

Abstract
Two-dimensional crystallization on lipid monolayers is a versatile tool to obtain structural information of proteins by electron microscopy. An inherent problem with this approach is to prepare samples in a way that preserves the crystalline order of the protein array and produces specimens that are sufficiently flat for high-resolution data collection at high tilt angles. As a test specimen to optimize the preparation of lipid monolayer crystals for electron microscopy imaging, we used the S-layer protein sbpA, a protein with potential for designing arrays of both biological and inorganic materials with engineered properties for a variety of nanotechnology applications. Sugar embedding is currently considered the best method to prepare two-dimensional crystals of membrane proteins reconstituted into lipid bilayers. We found that using a loop to transfer lipid monolayer crystals to an electron microscopy grid followed by embedding in trehalose and quick-freezing in liquid ethane also yielded the highest resolution images for sbpA lipid monolayer crystals. Using images of specimens prepared in this way we could calculate a projection map of sbpA at 7 Å resolution, one of the highest resolution projection structures obtained with lipid monolayer crystals to date.
Keywords: electron crystallography, S-layer protein, lipid monolayers, sugar embedding
1. Introduction
Fromherz was the first to demonstrate that soluble proteins can form ordered arrays on the surface of a lipid monolayer at the air/water interface and that these arrays can be imaged by electron microscopy (EM) (Fromherz, 1971). Subsequently, the lipid monolayer crystallization method was used to make soluble proteins amenable to structural studies by electron crystallography (Uzgiris and Kornberg, 1983). The underlying principle of array formation is that association of a target protein with the lipid monolayer leads to concentration and partial alignment of the protein. Since the lipid monolayer is in its fluid phase, the lipids, and hence the associated proteins, can diffuse in the plane of the monolayer, allowing the proteins to interact with each other and under favorable conditions to form regular arrays. The lipid monolayer is usually formed with neutral lipids spiked either with charged lipids to induce association of proteins by electrostatic interactions (Darst et al., 1988; Mosser et al., 1991; Taylor and Taylor, 1993; Taylor and Taylor, 1999) or with functionalized lipids, such as lipids containing Ni-NTA headgroups that specifically recruit His-tagged proteins to the monolayer (Kubalek et al., 1994). Once introduced, the method was subsequently adapted for use with membrane proteins (Lévy et al., 1999; Lebeau et al., 2001) and for the assembly of protein complexes (Celia et al., 1999; Kelly and Taylor, 2005; Kelly et al., 2006).
Although it was possible to record high-resolution (∼3 Å) EM data of streptavidin crystals on a lipid monolayer (Kubalek et al., 1991; Avila-Sakar and Chiu, 1996), a persistent problem in the use of this technique lies in the transfer of lipid monolayer crystals to an EM grid without deteriorating the order of the protein arrays. In addition, lipid monolayer samples are often not flat, and to date no high-resolution images have been reported for tilted lipid monolayer crystals. A reliable protocol to reproducibly prepare specimens for high-resolution EM data collection would thus have the potential to boost interest in lipid monolayer crystallization for structural studies of soluble proteins.
Once crystals have formed on the lipid monolayer, often using a Teflon crystallization block (Fig. 1A), they can be transferred to an EM grid using the direct transfer method (Fig. 1B, C), historically referred to as the Langmuir-Schaefer transfer method (Langmuir and Schaefer, 1938). For this technique, a hydrophobic carbon film mounted on an EM grid is brought into direct contact with the hydrophobic tails of the lipids in the monolayer (Fig. 1B). When the grid is lifted off, the monolayer and hence the associated crystals adhere to the carbon film and are thus transferred to the grid (Fig. 1C). The crystals on the grid can then be blotted and either negatively stained or vitrified for subsequent observation in the electron microscope. However, touching a lipid monolayer with a carbon film imparts mechanical force that tends to distort the attached crystals (Brisson et al., 1999). Thus, a holey grid is often used in conjunction with direct transfer and specimen vitrification. Images are then taken in regions without carbon film, where the crystals should be least affected. Although variations of this technique have yielded both the highest resolution lipid monolayer crystals to date (Kubalek et al., 1991; Avila-Sakar and Chiu, 1996) as well as three-dimensional (3D) reconstructions at lower resolutions (Tang et al., 2001; Wendt et al., 2001; Liu et al., 2003; Liu et al., 2004; Kelly et al., 2006; Liu et al., 2006), high-resolution 3D structures have proved difficult to obtain in this manner. An alternative approach to the direct transfer method is the loop transfer technique, in which a wire loop is used to lift off the lipid monolayer and to deposit it on an EM grid (Fig. 1D, E). This method was initially developed with holey carbon grids (Asturias and Kornberg, 1995), but a continuous carbon support film can also be used. In contrast to the direct method, when loop transfer is used, the protein array rather than the lipid monolayer makes contact with the carbon film (Fig. 1E), leading to a more robust interaction between the protein crystal and the carbon support film.
Figure 1. The direct and loop transfer methods.
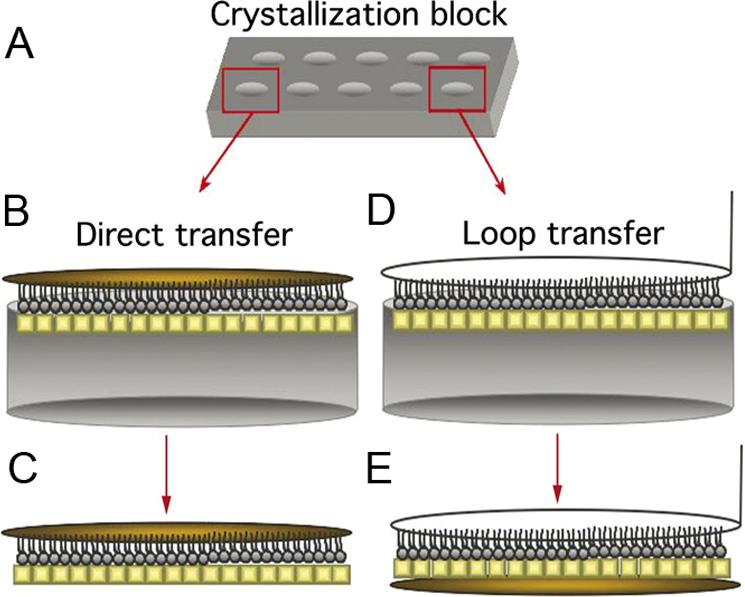
(A) Teflon block used to grow 2D crystals on lipid monolayers. (B, C) Direct transfer method, after which the lipid monolayer is in contact with the carbon film. (D, E) Loop transfer method, after which the protein crystal is in contact with the carbon film. For (B – E) Protein crystals are in yellow while the EM grid is colored gold.
