions of fibrosis were also seen (Fig. 4 d). Fibrosis was not seen in gsg −/− hearts at 4 and 8 wk of age (data not shown). Of a cohort of ten gsg− /− mice, five died before 5 mo of age. Postmortem analysis of these gsg −/− mice revealed areas of ventricular fibrosis that were visible on gross examination (data not shown).
Figure 4.
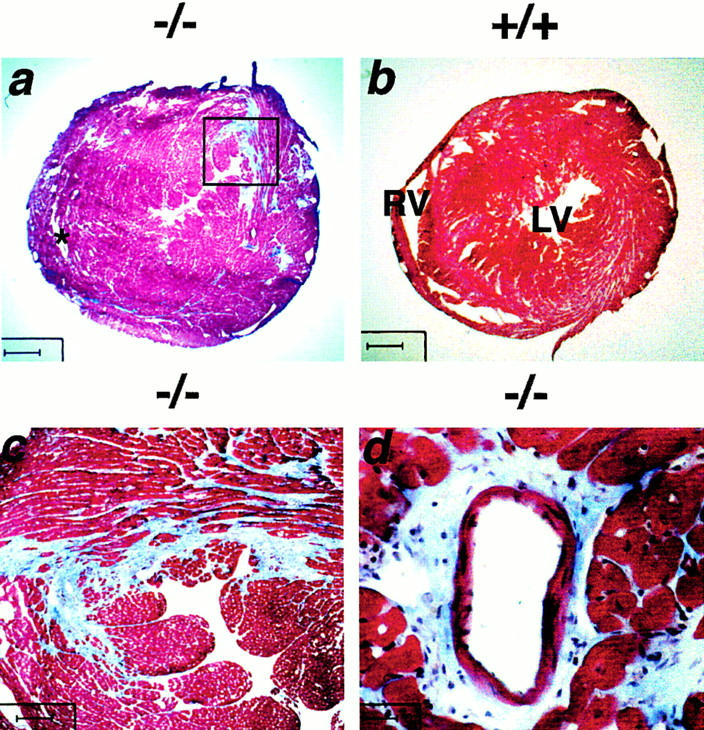
Cardiomyopathy in gsg −/−mice. (a and b) Masson trichrome–stained hearts taken from 20-wk-old littermates. Bar, 625 μm. LV, left ventricle; RV, right ventricle. (a) A representative gsg −/− heart with significant increases in right and left ventricular wall thickness. (c) The region within the box of a. The gsg− /− heart shown in a shows marked fibrosis, seen as blue staining. Wall thickness of the right ventricle is markedly increased (a); the right ventricular cavity is marked with an asterisk. (c) Bar, 100 μm. (d) A higher magnification view of perivascular fibrosis is seen. (d) Bar, 25 μm.
Sarcoglycan Loss Causes Muscular Dystrophy Without Alteration in Dystrophin
Immunostaining of γ-sarcoglycan–deficient and dystrophin-deficient skeletal muscle is shown in Fig. 5 a. mdx muscle showed a marked secondary deficiency of β-, γ-, and δ-sarcoglycan in addition to the primary defect of dystrophin loss. In gsg −/− muscle, dystrophin staining is intact, but β- and δ-sarcoglycan show severe secondary reduction at the sarcolemma. In mdx muscle, revertant fibers are commonly seen that reexpress dystrophin. Dystrophin-positive revertant fibers in mdx also reexpress sarcoglycan as shown for δ-sarcoglycan (Fig. 5 a, bottom middle). No such revertant phenomenon is seen in gsg −/− muscle. Partial retention of α-sarcoglycan at the sarcolemma was seen (Fig. 5 b, top). Immunoblotting for α-sarcoglycan shows that 10–30% of α-sarcoglycan is expressed in gsg −/− muscle (Fig. 5 c). Normal levels of ε-sarcoglycan are present in gsg −/− muscle, yet ε-sarcoglycan is completely absent in mdx muscle. β-dystroglycan and laminin-α2 (merosin) staining was intact at the sarcolemma of gsg −/− muscle. In muscle lacking dystrophin, the dystrophin-related protein utrophin is upregulated to compensate for the lack of dystrophin protein (Helliwell et al., 1992; Karpati et al., 1993; Love et al., 1989). Utrophin staining in gsg −/− muscle is unchanged from wild-type controls, consistent with a normal dystrophin content in γ-sarcoglycan–deficient animals. Integrin-β1 was also unchanged in gsg −/− muscle.
