

Abstract
Insulin stimulates adipose cells both to secrete proteins and to translocate the GLUT4 glucose transporter from an intracellular compartment to the plasma membrane. We demonstrate that whereas insulin stimulation of 3T3-L1 adipocytes has no effect on secretion of the α3 chain of type VI collagen, secretion of the protein hormone adipocyte complement related protein of 30 kD (ACRP30) is markedly enhanced. Like GLUT4, regulated exocytosis of ACRP30 appears to require phosphatidylinositol-3-kinase activity, since insulin-stimulated ACRP30 secretion is blocked by pharmacologic inhibitors of this enzyme. Thus, 3T3-L1 adipocytes possess a regulated secretory compartment containing ACRP30. Whether GLUT4 recycles to such a compartment has been controversial. We present deconvolution immunofluorescence microscopy data demonstrating that the subcellular distributions of ACRP30 and GLUT4 are distinct and nonoverlapping; in contrast, those of GLUT4 and the transferrin receptor overlap. Together with supporting evidence that GLUT4 does not recycle to a secretory compartment via the trans-Golgi network, we conclude that there are at least two compartments that undergo insulin-stimulated exocytosis in 3T3-L1 adipocytes: one for ACRP30 secretion and one for GLUT4 translocation.
Keywords: exocytosis, monosaccharide transport proteins, insulin, adipose tissue, secretion
Adipocytes function as endocrine cells, and are the exclusive source of several serum proteins including leptin, adipsin (equivalent to complement factor D), and adipocyte complement related protein of 30 kD (ACRP30)1 (also called adipoQ) (Kitagawa et al. 1989; Zhang et al. 1994; Scherer et al. 1995; Hu et al. 1996). Of these, leptin has received the most attention because of its clear role in regulating body weight. ACRP30 likely also plays an important role in energy homeostasis, since it is dysregulated in obesity and has close structural homology to TNF-α, another protein secreted by adipocytes and implicated in insulin resistance (Hu et al. 1996; Uysal et al. 1997; Shapiro and Scherer 1998). Secretion of ACRP30 from 3T3-L1 adipocytes, like that of adipsin and leptin, is enhanced by insulin stimulation (Kitagawa et al. 1989; Scherer et al. 1995; Barr et al. 1997; Bradley and Cheatham 1999). Importantly, it has not been determined whether this effect of insulin is mediated by a regulated secretory compartment, or if insulin instead nonspecifically accelerates the entire secretory pathway. In the case of leptin, insulin appears to acutely stimulate export from the endoplasmic reticulum (ER) of isolated rat adipocytes (Barr et al. 1997). Yet, whether this effect is solely responsible for the insulin-mediated enhancement of leptin secretion remains unknown.
Insulin also regulates intracellular trafficking of the GLUT4 glucose transporter in adipose and muscle. This regulation is of central importance in glucose homeostasis, since it is primarily the presence of GLUT4 in the plasma membrane that determines glucose utilization in these tissues (Kahn 1996; Stenbit et al. 1997). Upon binding of insulin to its receptor, the rate of GLUT4 exocytosis increases with little or no decrease in the rate of GLUT4 endocytosis, resulting in a net shift in the subcellular distribution of GLUT4 to the plasma membrane (Satoh et al. 1993; Yang and Holman 1993). Once in the plasma membrane, GLUT4 facilitates diffusion of glucose into the cell, resulting in a 20–30-fold increase in the rate of glucose uptake in the presence of insulin. The effect of insulin on GLUT4 trafficking is mediated, at least in part, by phosphatidylinositol-3-kinase (PI-3 kinase), but the downstream effectors of this enzyme, as well as the subcellular compartment(s) that are mobilized, are poorly defined (Rea and James 1997; Jiang et al. 1998).
Several investigators have attempted to determine whether or not the insulin-stimulatable GLUT4 compartment is part of a regulated pathway for protein secretion: is the compartment more analogous to endosomally derived synaptic vesicles, or to biosynthetically derived secretory vesicles? The latter possibility is consistent with the finding that GLUT4 is present in the trans-Golgi network (TGN), the site where most secretory vesicles form, and that it is depleted from this compartment after insulin stimulation (Slot et al. 1991; Rindler 1992). Indeed, when exogenously expressed in differentiated PC12 neuroendocrine cells, GLUT4 was concentrated in large dense core vesicles, characteristic of a specialized secretory compartment, as well as in early and late endosomes (Hudson et al. 1993). In contrast, other investigators working with the same cell type found that exogenously expressed GLUT4 was targeted to small vesicles, distinct from both large dense core vesicles and small synaptic vesicles, as examined by both subcellular fractionation and electron microscopy (Herman et al. 1994). This compartment was mobilized by insulin stimulation and appeared to be present in several cell types, suggesting that it is not part of a specialized secretory pathway. Similar results were found in insulinoma cells, where exogenously expressed GLUT4 was targeted to vesicles distinct from both insulin-containing secretory granules and synaptic-like vesicles (Thorens and Roth 1996).
In the above studies, the GLUT4 cDNA was transfected into insulinoma cells or PC12 neuroendocrine cells, chosen because they have well-characterized pathways for regulated secretion or for synaptic vesicle recycling. Because targeting may have been affected either by overexpression of GLUT4 protein or by expression in a non-native cell type, Slot et al. 1997 examined endogenous GLUT4 in cardiac muscle. Electron microscopy indicated that 50–60% of endogenous GLUT4 is targeted to secretory granules in cardiac myocytes, as assessed by costaining for atrial natriuretic factor, and suggested that a large proportion of GLUT4 enters a regulated secretory pathway at the TGN. However, insulin stimulation failed to cause demonstrable atrial natriuretic factor release, though it significantly increased 2-deoxyglucose uptake. Other investigators found that in rat adipocytes, wortmannin blocks insulin-stimulated GLUT4 trafficking at a step apparently distal to segregation from late endosomal and TGN markers, suggesting that GLUT4 does not recycle through these compartments (Malide and Cushman 1997). Thus, conflicting data have been reported as to whether insulin stimulates GLUT4 exocytosis via a regulated secretory compartment, or if GLUT4 recycling occurs independently of insulin-stimulated protein secretion.
To determine if a regulated secretory compartment exists in 3T3-L1 adipocytes, we compared secretion of ACRP30 with that of the α3 subunit of mouse type VI collagen. Previous work in our laboratory has shown that ACRP30 is highly enriched in adipose tissue, that its expression is induced during 3T3-L1 adipocyte differentiation, and that it undergoes basal as well as insulin-stimulated secretion (Scherer et al. 1998). We hypothesized that if a regulated secretory compartment exists in these cells, then ACRP30 but not α3 (VI) collagen might be targeted to this compartment. Consistent with our prediction, pulse-chase experiments demonstrate that secretion of ACRP30, but not α3 (VI) collagen, is enhanced by insulin. Secretion of ACRP30, but not α3 (VI) collagen, is also stimulated by a calcium ionophore (A23187); this effect is observed late in the chase period and is less dramatic than that of insulin. We also demonstrate that insulin-stimulated exocytosis of the ACRP30 compartment is likely mediated by PI-3 kinase, since inhibitors of this enzyme block insulin-stimulated, but not basal ACRP30 secretion. Because much leptin appears to reside in the ER in primary rat adipocytes (Barr et al. 1997), we costained ACRP30 and GRP94, a resident protein of the ER. We observed partial colocalization of ACRP30 with this protein; the proportion of ACRP30 that does not overlap GRP94 is presumably in a more distal compartment of a regulated secretory pathway. These data indicate that an insulin-regulated secretory compartment is present in 3T3-L1 adipocytes, and that ACRP30 but not α3 (VI) collagen is targeted to this compartment.
We next performed deconvolution immunofluorescence microscopy to determine if GLUT4 recycles to the regulated exocytic compartment containing ACRP30. Our data demonstrate that whereas the subcellular distributions of GLUT4 and transferrin receptor (TfnR) overlap, those of ACRP30 and GLUT4 are distinct and nonoverlapping in both unstimulated and insulin-stimulated cells. To support our conclusion that GLUT4 does not recycle to a secretory pathway, we confirm that GLUT4 in the perinuclear region does not colocalize with a Golgi marker (β-COP), and is distinct from a TGN marker (γ-adaptin), with no apparent difference between unstimulated and insulin-stimulated cells. We conclude that there are at least two compartments that undergo insulin-regulated exocytosis in 3T3-L1 adipocytes: one for ACRP30 secretion and one for GLUT4 translocation.
