

Abstract
It is generally believed that amyloid β peptides (Aβ) are the key mediators of Alzheimer’s disease. Therapeutic interventions have been directed toward impairing the synthesis or accelerating the clearance of Aβ. An equilibrium between blood and brain Aβ exists mediated by carriers that transport Aβ across the blood brain barrier. Passive immunotherapy has been shown to be effective in mouse models of AD, where the plasma borne antibody binds plasma Aβ causing an efflux of Aβ from the brain. As an alternative to passive immunotherapy we have considered the use of Aβ-degrading peptidases to lower plasma Aβ levels. Here we compare the ability of three Aβ-degrading peptidases to degrading Aβ. Biotinylated peptidases were coupled to the surface of biotinylated erythrocytes via streptavidin. These erythrocyte-bound peptidases degrade Aβ peptide in plasma. Thus, peptidases bound to or expressed on the surface of erythroid cells represent an alternative to passive immunotherapy.
1. Introduction
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder of the elderly affecting over 5 million individuals. It is generally believed that amyloid βpeptides (Aβ) are the key mediators of the disease. These amyloid β peptides are generated through a minor metabolic pathway involving the proteolytic processing of the amyloid precursor protein (APP) [38, 39] by proteases collectively known as secretases. Oligomers formed by the aggregation of Aβ appear to be the causative agent leading first to synaptic damage and then to cognitive decline [13, 14, 44].
Within the CNS the accumulation of Aβ is dependent upon the relative rates of synthesis and clearance. A considerable effort has been expended on attaining the goal of decreasing the rate of Aβsynthesis by inhibiting the secretases responsible for its formation. More recently, studies have explored the strategy of increasing Aβ clearance as a therapeutic approach for AD. These have largely focused on immunotherapeutic approaches (either passive or active immunization) to bind to and then clear Aβ [7, 35, 37, 45]. Targeted upregulation of the Aβ-degrading peptidases that hydrolytically cleave Aβ has also received some, albeit less, attention [18, 22].
It has been shown that there is an efflux of soluble Aβ from the brain, across the blood brain barrier, and into the plasma mediated by carrier proteins and receptors. These include apolipoprotein E (apoE), α2-macroglobulin (α2M), and the low-density lipoprotein receptor-related protein (LRP) [6, 8, 10, 40, 46]. Conversely the receptor for advanced glycation end products (RAGE) [5] transports Aβ into the brain from plasma.
DeMattos et al. [7] took advantage of the fact that Aβ is transported from the brain into plasma by injecting an anti-Aβ antibody into the blood of hAPP transgenic mice. This peripheral administration of an anti-Aβ monoclonal antibody resulted in a 1,000-fold increase in plasma Aβ levels and a marked reduction in the deposition of Aβ in the brain. The mechanism responsible for the lowering of brain Aβ was believed to involve the binding of plasma Aβ in immune complexes, thus preventing its re-entry into the brain and creating a unidirectional efflux of Aβ from the brain to the periphery. This phenomenon was termed the “sink effect”. Matsuoka et al. [30] obtained similar results using two Aβ-binding compounds, ganglioside GM1 and gelsolin, to bind plasma Aβ in hAPP transgenic mice. Ganglioside GM1 and gelsolin both led to a lowering of brain Aβ by 50% or more.
A potential problem associated with passive immunization and the use of Aβ-binding compounds is that they result I the accumulation of relatively large amounts of Aβ in plasma. There have been differing reports as to whether passive immunization increases cerebral amyloid angiopathy and causes microhemorrhaging. Wilcock et al. [45] reported that passive immunization increased vascular amyloid staining in old Tg2576 hAPP mice. In addition, Racke et al. and Pfeifer et al. [35, 37] reported that passive immunization with anti-Aβ monoclonal antibodies exacerbated cerebral amyloid angiopathy-related microhemorrhage in hAPP Tg mice. In contrast, these effects were not seen in the passive immunization study of Levites et al. [23, 24].
In this study, we have investigated yet another approach, that of using Aβ-degrading peptidases to hydrolyze plasma Aβ. We have examined the ability of three Aβ-degrading enzymes, neprilysin (NEP) [16], insulysin (IDE) [36], and metalloprotease-1 (MP-1) [12] coupled to erythrocytes to degrade Aβ both in vitro and in vivo. Expression of Aβ-degrading enzymes in the periphery represents a novel way to lower plasma Aβlevels by hydrolytic cleavage rather than by forming Aβ complexes, and the use of erythrocyte coupling as a drug delivery system has been shown to be a promising therapeutic avenue [9, 26, 33]. Although brain expression of Aβ-degrading enzymes as a way to lower brain Aβ levels has been used successfully in a number of studies [15, 18, 22, 29] peripheral expression has not been explored to any great extent. Further, the peripheral overexpression of an Aβ-degrading enzyme, if shown to be efficacious at clearing brain amyloid, is much more desirable from a therapeutic perspective. Here we demonstrate that three Aβ-degrading enzymes can be coupled to erythrocytes and degrade plasma Aβ in vivo, thus identifying peripheral expression of Aβ-degrading enzymes as a potentially novel therapy for treating AD.
2. Materials and Methods
2.1. Peptidase activity assays
NEP activity was measured using the fluorogenic substrate glutaryl-Ala-Ala-Phe-4-methoxy-β-naphthylamide, (GAA, Sigma) [25]. Reaction mixtures contained 1 mM GAA, 1 μg of purified recombinant human puromycin sensitive aminopeptidase [43] and a source of NEP in 200 μl of 20 mM MES buffer, pH 6.5. The reaction was followed on a Spectra Max Gemini XS fluorescent plate reader (Molecular. Devices) at an emission wavelength of 425 nm and an excitation wavelength of 340 nm. IDE activity was assayed with the fluorogenic substrate Abz-GGFLRKHGQ-EDDnp (Abz-RK) as previously described [41]. In brief, IDE was added to reaction mixtures containing 10 μM Abz-RK in 50 mM Tris-HCl buffer at pH 7.4 and the increase in fluorescence followed on a Spectra Max Gemini XS fluorescent plate reader at an emission wavelength of 419 nm and an excitation wavelength of 318 nm. The Abz-RK fluorogenic substrate was synthesized as previously described [4]. MP-1 activity was measured in a similar manner to IDE using the fluorogenic substrate Abz-GGFLRRGQ-EDDnp (Abz-RR). Reaction mixtures contained 4 μM Abz-RR in 20 mM Tris-HCl buffer pH 7.4 and the increase in fluorescence was assayed under the same conditions used for Abz-RK [3, 21].
Protein concentrations were determined using the BCA protein assay kit (Pierce).