In a related field, electron crystallography of conventional two-dimensional (2D) crystals, i.e., membrane proteins reconstituted into lipid bilayers, a number of high-resolution structures have now been determined (Henderson et al., 1990; Kühlbrandt et al., 1994; Kimura et al., 1997; Murata et al., 2000; Gonen et al., 2005; Hiroaki et al., 2006; Holm et al., 2006). Advances in specimen preparation, in particular the embedding of the specimen in sugar solutions (Unwin and Henderson, 1975; Jap et al., 1990; Wang and Kühlbrandt, 1991; Hirai et al., 1999; Gyobu et al., 2004), have played a crucial role in the success of this method. It was thus of interest to us, whether sugar embedding could be adapted to the preparation of lipid monolayer crystals and how it would compare to the conventional preparation methods currently used for lipid monolayer crystals. As a test specimen to study specimen preparation methods, we selected the S-layer protein sbpA. S-layer proteins have been historically used for electron crystallographic studies due to their inherent propensity to self-assemble into ordered arrays (for a review, see Baumeister et al., 1988). In particular, we chose the protein sbpA from Bacillus sphaericus, because it is easy to purify from the cell wall of native bacteria (Schuster et al., 2005) and it has been shown to form 2D crystals (Pum and Sleytr, 1994) in a calcium-dependent manner (Pum and Sleytr, 1995). Native and recombinant S-layer proteins have also recently been used as building blocks to design two-dimensional scaffolds with engineered properties for various nanotechnology applications (Sleytr et al., 2003), raising interest in determining their structure. However, structural studies of sbpA crystals have so far not exceeded a resolution of about 20 Å (Lepault and Pitt, 1984; Lepault et al., 1986). By systematically testing various specimen preparation protocols, we found that we could produce the best specimens by transferring the lipid monolayer crystals to a continuous carbon film using loop transfer followed by embedding the sample in trehalose and quick-freezing in liquid ethane. With images of specimens prepared in this way, we could calculate a projection map of sbpA at a resolution of 7 Å. This is one of the highest resolution maps obtained with 2D crystals grown on lipid monolayers to date. At this point the resolution may no longer be limited by the specimen preparation technique but by the size and order of the sbpA arrays themselves.
2. Materials and Methods
2.1. Purification of sbpA
sbpA was purified as described (Schuster et al., 2005) with minor modifications. Briefly, Bacillus sphaericus (ATCC number 4525) was grown at 32°C in SVIII medium (50 mM Hepes, pH 7.2, 7 mM K2HPO4, 10 g/l peptone, 5 g/l yeast extract, 5 g/l meat extract, 0.2 mM MgSO4, 1.8 mM sucrose, 17 mM glucose). The cells were lysed by sonication in 50 mM Tris, pH 7.2, and the cell walls were isolated by centrifugation at 16,000g for 15 minutes. The pellet was resuspended in Buffer A (0.75% Triton X-100 in 50 mM Tris, pH 7.2) with a Tissue Tearor (BioSpec Products, Bartlesville, OK) and centrifuged at 28,000g for 10 minutes. The cell walls were washed three more times in Buffer A. sbpA (1.5 to 2.0 mg/ml) was released from the cell walls by unfolding the protein in Buffer B (50 mM Tris, pH 7.2, 5 M guanidine HCl) for 30 minutes at room temperature with stirring. The cell walls were removed by centrifugation at 100,000g for 45 minutes. sbpA in the supernatant was refolded by dialysis against distilled water and aggregates were removed by centrifugation at 100,000g for 30 minutes.
2.2. Lipid monolayer crystallization of sbpA
1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was purchased from Avanti Polar lipids (Alabaster, AL) and didodecyldimethylammonium bromide (DDMA) was purchased from Acros Organics (Geel, Belgium). To set up monolayer crystallization trials Teflon blocks with 10 − 18 wells were used, each well being 5 mm in diameter and ∼1 mm in depth. The total volume of sample placed into each well was ∼25 μl. (Fig. 1A). For crystallization, 23.1 μl of crystallization buffer containing 20 mM Tris, pH 8, 100 mM NaCl and 50 mM CaCl2 was added to the wells. Lipid solution was prepared by mixing 20% DDMA (1 mg/ml) in chloroform with 55% DMPC (1 mg/ml) in chloroform with 25% chloroform. 1 μl of this lipid mixture was added on top of the protein solution in each well. After approximately 15 minutes of incubation at room temperature, a Hamilton syringe was used to inject 1.9 μl of sbpA protein in deionized water (1 mg/ml) into the aqueous sub-phase to initiate crystallization. The crystallization blocks were incubated in a sealed humid environment at room temperature. While crystals formed within two hours of incubation and were stable for at least two months, samples for cryo-EM experiments were incubated for one week before specimens were prepared.
2.3. Lipid monolayer transfer techniques
Lipid monolayer crystals were transferred to continuous carbon film mounted on copper EM grids (400 mesh, Ted Pella, Redding, CA) or to holey carbon grids (400 mesh, 2/4 Quantifoil Micro Tools GmbH, Germany) using both the direct transfer (Uzgiris and Kornberg, 1983) and loop transfer (Asturias and Kornberg, 1995) methods. For direct transfer, an EM grid (without prior glow discharging) was carefully placed on top of the lipid monolayer sample in the crystallization block and gently picked up again with a pair of forceps. The grid was blotted with Whatman #1 filter paper (Whatman International Ltd, Middlesex, England) and either negatively stained with 1% uranyl acetate or plunge-frozen in either liquid nitrogen or liquid ethane. For the loop transfer, a thin platinum loop with a diameter of 3.5 mm (Ernest F. Fullam, Inc., Latham, NY) was brought gently into contact with the lipid monolayer sample in the crystallization block. The loop was then lifted up and the sample applied to a glow-discharged carbon film mounted on an EM grid. The grid was blotted and negatively stained or plunge-frozen as described above.
2.4. Sample preparation
We used six methods to prepare frozen-hydrated specimens of lipid monolayer sbpA 2D crystals. Direct transfer of 2D crystals to (1) holey and (2) continuous carbon grids, followed by blotting for 10 seconds and plunge-freezing into liquid ethane; loop transfer to (3) holey and (4) continuous carbon grids, followed by blotting for 10 seconds and plunge-freezing into liquid ethane; loop transfer to continuous carbon grids, followed by embedding with 5% trehalose solution and plunge-freezing either into (5) liquid nitrogen or (6) liquid ethane.