Materials and Methods
Antibodies and Reagents
A rabbit polyclonal antibody raised against the carboxy terminus of GLUT4 (MC2A) (Charron et al. 1989) was a gift of Dr. Maureen Charron (Albert Einstein College of Medicine, Bronx, NY). A goat polyclonal carboxy-terminal GLUT4 antibody was purchased from Santa Cruz Biotechnology. Rabbit ACRP30 and α3 (VI) collagen antisera (Scherer et al. 1995, Scherer et al. 1998) were a gift of Dr. Philipp Scherer (Albert Einstein College of Medicine). A rat TfnR mAb was purchased from PharMingen. Rabbit antiserum to β-COP and rat anti-GRP94 mAb antibody (clone 9G10) were from Affinity Bioreagents. A mouse mAb to adaptin-γ was purchased from Transduction Laboratories. Normal goat and donkey sera and fluorophore-conjugated secondary antibodies cross-adsorbed against the relevant species-specific IgGs were purchased from Jackson ImmunoResearch, Inc. Paraformaldehyde (16% solution) was purchased from Electron Microscopy Sciences.
Cell Culture
Murine 3T3-L1 fibroblasts were kindly provided by E. Santos (National Institutes of Health, Bethesda, MD) and were cultured in DME containing 10% fetal calf serum (Benito et al. 1991). Differentiation was induced as described (Frost and Lane 1985). Appropriate differentiation was confirmed by noting accumulation of lipid droplets, and cells were used 8–12 d after induction of differentiation.
Pulse-chase Experiments and Immunoprecipitations
The protocol used was modified from Scherer et al. 1995. 3T3-L1 adipocytes grown and differentiated in 10-cm tissue culture dishes were starved in DME for a total of at least 3 h. During the final hour of serum starvation, the medium was changed to DME lacking cysteine and methionine (ICN Radiochemicals). For some experiments, 100 nM wortmannin (Sigma Chemical Co.), 10 μM LY294002 (Calbiochem), or 10 ng/ml rapamycin (Calbiochem) was added during the last 45 min of starvation. Cells were then pulse-labeled for 15 min in the same medium supplemented with 0.5–0.7 mCi/ml Express Protein Labeling reagent, a mixture of 35S-labeled cysteine and methionine (1,000 Ci/mmol; DuPont/New England Nuclear). Cells were then washed twice with DME containing unlabeled cysteine and methionine, and then incubated during a 2-h chase period at 37°C in DME containing 300 μM cycloheximide to prevent further protein synthesis, with or without 160 nM insulin or 200 μM A23187 (Calbiochem). Cells that were pretreated with wortmannin, LY294002, or rapamycin were maintained in the presence of these drugs during the chase period. At 30-min intervals, the medium was collected from each plate and was replaced with identical fresh medium. Samples were kept on ice until all were collected, then insoluble debris was removed by centrifugation (15,000 g, 10 min, 4°C). Samples were precleared by incubation with 60 μl protein A–Sepharose for 30–60 min at 4°C. Immunoprecipitations each used 6 μl of antisera directed against ACRP30 or α3 (VI) collagen, and were allowed to proceed for a minimum of 4 h at 4°C, after which 100 μl protein A–Sepharose was added and the incubations were continued for an additional 45 min. ACRP30 and α3 (VI) collagen were immunoprecipitated sequentially from the same samples. Immunoprecipitates were washed five times in TNET (1% Triton X-100, 150 mM NaCl, 20 mM Tris, pH 8.0, 2 mM EDTA), electrophoresed on SDS-PAGE, and the dried gels were fixed, treated with sodium salicylate or Enhance (Amersham), exposed, and quantified using a Fuji PhosphorImager. Experiments were performed at least twice, with similar results each time.
Immunofluorescence Microscopy
3T3-L1 adipocytes were reseeded 1 d before fixation to either coverslips or to the wells of teflon-coated microscope slides (Cel-Line Associates, Inc.). Cells were serum starved in DME for a minimum of 3 h, then stimulated or not with 160 nM insulin for 12 min. Cells were washed with cold PBS containing 0.9 mM Ca++ and 0.5 mM Mg++ (PBS++). Fixation was with 3–4% paraformaldehyde in PBS for 45 min at room temperature, followed by permeabilization with 0.2% Triton X-100 in PBS++ at 4°C. In some instances, cells were washed with PBS++ and stored before staining at 4°C in PBS++, 2% BSA, and sodium azide. After washing the cells again with PBS++, nonspecific antibody binding was blocked with PBS++ containing 2% BSA and 4% goat or donkey serum (blocking buffer), as appropriate to the source of the secondary antibody, for 30 min. In experiments using the goat GLUT4 antisera, only donkey secondary antibodies were used. Primary antibodies were used at dilutions ranging from 1:200 to 1:500 in blocking buffer. Incubation with primary antibodies was for 60–120 min at 25°C or 37°C in a humidified chamber. The cells were again washed with PBS++, and then incubated with FITC- or Cy3-conjugated goat or donkey secondary antibodies at dilutions of 1:200 to 1:400 in blocking buffer. Incubation with secondary antibodies was for 30–45 min at 25°C or 37°C in a humidified chamber. The cells were rinsed twice with PBS++ and then washed at least three times, for 10 min each, with PBS++. Coverslips were mounted using Vectashield (Vector Laboratories). For all microscopy experiments, controls were done to demonstrate that the binding of secondary antibodies was specific for the intended primary antibody. Additionally, the specificity of the rabbit ACRP30 and rabbit and goat GLUT4 antisera was shown in peptide competition experiments (data not shown).
Deconvolution microscopy was performed using a CELLscan system by Scanalytics, Inc., attached to either an Olympus or a Nikon Eclipse E-800 microscope. Both microscopes were equipped with an excitation filter wheel and a triple bandpass cube containing a dichroic mirror and emission filter, ensuring registration of two or three color images in both the X–Y plane and along the Z-axis. Empirical point spread functions were obtained immediately before image acquisition by using 0.1-μm diam latex beads fluorescent at the appropriate wavelengths (Molecular Probes). Beads were diluted in PBS and briefly sonicated to disrupt aggregates, and were then by mounted on microscope slides using Vectashield under conditions identical to those for 3T3-L1 cells. For both point spread function and image acquisition, through-focusing was done using the 100×/1.4 NA objective and acquiring data at 250-nm intervals. For cross-sectional images of cells, at least nine planes of raw image data were obtained so as to optimize reconstruction of the center plane image. Deconvolution was done on a CSPI high speed processor, and both raw and reconstructed images were acquired with 12-bit/pixel resolution. Under these conditions, we estimate lateral resolution at 180 nm. Images were pseudocolored and merged using Photoshop (Adobe Systems, Inc.); final pixel depth is 8 bits/channel.
Results
Insulin Stimulates Secretion of ACRP30, But Not α3 (VI) Collagen
To determine if the insulin-stimulated enhancement of ACRP30 secretion is mediated by a regulated secretory compartment, or if it represents a nonspecific acceleration of the entire secretory pathway, we compared secretion of ACRP30 and α3 (VI) collagen in a pulse-chase experiment. Previous work done in our laboratory using a complex antisera raised against many proteins secreted from 3T3-L1 adipocytes demonstrated that insulin enhances secretion of some, but not all, of these proteins (Scherer et al. 1998; Scherer, P.E., and H.F. Lodish, unpublished results). We hypothesized that one specific protein that does not participate in an insulin-regulated secretory compartment might be α3 (VI) collagen (Scherer et al. 1998). Accordingly, we followed secretion into the media of a discrete population of newly synthesized protein, synthesized during a short pulse of labeled amino acids and chased in the presence of cycloheximide and in the presence or absence of insulin or a calcium ionophore, A23187. The media were collected at intervals throughout the chase period, and α3 (VI) collagen and ACRP30 were immunoprecipitated sequentially from the same samples. Fig. 1 A shows a representative experiment; in Fig. 1 B these data are used to plot the cumulative amount of each protein secreted. Whereas insulin clearly enhances ACRP30 secretion, there is no effect on α3 (VI) collagen secretion. The insulin-stimulated increase in ACRP30 secretion is most marked during the early part of the chase period, consistent with an effect of insulin to accelerate the early part of the secretory pathway. Yet, insulin's effect may not be limited to this part of the secretory pathway, since ACRP30 secretion is increased even at later time points. A23187 has a minimal effect early in the chase period, but enhances ACRP30 secretion, and not α3 (VI) collagen secretion, after 90 min. This result is consistent with an effect late in the secretory pathway, involving a compartment to which ACRP30, but not α3 (VI) collagen, is targeted. Thus, the data indicate that the insulin-stimulated enhancement of ACRP30 secretion is not due to nonspecific acceleration of the entire secretory pathway. Rather, ACRP30 is targeted to a regulated secretory compartment in 3T3-L1 adipocytes, and α3 (VI) collagen is excluded from this compartment.