2.2. Preparation of biotinylated and erythrocyte-conjugated peptidases
A secreted form of NEP was expressed in HEK 293 cells transduced with lentivirus expressing the extracellular domain of NEP fused to the pepsin secretion signal [28]. Cells were expressed for 4 to 5 days in serum-free media. NEP was purified to near homogeneity from the culture supernatant by ion-exchange chromatography on a Pharmacia Mono Q column. Recombinant His-tagged IDE and His-tagged MP-1 were expressed from baculovirus infected Sf9 cells and purified by chromatography on a His-select nickel column (Sigma). The purity of each peptidase was shown to be greater than 90% as determined by SDS-PAGE. Peptidases were biotinylated by incubation with biotin N-hydroxysuccinimide (biotin-NHS) Vector Laboratories) as described by the manufacture. Briefly, 1 mg/ml of peptidase was incubated with 0.1 mg/ml biotin-NHS in 1 ml of 100 mM HEPES buffer, pH 8.0, for 1 h at room temperature. Excess biotin was removed by overnight dialysis at 4oC against phosphate buffered saline (PBS). The number of biotin molecules bound to NEP, IDE and MP-1 was determined using the Quant Tag Biotin kit (Vector laboratories).
Mouse erythrocytes were biotinylation with biotin-NHS as described by Muzykantov et al. [34]. Briefly, fresh mouse erythrocytes were washed 3 times with PBS and incubated with 50 μM biotin-NHS in PBS for 1 h at room temperature. After the cells were washed three times with PBS, erythrocytes labeled with biotin (b-erythrocytes) were incubated with streptavidin (SA) for 30 min to form the complex SA-b-erythrocytes and then washed 4 times with PBS.
The complex between peptidase and erythrocyte was formed by incubation of SA-b-erythrocytes with biotinylated peptidase at room temperature for 30 min. A 10% suspension of SA-b-erythrocytes in 250 μl PBS was centrifuged at 1000 rpm in a Sorvall RT6000B centrifuge for 10 min and resuspended in 50 μl PBS. Biotinylated peptidase (100 ng) was then incubated in 100 μl PBS buffer with 50 μl SA-b-erythrocytes at room temperature for 1 hr. The supernatant along with biotinylated peptidase corresponding to 0, 10, 50, and 100% input was subjected to Western blot analysis and the peptidase band corresponding to unbound protein was quantitated using ImageQuant software. Using native non-biotinylated erythrocytes as a control we did not detect any non-specific binding of biotinylated peptide. The difference between the amount of peptidase initially incubated with erythrocytes and the amount of peptidase detected in the supernatant was used to calculate the amount of bound peptidase.
2.3. Aβ Degradation assay
Degradation of Aβ1–40 was measured by the incubation of free or erythrocyte conjugated peptidase (NEP, IDE or MP-1) with 250 pM Aβ1–40 (California Peptide Research, Inc.) in 100 μl of PBS for 60 min. at 37oC. Phosphoramidon at 200 μM was used to inhibit NEP activity, while 2 mM 1–10-phenanthroline (Sigma) was used to inhibit IDE and MP-1 activity. After centrifugation, an aliquot of the supernatant were subjected to an Aβ1–40 sandwich ELISA as prescribed previously [31]. Ab9 (anti-Aβ1–6) was used as the capture antibody and 4G8 (anti-Aβ17–24) as the detection antibody.
Aβ1–40 degradation was also measured by following the disappearance of Aβ1–40 by HPLC using a Vydac C4 column and a linear gradient from 0.1% trifluoroacetic acid (TFA) in 95% water, 5% acetonitrile to 0.1% trifluoroacetic acid in 50% water, 50% acetonitrile. Aβ1–40 was detected at 214 nm and quantitated from the peak area.
Insulin degradation by peptidases
Insulin degradation was followed by HPLC. Briefly, 100 ng of IDE, NEP or MP-1 was incubated with 10 μM insulin in 100 μl PBS for 15 min at 37oC. The reaction was stopped and insulin cleaved to it’s A and B chains by adding 200 μl of 10 mM DTT prepared in 100 mM NH4HCO3 and incubating for 60 min at 56oC. To the mixture was then added 200 μl of 55 mM iodoacetamide (in 100 mM NH4HCO3) and incubation was continued for 45 min at room temperature in the dark. The samples were then dried on a Speed Vac and redissolved in 150 μl of 20 mM Tris buffer, pH 7.4, and then 15 μl of 5% TFA. The A and B chains were separated on a Vydac C4 column using a gradient from 0.1% TFA in 95% water, 5% acetonitrile to 0.1% TFA in 50% water, 50% acetonitrile. Insulin A and B chains detected at 214 nm and quantitated by measuring their peak areas. By testing three replicates of insulin the reproducibility was found to be ± 3% for insulin A chain and ± 4 % for insulin B chain.
2.4. Flow cytometry analysis of peptidases labeled erythrocytes
The fraction of erythrocytes containing bound peptidase was evaluated by flow cytometry. Anti-NEP antibody (anti-CD10, Dako), a rabbit polyclonal anti-rat IDE made in our laboratory, and a rabbit anti-MP-1 peptide sequence (753AEMTDIKPILRKLPRIK770K) made in our laboratory were used fore this analysis. Peptidase containing erythrocytes were incubated with primary antibody for 1 h, after which time they were washed three times with cold PBS, and then incubated with FITC labeled goat anti mouse or FITC labeled goat anti-rabbit antibody (Vector Laboratories, Inc). The peptidase containing erythrocytes were then washed three times with cold PBS and analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
2.5. In vivo degradation of Aβby Aβ-degrading peptidases
Peptidases-coupled erythrocytes and control erythrocytes in PBS were injected intravenously via the tail vein into mice (n=4 for each treatment). Five minutes latter a bolus of 1 μg of Aβ1–40 in PBS was injected into the subarachnoid space of the cisterna magna. At time points of 5, 15, 30, 45, and 60 min following Aβ injection blood samples (50 μl) were collected from the retro-orbital sinus into tubes containing 100 units/ml heparin. The blood samples were then centrifuged at 2,000 rpm in a microfuge and the supernatant taken for Aβ measurements by sandwich ELISA.
3. Results
As a prelude to utilizing neuropeptidases as a method to lower brain amyloid peptide (Aβ) levels we compared the in vitro activity of three Aβ degrading enzymes, insulysin (insulin degrading enzyme, IDE), neprilysin (neutral endopeptidase, NEP), and metalloprotease 1 (pitrilysin, MP-1). As shown in Fig. 1, all three enzymes exhibited the expected Aβ-degrading activity, with IDE showing the highest activity. Using 25 or 100 ng of protein, IDE hydrolyzed ∼95% and 98% of Aβ1–40, respectively, after 1 hr incubation at 37oC, indicating that the maximal rate was achieved at or below the 25 ng level. A slower hydrolysis rate was seen with NEP where 25 and 100 ng of enzyme degraded ∼70% and 95% Aβ1–40 peptide, respectively. The activity of MP-1 toward Aβ1–40 was somewhat slower than IDE or NEP with ∼15% degradation for 25 ng of MP-1 and 70% for 100 ng of MP-1 over the 1 h time course. We also measured Aβ1–40, cleavage at a higher concentration of 10 μM by HPLC. At this higher Aβ1–40, concentration the activity of NEP is 0.05 nmol Aβ1–40, cleaved/min/μg, for IDE a rate of 0.13 nmol Aβ1–40, cleaved/min/μg was obtained, while for MP-1 the rate was 0.04 nmol Aβ1–40, cleaved/min/μg. Thus at the higher peptide concentration the activities become more nearly equal suggesting IDE has a lower Km for Aβ1–40.