2.5. Data collection
Negatively stained specimens were imaged in a T12 electron microscope (FEI, Hillsboro, OR) equipped with a LaB6 filament and operated at an acceleration voltage of 120 kV. Images were taken on a Gatan Ultrascan 894 2K×2K CCD camera. Grids of frozen-hydrated sbpA crystals were transferred into a F20 electron microscope (FEI, Hillsboro, OR) equipped with a field emission gun using an Oxford cryo-specimen holder, maintaining a temperature of −180°C. Samples were examined at an acceleration voltage of 200 kV and images of crystals were recorded at a magnification of 50,000× using low-dose procedures. Images were recorded either with a Gatan MegaScan 794 2K×2K CCD camera (for general characterization of the samples) or on Kodak SO-163 film (for quantitative analyses). Films were developed for 12 minutes with full-strength Kodak D-19 developer at 20°C.
2.6. Image processing
An optical diffractometer was used to select for drift-free images and to identify the best diffracting regions of the imaged sbpA 2D crystals. Micrograph areas (5000 × 5000 pixels) of sbpA 2D crystals were digitized with a Zeiss SCAI scanner (Carl Zeiss Inc., Oberkochen, Germany) using a step size of 7 μm and processed with the 2dx software package (Gipson et al., 2007), which included lattice unbending and correction for the contrast transfer function (CTF). The 5 best images, ranging in defocus from −0.8 to −2 μm, were merged and used to calculate a projection map with imposed p4 symmetry using the MRC (Crowther et al., 1996) and CCP4 (Collaborative Computational Project, Number 4, 1994) software packages. A negative temperature factor of −200 was applied to the projection map to enhance the high-resolution Fourier terms.
3. Results
3.1. 2D crystallization of sbpA
Ca2+ has been shown to induce sbpA to form regular arrays (Pum and Sleytr, 1995). We could reproduce this array formation with sbpA purified from the cell wall of Bacillus sphaericus (Fig. 2A). When protein was added to Ca2+-free crystallization buffer, no sbpA arrays formed in the sample (data not shown), but when protein was added to Ca2+-containing crystallization buffer small sbpA arrays appeared within two hours (Fig. 2B). While these arrays grew in size over time (Fig. 2C), the crystal order did not improve with time (compare Fourier transforms shown as insets in Figs. 2B and C). Since sbpA has been shown to form regular arrays on positively charged lipid monolayers (Pum and Sleytr, 1994), we investigated whether the presence of a monolayer would induce the formation of better ordered 2D crystals. As before, initial sbpA arrays formed on lipid monolayers within two hours of Ca2+ addition (Fig. 2D). However, in contrast to sbpA arrays formed in solution, the 2D crystals formed on the lipid monolayer not only grew in size over time (Fig. 2E) but also showed a marked improvement in crystal order (compare Fourier transforms shown as insets in Figs. 2D and E). These results showed that 2D crystals formed on monolayers are of better quality and thus better suited for structural studies of sbpA than 2D crystals grown in solution.
Figure 2. Negatively stained sbpA crystals formed in solution and on a lipid monolayer.

(A) SDS PAGE gradient gel showing purified sbpA used for crystallization. Lane 1: Mark 12 protein standard (Invitrogen Corporation, Carlsbad, CA); lane 2: sbpA (MW ∼120 kDa). (B) sbpA crystals formed in solution within 2 hours after addition of 50 mM CaCl2. (C) After incubation for 24 hours, the sbpA crystals grew larger but the order of the sbpA arrays did not improve significantly (compare Fourier transforms shown as insets in panels B and C). (D) sbpA crystals also formed within 2 hours of incubation with CaCl2 on lipid monolayers. (E) After a 24 hour incubation, the crystals grew not only bigger but also improved in order (compare Fourier transforms shown as insets in panels D and E). Scale bars in the images are 100 nm; scale bars in the Fourier transform are (6.5 nm)−1.
3.2. Direct and loop transfer of sbpA crystals onto EM grids
We used negative staining to assess specimens prepared by the direct and loop transfer methods. For these experiments grids covered with a continuous carbon film were used. Representative images of crystals prepared with 1% uranyl acetate recorded at low magnification (8,700×) show that large crystalline arrays of sbpA were present on the grid after direct transfer (Fig. 3A) as well as after loop transfer (Fig. 3B). Upon examining several grids prepared by the two transfer methods, we found that independent of the transfer method typically ∼80% of the grid squares contained crystals. Grids prepared using the direct transfer method had, however, a greater number of smaller, broken crystals. A typical grid square contained between 5 to 8 crystals, which varied in size from 0.5 to 2 μm. This transfer method also tended to produce specimens, in which crystals were stacked on top of each other, resulting in several overlaid diffraction patterns in Fourier transforms of the images (data not shown). Grids prepared using the loop transfer showed about the same level of crystal coverage, but the crystals were in general less broken, as expected with this more gentle transfer technique. We typically found 2 to 4 crystals per grid square, which ranged in size from 3 to 5 μm. At higher magnification (52,000×) the square lattice formed by sbpA is clearly seen in negatively stained samples prepared by either method (Figs. 3C and D), and Fourier transforms (insets in Figs. 3C and D) showed no difference in crystal order (number of diffraction orders in Fourier transforms) to the resolution limit introduced by the stain (∼20 Å). Our results thus suggest that the direct and loop transfer methods are equally efficient in adsorbing sbpA 2D crystals to continuous carbon grids, without loss of crystal order to a resolution of at least 20 Å. The loop method appears, however, to induce less mechanical stress on the crystals during the transfer as indicated by the larger size of the observed crystals.
Figure 3. Negatively stained sbpA monolayer crystals prepared with the direct and loop transfer methods.

(A) Low magnification image (8,700×) and (C) high magnification image (52,000×) of sbpA monolayer crystals prepared with the direct transfer method. (B) Low magnification image (8,700×) and (D) high magnification image (52,000×) of sbpA monolayer crystals prepared using the loop transfer method. The insets in panels C and D show the Fourier transforms of the crystals. Scale bars in the images are 100 nm; scale bars in the Fourier transform are (6.5 nm)−1.
3.3. Frozen-hydrated sbpA crystals on holey carbon film
In an attempt to obtain higher resolution information, we prepared frozen-hydrated specimens of the sbpA crystals using holey carbon film according to the protocol introduced by Kubalek et al. (1991). We used the direct (Fig. 4A) and the loop transfer method (Fig. 4B) (3 grids per method) to screen for lipid monolayer crystals of sbpA vitrified on holey carbon films. For each grid, ∼50 crystals were imaged on CCD camera, none of which diffracted beyond a resolution of 25 Å (Fig. 4, insets).