Figure 1.

Insulin stimulates ACRP30 secretion from 3T3-L1 adipocytes, but does not stimulate α3 (VI) collagen secretion. Three 10-cm plates of 3T3-L1 adipocytes were serum-starved and total cellular protein was labeled with a 15-min pulse of 35S-labeled cysteine and methionine. Cells were washed and then chased in serum-free media containing cycloheximide to inhibit further protein synthesis. Additionally during the chase period, cells were maintained in the presence or absence of 160 nM insulin or 200 μM A23187. The media were collected and replaced at 30-min intervals, and the appearance of labeled ACRP30 or α3 (VI) collagen in the media was assessed by sequential immunoprecipitations. The immunoprecipitates were analyzed SDS-PAGE, and bands were detected and quantified using a PhosphorImager. A shows the bands corresponding to ACRP30 and α3 (VI) collagen for each condition. In B, these bands were quantified and the cumulative secretion into the media is plotted for each protein. The total amount of protein secreted by unstimulated cells is taken as 100% and all other data are plotted relative to this amount.
Inhibitors of PI-3 Kinase Block Insulin-stimulated, But Not Basal, ACRP30 Secretion
PI-3 kinase activity is stimulated by insulin, and has been implicated in the insulin signal transduction pathway leading to GLUT4 exocytosis. To determine if PI-3 kinase activation is also required for insulin-stimulated ACRP30 secretion, we performed pulse-chase experiments similar to those described in Fig. 1, but in the presence or absence of two pharmacologic inhibitors of PI-3 kinase, wortmannin and LY294002. Fig. 2 plots cumulative ACRP30 secretion, and demonstrates that ACRP30 secretion is enhanced by insulin in cells left untreated or in the presence of rapamycin, used here as a negative control, and that this effect is absent in the presence of wortmannin or LY294002. Like the results shown in Fig. 1, the increased secretion is especially marked during the first 30 min of the chase period in the untreated and rapamycin-treated cells. In the presence of wortmannin, ACRP30 secretion at the 30-min time point is not significantly enhanced by insulin, and at subsequent time points the cumulative secretion in the presence of insulin is even less than in the absence of insulin. Similarly, LY294002 completely abolishes the effect of insulin, so that cumulative ACRP30 secretion is essentially identical in the presence or absence of insulin in cells treated with this drug. No definite effect is seen on basal ACRP30 secretion under any of the conditions tested. Thus, PI-3 kinase inhibitors specifically block insulin-stimulated exocytosis of ACRP30. Together with the data presented in Fig. 1, we conclude that ACRP30 is targeted to a regulated secretory compartment, the exocytosis of which is stimulated by insulin through a PI-3 kinase activity.
Figure 2.

PI-3 kinase inhibitors block insulin-stimulated, but not basal, ACRP30 secretion. Eight 10-cm plates of 3T3-L1 adipocytes were serum-starved and then treated or not with wortmannin, LY294002, or rapamycin at the indicated concentrations for 45 min. As in Fig. 1, total cellular protein was labeled with a 15-min pulse of 35S-labeled cysteine and methionine, followed by inhibition of further protein synthesis with cycloheximide. During the chase period, cells were maintained in the presence or absence of 160 nM insulin and the continued presence or absence of inhibitors, and the medium was collected and replaced at 30-min intervals. ACRP30 was quantitatively immunoprecipitated from the media collected at each time point. The immunoprecipitates were separated by SDS-PAGE and the amount of ACRP30 present at each data point was quantified using a PhosphorImager. Cumulative ACRP30 is plotted for cells chased in the absence (open squares) or presence (filled diamonds) of insulin. The total amount of protein secreted by the untreated cells in the presence of insulin is taken as 100%; all other data are plotted relative to this amount.
ACRP30 Immunostaining Partially Overlaps with That of GRP94, a Resident of the ER
It has been reported that leptin largely colocalizes with calnexin, a marker for the ER, in isolated rat adipocytes (Barr et al. 1997). To determine whether this is also true of ACRP30 in 3T3-L1 adipocytes, we used deconvolution immunofluorescence microscopy to obtain cross-sectional images of cells stained for ACRP30 and GRP94. GRP94 (also called endoplasmin) is a well-characterized lumenal protein of the ER that contains a carboxy-terminal KDEL motif, and that functions in concert with GRP78/BiP to assist in protein folding (Koch et al. 1986; Gething and Sambrook 1992; Melnick et al. 1992, Melnick et al. 1994). As shown in Fig. 3, some but not all ACRP30 staining overlaps with that of GRP94. GRP94 staining (shown in red) is reticular with some pronounced punctae, and is present throughout the cytoplasm in both unstimulated (Fig. 3 a) and insulin-stimulated (Fig. 3 d) cells. ACRP30 staining (Fig. 3b and Fig. e, green) is mostly punctate, but has a reticular component as well. In unstimulated cells, there is moderate overlap of these two proteins (Fig. 3 c, yellow), though it is also clear that some ACRP30 staining does not overlap with GRP94 staining (Fig. 3 c, green). ACRP30 expression was heterogeneous, in that it was commonly observed that not all cells that had differentiated by morphological criteria (as assessed by the presence of lipid droplets) stained for ACRP30 on immunofluorescence microscopy. The staining for GRP94 (red), but not ACRP30, present in the lower left quadrant of Fig. 3 c represents an adjacent cell not expressing ACRP30; there is very little staining for GRP94 by itself (i.e., not overlapping ACRP30) in the remainder of the image. In comparison to unstimulated cells, insulin-stimulated cells have more prominent staining for GRP94 that does not overlap with ACRP30 (Fig. 3 f, red), though some overlap also remains (yellow). These findings are subtle, but are observed consistently in the highest quality images. Thus, the data suggest that insulin stimulates movement of ACRP30 out of the ER. More significantly, a proportion of ACRP30 does not overlap with GRP94, and is presumably in a more distal compartment of a regulated secretory pathway.
Figure 3.

ACRP30 and GRP94 staining partially overlap. 3T3-L1 adipocytes were costained for ACRP30 (shown in green) and GRP94 (shown in red), and optical cross-sections were acquired by deconvolution immunofluorescence microscopy. To demonstrate a large amount of staining for both proteins, the images presented are merged stacks of nine optical cross-sections spanning a total of 2.25 μm perpendicular to the image plane; a similar degree of overlap was observed in individual cross-sections. GRP94 staining is shown in a and d; ACRP30 staining is shown in b and e. Merged images are shown in c and f. a–c show staining of unstimulated 3T3-L1 adipocytes, and d–f show staining of insulin-stimulated 3T3-L1 adipocytes. Bar, 10 μm.