Figure 1. The degradation of Aβ1–40 by NEP, IDE and MP-1.
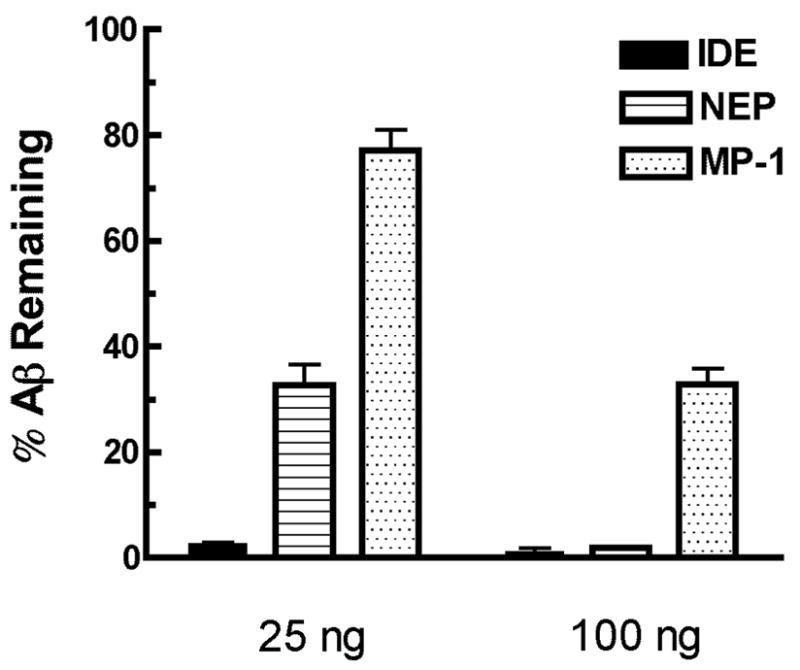
The ability of 25 or 100 ng of NEP, IDE or MP-1 to degrade 250 pM Aβ1–40 was measured by determining the amount of Aβ1–40 remaining after a 1 h incubation at 37°C by sandwich ELISA.
The error-bars presented SEM (n = 3).
** p< 0.05 for IDE vs NEP and for NEP vs MP-1 at the dose of 25 ng.
*** p<0.001 IDE vs MP-1 at the dose of 25 ng.; IDE or NEP vs MP-1 at the dose of 100 ng.
The IDE dose of 25 ng was maximal with no significant difference between the doses of 25 and 100 ng observed.
We also measured the hydrolysis of intact insulin by these three peptidases. In this case we incubated the peptidase with intact insulin and then reduced the insulin to its respective A and B chains which were measured by HPLC. As shown in Fig 2, IDE (0.1 μg) was able to cleave 13% of the insulin A chain and 29% of the insulin B chain. The same amount of NEP showed no measurable cleavage of either the insulin A or B chains, while MP-1 cleaved 14% of the insulin A chain and 10% of the B chain.
Figure 2. Cleavage of insulin by NEP, IDE and MP-1.
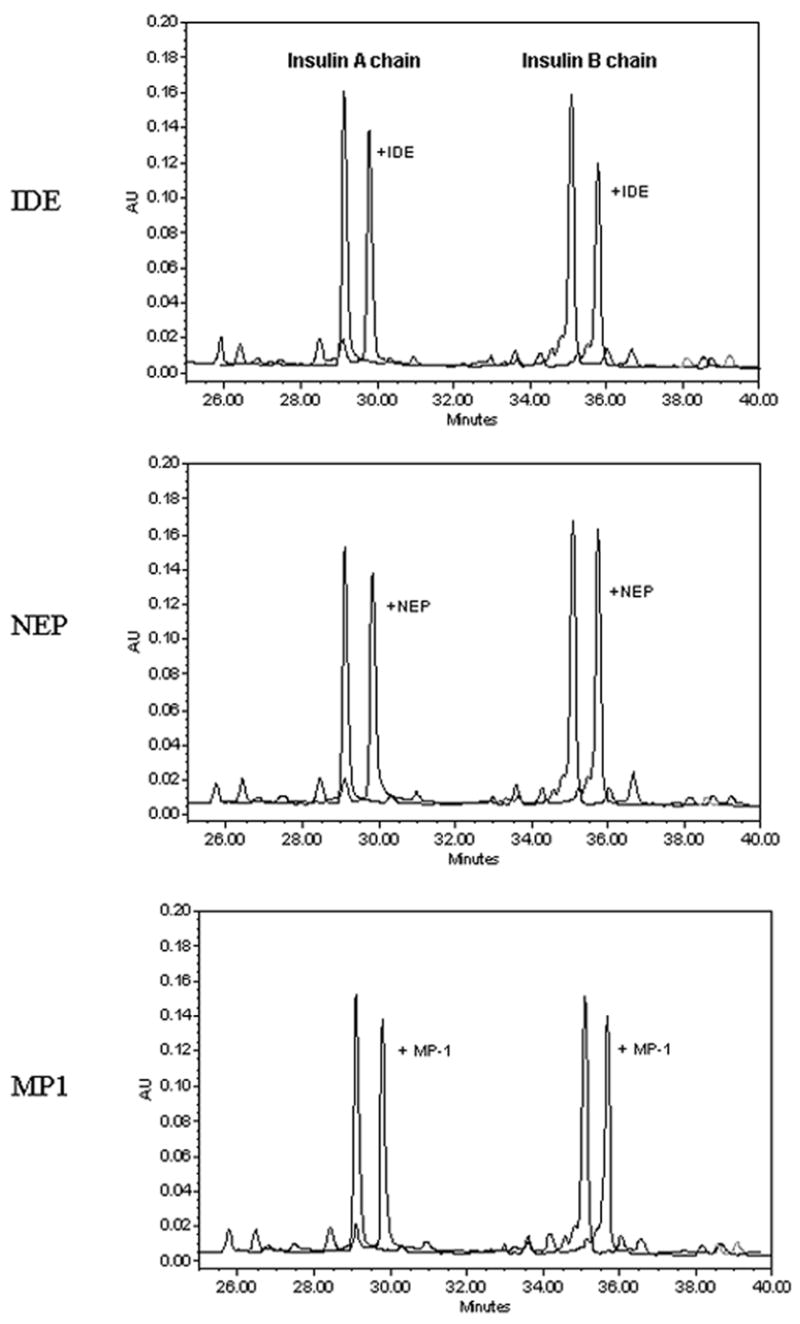
IDE, NEP or MP-1 (100 ng) was incubated with 10 μM insulin in 100 μl PBS for 15 min at 37oC. The reaction was stopped and insulin cleaved to its A and B chains as indicated in Methods. The A and B chains were separated on a Vydac C4 HPLC column with detection at 214 nm and quantitation by measuring peak areas. Shown is the HPLC profile of insulin A and B chains in the presence or absence of added peptidase. The profiles have been offset for ease of comparison.