Figure 4. Images of frozen-hydrated sbpA monolayer crystals prepared on holey carbon grids.

(A) sbpA crystals transferred to Quantifoil grids using the direct method. (B) sbpA crystals transferred to Quantifoil grids using the loop method. The Fourier transforms of these representative images shown as insets in panels A and B do not show diffraction spots beyond a resolution of 25 Å. Scale bars in the images are 100 nm; scale bars in the Fourier transform are (6.5 nm)−1.
3.4. Frozen-hydrated sbpA crystals on continuous carbon film
We next tested whether continuous carbon film would be a better option for vitrifying sbpA crystals formed on lipid monolayers. Images taken from crystals transferred to the EM grid using the direct method also showed diffraction spots to about 25 Å, similar to images taken from crystals on holey carbon film. Using the loop rather than the direct method to transfer crystals to continuous carbon grids was an improvement as some images showed diffraction spots close to 15 Å resolution. To quantify this improvement, we prepared three grids of vitrified sbpA crystals on continuous carbon film either by direct or loop transfer. We collected 18 images from each of the six grids on film and inspected their diffraction patterns on an optical laser bench. Overall, 69% of the images taken from crystals prepared with the direct transfer did not diffract, and only 7% of the images showed diffraction spots in the resolution range from 30 to 26 Å (Fig. 5A). In the case of images taken from crystals prepared using the loop transfer, 39% of the images still did not show any diffraction, but 33% of the images showed diffraction spots in the resolution range from 30 to 16 Å and 7% of the images showed diffraction in the resolution range from 20 to 16 Å (Fig. 5B). Using loop transfer in combination with continuous carbon film thus resulted in a modest improvement in the preservation of the sbpA crystals.
Figure 5. Resolution of images taken from crystals prepared on continuous carbon grids by the indicated methods.

For each preparation method, 3 grids were prepared and 18 images of sbpA crystals were taken from each grid (indicated by different grey shades). The resolution of the imaged crystals was assessed by the highest diffraction spots seen by optical diffraction. The graphs show the number of images taken from each grid that showed diffraction spots in the indicated resolution ranges.
3.5. Trehalose-embedded sbpA crystals
Sugar-embedding, especially in trehalose (Jap et al., 1990; Hirai et al., 1999; Gyobu et al., 2004), is currently considered the best preparation method for 2D crystals formed by reconstitution of membrane proteins into lipid bilayers. We therefore wanted to test whether sugar embedding could be adapted for the use with sbpA 2D crystals formed on lipid monolayers. To embed sbpA crystals in sugar we transferred the crystals to grids covered with a continuous carbon support film and allowed them to adsorb for 2 minutes. We then added 2 μl of 5% trehalose solution either to the front side of the grid (carbon-coated side), to which the crystals were adsorbed, or to the back side of the grid (grid-bar side). The grids were blotted and frozen in liquid nitrogen or in liquid ethane. All attempts to produce trehalose-embedded specimens using the direct transfer method failed. This is most likely due to the fact that crystals directly transferred to a grid interact with the carbon film through the hydrophobic tails of the lipid monolayer (Fig. 1C). This interaction appears to be too weak to withstand the additional treatment of the grid with sugar solution, presumably causing the crystals to be sucked away during blotting or to be ripped off the grid during plunge-freezing.
We then transferred the crystals to continuous carbon grids using the loop method, in which case it is the protein array that makes contact with the carbon film (Fig. 1E). When we applied sugar to the front side of the grid (carbon-coated side), many of the crystals were covered with small lipid vesicles (similar to what can be seen in Fig. 3B), which caused distortions in the sbpA arrays. We found that the best way to prepare sugar embedded specimens was to use the loop method to transfer the crystals to a glow-discharged grid covered with a continuous carbon film, allow the crystals to adsorb for 2 minutes, add 2 μl of 5% trehalose solution onto the back side of the grid, the grid-bar side, (spreading the drop evenly so that the trehalose can diffuse into the specimen and embed the crystals), blot the grid from the back side for 10 seconds with Whatman #1 paper before freezing either in liquid nitrogen or liquid ethane. The quality of specimen preservation was quantified as described above by taking 18 images from 3 different grids and assessing the resolution by optical diffraction. Freezing sugar embedded samples in liquid nitrogen did not produce significantly better results than vitrification in the absence of trehalose. 26% of the images did not show any diffraction and the best images (17%) only showed diffraction spots to a resolution of about 21 Å (Fig. 5C). Freezing sugar embedded samples in liquid ethane dramatically improved the preservation of the crystals. Only 22% of the images did not show any diffraction, the majority of images (72%) showed spots to a resolution better than 20 Å and 20% of the images showed diffraction spots to a resolution better than 11 Å (Fig. 5D).
3.6. Projection map of spbA at 7 Å resolution
Low-dose images of lipid monolayer crystals of sbpA, transferred to a continuous carbon grid with the loop technique and embedded in 5% trehalose before freezing in liquid ethane, were used to calculate a projection map. The quality of the images and the imaged crystals was assessed by optical diffraction. The best ordered crystalline areas (showing diffraction spots beyond a resolution of 10 Å) in high-quality images (with no signs of specimen drift or charging) were selected for further image processing. After unbending and correction for the contrast transfer function (CTF), CTF plots typically showed good completeness of diffraction data to ∼7 Å resolution, and occasionally IQ 3 and 4 spots to a resolution of about 4 Å (Fig. 6A). The sbpA 2D crystals have unit cell dimensions of a = b = 133 ± 0.5 Å and γ=90° (n = 8) and display p4 symmetry, determined using the program ALLSPACE (Valpuesta et al., 1994) (Table 1). The amplitude and phase data from the best 5 images were merged to 7 Å resolution (Table 2) while applying p4 symmetry. The final projection map of trehalose-embedded sbpA is shown in Figure 6B. The projection map shows that a unit cell of the sbpA 2D crystals (outlined in black in Fig. 6B) contains a single tetramer, as previously described for native cell walls (Lepault and Pitt, 1984; Lepault et al., 1986).
Figure 6. Electron crystallographic analysis of trehalose-embedded sbpA lipid monolayer crystals.