GLUT4 and ACRP30 Do Not Colocalize within 3T3-L1 Adipocytes
To determine if GLUT4 recycles to the insulin-regulated secretory pathway containing ACRP30, we employed deconvolution immunofluorescence microscopy to obtain cross-sectional images of GLUT4 and ACRP30 in 3T3-L1 adipocytes. Fig. 4 presents representative images of adipocytes stained for GLUT4 (shown in red) and ACRP30 (shown in green). The staining for GLUT4 is punctate, consistent with a vesicular compartment, with prominent perinuclear staining that is in a more tubulovesicular pattern (Fig. 4, a, d, and g). This distribution has been noted previously by others, and is characteristic of GLUT4 (Piper et al. 1991; Smith et al. 1991). ACRP30 staining, too, is mainly punctate, but also has a reticular component and is present more equally throughout the cytoplasm, with less perinuclear accumulation (Fig. 4b, Fig. e, and Fig. h). The merged images (Fig. 4c, Fig. f, and Fig. i) demonstrate that there is essentially no overlap (yellow) in the subcellular distribution of the two proteins. This is the case for both unstimulated and insulin-stimulated cells (Fig. 4, a–f and g–i, respectively). Indeed, because the staining for these two proteins is so distinct, we have presented views through several cross-sections in Fig. 4 and still see no significant overlap. Insulin stimulation (160 nM, 12 min) of cells before fixation clearly results in accumulation of GLUT4 at the plasma membrane (Fig. 4 g, arrowheads), but did not cause any obvious change in the pattern of ACRP30 staining. Thus, GLUT4 does not appear to participate in the regulated secretory compartment that contains ACRP30.
Figure 4.
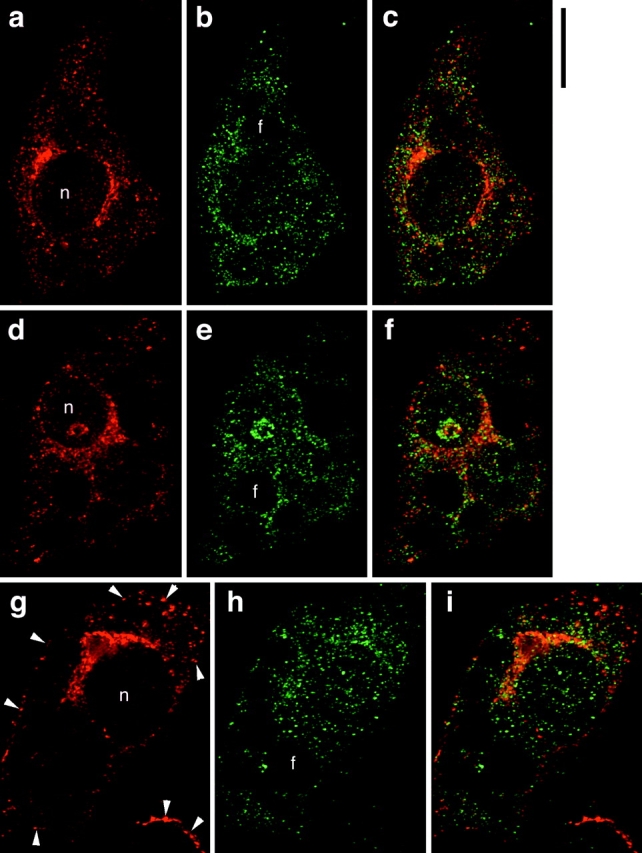
ACRP30 and GLUT4 do not colocalize in 3T3-L1 adipocytes. Immunofluorescence of GLUT4 (red) and ACRP30 (green) was performed on 3T3-L1 adipocytes, and optical cross-sections were acquired by deconvolution microscopy. The images presented are views through nine optical cross-sections. GLUT4 staining is shown in a, d, and g; ACRP30 staining is shown in b, e, and h. Merged images are shown in c, f, and i. a–f show staining of unstimulated cells, and g–i show staining of insulin-stimulated cells. Arrowheads highlight increased plasma membrane localization of GLUT4 in cells stimulated with insulin. n, nucleus. f, lipid. Bar, 10 μm.
TfnR Immunostaining Significantly Overlaps with That of GLUT4, But Not ACRP30
Fig. 5 shows immunofluorescence images of cells stained with GLUT4 (in red) and TfnR (in green). Both GLUT4 (Fig. 5, a and d) and TfnR (Fig. 5b and Fig. e) are present in a punctate pattern at the periphery of the cell and in a more tubulovesicular pattern adjacent to the nucleus. The merged images (Fig. 5c and Fig. f) demonstrate that there is significant colocalization (yellow) of the two proteins, with perhaps half of each colocalizing with the other. This colocalization is present in the periphery of the cell as well as in the perinuclear region. In cells stimulated with insulin (Fig. 5, d–f), there is more prominent staining of both GLUT4 and TfnR at the plasma membrane (arrowheads, Fig. 5d and Fig. e). Of note, neither the perinuclear GLUT4 nor the peripheral GLUT4 overlaps completely with TfnR. After insulin stimulation, prominent GLUT4 staining remains in the perinuclear region, whereas TfnR staining is somewhat less marked (Fig. 5 f). Though this effect of insulin was not seen in all images, it may represent participation of TfnR, but not GLUT4, in a perinuclear recycling endosome.
Figure 5.
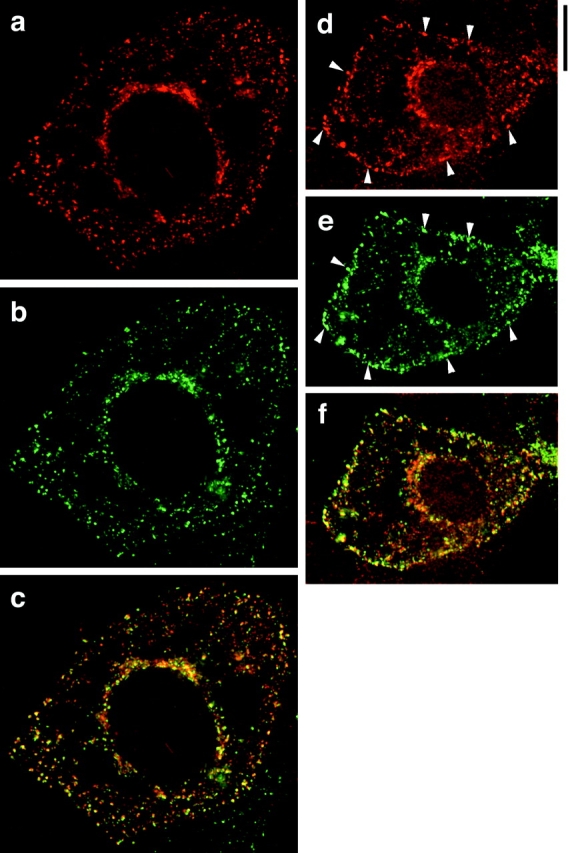
GLUT4 and TfnR overlap. Immunofluorescence of GLUT4 (red) and TfnR (green) was performed on 3T3-L1 adipoctes. As in Fig. 4, each image is a merged composite of nine adjacent optical cross-sections; a similar degree of overlap was seen in individual cross-sectional images. GLUT4 staining is shown in a and d; TfnR staining is shown in b and e. Merged images are shown in c and f. a–c show staining of unstimulated cells, and d–f show staining of insulin-stimulated cells. Arrowheads highlight increased plasma membrane localization of both GLUT4 and TfnR after insulin stimulation. Bar, 10 μm.
As an additional control experiment, we stained cells for TfnR and ACRP30. As shown in Fig. 6, there is essentially no overlap in the distribution of these two proteins. ACRP30 (in red) is once again seen to have punctate and reticular components in both unstimulated (Fig. 6 a) and insulin-stimulated (Fig. 6 d) cells. The apparent perinuclear staining in Fig. 6 d is due, at least in part, to the presence of several lipid droplets throughout the periphery of the cell. TfnR staining (Fig. 6b and Fig. e, green) is present in a more tubulovesicular pattern. There is some TfnR present at the plasma membrane after insulin stimulation (Fig. 6 e), though this is less marked than in Fig. 5. The merged images demonstrate very little overlap (yellow) in the subcellular distributions of these proteins, in both unstimulated and insulin-stimulated cells (Fig. 6c and Fig. f, respectively), corroborating the data shown in Fig. 4 and Fig. 5. We conclude that GLUT4 and TfnR are targeted to compartments that are distinct from those containing ACRP30.
Figure 6.

ACRP30 and TfnR do not colocalize. Immunofluorescence of 3T3-L1 adipocytes stained for ACRP30 (red) and TfnR (green). Each image is the merged stack of nine optical cross-sections, as in Fig. 4 and Fig. 5. ACRP30 staining is shown in a and d; TfnR staining is shown in b and e. Merged images are shown in c and f. a–c show staining of unstimulated 3T3-L1 adipocytes, and d–f show staining of insulin-stimulated 3T3-L1 adipocytes. Bar, 10 μm.