In applying the use of these peptidases to create a peripheral “sink” for Aβ, we conducted preliminary experiments and found that the addition of neprilysin to blood led to a rapid loss of its activity. We then considered coupling neprilysin and the other Aβ degrading enzymes to erythrocytes through a biotin-streptavidin linkage as described by Muzykantov et al. [34]. Therefore the effect of biotinylation on peptidase activity was first determined by comparing the activity of the derivatized and underivatized peptidases using synthetic substrates. We initially compared each peptidase after reaction with a standard amount of biotin N-hydroxysuccinimide (biotin-NHS), 0.1 mg/ml, for 1 h at room temperature. Biotinylation was verified by subjecting the peptidases to Western blot analysis before and after biotinylation using specific antisera for each peptidase and HRP-conjugated streptavidin for detecting biotin. As shown in Table 1 biotinylation of NEP and IDE only slightly affected their activity, however under these conditions MP-1 activity was reduced by 70 %. By reducing the amount of biotin NHS used in the biotinylation reaction from 0.1 mg/ml to 0.05 mg/ml the activity of MP-1 decreased only 30%. Thus all three peptidases could be biotinylated with a loss of only 30% or less activity.
Table 1. The effect of biotinylation on the activity of NEP, IDE and MP-1.
1 mg/ml NEP, IDE and MP-1 were incubated with 0.1mg/ml biotin-NHS in 30 μl 200 mM pH8.0 HEPES buffer at room temperature for 1 hr. The substrates for NEP, IDE and MP-1 are Glutaryl-Ala-Ala-Phe-4-methoxy-β-naphthylamide, Abz-GGFLRKHGQ-EDDnp and Abz-GGFLRRGQ-EDDnp, respectively.
| Biotin− | Biotin+ | |
|---|---|---|
| NEP | 1.00 (0.56 nmols/min/μg) | 0.79 (0.44 nmols/min/μg |
| IDE | 1.00 (7.75 nmols/min/μg) | 0.73 (5.63 nmols/min/μg) |
| MP-1 | 1.00 (0.06 nmols/min/μg) | 0.3 (0.018 nmols/min/μg) |
| *0.68 (0.041 nmols/min/μg) |
Biotinylation at 0.05mg/ml
In order to couple the biotinylated peptidases to erythrocytes we isolated and biotinylated mouse erythrocytes according to the method of Muzykantov et al.[34]. It has previously been observed that there is a limit to which erythrocytes can be biotinylated after which they become unstable [34]. We therefore tested the stability of erythrocytes modified at concentrations of biotin N-hydroxysuccinimide of 25, 50 and 75 mM by reconstituting the biotinylated erythrocytes in fresh mouse serum. As a measure of biotinylated erythrocyte stability and streptavidin modified biotinylated erythrocyte stability we incubated the modified erythrocytes for 1 h at 37°C and followed hemoglobin release in the supernatant spectroscopically at 405 nm. We found no significant lysis of erythrocytes at all three levels for biotinylation and conjugating streptavidin did not affect stability. We therefore used 50 μM biotin N-hydroxysuccinimide for biotinylating erythrocytes.
We coupled 100 ng of biotinylated peptidase to 5.0×108 streptavidin conjugated biotinylated erythrocytes (SA-b-erythrocytes) in 100 μl of PBS buffer by incubation at room temperature for 1 hr. The extent of coupling of the peptidase to SA-b-erythrocytes was determined by Western blot analysis of the supernatant of the coupling reaction after removal of the erythrocytes by centrifugation. Under these conditions 74 ng (74%) NEP, 55 ng (55%) of IDE, and 42 ng (42%) of MP-1 were bound to erythrocytes. We found that the bound peptidase did not wash off the erythrocyte as no appreciable peptidase activity was detected in the supernatant of peptidase-containing erythrocytes washed five times with PBS. The majority of the erythrocytes contained bound peptidase as determined by flow cytometry, Fig. 3. The peptidase positive erythrocyte population corresponded to ∼94% for NEP-erythrocytes, ∼97% for IDE-erythrocytes, and ∼86% for MP-1-erythrocytes. Based on the amount of biotinylated peptidase coupled to erythrocytes it could be calculated that NEP was linked to erythrocytes at a concentration of 1.5×10−4 pg/erythrocyte, that IDE was linked at 1.1×10−4 pg/erythrocyte and that MP-1 was linked at 8.6×10−5 pg/erythrocyte.
Figure 3. Flow cytometry analysis of the binding of biotinylated NEP, IDE and MP-1 to biotinylated erythrocytes modified with streptavidin.