(A) CTF plot of a representative image of a trehalose-embedded 2D crystal. The circles represent CTF zero transitions and the boxed numbers depict the IQ values of the individual reflections as defined by Henderson et al. (1986). Scale bar is (1.5 nm)−1. (B) p4-symmetrized projection map of sbpA at 7 Å resolution. A unit cell with a side length of a = b = 133 Å is outlined in black. The major (M), arm (A) and minor (m) domains of two sbpA subunits are labeled. The light grey circles show the arrangement of the domains if the monomer adopted a compact organization in the 2D arrays and the dark grey circles the arrangement of the domains if the monomer adopted an extended organization. Nearest and second-nearest neighboring sbpA tetramers with respect to the central tetramer are labeled by “1” and “2”, respectively.
Table 1.
Internal phase residuals of all possible two-sided plane groups using a representative image.
| Two-sided plane group | Phase residual (degrees)a | Number of comparisons | Target residual (degrees)b |
|---|---|---|---|
| p1 | 27.8c | 434 | |
| p2 | 40.8d | 217 | 40.8 |
| p12_b | 73.7 | 113 | 29.3 |
| p12_a | 74.0 | 111 | 29.0 |
| p121_b | 76.6 | 113 | 29.3 |
| p121_a | 72.4 | 111 | 29.0 |
| c12_b | 73.7 | 113 | 29.3 |
| c12_a | 74.0 | 111 | 29.0 |
| p222 | 59.0 | 441 | 34.2 |
| p2221_b | 58.4 | 441 | 34.2 |
| p2221_a | 62.2 | 441 | 34.2 |
| p22121 | 69.3 | 441 | 34.2 |
| c222 | 59.0 | 441 | 34.2 |
| p4 | 30.0d | 525 | 33.2 |
| p422 | 51.3 | 998 | 30.6 |
| p4212 | 61.6 | 998 | 30.6 |
Internal phase residuals were determined using the program ALLSPACE (Valpuesta et al., 1994) from spots of IQ1 to IQ5 to 7 Å resolution. Only plane groups compatible with the sbpA lattice are shown.
Phase residual versus other spots (90° random).
Target residual based on statistics taking Friedel weight into account.
Note that in space group p1 no phase comparison is possible, so the numbers given here are theoretical phase residuals based on the signal-to-noise ratio of the observed diffraction spots.
Within 20% of target residual.
Table 2.
Phase residuals in resolution shells.
| Resolution shell (Å) | Number of phases | Mean value of Δαc (deg.) | Standard error (deg.) | Mean figure of merit | |
|---|---|---|---|---|---|
| From | To | ||||
| 200.0 | 20.0 | 34 | 12.7 | 2.1 | 0.95 |
| 20.0 | 12.0 | 62 | 22.7 | 2.9 | 0.86 |
| 12.0 | 9.0 | 74 | 25.0 | 2.5 | 0.82 |
| 9.0 | 7.0 | 112 | 36.5 | 2.3 | 0.65 |
| 7.0 | 6.0 | 105 | 43.6 | 2.5 | 0.52 |
Δαc, Difference between the symmetry-imposed phase of 0° and 180° and the observed combined phase. 45° is random.
The sbpA tetramer has a complex projection structure with each subunit consisting of three domains as described previously (Lepault and Pitt, 1984; Lepault et al., 1986). The four major domains (labeled M in Fig. 6B) form a square and constitute the core of the sbpA tetramer. The four major domains surround a square low-density area in the center of the tetramer. Four smaller arm domains (labeled A in Fig. 6B) extend from the sides of the core tetramer. The arm domains make contact with the major and arm domains of one subunit from each of the four neighboring tetramers. Each sbpA tetramer thus forms strong connections to the four nearest neighboring sbpA tetramers (labeled “1” in Fig. 6B) through interactions involving the major and arm domains. The minor domain of the sbpA subunit (labeled m in Fig. 6B) does not show an obvious connection to the major and arm domains. The light and dark grey circles thus indicate the two possibilities how the three domains may be arranged in an sbpA subunit. Minor domains originating from four different sbpA tetramers also form a tetramer along the four-fold axis at the corners of each unit cell (Fig. 6B). Each minor domain thus mediates connections with two of the nearest neighboring sbpA tetramers and a second-nearest neighboring sbpA tetramer (labeled “2” in Fig. 6B).
4. Discussion
Advances in specimen preservation have played a pivotal role in obtaining high-resolution structures for membrane proteins by electron crystallography of 2D crystals. Sugar embedding was originally introduced when Unwin and Henderson preserved purple membranes in glucose for electron microscopic imaging (Unwin and Henderson, 1975), resulting in the first 3D density map of a membrane protein (Henderson and Unwin, 1975) and later in the first atomic model of a protein based on electron crystallographic data (Henderson et al., 1990). The sugar serves both as non-volatile replacement for water and as cryo-protectant in the freezing of the crystals. In addition, because sugar embedding involves much more extensive drying of the sample as compared to sample vitrification, specimens prepared in sugar tend to be flatter and better suited for collecting data from tilted specimens. Tannic acid was subsequently used to preserve 2D crystals of plant light-harvesting complex II (Kühlbrandt et al., 1994; Wang and Kühlbrandt, 1991) and trehalose has been used to preserve a number of 2D crystals (e.g., Jap et al., 1990; Hirai et al., 1999; Murata et al., 2000). Trehalose is the sugar expressed by organisms in response to cold shock (Kandror et al., 2002) and it has characteristics that make it particularly well suited for the preservation of protein structure upon freezing (De Carlo et al., 1999; Hirai et al., 1999). The main purpose of this study was thus to evaluate whether trehalose embedding could be adapted for specimen preparation of 2D crystals grown on lipid monolayers and whether it would preserve the structure better than the currently used techniques.
Sugar embedding has been used before to prepare specimens of streptavidin 2D crystals grown on lipid monolayers. Kubalek et al. (1991) used the direct transfer method to apply streptavidin crystals to reticulated carbon grids and used 1% glucose in combination with freezing in liquid nitrogen to preserve the crystals. Electron diffraction patterns of untilted crystals prepared in this way showed diffraction spots to a resolution of 2.8 Å. Avila-Sakar and Chiu (1996) also prepared streptavidin lipid monolayer crystals by direct transfer to reticulated carbon grids, but they froze the grids in liquid ethane without prior addition of sugar. Using this preparation method the authors could collect electron diffraction patterns and images that allowed them to calculate a projection map at 3 Å resolution. The authors also reported that the vitrified crystals were not sufficiently flat to collect data from tilted specimens, but that they could record high-resolution electron diffraction patterns from glucose-embedded crystals at a tilt angle of 50° (Avila-Sakar and Chiu, 1996). These studies thus not only report the highest resolution data obtained with lipid monolayer crystals to date, but also suggest that sugar embedding may indeed also be the best specimen preparation procedure for lipid monolayer crystals to collect data from tilted specimens.