The Subcellular Distribution of GLUT4 Does Not Overlap with That of β-COP, and Is Mostly Separate from That of γ-Adaptin
If GLUT4 does not recycle to a regulated secretory compartment, then GLUT4 in the perinuclear region likely does not represent recycling to the Golgi complex or TGN. We sought to confirm this in 3T3-L1 adipocytes by microscopy of cells costained for GLUT4 and Golgi markers. We first used an antibody to detect endogenous β-COP, a component of the nonclathrin COPI vesicle coat that has been localized previously to the Golgi complex (Duden et al. 1991). Fig. 7 demonstrates that the distributions of GLUT4 and β-COP are closely apposed, but do not overlap either in unstimulated or in insulin-stimulated cells. GLUT4 (in red) is present in unstimulated cells (Fig. 7 a) and in insulin-stimulated cells (Fig. 7 d) in its characteristic perinuclear location. Compared with Fig. 4 and Fig. 5, less plasma membrane GLUT4 staining is seen in the insulin-treated cells. This is in part due to cell-to-cell variability; also, data from only one optical cross-section are shown because views through several cross-sections resulted in the artifactual appearance of overlap due to staining of GLUT4 and β-COP in different planes. β-COP staining (in green) is also perinuclear, and is not markedly different in basal (Fig. 7 b) and in insulin-treated cells (Fig. 7 e). The merged images clearly show that there is essentially no overlap (yellow) in the distributions of GLUT4 and β-COP staining in either unstimulated (Fig. 7 c) or in insulin-stimulated (Fig. 7 f) cells.
Figure 7.

Perinuclear GLUT4 and β-COP staining do not overlap. Immunofluorescence of 3T3-L1 adipocytes stained for GLUT4 (red) and β-COP (green) was performed and optical cross-sections were acquired by deconvolution microscopy. The images shown are the center cross-sections from the reconstructed stack of images. GLUT4 staining is shown in a and d; β-COP staining is shown in b and e. Merged images are shown in c and f. a–c show staining of unstimulated 3T3-L1 adipocytes, and d–f show staining of insulin-stimulated 3T3-L1 adipocytes. Bar, 10 μm.
We next stained cells for GLUT4 and γ-adaptin, a component of the AP-1 adaptor complex involved in ADP-ribosylation factor–dependent budding of clathrin-coated vesicles from the TGN (Ahle et al. 1988; Robinson 1990; Stamnes and Rothman 1993; Traub et al. 1993; Seaman et al. 1996). Single optical cross-sections of unstimulated and insulin-stimulated 3T3-L1 adipocytes are shown in Fig. 8; whereas there is partial overlap of GLUT4 and γ-adaptin staining, the overall impression is that these compartments are distinct. GLUT4 is shown in red and can more readily be detected on the plasma membrane of insulin-stimulated cells (Fig. 8 d, arrowheads) than in unstimulated cells (Fig. 8 a). As noted for the other images of GLUT4 presented above, no qualitative differences are observed in the perinuclear GLUT4 distribution before and after insulin stimulation. Similarly, γ-adaptin staining (in green) is perinuclear and tubulovesicular in nature, and is not noticeably different in unstimulated (Fig. 8 b) and in insulin-stimulated (Fig. 8 e) cells. The merged images (Fig. 8c and Fig. f) show very little overlap (yellow); most staining is exclusively for either GLUT4 or for γ-adaptin. Review of several images revealed no difference between unstimulated and insulin-stimulated cells in the degree of overlap (compare Fig. 8c and Fig. f). Thus, whereas the possibility of GLUT4 recycling to the TGN cannot be excluded, the perinuclear GLUT4 and γ-adaptin staining patterns are clearly not identical, and the data support the notion that GLUT4 does not rejoin the secretory pathway at the level of the Golgi complex or TGN.
Figure 8.

Perinuclear GLUT4 and γ-adaptin staining are mostly separate. 3T3-L1 adipocytes were stained for GLUT4 (red) and γ-adaptin (green) before deconvolution microscopy. The images shown are the center cross-sections from the deconvolved set of images. GLUT4 staining is shown in a and d; γ-adaptin staining is shown in b and e. Merged images are shown in c and f. a–c show staining of unstimulated 3T3-L1 adipocytes, and d–f show staining of insulin-stimulated 3T3-L1 adipocytes. Arrowheads indicate plasma membrane localization of GLUT4 in cells treated with insulin. Bar, 10 μm.
Discussion
We have shown that insulin stimulates exocytosis of a regulated secretory compartment, containing ACRP30, in 3T3-L1 adipocytes. This compartment is distinct from that containing α3 (VI) collagen, because insulin has no effect on secretion of this protein. Therefore, it is not the case that insulin enhances ACRP30 secretion merely by accelerating the entire constitutive secretory pathway. Our observation that a calcium ionophore also stimulates ACRP30 secretion, and not α3 (VI) collagen secretion, supports this inference. Thus, we propose that a portion of ACRP30 is sorted into regulated secretory vesicles whose exocytosis is stimulated by insulin, and that the remainder is sorted into vesicles that undergo constitutive exocytosis. We propose that the latter population accounts for secretion of ACRP30 in the absence of insulin, and also contains α3 (VI) collagen and other proteins whose secretion is not enhanced by insulin. Partial sorting of protein hormones into regulated secretory vesicles has been observed in other types of cultured cells (Moore et al. 1983; Sambanis et al. 1991).
We also present immunofluorescence microscopy data demonstrating that whereas some ACRP30 staining overlaps with that of GRP94, and is presumably in the ER, additional ACRP30 staining does not overlap this marker and likely represents protein in peripheral storage vesicles. Insulin apparently accelerates movement of ACRP30 out of the ER, because staining for GRP94 that did not overlap with ACRP30 was only observed in cells that had been stimulated with insulin before fixation. Stimulation of ACRP30 export from the ER may account for observation that the insulin-stimulated enhancement of ACRP30 secretion is most marked in the early part of the chase period. Such export would have to be selective, since insulin did not increase α3 (VI) collagen secretion. Yet, this may not be the only site of insulin action: our observation that insulin also increases ACRP30 secretion late in the chase period, combined with our immunofluorescence data showing that some ACRP30 is in peripheral vesicles, suggests that it mobilizes a pool of regulated secretory vesicles in the periphery.
Our proposal that insulin stimulates exocytosis of regulated secretory vesicles is in contrast to that of Barr et al. 1997, who found no light microscopic evidence that leptin is targeted to such vesicles in rat adipose cells. These investigators found that the vast majority of leptin staining coincided with a marker for the ER, and that insulin stimulated export from this compartment. In this respect, our data that insulin stimulates ACRP30 export from the ER result is similar. It is possible that the presence of a small proportion of leptin in regulated vesicles in the periphery could have gone undetected by Barr et al. 1997, especially given the morphological challenge presented by primary adipocytes. Moreover, whereas insulin accelerates secretion of leptin and adipsin as well as ACRP30, it is not known if all of these proteins share a common exocytic pathway (Kitagawa et al. 1989; Scherer et al. 1995; Barr et al. 1997; Bradley and Cheatham 1999). Finally, these different interpretations may reflect actual differences in the cells used for experiments.
Our work demonstrates that insulin-stimulated enhancement of ACRP30 secretion is blocked by pharmacologic PI-3 kinase inhibitors, suggesting that PI-3 kinase activation is necessary for insulin stimulation of ACRP30 secretion. PI-3 kinase has been previously implicated in the insulin signal transduction pathway leading to GLUT4 exocytosis, and treatment of intact fat or muscle cells with LY294002 or wortmannin blocks insulin-stimulated translocation of GLUT4 to the plasma membrane (Cheatham et al. 1994; Clarke et al. 1994; Le Marchand-Brustel et al. 1995). Rapamycin, which we used as a negative control, acts downstream of PI-3 kinase to block insulin-stimulated p70 S6 kinase activation, but has no effect on glucose transport in 3T3-L1 adipocytes (Fingar et al. 1993; Monfar et al. 1995; Weng et al. 1995). Though the specificity of wortmannin and LY294002 has been questioned (Cross et al. 1995; Ferby et al. 1996; Balla et al. 1997), recent work demonstrates that the effect of wortmannin on insulin-stimulated hexose uptake is largely reversed by membrane-permeant esters of phosphatidylinositol-3,4,5-trisphosphate, a product of PI-3 kinase activity (Jiang et al. 1998). Therefore, the notion that PI-3 kinase activation is necessary for insulin-triggered GLUT4 exocytosis is supported, and it is through inhibition of this activity that wortmannin and LY294002 block insulin-stimulated GLUT4 trafficking. By extension, a strong argument is made that it is through inhibition of PI-3 kinase activity that these drugs block the effect of insulin to augment ACRP30 secretion.