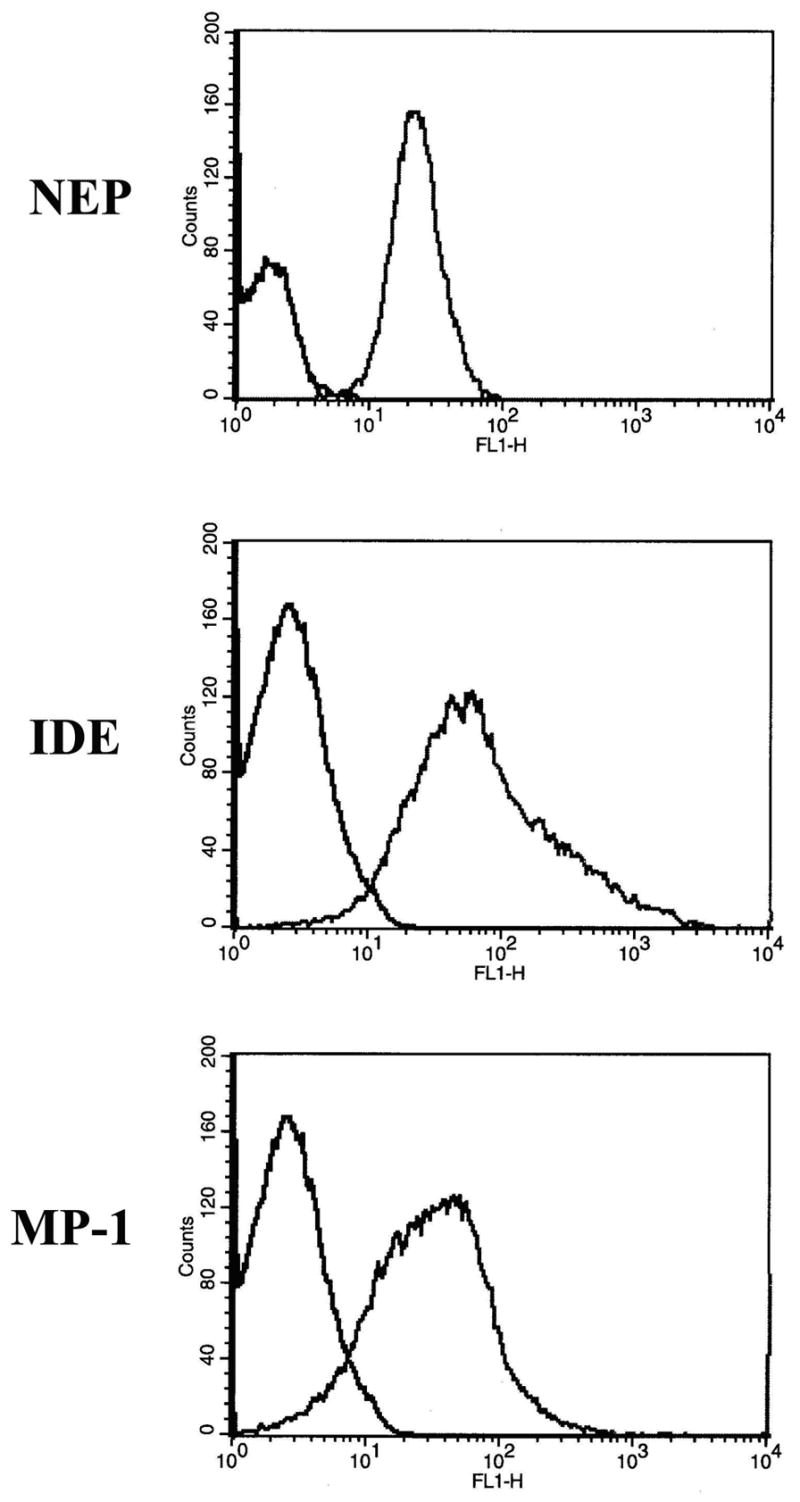
The biotinylated peptidases were couple to biotinylated erythrocytes via streptavidin and then subjected to FACS analysis using FITC conjugated goat anti-mouse for NEP or goat anti-rabbit for IDE or MP-1. Control erythrocytes are represented by the left curves while erythrocytes containing bound peptidase are shown by the curves to the right. FACS analysis shows that the percentages of erythrocytes containing NEP, IDE or MP-1 are 94%, 97% and 86%, respectively.
The activity of peptidase-coupled erythrocytes toward Aβ was evaluated by incubation of 250 pM Aβ1–40 with 5×107 or 2.5×108 peptidase-containing erythrocytes at 37oC for 1 h and measuring the Aβ remaining by sandwich ELISA (Fig 4). Under these conditions NEP-containing erythrocytes degraded 60% and 85% of the Aβ1–40, respectively. Using the same conditions and the same number of IDE-containing erythrocytes 80% and 87% of the Aβ1–40 was hydrolyzed. With MP-1-containing erythrocytes 20% and 75% of the Aβ1–40 was hydrolyzed. Since each of the peptidases has nearly the same molecular weight we can normalize these rates based on the amount of bound peptidase. In terms of the ability to degrade Aβ if we assign IDE bound to erythrocytes a relative activity of 1, we can estimate that on a molar basis erythrocyte-bound NEP is ∼55% as effective as IDE and erythrocyte-bound MP-1 is ∼38% as effective as IDE. This parallels the relative activities of the peptidases in solution. Thus these erythrocyte-bound biotinylated peptidases are able to efficiently hydrolyze Aβ peptide.
Figure 4. The degradation of Aβ1–40 by peptidase-coupled erythrocytes.
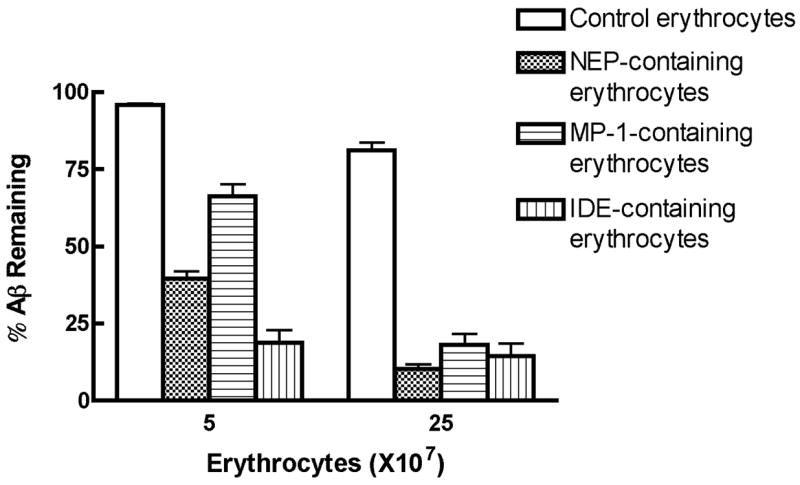
Peptidases bound to erythrocytes were incubated with Aβ1–40 (250 pM) at 5×107 or 2.5×107 peptidase-coupled erythrocytes in PBS buffer at 37°C for 1 h. The Aβ1–40 remaining was assayed by sandwich ELISA.
Error-bars represent the SEM.
*** p<0.001, IDE-coupled, NEP-coupled, or MP-1-coupled erythrocytes vs control erythrocytes; IDE-coupled or NEP-coupled vs MP-1-coupled erythrocytes at 5×107 cells or 25×107 cells, n=3.
No significant difference between IDE-coupled and NEP-coupled erythrocytes was observed.