Figure 5.

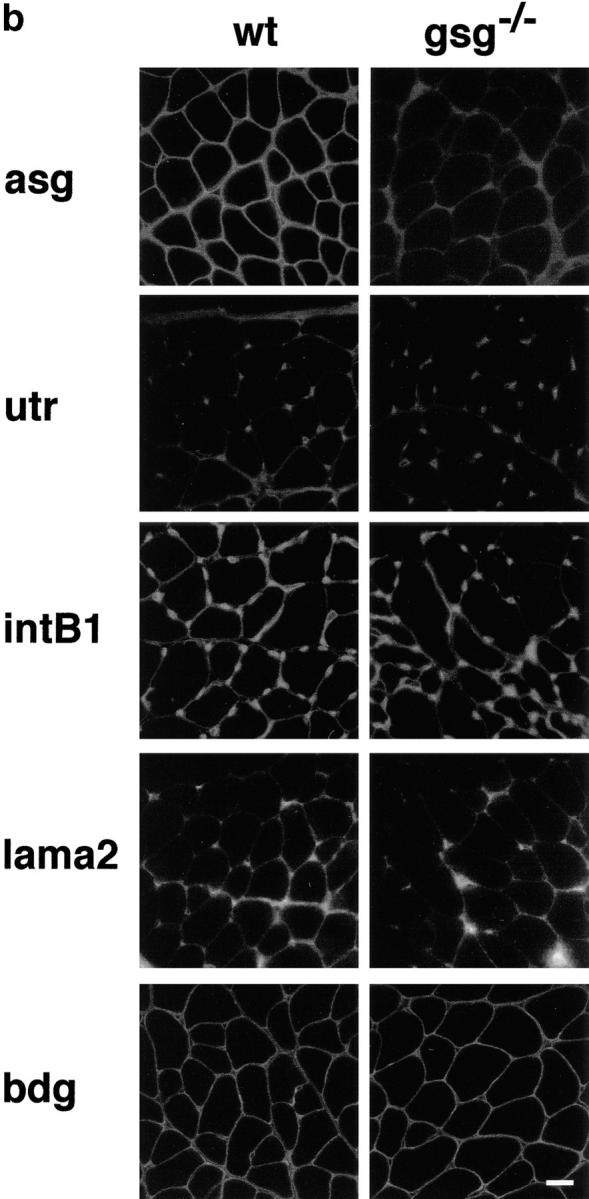
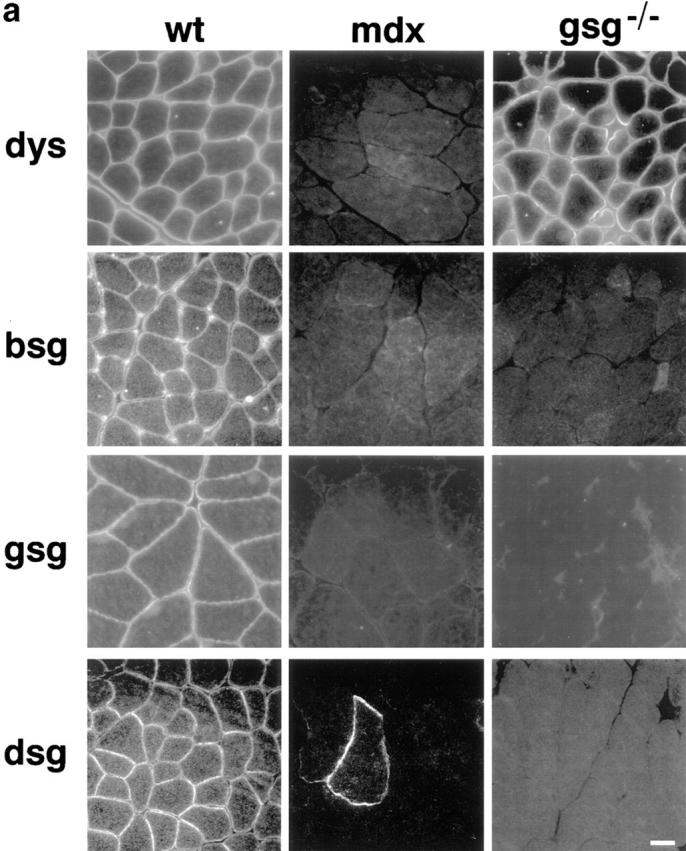
Immunostaining for components of the dystrophin–glycoprotein complex (DGC) in mdx and gsg −/− muscle. (a) Immunostaining for dystrophin (dys), β-sarcoglycan (bsg), γ-sarcoglycan (gsg), and δ-sarcoglycan (dsg). β-, γ, and δ-sarcoglycan are absent in both dystrophin-deficient muscle (mdx) and γ-sarcoglycan deficient muscle (gsg −/−). Revertant fibers that regain the ability to express dystrophin are commonly seen in mdx muscle. These revertant fibers also regain the ability to express sarcoglycan (shown for δ-sarcoglcyan). Dystrophin remains present at the membrane in gsg −/− muscle. (b) Immunostaining for α-sarcoglycan (asg), utrophin (utr), integrin-β1 (intB1) laminin-α2 (merosin, lama2) and β-dystroglycan (bdg) in normal and gsg −/− muscle. The dystrophin-related protein utrophin is also normal at the membrane of gsg −/− muscle as is laminin-α2 (merosin) and dystroglycan. Bar, 25 μm. (c) Immunoblotting with α-sarcoglycan and ε-sarcoglycan antibodies in gsg −/− and mdx muscle. α-sarcoglycan is partially retained in gsg−/− muscle. ε-sarcoglycan is completely absent from mdx muscle, while it is normal in gsg −/− muscle. (Lanes 1–3) Immunoblotting with anti-α-sarcoglycan monoclonal antibody. Lane 1, normal muscle; lane 2, gsg −/− muscle; lane 3, mdx muscle.
Membrane Defects in Muscular Dystrophy
Evans blue dye (EBD) is a small molecular mass tracer that tightly complexes with serum albumin. Normal muscle fibers are impermeable to the EBD–albumin complex, while mdx muscle fibers are positively stained, reflecting a loss of membrane integrity (Matsuda et al., 1995; Straub et al., 1997). EBD was injected into mutant and normal mice to determine the extent sarcolemmal disruption as a result of sarcoglycan deficiency. No EBD staining was seen in gsg +/+ and gsg +/−animals at any age (data not shown). In contrast, 2-wk-old gsg −/− mice showed EBD staining of the diaphragm, shoulder girdle, and pectoralis major muscles on