Similar to our results with ACRP30, it has been shown very recently that the PI-3 kinase inhibitor, LY294002, blocks insulin-stimulated leptin secretion from rat adipocytes (Bradley and Cheatham 1999). These investigators also found that rapamycin decreased insulin-stimulated leptin secretion, though not as completely as LY294002; in contrast, we observed no effect of rapamycin on insulin-stimulated ACRP30 secretion. Aside from the observation that ACRP30 and leptin may not share a common regulated secretory pathway, this apparent contradiction can be resolved because our experiments using rapamycin were done in the presence of cycloheximide, whereas those of Bradley and Cheatham 1999 were not. Thus, as pointed out by these authors, it may be the case that the major effect of rapamycin was to prevent insulin-stimulated mRNA translation.
We have shown that there is no overlap in the subcellular distributions of GLUT4 and ACRP30 in unstimulated and in insulin-stimulated 3T3-L1 adipocytes. Together with our result that ACRP30 participates in a regulated secretory compartment, we conclude that GLUT4 does not recycle to a regulated secretory compartment, as defined by ACRP30. Important control experiments demonstrate that the subcellular distributions of GLUT4 and TfnR overlap substantially, whereas those of TfnR and ACRP30 are distinct and nonoverlapping. Further support that GLUT4 does not recycle to a regulated secretory pathway are our findings that perinuclear GLUT4 does not colocalize with a Golgi marker (β-COP) and is mostly distinct from that of a TGN marker (γ-adaptin). Thus, these parts of the secretory pathway are not major sites to which GLUT4 is distributed, either in the absence of insulin or after insulin stimulation.
The question of whether GLUT4 recycles to a regulated secretory pathway, or is instead targeted to synaptic-like vesicles, has been controversial. Conflicting results may have been obtained because previous studies relied on exogenously expressed GLUT4 in cells that do not normally take up glucose when stimulated by insulin, or were conducted using tissue that does not have a secretory pathway that is regulated by insulin (Hudson et al. 1993; Herman et al. 1994; Thorens and Roth 1996; Slot et al. 1997). We have addressed these concerns by studying endogenous GLUT4 in a well established adipose cell culture system, by showing that ACRP30 participates in a regulated secretory compartment in these cells, and by comparing the subcellular distributions of ACRP30 and GLUT4. Our conclusion that GLUT4 does not recycle to a secretory compartment is similar to that of Malide and Cushman 1997, who showed that in primary adipocytes wortmannin disrupts endocytic trafficking of GLUT4, but has no effect on trafficking of late endosomal or TGN markers. Their data suggest that GLUT4 does not recycle through the late endosome to the Golgi complex. Yet, this conclusion rests on the assumption that if this were the case, GLUT4 would traffic together with these markers. Additionally, proteins such as TGN38 recycle from the TGN to the plasma membrane quite slowly, and relatively short term wortmannin treatment might not appreciably alter their distribution (Ghosh et al. 1998). By examining the distribution of GLUT4 relative to a protein that participates in regulated secretion, we have avoided these potential pitfalls.
We observed significant overlap in the subcellular distributions of GLUT4 and TfnR. Other investigators have also noted overlap of GLUT4 and endosomal markers, including TfnR, yet contradictory data have been reported (Tanner and Lienhard 1989; Hudson et al. 1992; Hanpeter and James 1995; Ralston and Ploug 1996; Malide et al. 1997). The variability in the literature may be explained, at least in skeletal muscle, by the recent observation that there are two distinct intracellular GLUT4 compartments: one that cofractionates with the early endosomal markers TfnR and annexin II, and a second compartment from which GLUT4 is depleted after insulin stimulation (Aledo et al. 1997). Likewise in 3T3-L1 adipocytes, experiments involving either immunoisolation of GLUT4 vesicles or ablation of transferrin (Tfn)–containing compartments determined the presence of distinct GLUT4 populations (Livingstone et al. 1996). In both of these reports, ∼40% of GLUT4 colocalized with the TfnR, and ∼40% of the TfnR colocalized with GLUT4, consistent with our data as well. Moreover, both reports suggest that insulin stimulates movement of GLUT4 from the TfnR-negative pool to the plasma membrane. Although it seems likely that the TfnR-positive GLUT4 compartment is the precursor of the TfnR-negative, insulin-regulated GLUT4 compartment, this has not been definitively established. Our data add that the TfnR-negative, insulin-regulated GLUT4 compartment is not identical to the insulin-regulated compartment for ACRP30 secretion.
We also found that perinuclear GLUT4 does not appear to rejoin the secretory pathway at the level of the Golgi complex. GLUT4 immunostaining does not overlap with that of β-COP, a protein found on the cis side of the Golgi complex in pancreatic acinar cells and on the lateral rims and trans face of the Golgi complex in spermatids (Oprins et al. 1993; Martinez-Menarguez et al. 1996). An additional pool of β-COP has been described on membranes of the TGN, though this pool appears not to participate in budding of transport vesicles (Griffiths et al. 1995). We also found that GLUT4 immunostaining is mostly separate from that of γ-adaptin; overall, the data show that the perinuclear GLUT4 compartment does not correspond to the TGN. Our interpretation is consistent with previous light microscopy data that in insulinoma cells, transfected GLUT4 did not significantly overlap with the TGN marker protein, TGN38 (Thorens and Roth 1996). Other reports have described minimal overlap of GLUT4 and TGN38 in rat adipose cells, and of GLUT4 and giantin in cultured myotubes (Ralston and Ploug 1996; Malide and Cushman 1997; Malide et al. 1997). Biochemical studies of 3T3-L1 adipocytes found that only 5–10% of low density microsomal GLUT4 was copurified by immunoadsorption of vesicles using an antiserum to TGN38; the copurified GLUT4 did not correspond to the insulin-regulated GLUT4 compartment within the low density microsomal fraction (Martin et al. 1994). Thus, our data concerning the perinuclear GLUT4 compartment are in agreement with other studies noting close association, but not identity, with the TGN.
In summary, we have shown that ACRP30 participates in an insulin-regulated secretory compartment in 3T3-L1 adipocytes, and that α3 (VI) collagen does not. Insulin appears to accelerate ACRP30 secretion at both early and late steps in its secretory pathway, possibly corresponding to export of ACRP30 from the ER and to exocytosis of regulated secretory vesicles containing this protein. Like GLUT4 exocytosis, insulin-stimulated ACRP30 secretion is blocked by inhibitors of PI-3 kinase. Finally, we show that GLUT4 does not recycle to the regulated secretory pathway containing ACRP30. Insulin-stimulated PI-3 kinase activity must therefore act through effectors present at multiple locations within the cell.
Acknowledgments
We would like to thank Drs. Maureen Charron and Philipp Scherer for kind gifts of antisera; Drs. Miyoung Chun, Barbara Panning, and Paul Matsudaira for advice on fluorescence microscopy; and Dr. Philipp Scherer for advice on the pulse-chase experiments. We thank Helen M. Lin for technical assistance, and Drs. Nai-Wen Chi and Andreas Stahl for comments on the manuscript.
This work was conducted using the W.M. Keck Foundation Biological Imaging Facility at the Whitehead Institute, and was supported by a National Institutes of Health Physician Scientist Award (DK02371) to J.S. Bogan, and by grants to H.F. Lodish from National Institutes of Health (DK47618) and from Pfizer Corporation.
