

Abstract
In cardiac muscle, the giant protein titin exists in different length isoforms expressed in the molecule's I-band region. Both isoforms, termed N2-A and N2-B, comprise stretches of Ig-like modules separated by the PEVK domain. Central I-band titin also contains isoform-specific Ig-motifs and nonmodular sequences, notably a longer insertion in N2-B. We investigated the elastic behavior of the I-band isoforms by using single-myofibril mechanics, immunofluorescence microscopy, and immunoelectron microscopy of rabbit cardiac sarcomeres stained with sequence-assigned antibodies. Moreover, we overexpressed constructs from the N2-B region in chick cardiac cells to search for possible structural properties of this cardiac-specific segment.
We found that cardiac titin contains three distinct elastic elements: poly-Ig regions, the PEVK domain, and the N2-B sequence insertion, which extends ∼60 nm at high physiological stretch. Recruitment of all three elements allows cardiac titin to extend fully reversibly at physiological sarcomere lengths, without the need to unfold Ig domains. Overexpressing the entire N2-B region or its NH2 terminus in cardiac myocytes greatly disrupted thin filament, but not thick filament structure. Our results strongly suggest that the NH2-terminal N2-B domains are necessary to stabilize thin filament integrity. N2-B–titin emerges as a unique region critical for both reversible extensibility and structural maintenance of cardiac myofibrils.
Keywords: heart muscle, connectin, elasticity, transfection, sarcomere
Titin (first described as connectin) is a giant sarcomeric protein of striated muscle with many diverse functions (Wang 1996; Maruyama 1997). To name three of the best known, (a) the filamentous titin molecules of >1 μm length provide binding sites for other myofibrillar proteins such as α-actinin and T-cap (telethonin) in the Z-disk, actin in the I-band near the Z-disk, and myosin, myosin-binding protein C (MyBP-C or C-protein)1, M-protein, and myomesin in the A-band (see reviews by Trinick 1996; Gregorio et al. 1999, and relevant citations therein). Thus, titin is a structural polypeptide that may well contain a molecular blueprint for sarcomere assembly. (b) The I-band region of titin is elastic and responsible for passive tension generation upon stretch of nonactivated striated muscle (Erickson 1997; Linke and Granzier 1998). Titin can therefore be viewed as a molecular spring. (c) The polypeptide contains, near its COOH-terminal end at the M-line, a kinase domain (Labeit et al. 1992), which may be active during muscle development (Mayans et al. 1998). Hence, titin may also be involved in intracellular signal transduction pathways (see Labeit et al. 1997). These important roles have made titin the target of an increasing number of biophysical and cell biological studies.
The structure of titin is highly modular. In the A-band, titin consists of two motif types, immunoglobulin-like (Ig) modules and fibronectin type-3 domains (Labeit et al. 1992); both types are arranged in super-repeat patterns. M-line titin and Z-disk titin contain no fibronectin modules but are composed of Ig domains interspersed with unique sequences (for a review, see Gregorio et al. 1999). The main section of I-band titin is made up of two principal, structurally distinct, regions (see Fig. 1), stretches of tandemly arranged Ig modules that adopt a stable β-sheet fold (Politou et al. 1995), separated by a unique sequence of elusive secondary structure, the PEVK domain (Labeit and Kolmerer 1995). Both regions exist in tissue-specific length variants, which results in the characteristic size of each titin isoform (Labeit and Kolmerer 1995). In addition to the differentially expressed Ig-repeats and PEVK domains, skeletal muscle titin contains a linker segment termed N2-A region, whereas cardiac-muscle titin coexpresses two different linker regions, N2-A and N2-B (see Fig. 1), at the sarcomere level (Linke et al. 1996). Details on the differential expression of N2-A/N2-B regions are not known. Interestingly, the N2-B region contains several Ig domains and sequence insertions unique to this isoform, including a 572-residue-long nonmodular sequence of heretofore unknown function. Investigating the functional significance of the structurally distinct regions in cardiac I-band titin was the focus of this study.
Figure 1.

Schematic view of the domain architecture of the elastic I-band titin in heart (after Labeit and Kolmerer 1995). Two different isoforms are expressed, N2-A and N2-B. In both isoforms a PEVK domain is flanked by stretches of tandemly arranged Ig-like modules. Unique to each isoform is the central I-band region (N2 region), which is made up of isoform-specific Ig domains and nonmodular sequences, notably a 572-residue insertion in N2-B. The epitope locations of the titin antibodies used in this study are indicated by the colored arrows. Note also the positions of the four recombinant N2-B–titin constructs prepared for transfection experiments: 1, entire N2-B region; 2, NH2-terminal N2-B (region bounded by Ig domains I16/17); 3, middle N2-B (unique sequence insertion); 4, COOH-terminal N2-B (Ig domains I18/19). FN3, fibronectin type-III. Dashed curves indicate missing sequence information.