gross examination (data not shown). By 4 wk of age, gsg −/− animals displayed significant staining in nearly all muscle groups examined. Bands of EBD-positive tissue gave the gsg −/− diaphragm a striking, radially streaked appearance not seen in wild-type littermate controls (Fig. 6 a). The adductor muscles of the leg were commonly EBD-positive, as were the pectoralis major, latissimus dorsi, rectus abdominus, external oblique, and triceps brachii muscles. Mutant muscles lacking macroscopic evidence of EBD uptake showed microscopic evidence of EBD-positive fiber clusters demonstrating the extensive nature of the dystrophic process in gsg −/− mice (Fig. 6 b).
Figure 6.

Vital staining with Evans blue dye (EBD) reveals disruption of membrane integrity in sarcoglycan-deficient muscle. Mice were injected with EBD and killed after 12 h. (a) Low-power magnification (5×) of diaphragm muscle from EBD-injected wild-type and mutant mice. Diaphragm muscle from gsg −/− mice exhibits segmental EBD uptake (seen as blue areas) while no EBD uptake is seen in wild-type diaphragm. (b) Regions of EBD uptake are seen as red cytoplasmic staining on fluorescence microscopy. No evidence of EBD microscopic staining is seen in gsg +/+ muscle (data not shown). (Left) a region of gsg −/− triceps muscle with marked EBD staining (black bar, 100 μm). (Right) A higher power view of gsg −/− gastrocnemius muscle showing EBD staining (white bar, 25 μm). (c) Apoptosis in γ-sarcoglycan–deficient muscle. TUNEL-labeling was performed on quadriceps muscle from a gsg −/− mouse. TUNEL-positive nuclei appear green. No TUNEL-positive nuclei were found in normal gsg +/+ myofibers (left). In contrast, myofibers with TUNEL-positive nuclei were seen commonly in gsg− /− muscle (right). (d) Counterstaining with an anti-dystrophin antibody (yellow-orange) demonstrates that TUNEL-positive nuclei are found within gsg− /− myofibers. Myofibers undergoing degeneration and demonstrating decreased membrane staining for dystrophin also show evidence of apoptosis.