In order to assess the ability of the erythrocyte-bound peptidases to degrade Aβ peptide in vivo we made use of the observation of DeMattos et al. [7] that injection of a bolus of Aβ into the subarachnoid space of the cisterna magna leads to its efflux from the brain into the blood, followed by a relatively slow clearance of the Aβ from the blood, Fig 5. We therefore injected the erythrocyte-containing peptidases into mice via the tail vein of mice and then 5 minutes latter injected a 1 μg bolus of Aβ1–40 into the subarachnoid space of the cisterna magna. We then measured the ability of the erythrocyte-bound peptidases to increase the rate of plasma Aβ1–40 clearance by sampling blood via the retrobulbar venous plexus and determining the Aβ concentration by sandwich ELISA. As shown in Fig. 5, the Aβ1–40 in plasma of mice administrated any of the three peptidase-containing erythrocytes was cleared more rapidly than in control mice. This is evident at the earliest time point we measured, 5 minutes. If we use the 30 minute time point to compare the effectiveness of each of the peptidases it can be seen that IDE is the most effective, followed by NEP, and then MP-1, which is the same order seen with the ability of either free peptidase or erythrocyte-bound peptidase to cleave Aβ1–40 in vitro. We also measured the stability of the peptidase-containing erythrocytes in blood during the course of this experiment. This was done by binding the fluorescent dye PKH26 to the peptidase-containing erythrocytes and following the loss of dye containing erythrocyte in blood by FACS analysis. We previously determined that PKH26 did not affect the clearance rate of underivatized erythrocytes. As shown in Fig 6 all three peptidase-modified erythrocytes were stable over the time course of the experiment. Figure 6 also shows that a small percentage of the erythrocyte population contained bound peptidase and that this small fraction was effective in lowering in Aβ1–40. Thus all three of these peptidases when present at low levels are effective in clearing Aβ from plasma in vivo.
Figure 5. The effect of peptidase-coupled erythrocytes on the clearance of Aβ1–40 from plasma.
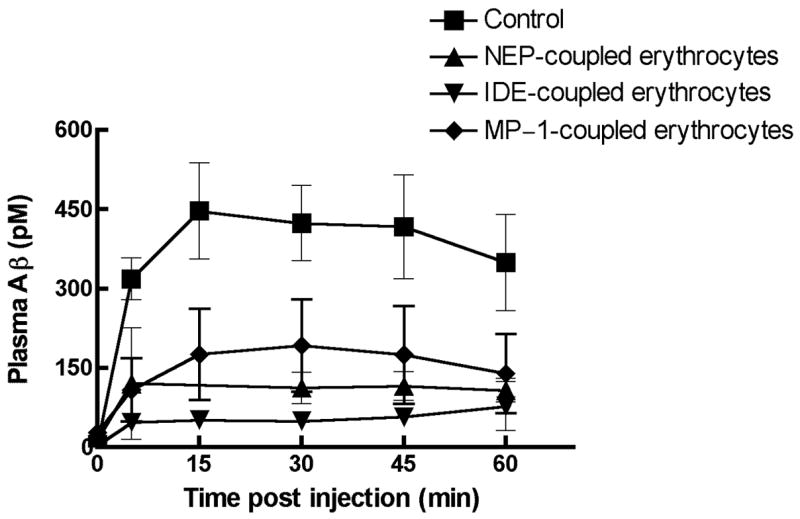
Peptidase-coupled erythrocytes were injected into mice intravenously, 5 min prior to a bolus injection of 1 μg of Aβ1–40 into the subarachnoid space of cisterna magna. At the indicated times post injection of the Aβ1–40 a 50 μl aliquot of blood was collected and assayed for Aβ1–40 by ELISA. Differences between various peptidase-coupled erythrocytes was calculated at the 30 min post Aβ1–40 injection time point.
Error-bars represent the SEM. (p<0.01, IDE- coupled vs NEP-coupled erythrocytes and p<0.05, IDE-coupled vs MP-1-coupled erythrocytes).
* p<0.05, MP-1-coupled vs control erythrocytes;
** p<0.01, NEP-coupled vs control erythrocytes,
*** p<0.001, IDE-coupled vs control erythrocytes. n=4.
Figure 6. Stability of peptidase coupled erythrocytes in plasma.
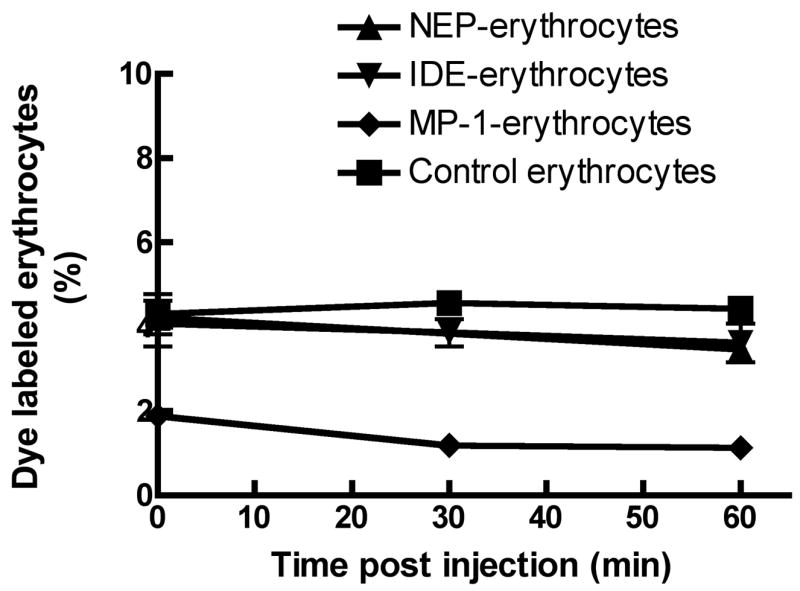
Erythrocytes (∼8×108/100 μl) containing bound peptidase were treated with the fluorescent dye PKH26 prior to injection into mouse blood via the tail vein. At the indicated times blood was collected and the amount of PKH26 containing erythrocytes determined by FACS. The level of 2 to 4% of the dye containing erythrocytes agrees with the initial dilution of 0.1 ml erythrocytes added to ∼2.5 ml erythrocytes per mouse.
4. Discussion
The finding of transport of Aβ peptides from the brain into plasma and vice versa mediated by receptors, such as LRP and RACE, provides a rationale for using peripheral Aβ clearance as an approach for lowering brain Aβ in AD. In this regard both passive Aβ immunotherapy and the use of Aβ-binding compounds have proven effective in transgenic mouse models of AD. Both passive Aβ immunotherapy and Aβ-binding compounds sequester Aβ in plasma generating a sink effect in which the decrease of free plasma Aβ results in a change in the equilibrium between blood and brain Aβ and drives the export of Aβ from brain into blood. Although anti-Aβ antibodies effectively decrease brain Aβ, they have been found to trigger meningoencephalitis and cerebrovascular hemorrhage in clinical trials or in animal models. Ganglioside GM1 and gelsolin are able to bind to Aβ and thus can sequester Aβ from brain to blood. However, GM1 also binds other plasma molecules such as cholesterol. Thus an alternative to sequestering Aβ in plasma is the use of Aβ-degrading peptidases, which instead of building up complexed Aβ in plasma, actually degrades the Aβ into innocuous peptides. Therefore, this approach may circumvent some of the potential problems associated with the peripheral sequestration of Aβ.