Our results differ from those obtained by Kubalek et al. (1991), as we were unable to produce sugar-embedded specimens using the direct transfer method for our sbpA lipid monolayer crystals. The difference may be explained by the fact that we used continuous carbon film rather than reticulated carbon film and the associated differences in the way the sugar was applied to the specimen. Instead, we used the loop transfer technique developed by Asturias and Kornberg (1995) to adsorb our monolayer crystals to continuous carbon grids. Since with this technique the protein array interacts with the carbon film rather than the hydrophobic lipid tails of the monolayer, the crystals are more firmly attached and can thus withstand the sugar embedding procedure. A surprising finding of our study is that trehalose embedding in combination with freezing in liquid nitrogen (Fig. 5C) did not improve the preservation of the sbpA crystals compared to vitrified samples (Fig. 5B), while trehalose-embedded specimens plunge-frozen in liquid ethane showed a significantly better preservation of the crystals (Fig. 5D). The reason for this difference is not clear, since the trehalose should function as cryo-protectant and make the freezing rate irrelevant for specimen preservation. Nevertheless, the difference between freezing the monolayer crystals in liquid nitrogen versus freezing them in liquid ethane was substantial and reproducible.
Our specimen preparation protocol, which consists of loop transfer of the monolayer crystals to continuous carbon grids, embedding with 5% trehalose and subsequent freezing in liquid ethane, allowed us to collect images and calculate a projection map of sbpA at 7 Å resolution (Fig. 6). This resolution is low compared to a resolution of 3 Å obtained with streptavidin monolayer crystals (Kubalek et al., 1991; Avila-Sakar and Chiu, 1996). Streptavidin is, however, a much smaller protein (MW 15 kDa) and forms highly ordered 2D crystals with a smaller unit cell (a = b = 82.3 Å). Streptavidin 2D crystals are therefore probably more rigid than 2D arrays formed by the larger sbpA tetramer and hence less susceptible to damage introduced by the specimen preparation procedure. This notion is supported by the fact that data of comparable resolution could be collected from vitrified (Avila-Sakar and Chiu, 1996) and glucose-embedded (Kubalek et al., 1991) streptavidin crystals. This is clearly not the case for sbpA crystals, since images of specimens prepared by direct transfer to holey carbon grids and subsequent freezing, in contrast to our images collected from trehalose-embedded crystals, never diffracted beyond a resolution of 25 Å. Although we cannot be certain, it is likely that the resolution of our sbpA projection map is not limited to a resolution of 7 Å by the specimen preparation method but by the order of the sbpA arrays themselves. We predict that images of streptavidin crystals prepared by the trehalose-embedding protocol presented in this manuscript would also diffract to at least 3 Å resolution, although this prediction needs experimental testing.
Our 7 Å projection map of sbpA is a substantial improvement compared to previous structural studies on sbpA arrays, which were all limited to a resolution of about 20 Å (Aebi et al., 1974; Lepault and Pitt, 1984; Lepault et al., 1986; Pum and Sleytr, 1994). Our higher-resolution projection map corroborates the previously published low-resolution maps (Aebi et al., 1974; Lepault and Pitt, 1984; Lepault et al., 1986). The first negatively stained projection map of sbpA already revealed the three domains of an sbpA subunit (Aebi et al., 1974), which were subsequently named major domain, arm domain and minor domain (Lepault and Pitt, 1984). These three domains became more clearly defined in a later 3D reconstruction from negatively stained sbpA crystals (Lepault et al., 1986). The complex 3D structure of the sbpA tetramer makes it difficult to interpret our projection map in terms of secondary structure elements despite the resolution of 7 Å, which should resolve α-helices. Only the minor domain shows distinct structural features and is resolved in our map into three circular densities (Fig. 6B). The diameter of these three densities of about 8 Å would be consistent with three helices forming a bundle and running perpendicular to the plane of the lipid monolayer.
Neither the previous low-resolution maps nor our 7 Å projection map reveal how the three domains are connected in an sbpA subunit. One possibility is that the three domains form a compact arrangement, where the major, arm and minor domains form a triangle (light grey circles in Fig. 6B). Alternatively, the three domains could form an extended, linear arrangement as indicated by the dark grey circles in Fig. 6B. Although the precise domain organization of sbpA is not clear, similar architectures are found in other tetragonal surface layers, as for example in the S-layer from Sporosarcina ureae (Engelhardt et al., 1986). Higher resolution information on the 3D structure will be needed, however, to elucidate the exact building plan of these tetragonal S-layers. Knowledge of the structure will be crucial in attempts to engineer recombinant S-layer proteins that form arrays with designed properties. S-layers have potential for use in the fabrication of biotemplated energy generating devices such as batteries (Long et al., 2004; Nam et al., 2006) and solar cells (Das et al., 2004), where precise engineering of the S-layer structure can create a scaffold for the growth and arrangement of nanomaterials. A 3D reconstruction at a resolution of 10 Å or better will require flat specimens that can be used to collect EM data from tilted specimens. Since sugar embedding produced flatter specimens of streptavidin crystals grown on lipid monolayers (Avila-Sakar and Chiu, 1996), the trehalose embedding technique presented here may lay the foundation to determine 3D structures of lipid monolayer crystals of sbpA as well as other proteins at resolutions better than 10 Å.