Myofiber Loss is Mediated by Apoptosis
To investigate the mechanism of myofiber degeneration in γ-sarcoglycan deficiency, TUNEL was performed on quadriceps muscle from 8-wk-old gsg +/+, gsg +/−, and gsg −/− mice. No TUNEL-positive myonuclei were detected in gsg +/+ and gsg +/− skeletal muscle (Fig. 6 c, left). In contrast, TUNEL-positive nuclei were commonly found in gsg −/− muscle (Fig. 6 c, right), indicating that apoptosis contributed to muscle degeneration in γ-sarcoglycan–deficient muscular dystrophy. TUNEL-positive nuclei were most commonly seen in degenerating areas of gsg −/− muscle, but were seen also in intact or partially degenerated muscle fibers (Fig. 6 d). The presence of TUNEL-positive myonuclei in intact, dystrophin-positive muscle fibers suggests that sarcoglycan deficiency leads to apoptosis as an early event in the dystrophic process.
Normal Sarcomeres, Neuromuscular Junctions, and Basement Membrane in γ-Sarcoglycan Deficiency
The sarcoglycans are found throughout the sarcolemma, and are not excluded at the neuromuscular junctions. The sarcomeres, membrane, and neuromuscular junctions of gsg −/− mice were examined by EM and compared with wild-type littermates. No ultrastructural differences were seen in the sarcomeres, membrane, or neuromuscular junctions of gsg −/− mice (Fig. 7), suggesting that membrane disruptions visualized by EBD staining are below the limit of resolution of EM. Gross ultrastructural abnormalities are not a feature of sarcoglycan deficiency.
Figure 7.
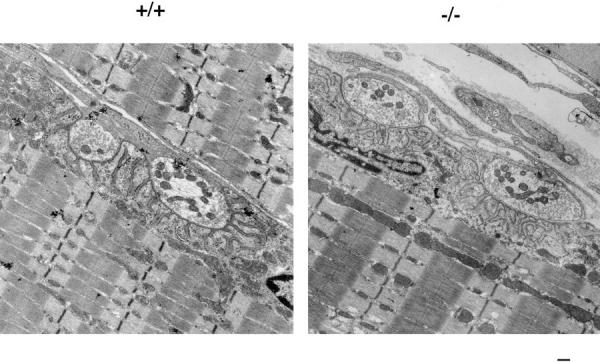
Ultrastructural analysis of gsg −/− and normal muscle. (5,000×) Neuromuscular junction, sarcomeres, and membrane from gsg −/− and wild-type muscle (gsg +/+) shows that neuromuscular junctions form normally with normal post synaptic folds. The sarcolemma is intact throughout its length, and the basement membrane is continuous and apparently normal at this level of ultrastructure. Sarcomere formation is normal in gsg −/− muscle. Bar, 200 nm.
Discussion
γ-Sarcoglycan is Critical for Cardiac and Skeletal Muscle
We have developed mice lacking γ-sarcoglycan through homologous recombination in embryonic stem cells. Unlike mdx mice that display a more mild phenotype than DMD patients, mice lacking γ-sarcoglycan develop a phenotype that closely parallels the human disease exhibiting the clinical and histopathologic features of LGMD, including pseudohypertrophy, elevated serum CK, degeneration, and regeneration of skeletal muscle. Furthermore, gsg −/− mice develop marked cardiomyopathy that is likely to play a role in their early death. The degree of left ventricular wall thickening is consistent with an intrinsic cardiomyopathy that is likely hypertrophic in nature. The presence of marked fibrosis in the posterior segment of the left ventricle likely arises from similar pathogenetic mechanisms that affect skeletal muscle in sarcoglycan deficiency. It is possible that wall thickening seen in the right ventricle of gsg −/− hearts arises in part as a secondary consequence of restrictive lung disease and elevated pulmonary vascular resistance. The marked dystrophic process that affects the respiratory muscles, particularly the diaphragm, may lead to a restrictive lung disease and hence, a secondary cardiomyopathy.