Footnotes
1.used in this paper: ACRP30, adipocyte complement related protein of 30 kD; GLUT, glucose transporter; PI-3 kinase, phosphatidylinositol-3-kinase; TfnR, transferrin receptor
References
- Ahle S., Mann A., Eichelsbacher U., Ungewickell E. Structural relationships between clathrin assembly proteins from the Golgi and the plasma membrane. EMBO (Eur. Mol. Biol. Organ.) J. 1988;7:919–929. doi: 10.1002/j.1460-2075.1988.tb02897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aledo J.C., Lavoie L., Volchuk A., Keller S.R., Klip A., Hundal H.S. Identification and characterization of two distinct intracellular GLUT4 pools in rat skeletal muscleevidence for an endosomal and an insulin-sensitive GLUT4 compartment. Biochem. J. 1997;325:727–732. doi: 10.1042/bj3250727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla T., Downing G.J., Jaffe H., Kim S., Zolyomi A., Catt K.J. Isolation and molecular cloning of wortmannin-sensitive bovine type III phosphatidylinositol 4-kinases. J. Biol. Chem. 1997;272:18358–18366. doi: 10.1074/jbc.272.29.18358. [DOI] [PubMed] [Google Scholar]
- Barr V.A., Malide D., Zarnowski M., Taylor S.I., Cushman S.W. Insulin stimulates both leptin secretion and production by rat white adipose tissue. Endocrinology. 1997;138:4463–4472. doi: 10.1210/endo.138.10.5451. [DOI] [PubMed] [Google Scholar]
- Benito M., Porras A., Nebreda A., Santos E. Differentiation of 3T3-L1 fibroblasts to adipocytes induced by transfection of ras oncogenes. Science. 1991;253:565–568. doi: 10.1126/science.1857988. [DOI] [PubMed] [Google Scholar]
- Bradley R.L., Cheatham B. Regulation of ob gene expression and leptin secretion by insulin and dexamethasone in rat adipocytes. Diabetes. 1999;48:272–278. doi: 10.2337/diabetes.48.2.272. [DOI] [PubMed] [Google Scholar]
- Charron M.J., Brosius F.C., III, Alper S.L., Lodish H.F. A glucose transport protein expressed predominately in insulin-responsive tissues. Proc. Natl. Acad. Sci. USA. 1989;86:2535–2539. doi: 10.1073/pnas.86.8.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham B., Vlahos C.J., Cheatham L., Wang L., Blenis J., Kahn C.R. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol. Cell. Biol. 1994;14:4902–4911. doi: 10.1128/mcb.14.7.4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J.F., Young P.W., Yonezawa K., Kasuga M., Holman G.D. Inhibition of the translocation of GLUT1 and GLUT4 in 3T3-L1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem. J. 1994;300:631–635. doi: 10.1042/bj3000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross M.J., Stewart A., Hodgkin M.N., Kerr D.J., Wakelam M.J. Wortmannin and its structural analogue demethoxyviridin inhibit stimulated phospholipase A2 activity in Swiss 3T3 cells. Wortmannin is not a specific inhibitor of phosphatidylinositol 3-kinase. J. Biol. Chem. 1995;270:25352–25355. doi: 10.1074/jbc.270.43.25352. [DOI] [PubMed] [Google Scholar]
- Duden R., Griffiths G., Frank R., Argos P., Kreis T.E. Beta-COP, a 110 kd protein associated with non-clathrin-coated vesicles and the Golgi complex, shows homology to beta-adaptin. Cell. 1991;64:649–665. doi: 10.1016/0092-8674(91)90248-w. [DOI] [PubMed] [Google Scholar]
- Ferby I.M., Waga I., Hoshino M., Kume K., Shimizu T. Wortmannin inhibits mitogen-activated protein kinase activation by platelet-activating factor through a mechanism independent of p85/p110-type phosphatidylinositol 3-kinase. J. Biol. Chem. 1996;271:11684–11688. doi: 10.1074/jbc.271.20.11684. [DOI] [PubMed] [Google Scholar]
- Fingar D.C., Hausdorff S.F., Blenis J., Birnbaum M.J. Dissociation of pp70 ribosomal protein S6 kinase from insulin-stimulated glucose transport in 3T3-L1 adipocytes. J. Biol. Chem. 1993;268:3005–3008. [PubMed] [Google Scholar]
- Frost S.C., Lane M.D. Evidence for the involvement of vicinal sulfhydryl groups in insulin-activated hexose transport by 3T3-L1 adipocytes. J. Biol. Chem. 1985;260:2646–2652. [PubMed] [Google Scholar]
- Gething M.J., Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Ghosh R.N., Mallet W.G., Soe T.T., McGraw T.E., Maxfield F.R. An endocytosed TGN38 chimeric protein is delivered to the TGN after trafficking through the endocytic recycling compartment in CHO cells. J. Cell Biol. 1998;142:923–936. doi: 10.1083/jcb.142.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths G., Pepperkok R., Locker J.K., Kreis T.E. Immunocytochemical localization of beta-COP to the ER-Golgi boundary and the TGN. J. Cell Sci. 1995;108:2839–2856. doi: 10.1242/jcs.108.8.2839. [DOI] [PubMed] [Google Scholar]
- Hanpeter D., James D.E. Characterization of the intracellular GLUT-4 compartment. Mol. Membr. Biol. 1995;12:263–269. doi: 10.3109/09687689509072426. [DOI] [PubMed] [Google Scholar]
- Herman G.A., Bonzelius F., Cieutat A.M., Kelly R.B. A distinct class of intracellular storage vesicles, identified by expression of the glucose transporter GLUT4. Proc. Natl. Acad. Sci. USA. 1994;91:12750–12754. doi: 10.1073/pnas.91.26.12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E., Liang P., Spiegelman B.M. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem. 1996;271:10697–10703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- Hudson A.W., Ruiz M., Birnbaum M.J. Isoform-specific subcellular targeting of glucose transporters in mouse fibroblasts. J. Cell Biol. 1992;116:785–797. doi: 10.1083/jcb.116.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson A.W., Fingar D.C., Seidner G.A., Griffiths G., Burke B., Birnbaum M.J. Targeting of the “insulin-responsive” glucose transporter (GLUT4) to the regulated secretory pathway in PC12 cells. J. Cell Biol. 1993;122:579–588. doi: 10.1083/jcb.122.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T., Sweeney G., Rudolf M.T., Klip A., Traynor-Kaplan A., Tsien R.Y. Membrane-permeant esters of phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998;273:11017–11024. doi: 10.1074/jbc.273.18.11017. [DOI] [PubMed] [Google Scholar]
- Kahn B.B. Lilly lecture 1995. Glucose transportpivotal step in insulin action. Diabetes. 1996;45:1644–1654. doi: 10.2337/diab.45.11.1644. [DOI] [PubMed] [Google Scholar]
- Kitagawa K., Rosen B.S., Spiegelman B.M., Lienhard G.E., Tanner L.I. Insulin stimulates the acute release of adipsin from 3T3-L1 adipocytes. Biochim. Biophys. Acta. 1989;1014:83–89. doi: 10.1016/0167-4889(89)90244-9. [DOI] [PubMed] [Google Scholar]
- Koch G., Smith M., Macer D., Webster P., Mortara R. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J. Cell Sci. 1986;86:217–232. doi: 10.1242/jcs.86.1.217. [DOI] [PubMed] [Google Scholar]
- Le Marchand-Brustel Y., Gautier N., Cormont M., Van Obberghen E. Wortmannin inhibits the action of insulin but not that of okadaic acid in skeletal musclecomparison with fat cells. Endocrinology. 1995;136:3564–3570. doi: 10.1210/endo.136.8.7628394. [DOI] [PubMed] [Google Scholar]
- Livingstone C., James D.E., Rice J.E., Hanpeter D., Gould G.W. Compartment ablation analysis of the insulin-responsive glucose transporter (GLUT4) in 3T3-L1 adipocytes. Biochem. J. 1996;315:487–495. doi: 10.1042/bj3150487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malide D., Cushman S.W. Morphological effects of wortmannin on the endosomal system and GLUT4-containing compartments in rat adipose cells. J. Cell Sci. 1997;110:2795–2806. doi: 10.1242/jcs.110.22.2795. [DOI] [PubMed] [Google Scholar]