In this study we have examined the ability of three known Aβ-degrading peptidases, NEP, IDE, and MP-1, to degrade Aβ when coupled to erythrocytes. Since plasma Aβ levels are normally very low and difficult to detect, we used a bolus injection of Aβ into the brain as described by DeMattos et al. [7], which was subsequently transported to plasma. An alternative could be to use APP transgenic mice in which the APP is expressed from the platelet-derived growth factor promoter. In that particular APP transgenic mouse model plasma Aβ levels are significant.
The results show that all three of the peptidases tested are effective in degrading Aβ in vivo. Under our assay conditions, IDE possessed the highest activity toward Aβ1–40, whether alone or coupled to erythrocytes. However, IDE also most efficiently degraded insulin, which may limit its application for use in the periphery. Although at a slower rate, MP-1 also cleaved insulin. The ability to hydrolyze insulin is not shared by NEP which may makes a better candidate. NEP has been considered a promising candidate for AD treatment, since it appears to be the major, or one of the major enzymes involved in Aβ clearance in the brain [11, 19, 20, 28].
NEP has other advantages over IDE and MP-1. NEP naturally exists on the plasma membrane with its active site facing outside the cell. This extracellular localization of the catalytic domain of NEP places it in an ideal location to hydrolyze extracellular Aβ. In contrast MP-1 is a mitochondrial enzyme, and IDE is predominantly intracellular. In addition NEP is normally expressed in the blood, albeit at low levels [1, 27, 42] thus reducing the likelihood it would be immunogenic. However, there are potential problems with NEP in that it can convert angiotensin I to angiotensin II and hydrolyze other vassoactive peptides. Although this normally occurs in the liver, there is the potential that NEP in the blood could have an adverse effect on blood pressure. Our preliminary studies suggest that expression of NEP in the blood can be accomplished where there is no effect on blood pressure, yet brain Aβ levels can be decreased.
Delivery of drugs or antigens to target cells or tissues using erythrocytes as a carrier has been previously investigated [26]. This approach has several advantages. The system can regulate the biocompatibility of the erythrocytes and molecules on or in the erythrocytes, and thus prolong the bioavailability of the molecules in blood. Erythrocytes can also be used as a reservoir to allow slow release of drug or protein at a sustained therapeutic level [2, 17, 32]. Murciano et al. [33] explored an innovative method in which tPA was coupled to the surface of erythrocytes and targeted the tPA-erythrocyte conjugate to blood clots. It was demonstrated that a low level of biotinylation of erythrocytes does not result in significant lysis by autologous plasma [33]. As noted, free NEP was not stable when added to blood, however when bound to erythrocytes this enzyme is stabilized, providing another advantage of using erythrocytes as a carrier.
The present study demonstrates the ability of Aβ-degrading peptidases to be coupled to erythrocytes in an active form and to retain the ability to lower plasma Aβ levels. Only a small fraction of the erythrocytes need to carry the Aβ-degrading peptidase to be effective. Future studies will be directed at testing erythrocytes as a carrier of Aβ-degrading peptidases in transgenic mouse models of AD.
Acknowledgments
This work was supported in part by grants DA 02243 from the National Institute on Drug Abuse, AG 24899 from the National Institute on Aging, NS 46517 from the National Institute on Neurological Disorders and Stroke, and P20 RR02017 from the NCRR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Yinxing Liu, Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, KY 40536, Department of Molecular and Cellular Biochemistry, University of Kentucky.
Hanjun Guan, Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, KY 40536, Department of Molecular and Cellular Biochemistry, University of Kentucky.
Tina L. Beckett, Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, KY 40536, Department of Molecular and Cellular Biochemistry, University of Kentucky
Maria Aparecida Juliano, Department of Biophysics, Escola Paulista de Medicina, Sao Paulo, Brazil..
Luiz Juliano, Department of Biophysics, Escola Paulista de Medicina, Sao Paulo, Brazil..
Eun Suk Song, Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, KY 40536, Department of Molecular and Cellular Biochemistry, University of Kentucky.
K. Martin Chow, Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, KY 40536, Department of Molecular and Cellular Biochemistry, University of Kentucky.
M. Paul Murphy, Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, KY 40536, Department of Molecular and Cellular Biochemistry, University of Kentucky.
Louis B. Hersh, Department of Molecular and Cellular Biochemistry, University of Kentucky, Lexington, KY 40536, Department of Molecular and Cellular Biochemistry, University of Kentucky
References
- 1.Almenoff J, Teirstein AS, Thornton JC, Orlowski M. Identification of a thermolysin-like metalloendopeptidase in serum: activity in normal subjects and in patients with sarcoidosis. The Journal of laboratory and clinical medicine. 1984;103:420–31. [PubMed] [Google Scholar]
- 2.Beutler E, Dale GL, Guinto DE, Kuhl W. Enzyme replacement therapy in Gaucher’s disease: preliminary clinical trial of a new enzyme preparation. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:4620–3. doi: 10.1073/pnas.74.10.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chow KM, Csuhai E, Juliano MA, St Pyrek J, Juliano L, Hersh LB. Studies on the subsite specificity of rat nardilysin (N-arginine dibasic convertase) The Journal of biological chemistry. 2000;275:19545–51. doi: 10.1074/jbc.M909020199. [DOI] [PubMed] [Google Scholar]
- 4.Csuhai E, Juliano MA, Pyrek JS, Harms AC, Juliano L, Hersh LB. New fluorogenic substrates for N-arginine dibasic convertase. Analytical biochemistry. 1999;269:149–54. doi: 10.1006/abio.1999.4033. [DOI] [PubMed] [Google Scholar]
- 5.Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nature medicine. 2003;9:907–13. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 6.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–44. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 7.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8850–5. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science (New York, NY. 2002;295:2264–7. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- 9.Dominici S, Laguardia ME, Serafini G, Chiarantini L, Fortini C, Tripiciano A, et al. Red blood cell-mediated delivery of recombinant HIV-1 Tat protein in mice induces anti-Tat neutralizing antibodies and CTL. Vaccine. 2003;21:2073–81. doi: 10.1016/s0264-410x(02)00746-6. [DOI] [PubMed] [Google Scholar]
- 10.Du Y, Ni B, Glinn M, Dodel RC, Bales KR, Zhang Z, et al. alpha2-Macroglobulin as a beta-amyloid peptide-binding plasma protein. Journal of neurochemistry. 1997;69:299–305. [PubMed] [Google Scholar]
- 11.Eckman EA, Adams SK, Troendle FJ, Stodola BA, Kahn MA, Fauq AH, et al. Regulation of steady-state beta-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. The Journal of biological chemistry. 2006;281:30471–8. doi: 10.1074/jbc.M605827200. [DOI] [PubMed] [Google Scholar]