The phenotype in mice lacking γ-sarcoglycan is similar to that seen in the Syrian hamster, BIO 14.6, that displays both cardiomyopathy and muscular dystrophy (Homburger et al., 1962). The BIO 14.6 hamster is a spontaneously arising mutant that has a large, not fully defined deletion that encompasses part of an alternative first exon of the hamster δ-sarcoglycan gene (Nigro et al., 1997; Sakamoto et al., 1997). Recently, membrane defects as observed by EBD staining were described in the BIO 14.6 hamster (Holt et al., 1998). The similar phenotype in gsg −/− mice and the BIO 14.6 hamster argues that both γ- and δ-sarcoglycan have critical and nonredundant roles for cardiac and skeletal muscle. The cardiomyopathic phenotype of gsg −/− mice is strikingly different from that seen in mdx mice since mdx mice display little to no cardiomyopathy. This result further suggests that the sarcoglycans, particularly γ- and δ-sarcoglycan, are critical for cardiac muscle function.
gsg −/− mice display a more severe skeletal muscle phenotype than mdx mice since gsg −/− mice show a greater serum CK elevation, early and more extensive fibrosis, and a higher percentage of centrally placed nuclei (McArdle et al., 1994; Deconinck et al., 1997). mdx mice show a period of intense muscle necrosis at 3–8 wk of age followed by a relative plateau period. gsg −/− mice show marked necrosis at 3–8 wk of age. Areas of necrosis were commonly seen in gsg −/− muscle from older animals, but regions of necrosis appeared less often than in 8-wk animals. Serum CK in gsg −/− animals peaks at 8 wk of age, but remain grossly elevated at 16 wk of age at levels twice that seen in mdx (data not shown).
gsg −/− mice die at a similar age as the recently described double knockout mice that lack both dystrophin and utrophin (Deconinck et al., 1997; Grady et al., 1997). However, gsg −/− mice do not show gross skeletal abnormalities such as kyphoscoliosis described in the double knockout mice.
Interactions Within the DGC
The absence of γ-sarcoglycan produces a nearly complete secondary reduction of both β- and δ-sarcoglycan. β- and δ-sarcoglycan mRNA was normally produced in gsg −/− myofibers (data not shown). Thus, γ-sarcoglycan is necessary for the proper assembly and membrane localization of β- and δ-sarcoglycan proteins. Curiously, α-sarcoglycan appears to be partially retained in gsg −/− muscle, suggesting that other components can stabilize α-sarcoglycan at the membrane. ε-sarcoglycan is completely absent in mdx muscle, while it is produced at normal levels in γ-sarcoglycan deficiency. Since ε-sarcoglycan expression mirrors dystrophin expression in mdx and gsg −/− muscle, ε-sarcoglycan may be more closely associated with dystrophin.
Most importantly, the normal placement of dystrophin, β-dystroglycan, and laminin-α2 (merosin) in γ-sarcoglycan–deficient muscle indicates that this cytoskeleton– membrane–ECM interaction is intact and unaffected by γ-sarcoglycan deficiency. The normal ultrastructural appearance of gsg −/− sarcomeres, basement membrane, and neuromuscular junctions may favor alternative cellular defects such as aberrant cell signaling as a mediator of muscular dystrophy. Sarcoglycan has the primary structure suggestive of a cell surface receptor. The cytoplasmic domain of γ-sarcoglycan has five tyrosine residues, and recent studies imply bidirectional signaling with integrins may involve the sarcoglycan subunits (Yoshida et al., 1998). Identifying the extracellular and intracellular ligands that normally bind the subunits of sarcoglycan will be an important step in defining any potential cytoskeleton– sarcoglycan–ECM complex or signaling cascade.
Sarcoglycan Deficiency Mediates Pathology in DMD
In DMD and mdx, primary dystrophin mutations lead to a secondary reduction or mislocalization of a number of dystrophin-associated proteins, including the sarcoglycan subunits (Ohlendieck and Campbell, 1992; Ohlendieck et al., 1993). In mice lacking γ-sarcoglycan, the normal content and location of dystrophin suggests that sarcoglycan deficiency is sufficient to cause muscular dystrophy. It is clear that in gsg −/− muscle, the presence of dystrophin is insufficient to stabilize sarcoglycan, and cannot prevent the dystrophic process in both cardiac and skeletal muscle. Given the macromolecular nature of the DGC, the absence of sarcoglycan may lead to abnormal interactions within the DGC and the presence of muscular dystrophy. Whether restoration of the sarcoglycan complex, specifically β-, γ-, and δ-sarcoglycan will be sufficient to prevent the dystrophic phenotype is unknown. Previous data suggested that restoration of the DGC is not sufficient to prevent the dystrophic phenotype (Cox et al., 1994; Greenberg et al., 1994). However, DGC analysis in those studies was before the discovery of β-, γ-, and δ-sarcoglycan. Since sarcoglycan deficiency is the common feature in both DMD and LGMD, it is a likely mediator of the dystrophic process in both these disorders. Moreover, inhibiting apoptosis or developing new strategies to stabilize the sarcoglycan subunits, particularly β-, γ-, and δ-sarcoglycan, are reasonable therapeutic approaches to the treatment of Duchenne and the Duchenne-like muscular dystrophies.