ACKNOWLEDGEMENTS
This work was supported by the Army Research Office Institute of Collaborative Biotechnologies (to AMB), the Army Research Office Institute of Soldier Nanotechnologies (to AMB), the David and Lucile Packard Foundation (to AMB), DARPA/ONR N00014-01-1-1060 (to TK), Semiconductor Research Corporation task ID 1267.0001 (to TK), a National Science Foundation Graduate Research Fellowship (to JN), a Vinton Hayes Graduate Research Fellowship (to JN), and a MIT/Merck Graduate Research Fellowship (to JN). The molecular EM facility at Harvard Medical School was established with a generous donation from the Giovanni Armenise Harvard Center for Structural Biology and is supported by National Institutes of Health Grant GM62580 (to TW). Any opinions, findings, conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aebi U, Smith PR, Dubochet J, Henry C, Kellenberger E. A study of the structure of the T-layer of Bacillus brevis. J. Supramol. Str. Cell. 1974;1:498–522. doi: 10.1002/jss.400010606. [DOI] [PubMed] [Google Scholar]
- Asturias FJ, Kornberg RD. A novel method for transfer of two-dimensional crystals from the air/water interface to specimen grids: EM sample preparation/lipid-layer crystallization. J. Struct. Biol. 1995;114:60–66. doi: 10.1006/jsbi.1995.1005. [DOI] [PubMed] [Google Scholar]
- Avila-Sakar AJ, Chiu W. Visualization of beta-sheets and side-chain clusters in two-dimensional periodic arrays of streptavidin on phospholipid monolayers by electron crystallography. Biophys. J. 1996;70:57–68. doi: 10.1016/S0006-3495(96)79597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister W, Wildhaber I, Engelhardt H. Bacterial surface proteins. Some structural, functional and evolutionary aspects. Biophys. Chem. 1988;29:39–49. doi: 10.1016/0301-4622(88)87023-6. [DOI] [PubMed] [Google Scholar]
- Brisson A, Bergsma-Schutter W, Oling F, Lambert O, Reviakine I. Two-dimensional crystallization of proteins on lipid monolayers at the air-water interface and transfer to an electron microscopy grid. J. Cryst. Growth. 1999;196:456–470. [Google Scholar]
- Celia H, Wilson-Kubalek E, Milligan RA, Teyton L. Structure and function of a membrane-bound murine MHC class I molecule. Proc. Natl. Acad. Sci. U.S.A. 1999;96:5634–5639. doi: 10.1073/pnas.96.10.5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. Number 4. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Henderson R, Smith JM. MRC image processing programs. J. Struct. Biol. 1996;116:9–16. doi: 10.1006/jsbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- Darst SA, Ribi HO, Pierce DW, Kornberg RD. Two-dimensional crystals of Escherichia coli RNA polymerase holoenzyme on positively charged lipid layers. J. Mol. Biol. 1988;203:269–273. doi: 10.1016/0022-2836(88)90107-6. [DOI] [PubMed] [Google Scholar]
- Das R, Kiley PJ, Segal M, Norville J, Yu AA, Wang L, Trammell SA, Reddick LE, Kumar R, Stellacci F, Lebedev N, Schnur J, Bruce BD, Zhang S, Baldo M. Integration of photosynthetic protein molecular complexes in solid-state electronic devices. Nano Letters. 2004;4:1079–1083. [Google Scholar]
- De Carlo S, Adrian M, Kalin P, Mayer JM, Dubochet J. Unexpected property of trehalose as observed by cryo-electron microscopy. J. Microsc. 1999;196:40–45. doi: 10.1046/j.1365-2818.1999.00591.x. [DOI] [PubMed] [Google Scholar]
- Engelhardt H, Saxton WO, Baumeister W. Three-dimensional structure of the tetragonal surface layer of Sporosarcina ureae. J. Bacteriol. 1986;168:309–317. doi: 10.1128/jb.168.1.309-317.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromherz P. Electron microscopic studies of lipid protein films. Nature. 1971;231:267–268. doi: 10.1038/231267a0. [DOI] [PubMed] [Google Scholar]
- Gipson B, Zeng X, Zhang ZY, Stahlberg H. 2dx – User-friendly image processing for 2D crystals. J. Struct. Biol. 2007;157:64–72. doi: 10.1016/j.jsb.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyobu N, Tani K, Hiroaki Y, Kamegawa A, Mitsuoka K, Fujiyoshi Y. Improved specimen preparation for cryo-electron microscopy using a symmetric carbon sandwich technique. J. Struct. Biol. 2004;146:325–333. doi: 10.1016/j.jsb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Henderson R, Unwin PNT. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- Henderson R, Baldwin JM, Downing KH, Lepault J, Zemlin F. Structure of purple membrane from Halobacterium halobium: recording, measurement and evaluation of electron micrographs at 3.5 Å resolution. . Ultramicroscopy. 1986;19:147–178. [Google Scholar]
- Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- Hirai T, Murata K, Mitsuoka K, Kimura Y, Fujiyoshi Y. Trehalose embedding technique for high-resolution electron crystallography: application to structural study on bacteriorhoposin. J. Electron Microsc. 1999;48:653–658. doi: 10.1093/oxfordjournals.jmicro.a023731. [DOI] [PubMed] [Google Scholar]
- Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, Mizoguchi A, Fujiyoshi Y. Implications of the aquaporin-4 structure on array formation and cell adhesion. J. Mol. Biol. 2006;355:628–639. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- Holm PJ, Bhakat P, Jegerschold C, Gyobu N, Mitsuoka K, Fujiyoshi Y, Morgenstern R, Hebert H. Structural basis for detoxification and oxidative stress protection in membranes. J. Mol. Biol. 2006;360:934–945. doi: 10.1016/j.jmb.2006.05.056. [DOI] [PubMed] [Google Scholar]
- Jap BK, Downing KH, Walian PJ. Structure of PhoE porin in projection at 3.5 Å resolution. J. Struct. Biol. 1990;103:57–63. doi: 10.1016/1047-8477(90)90086-r. [DOI] [PubMed] [Google Scholar]