- Malide D., Dwyer N.K., Blanchette-Mackie E.J., Cushman S.W. Immunocytochemical evidence that GLUT4 resides in a specialized translocation post-endosomal VAMP2-positive compartment in rat adipose cells in the absence of insulin. J. Histochem. Cytochem. 1997;45:1083–1096. doi: 10.1177/002215549704500806. [DOI] [PubMed] [Google Scholar]
- Martin S., Reaves B., Banting G., Gould G.W. Analysis of the co-localization of the insulin-responsive glucose transporter (GLUT4) and the trans Golgi network marker TGN38 within 3T3-L1 adipocytes. Biochem. J. 1994;300:743–749. doi: 10.1042/bj3000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Menarguez J.A., Geuze H.J., Ballesta J. Identification of two types of beta-COP vesicles in the Golgi complex of rat spermatids. Eur. J. Cell Biol. 1996;71:137–143. [PubMed] [Google Scholar]
- Melnick J., Aviel S., Argon Y. The endoplasmic reticulum stress protein GRP94, in addition to BiP, associates with unassembled immunoglobulin chains. J. Biol. Chem. 1992;267:21303–21306. [PubMed] [Google Scholar]
- Melnick J., Dul J.L., Argon Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature. 1994;370:373–375. doi: 10.1038/370373a0. [DOI] [PubMed] [Google Scholar]
- Monfar M., Lemon K.P., Grammer T.C., Cheatham L., Chung J., Vlahos C.J., Blenis J. Activation of pp70/85 S6 kinases in interleukin-2-responsive lymphoid cells is mediated by phosphatidylinositol 3-kinase and inhibited by cyclic AMP. Mol. Cell. Biol. 1995;15:326–337. doi: 10.1128/mcb.15.1.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore H.P., Gumbiner B., Kelly R.B. Chloroquine diverts ACTH from a regulated to a constitutive secretory pathway in AtT-20 cells. Nature. 1983;302:434–436. doi: 10.1038/302434a0. [DOI] [PubMed] [Google Scholar]
- Oprins A., Duden R., Kreis T.E., Geuze H.J., Slot J.W. Beta-COP localizes mainly to the cis-Golgi side in exocrine pancreas. J. Cell Biol. 1993;121:49–59. doi: 10.1083/jcb.121.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper R.C., Hess L.J., James D.E. Differential sorting of two glucose transporters expressed in insulin-sensitive cells. Am. J. Physiol. 1991;260:C570–C580. doi: 10.1152/ajpcell.1991.260.3.C570. [DOI] [PubMed] [Google Scholar]
- Ralston E., Ploug T. GLUT4 in cultured skeletal myotubes is segregated from the transferrin receptor and stored in vesicles associated with TGN. J. Cell Sci. 1996;109:2967–2978. doi: 10.1242/jcs.109.13.2967. [DOI] [PubMed] [Google Scholar]
- Rea S., James D.E. Moving GLUT4The biogenesis and trafficking of GLUT4 storage vesicles. Diabetes. 1997;46:1667–1677. doi: 10.2337/diab.46.11.1667. [DOI] [PubMed] [Google Scholar]
- Rindler M.J. Biogenesis of storage granules and vesicles. Curr. Opin. Cell Biol. 1992;4:616–622. doi: 10.1016/0955-0674(92)90080-v. [DOI] [PubMed] [Google Scholar]
- Robinson M.S. Cloning and expression of gamma-adaptin, a component of clathrin-coated vesicles associated with the Golgi apparatus. J. Cell Biol. 1990;111:2319–2326. doi: 10.1083/jcb.111.6.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambanis A., Lodish H.F., Stephanopoulos G. A model of secretory protein trafficking in recombinant AtT-20 cells. Biotechnol. Bioeng. 1991;38:280–295. doi: 10.1002/bit.260380310. [DOI] [PubMed] [Google Scholar]
- Satoh S., Nishimura H., Clark A.E., Kozka I.J., Vannucci S.J., Simpson I.A., Quon M.J., Cushman S.W., Holman G.D. Use of bismannose photolabel to elucidate insulin-regulated GLUT4 subcellular trafficking kinetics in rat adipose cells. Evidence that exocytosis is a critical site of hormone action. J. Biol. Chem. 1993;268:17820–17829. [PubMed] [Google Scholar]
- Scherer P.E., Williams S., Fogliano M., Baldini G., Lodish H.F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- Scherer P.E., Bickel P.E., Kotler M., Lodish H.F. Cloning of cell-specific secreted and surface proteins by subtractive antibody screening. Nat. Biotechnol. 1998;16:581–586. doi: 10.1038/nbt0698-581. [DOI] [PubMed] [Google Scholar]
- Seaman M.N.J., Sowerby P.J., Robinson M.S. Cytosolic and membrane-associated proteins involved in the recruitment of AP-1 adaptors onto the trans-Golgi network. J. Biol. Chem. 1996;271:25446–25451. doi: 10.1074/jbc.271.41.25446. [DOI] [PubMed] [Google Scholar]
- Shapiro L., Scherer P.E. The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Curr. Biol. 1998;8:335–338. doi: 10.1016/s0960-9822(98)70133-2. [DOI] [PubMed] [Google Scholar]
- Slot J.W., Geuze H.J., Gigengack S., Lienhard G.E., James D.E. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 1991;113:123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot J.W., Garruti G., Martin S., Oorschot V., Posthuma G., Kraegen E.W., Laybutt R., Thibault G., James D.E. Glucose transporter (GLUT-4) is targeted to secretory granules in rat atrial cardiomyocytes. J. Cell Biol. 1997;137:1243–1254. doi: 10.1083/jcb.137.6.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R.M., Charron M.J., Shah N., Lodish H.F., Jarett L. Immunoelectron microscopic demonstration of insulin-stimulated translocation of glucose transporters to the plasma membrane of isolated rat adipocytes and masking of the carboxyl-terminal epitope of intracellular GLUT4. Proc. Natl. Acad. Sci. USA. 1991;88:6893–6897. doi: 10.1073/pnas.88.15.6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamnes M.A., Rothman J.E. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell. 1993;73:999–1005. doi: 10.1016/0092-8674(93)90277-w. [DOI] [PubMed] [Google Scholar]
- Stenbit A.E., Tsao T.S., Li J., Burcelin R., Geenen D.L., Factor S.M., Houseknecht K., Katz E.B., Charron M.J. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat. Med. 1997;3:1096–1101. doi: 10.1038/nm1097-1096. [DOI] [PubMed] [Google Scholar]
- Tanner L.I., Lienhard G.E. Localization of transferrin receptors and insulin-like growth factor II receptors in vesicles from 3T3-L1 adipocytes that contain intracellular glucose transporters. J. Cell Biol. 1989;108:1537–1545. doi: 10.1083/jcb.108.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B., Roth J. Intracellular targeting of GLUT4 in transfected insulinoma cellsevidence for association with constitutively recycling vesicles distinct from synaptophysin and insulin vesicles. J. Cell Sci. 1996;109:1311–1323. doi: 10.1242/jcs.109.6.1311. [DOI] [PubMed] [Google Scholar]
- Traub L.M., Ostrom J.A., Kornfeld S. Biochemical dissection of AP-1 recruitment onto Golgi membranes. J. Cell Biol. 1993;123:561–573. doi: 10.1083/jcb.123.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uysal K.T., Wiesbrock S.M., Marino M.W., Hotamisligil G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- Weng Q.P., Andrabi K., Klippel A., Kozlowski M.T., Williams L.T., Avruch J. Phosphatidylinositol 3-kinase signals activation of p70 S6 kinase in situ through site-specific p70 phosphorylation. Proc. Natl. Acad. Sci. USA. 1995;92:5744–5748. doi: 10.1073/pnas.92.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Holman G.D. Comparison of GLUT4 and GLUT1 subcellular trafficking in basal and insulin-stimulated 3T3-L1 cells. J. Biol. Chem. 1993;268:4600–4603. [PubMed] [Google Scholar]
- Zhang Y., Proenca R., Maffei M., Barone M., Leopold L., Friedman J.M. Positional cloning of the mouse obese gene and its human homologue [published erratum appears in Nature. 1995. 374:479] Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]