Acknowledgments
We thank Celeste Simon, Renée Hackenmiller, and Jeff Leiden for advice and comments, and Lisa Gottschalk for her help preparing figures.
Abbreviations used in this paper
- DMD
Duchenne muscular dystrophy
- LGMD
limb girdle muscular dystrophy
- CK
creatine kinase
- EBD
Evans blue dye
- DGC
dystrophin-glycoprotein complex
- ECM
extracellular matrix
Footnotes
A.A. Hack is supported by the Medical Scientist Training Program and the Training Program in Development Biology. E.M. McNally is supported by the National Institues of Health, Muscular Dystrophy Association, Heart Research Foundation, and a Research Resources award from the Howard Hughes Medical Institute to the University of Chicago Biological Sciences Division.
Address all correspondence to Elizabeth M. McNally, M.D., Ph.D., Department of Medicine, Section of Cardiology, 5841 S. Maryland Ave., MC 6088, The University of Chicago, Chicago, IL 60637. Tel.: (773) 702-2672. Fax: (773) 702-2681. E-mail: emcnally@medicine.bsd.uchicago.edu
References
- Bönnemann CG, McNally EM, Kunkel LM. Beyond dystrophin: current progress in the muscular dystrophies. Curr Opin Pediatrics. 1996;8:569–582. [PubMed] [Google Scholar]
- Bönnemann CG, Passos-Bueno MR, McNally EM, Vainzof M, de Sa E, Moreira, Marie SK, Pavanello RC, Noguchi S, Ozawa E, Zatz M, Kunkel LM. Genomic screening for beta-sarcoglycan gene mutations: missense mutations may cause severe limb-girdle muscular dystrophy type 2E (LGMD 2E) Am J Hum Genet. 1996;59:1040–1047. doi: 10.1093/hmg/5.12.1953. [DOI] [PubMed] [Google Scholar]
- Burghes AH, Logan C, Hu X, Belfall B, Worton RG, Ray PN. A cDNA clone from the Duchenne/Becker muscular dystrophy gene. Nature. 1987;328:434–437. doi: 10.1038/328434a0. [DOI] [PubMed] [Google Scholar]
- Campbell KP. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- Cox GA, Sunada Y, Campbell KP, Chamberlain JS. Dp71 can restore the dystrophin-associated glycoprotein complex in muscle but fails to prevent dystrophy. Nat Genet. 1994;8:333–339. doi: 10.1038/ng1294-333. [DOI] [PubMed] [Google Scholar]
- Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- Dubowitz, V. 1995. Muscle Disorders in Childhood. First edition. W.B. Saunders Co., Ltd., London. 19–50.
- Duggan DJ, Gorospe JR, Fanin M, Hoffman EP, Angelini C. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med. 1997;336:618–624. doi: 10.1056/NEJM199702273360904. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger AJ, Feng G, Sanes JR. Epsilon-sarcoglycan, a broadly expressed homologue of the gene mutated in limb-girdle muscular dystrophy 2D. J Biol Chem. 1997;272:32534–32538. doi: 10.1074/jbc.272.51.32534. [DOI] [PubMed] [Google Scholar]
- Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- Greenberg DS, Sunada Y, Campbell KP, Yaffe D, Nudel U. Exogenous Dp71 restores the levels of dystrophin associated proteins but does not alleviate muscle damage in mdx mice. Nat Genet. 1994;8:340–344. doi: 10.1038/ng1294-340. [DOI] [PubMed] [Google Scholar]
- Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, Tomé FM, Schwartz K, Fardeau M, Tryggvason K, Guicheney P. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin- deficient congenital muscular dystrophy. Nat Genet. 1995;11:216–218. doi: 10.1038/ng1095-216. [DOI] [PubMed] [Google Scholar]
- Helliwell TR, Man NT, Morris GE, Davies KE. The dystrophin-related protein, utrophin, is expressed on the sarcolemma of regenerating human skeletal muscle fibres in dystrophies and inflammatory myopathies. Neuromusc Dis. 1992;2:177–184. doi: 10.1016/0960-8966(92)90004-p. [DOI] [PubMed] [Google Scholar]
- Holt KH, Lim LE, Straub V, Venzke DP, Duclos F, Anderson RD, Davidson BL, Campbell KP. Functional rescue of the sarcoglycan complex in the BIO 14.6 hamster using δ-sarcoglycan gene transfer. Mol Cell. 1998;1:841–848. doi: 10.1016/s1097-2765(00)80083-0. [DOI] [PubMed] [Google Scholar]