- Kandror O, DeLeon A, Goldberg AL. Trehalose synthesis is induced upon exposure of Escherichia coli to cold and is essential for viability at low temperatures. Proc. Natl. Acad. Sci. U.S.A. 2002;99:9727–9732. doi: 10.1073/pnas.142314099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DF, Taylor KA. Identification of the β1-integrin binding site on α-actinin by cryoelectron microscopy. J. Struct. Biol. 2005;149:290–302. doi: 10.1016/j.jsb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Kelly DF, Taylor DW, Bakolitsa C, Bobkov AA, Bankston L, Liddington RC, Taylor KA. Structure of the α-actinin-vinculin head domain complex determined by cryo-electron microscopy. J. Mol. Biol. 2006;357:562–573. doi: 10.1016/j.jmb.2005.12.076. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Vassylyev DG, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T, Fujiyoshi Y. Surface of bacteriorhodopsin revealed by high-resolution electron crystallography. Nature. 1997;389:206–211. doi: 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- Kubalek EW, Kornberg RD, Darst SA. Improved transfer of two-dimensional crystals from the air/water interface to specimen support grids for high-resolution analysis by electron microscopy. Ultramicroscopy. 1991;35:295–304. doi: 10.1016/0304-3991(91)90082-h. [DOI] [PubMed] [Google Scholar]
- Kubalek EW, Le Grice SF, Brown PO. Two-dimensional crystallization of histidine-tagged, HIV-1 reverse transcriptase promoted by a novel nickel-chelating lipid. J. Struct. Biol. 1994;113:117–123. doi: 10.1006/jsbi.1994.1039. [DOI] [PubMed] [Google Scholar]
- Kühlbrandt W, Wang DN, Fujiyoshi Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- Langmuir I, Schaefer VJ. Activities of urease and pepsin monolayers. J. Am. Chem. Soc. 1938;60:1351–1360. [Google Scholar]
- Lebeau L, Lach F, Venien-Bryan C, Renault A, Dietrich J, Jahn T, Palmgren MG, Kühlbrandt W, Mioskowski C. Two-dimensional crystallization of a membrane protein on a detergent-resistant lipid monolayer. J. Mol. Biol. 2001;308:639–647. doi: 10.1006/jmbi.2001.4629. [DOI] [PubMed] [Google Scholar]
- Lepault J, Pitt T. Projected structure of unstained, frozen-hydrated T-layer of Bacillus brevis. EMBO J. 1984;3:101–105. doi: 10.1002/j.1460-2075.1984.tb01768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepault J, Martin N, Leonard K. Three-dimensional structure of the T-layer of Bacillus sphaericus P-1. J. Bacteriol. 1986;168:303–308. doi: 10.1128/jb.168.1.303-308.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévy D, Mosser G, Lambert O, Moeck GS, Bald D, Rigaud JL. Two-dimensional crystallization on lipid layer: a successful approach for membrane proteins. J. Struct. Biol. 1999;127:44–52. doi: 10.1006/jsbi.1999.4155. [DOI] [PubMed] [Google Scholar]
- Liu J, Taylor DW, Taylor KA. A 3-D reconstruction of smooth muscle α-actinin by cryo-EM reveals two different conformations at the actin-binding region. J. Mol. Biol. 2004;338:115–125. doi: 10.1016/j.jmb.2004.02.034. [DOI] [PubMed] [Google Scholar]
- Liu J, Wendt T, Taylor D, Taylor K. Refined model of the 10 S conformation of smooth muscle myosin by cryo-electron microscopy 3D image reconstruction. J. Mol. Biol. 2003;329:963–972. doi: 10.1016/s0022-2836(03)00516-3. [DOI] [PubMed] [Google Scholar]
- Liu J, Taylor DW, Krementsova EB, Trybus KM, Taylor KA. Three-dimensional structure of the myosin V inhibited state by cryo-electron tomography. Nature. 2006;442:208–211. doi: 10.1038/nature04719. [DOI] [PubMed] [Google Scholar]
- Long JW, Dunn B, Rolison DR, White HS. Three-dimensional battery architectures. Chem. Rev. 2004;104:4463–4492. doi: 10.1021/cr020740l. [DOI] [PubMed] [Google Scholar]
- Mosser G, Ravanat C, Freyssinet JM, Brisson A. Sub-domain structure of lipid-bound annexin-V resolved by electron image analysis. J. Mol. Biol. 1991;217:241–245. doi: 10.1016/0022-2836(91)90538-h. [DOI] [PubMed] [Google Scholar]
- Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- Nam KT, Kim D-W, Yoo PJ, Chiang C-Y, Meethong N, Hammond PT, Chiang Y-M, Belcher AM. Virus-enabled synthesis and assembly of nanowires for lithium ion battery electrodes. Science. 2006;312:885–888. doi: 10.1126/science.1122716. [DOI] [PubMed] [Google Scholar]
- Pum D, Sleytr UB. Large-scale reconstitution of crystalline bacterial surface-layer proteins at the air-water-interface and on lipid films. Thin Solid Films. 1994;244:882–886. [Google Scholar]
- Pum D, Sleytr UB. Anisotropic crystal growth of the S-layer of Bacillus sphaericus CCM 2177 at the air/water interface. Colloid. Surface. A. 1995;102:99–104. [Google Scholar]
- Schuster B, Györvary E, Pum D, Sleytr U. Nanotechnology With S-layer Proteins. In: Vo-Dinh T, editor. Protein Nanotechnology: Protocols, Instrumentation, and Applications. Humana Press; Totowa, New Jersey: 2005. pp. 101–124. [Google Scholar]
- Sleytr UB, Schuster B, Pum D. Nanotechnology and biomimetics with 2-D protein crystals. IEEE Eng. Med. Biol. 2003;22:140–150. doi: 10.1109/memb.2003.1213637. [DOI] [PubMed] [Google Scholar]
- Tang J, Taylor DW, Taylor KA. The three-dimensional structure of α-actinin obtained by cryoelectron microscopy suggests a model for Ca2+-dependent actin binding. J. Mol. Biol. 2001;310:845–858. doi: 10.1006/jmbi.2001.4789. [DOI] [PubMed] [Google Scholar]
- Taylor KA, Taylor DW. Projection image of smooth muscle α-actinin from two-dimensional crystals formed on positively charged lipid layers. J. Mol. Biol. 1993;230:196–205. doi: 10.1006/jmbi.1993.1136. [DOI] [PubMed] [Google Scholar]
- Taylor KA, Taylor DW. Structural studies of cytoskeletal protein arrays formed on lipid monolayers. J. Struct. Biol. 1999;128:75–81. doi: 10.1006/jsbi.1999.4167. [DOI] [PubMed] [Google Scholar]
- Unwin PNT, Henderson R. Molecular structure determination by electron microscopy of unstained crystalline specimens. J. Mol. Biol. 1975;94:425–440. doi: 10.1016/0022-2836(75)90212-0. [DOI] [PubMed] [Google Scholar]
- Uzgiris EE, Kornberg RD. Two-dimensional crystallization technique for imaging macromolecules, with application to antigen-antibody-complement complexes. Nature. 1983;301:125–129. doi: 10.1038/301125a0. [DOI] [PubMed] [Google Scholar]
- Valpuesta J, Carrascosa J, Henderson R. Analysis of electron microscope images and electron diffraction patterns of thin crystals of Ø29 connectors in ice. J. Mol. Biol. 1994;240:281–287. doi: 10.1006/jmbi.1994.1445. [DOI] [PubMed] [Google Scholar]
- Wang DN, Kühlbrandt W. High-resolution electron crystallography of light-harvesting chlorophyll a/b-protein complex in three different media. J. Mol. Biol. 1991;217:691–699. doi: 10.1016/0022-2836(91)90526-c. [DOI] [PubMed] [Google Scholar]
- Wendt T, Taylor D, Trybus KM, Taylor K. Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc. Natl. Acad. Sci. U.S.A. 2001;98:4361–4366. doi: 10.1073/pnas.071051098. [DOI] [PMC free article] [PubMed] [Google Scholar]