- 12.Falkevall A, Alikhani N, Bhushan S, Pavlov PF, Busch K, Johnson KA, et al. Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome, PreP. The Journal of biological chemistry. 2006;281:29096–104. doi: 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- 13.Glabe CC. Amyloid accumulation and pathogenesis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Abeta. Sub-cellular biochemistry. 2005;38:167–77. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- 14.Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiology of aging. 2006;27:570–5. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 15.Hong CS, Goins WF, Goss JR, Burton EA, Glorioso JC. Herpes simplex virus RNAi and neprilysin gene transfer vectors reduce accumulation of Alzheimer’s disease-related amyloid-beta peptide in vivo. Gene therapy. 2006;13:1068–79. doi: 10.1038/sj.gt.3302719. [DOI] [PubMed] [Google Scholar]
- 16.Howell S, Boileau G, Crine P. Neutral endopeptidase (EC 3.4.24.11): constructed molecular forms show new angles of an old enzyme Biochemistry and cell biology. Biochimie et biologie cellulaire. 1994;72:67–9. doi: 10.1139/o94-012. [DOI] [PubMed] [Google Scholar]
- 17.Ihler GM, Glew RH, Schnure FW. Enzyme loading of erythrocytes. Proceedings of the National Academy of Sciences of the United States of America. 1973;70:2663–6. doi: 10.1073/pnas.70.9.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwata N, Mizukami H, Shirotani K, Takaki Y, Muramatsu S, Lu B, et al. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-beta peptide in mouse brain. J Neurosci. 2004;24:991–8. doi: 10.1523/JNEUROSCI.4792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, et al. Metabolic regulation of brain Abeta by neprilysin. Science (New York, NY. 2001;292:1550–2. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 20.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nature medicine. 2000;6:143–50. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 21.Chow KMEC, Juliano MA, Pyrek JS, Juliano L, Hersh LB. Studies on the subsite specificity of rat nardilysin (N-arginine dibasic convertase) J Biol Chem. 2000;275:19545–19551. doi: 10.1074/jbc.M909020199. [DOI] [PubMed] [Google Scholar]
- 22.Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–93. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- 23.Levites Y, Das P, Price RW, Rochette MJ, Kostura LA, McGowan EM, et al. Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. The Journal of clinical investigation. 2006;116:193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levites YDP, Price RW, Rochette MJ, Kostura LA, McGowan EM, Murphy MP, Golde TE. Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J Clin Invest. 2006 Jan;116(1):193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C, Hersh LB. Neprilysin: assay methods, purification, and characterization. Methods Enzymol. 1995;248:253–263. doi: 10.1016/0076-6879(95)48018-8. [DOI] [PubMed] [Google Scholar]
- 26.Magnani M, Rossi L, Fraternale A, Bianchi M, Antonelli A, Crinelli R, et al. Erythrocyte-mediated delivery of drugs, peptides and modified oligonucleotides. Gene therapy. 2002;9:749–51. doi: 10.1038/sj.gt.3301758. [DOI] [PubMed] [Google Scholar]
- 27.Mari B, Checler F, Ponzio G, Peyron JF, Manie S, Farahifar D, et al. Jurkat T cells express a functional neutral endopeptidase activity (CALLA) involved in T cell activation. The EMBO journal. 1992;11:3875–85. doi: 10.1002/j.1460-2075.1992.tb05480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marr RA, Guan H, Rockenstein E, Kindy M, Gage FH, Verma I, et al. Neprilysin regulates amyloid Beta peptide levels. J Mol Neurosci. 2004;22:5–11. doi: 10.1385/JMN:22:1-2:5. [DOI] [PubMed] [Google Scholar]
- 29.Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, et al. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci. 2003;23:1992–6. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, et al. Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to beta-amyloid. J Neurosci. 2003;23:29–33. doi: 10.1523/JNEUROSCI.23-01-00029.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–9. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Millan CG, Marinero ML, Castaneda AZ, Lanao JM. Drug, enzyme and peptide delivery using erythrocytes as carriers. J Control Release. 2004;95:27–49. doi: 10.1016/j.jconrel.2003.11.018. [DOI] [PubMed] [Google Scholar]
- 33.Murciano JC, Medinilla S, Eslin D, Atochina E, Cines DB, Muzykantov VR. Prophylactic fibrinolysis through selective dissolution of nascent clots by tPA-carrying erythrocytes. Nature biotechnology. 2003;21:891–6. doi: 10.1038/nbt846. [DOI] [PubMed] [Google Scholar]
- 34.Muzykantov VR, Murciano JC, Taylor RP, Atochina EN, Herraez A. Regulation of the complement-mediated elimination of red blood cells modified with biotin and streptavidin. Analytical biochemistry. 1996;241:109–19. doi: 10.1006/abio.1996.0384. [DOI] [PubMed] [Google Scholar]
- 35.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, et al. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science (New York, NY) 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- 36.Qiu WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiology of aging. 2006;27:190–8. doi: 10.1016/j.neurobiolaging.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 37.Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25:629–36. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selkoe DJ. Amyloid beta-protein precursor: new clues to the genesis of Alzheimer’s disease. Current opinion in neurobiology. 1994;4:708–16. doi: 10.1016/0959-4388(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 39.Selkoe DJ. Normal and abnormal biology of the beta-amyloid precursor protein. Annual review of neuroscience. 1994;17:489–517. doi: 10.1146/annurev.ne.17.030194.002421. [DOI] [PubMed] [Google Scholar]
- 40.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. The Journal of clinical investigation. 2000;106:1489–99. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song ES, Juliano MA, Juliano L, Hersh LB. Substrate activation of insulin-degrading enzyme (insulysin) A potential target for drug development The Journal of biological chemistry. 2003;278:49789–94. doi: 10.1074/jbc.M308983200. [DOI] [PubMed] [Google Scholar]
- 42.Spillantini MG, Sicuteri F, Salmon S, Malfroy B. Characterization of endopeptidase 3.4.24.11 (“enkephalinase”) activity in human plasma and cerebrospinal fluid. Biochemical pharmacology. 1990;39:1353–6. doi: 10.1016/0006-2952(90)90012-a. [DOI] [PubMed] [Google Scholar]
- 43.Thompson MW, Hersh LB. Analysis of conserved residues of the human puromycin-sensitive aminopeptidase. Peptides. 2003;24:1359–65. doi: 10.1016/j.peptides.2003.07.012. [DOI] [PubMed] [Google Scholar]
- 44.Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein and peptide letters. 2004;11:213–28. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 45.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. Journal of neuroinflammation [electronic resource] 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang DS, Smith JD, Zhou Z, Gandy SE, Martins RN. Characterization of the binding of amyloid-beta peptide to cell culture-derived native apolipoprotein E2, E3, and E4 isoforms and to isoforms from human plasma. Journal of neurochemistry. 1997;68:721–5. doi: 10.1046/j.1471-4159.1997.68020721.x. [DOI] [PubMed] [Google Scholar]