- Homburger F, Baker JR, Nixon CW, Whitney R. Primary, generalized polymyopathy and cardiac necrosis in an inbred line of Syrian hamsters. Med Exp. 1962;6:339–345. doi: 10.1001/archinte.1962.03620230106015. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- Karpati G, Carpenter S, Morris GE, Davies KE, Guerin C, Holland P. Localization and quantitation of the chromosome 6-encoded dystrophin-related protein in normal and pathological human muscle. J Neuropath Exp Neurol. 1993;52:119–128. doi: 10.1097/00005072-199303000-00004. [DOI] [PubMed] [Google Scholar]
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- Lidov HG, Byers TJ, Watkins SC, Kunkel LM. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature. 1990;348:725–728. doi: 10.1038/348725a0. [DOI] [PubMed] [Google Scholar]
- Lim LE, Duclos F, Broux O, Bourg N, Sunada Y, Allamand V, Meyer J, Richard I, Moomaw C, Slaughter C, et al. Beta-sarcoglycan: characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet. 1995;11:257–265. doi: 10.1038/ng1195-257. [DOI] [PubMed] [Google Scholar]
- Love DR, Hill DF, Dickson G, Spurr NK, Byth BC, Marsden RF, Walsh FS, Edwards YH, Davies KE. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature. 1989;339:55–58. doi: 10.1038/339055a0. [DOI] [PubMed] [Google Scholar]
- Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem. 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- McArdle A, Edwards RHT, Jackson MJ. Time course of changes in plasma membrane permeability in the dystrophin-deficient mdxmouse. Muscle Nerve. 1994;17:1378–1384. doi: 10.1002/mus.880171206. [DOI] [PubMed] [Google Scholar]
- McNally EM, Duggan D, Gorospe JR, Bönnemann CG, Fanin M, Pegoraro E, Lidov HG, Noguchi S, Ozawa E, Finkel RS, et al. Mutations that disrupt the carboxyl-terminus of gamma-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996;5:1841–1847. doi: 10.1093/hmg/5.11.1841. [DOI] [PubMed] [Google Scholar]
- McNally EM, Ly CT, Kunkel LM. Human epsilon sarcoglycan is highly related to alpha-sarcoglycan (adhalin), the limb girdle muscular dystrophy type 2D gene. FEBS Lett. 1998;422:27–32. doi: 10.1016/s0014-5793(97)01593-7. [DOI] [PubMed] [Google Scholar]
- Monaco AP, Neve RL, Colletti FC, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646–650. doi: 10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- Nicholson LV, Davison K, Falkous G, Harwood C, O'Donnell E, Slater CR, Harris JB. Dystrophin in skeletal muscle. I. Western blot analysis using a monoclonal antibody. J Neurol Sci. 1989;94:125–136. doi: 10.1016/0022-510x(89)90223-2. [DOI] [PubMed] [Google Scholar]
- Nigro V, Piluso G, Belsito A, Politano L, Puca AA, Papparella S, Rossi E, Viglietto G, Esposito MG, Abbondanza C, et al. Identification of a novel sarcoglycan gene at 5q33 encoding a sarcolemmal 35 kDa glycoprotein. Hum Mol Genet. 1996;5:1179–1186. doi: 10.1093/hmg/5.8.1179. [DOI] [PubMed] [Google Scholar]
- Nigro V, Okazaki Y, Belsito A, Piluso G, Matsuda Y, Politano L, Nigro G, Ventura C, Abbondanza C, Molinari AM, et al. Identification of the Syrian hamster cardiomyopathy gene. Hum Mol Genet. 1997;6:601–607. doi: 10.1093/hmg/6.4.601. [DOI] [PubMed] [Google Scholar]
- Nissinen M, Helbling-Leclerc A, Zhang X, Evangelista T, Topaloglu H, Cruaud C, Weissenbach J, Fardeau M, Tomé FM, et al. Substitution of a conserved cysteine-996 in a cysteine-rich motif of the laminin alpha2-chain in congenital muscular dystrophy with partial deficiency of the protein. Am J Hum Genet. 1996;58:1177–1184. [PMC free article] [PubMed] [Google Scholar]
- Noguchi S, McNally EM, Ben K, Othmane, Hagiwara Y, Mizuno Y, Yoshida M, Yamamoto H, Bönnemann CG, Gussoni E, et al. Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270:819–822. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- Ohlendieck K, Campbell KP. Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol. 1992;115:1685–1694. doi: 10.1083/jcb.115.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlendieck K, Matsumura K, Ionasescu VV, Towbin JA, Bosch EP, Weinstein SL, Sernett SW, Campbell KP. Duchenne muscular dystrophy: deficiency of dystrophin-associated proteins in the sarcolemma. Neurology. 1993;43:795–800. doi: 10.1212/wnl.43.4.795. [DOI] [PubMed] [Google Scholar]
- Ozawa E, Yoshida M, Suzuki A, Mizuno Y, Hagiwara Y, Noguchi S. Dystrophin-associated proteins in muscular dystrophy. Hum Mol Genet. 1995;4:1711–1716. doi: 10.1093/hmg/4.suppl_1.1711. [DOI] [PubMed] [Google Scholar]
- Roberds SL, Anderson RD, Ibraghimov-Beskrovnaya O, Campbell KP. Primary structure and muscle-specific expression of the 50-kDa dystrophin-associated glycoprotein (adhalin) J Biol Chem. 1993;268:23739–23742. [PubMed] [Google Scholar]
- Rybakova IN, Amann KJ, Ervasti JM. A new model for the interaction of dystrophin with F-actin. J Cell Biol. 1996;135:661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto A, Ono K, Abe M, Jasmin G, Eki T, Murakami Y, Masaki T, Toyo-oka T, Hanaoka F. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, delta-sarcoglycan, in hamster: an animal model of disrupted dystrophin-associated glycoprotein complex. Proc Natl Acad Sci USA. 1997;94:13873–13878. doi: 10.1073/pnas.94.25.13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott E, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Geng Y, Ryder C, Barnard E, Darlison M, Barnard P. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- Straub V, Campbell KP. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr Opin Neurol. 1997;10:168–175. doi: 10.1097/00019052-199704000-00016. [DOI] [PubMed] [Google Scholar]
- Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunada Y, Bernier SM, Kozak CA, Yamada Y, Campbell KP. Deficiency of merosin in dystrophic dy mice and genetic linkage of laminin M chain gene to dy locus. J Biol Chem. 1994;269:13729–13732. [PubMed] [Google Scholar]
- Tybulewicz V, Crawford C, Jackson P, Bronson R, Mulligan R. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Vainzof M, Passos-Bueno MR, Canovas M, Moreira ES, Pavanello RC, Marie SK, Anderson LV, Bonnemann CG, McNally EM, Nigro V, et al. The sarcoglycan complex in the six autosomal recessive limb-girdle muscular dystrophies. Hum Mol Genet. 1996;5:1963–1969. doi: 10.1093/hmg/5.12.1963. [DOI] [PubMed] [Google Scholar]
- Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, Ibraghimov-Beskrovnaya O, Campbell KP. Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1-null mice. Hum Mol Genet. 1997;6:831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- Xu H, Christmas P, Wu XR, Wewer UM, Engvall E. Defective muscle basement membrane and lack of M-laminin in the dystrophic dy/dy mouse. Proc Natl Acad Sci USA. 1994;91:5572–5576. doi: 10.1073/pnas.91.12.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Shimizu T, Tanaka T, Campbell KP, Matsumura K. Dystroglycan is a binding protein of laminin and merosin in peripheral nerve. FEBS Lett. 1994;352:49–53. doi: 10.1016/0014-5793(94)00917-1. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Ozawa E. Glycoprotein complex anchoring dystrophin to sarcolemma. J Biochem. 1990;108:748–752. doi: 10.1093/oxfordjournals.jbchem.a123276. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Suzuki A, Yamamoto H, Noguchi S, Mizuno Y, Ozawa E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl beta-D-glucoside. Eur J Biochem. 1994;222:1055–1061. doi: 10.1111/j.1432-1033.1994.tb18958.x. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Pan Y, Hanada H, Iwata Y, Shigekawa M. Bidirectional Signaling between Sarcoglycans and the Integrin Adhesion System in Cultured L6 Myocytes. J Biol Chem. 1998;273:1583–1590. doi: 10.1074/jbc.273.3.1583. [DOI] [PubMed] [Google Scholar]