Because of the coexpression of N2-A and N2-B isoforms in cardiac muscle, the role of I-band titin is less well defined in this tissue type than in skeletal muscle. It is important from a physiological point of view to uncover the function of titin in the heart, whose elastic properties in diastole are highly relevant for the muscle's overall mechanical performance. What appears to be clear at this point is that in cardiac titin, both the poly-Ig segments and the PEVK region represent extensible elements contributing to myofibrillar elasticity (Linke et al. 1996; Granzier et al. 1997). However, previous approaches could not resolve isoform-specific features: to distinguish the mechanical properties of the two I-band titins, isoform-specific antibodies are needed. In this study, we investigated the elasticity of each cardiac titin segment using a multifaceted approach. We employed single myofibril mechanics in combination with immunofluorescence microscopy and immuno-EM on cardiac sarcomeres stained with a panel of sequence-assigned antibodies. Thus, a detailed picture of cardiac titin extension was obtained. We confirm that the Ig-segments and the PEVK domain contribute to titin elasticity, but we also find that N2-B–titin recruits a third spring element, the long insertion in the middle I-band region (Fig. 1). Thereby, cardiac titin stretches reversibly even to long physiological sarcomere lengths (SL) without requiring Ig domains to unfold.
A related and equally important issue is whether or not cardiac titin filaments run along the I-band independently of one another and of other myofibrillar proteins. One report suggested that I-band titin may be laterally associated with thin filaments (Funatsu et al. 1993). Indeed, actin can interact with titin in vitro (e.g., Soteriou et al. 1993; Jin 1995). On the other hand, titin was confirmed to bind to actin filaments only near the Z-disks of cardiac myofibrils (Linke et al. 1997; Trombitas and Granzier 1997). To analyze whether particular I-band regions of titin, which would by proximity bind to the sides of the thin filaments, might play a role in thin filament stability, we performed transfection experiments on primary cultures of chick cardiac cells. Overexpression of recombinant titin constructs comprising the entire N2-B region or only its NH2 terminus resulted in strong disruption of thin filament, but not thick filament, structure. This indicates that the N2-B–titin region bounded by the Ig domains I16 and I17 contributes either directly or indirectly to the stabilization of thin filaments. Future studies will focus on the identification of the putative I-band association with a thin filament component(s), how this interaction may be involved in myofibril assembly, and whether the interaction affects cardiac titin elasticity.
Materials and Methods
Antibodies to Cardiac Titin
For titin antibody epitope positions, see Fig. 1. The following monoclonal antibodies were used: T12 (purchased from Boehringer-Mannheim Biochemicals; Fürst et al. 1988), I18, S54/56 (Gautel et al. 1996), and I17. I17 was prepared according to the nomenclature and domain boundaries of the Ig domain I17 as in Labeit and Kolmerer 1995. The cDNA fragment coding for I17 was subcloned into a modified pET vector (Labeit et al. 1992), and His6-tagged protein was expressed and purified as described previously (Politou et al. 1994). BALB/c mice were immunized in a standardized scheme with the purified I17 titin using titerMax adjuvans (Vaxcel), and antibodies were generated as described (Gautel et al. 1996). The antibody was found to be specific for cardiac titin, recognizing a single defined epitope in the assay with recombinant titin domains (data not shown).
The following affinity-purified polyclonal antibodies were used: I20/22 (Linke et al. 1998a), MIR (Linke et al. 1996), and N2B. For N2B, base pairs 11,551–11,928 of the human cardiac titin cDNA entry (EMBL accession number X90568) were isolated by PCR (Saiki et al. 1985). The fragments were subcloned into modified pET9D vectors (Studier and Moffat 1991) that expressed their insert sequences as fusions with NH2-terminal His6 tags. The recombinant peptides were purified from the soluble fractions by nickel chelate affinity chromatography on NTA (Ni-NTA) resins as specified by the manufacturer (Qiagen). Antibodies were raised in rabbits by Eurogentec and the specific IgG fraction was isolated by affinity chromatography. Western blot analysis of extracts of cardiac muscle was used to verify the specificity of the antibodies (data not shown). The antibody is directed against an epitope within a 162-residue-long sequence near the NH2 terminus of the middle N2-B region (see Fig. 1).
Preparation of Single Cardiac Myofibrils
Myofibrils were isolated from freshly excised rabbit left ventricle as described (Linke et al. 1996). In brief, trabeculae were dissected, tied at both ends to a thin glass rod, and skinned in ice-cold rigor solution containing 0.5% Triton X-100 for a minimum of 4 h. The skinned strips were minced and homogenized in rigor buffer. A drop of the suspension was placed on a coverslip, and a single myofibril (sometimes a doublet) adhering lightly to the glass surface was picked up under a Zeiss Axiovert 135 inverted microscope with the help of water-hydraulic micromanipulators (Narishige) by two glass needle tips coated with water-curing silicone adhesive. Experiments were performed at room temperature in relaxing solution of 200 mM ionic strength, pH 7.1. All solutions were supplemented with the protease inhibitor leupeptin to minimize titin degradation (Linke et al. 1997).
Immunofluorescence Microscopy on Stretched Myofibrils
For immunofluorescence microscopy, a myofibril was stretched in relaxing buffer to a desired SL and was labeled with primary titin antibody and Cy3-conjugated secondary IgG (anti–rabbit or anti–mouse; Rockland). Sometimes, a stretched myofibril was fixed in 3.5% paraformaldehyde solution for 20 min before the staining procedure. Using both approaches, identical results were obtained, although fixation appeared to decrease fluorescence intensity. Control measurements with secondary antibody only showed no fluorescence. Antisera were used at the following concentrations (antibody/relaxing buffer): T12, 1:50; I17, 1:5 or undiluted; N2B, 1:5 or 1:10; I18, 1:5 or 1:10; S54/56, 1:20; I20/22, 1:50; MIR, 1:100; secondary antibodies, 1:50. For most experiments the incubation time was 20 min for both primary and secondary antibodies. Images were recorded in the epifluorescence mode of the microscope (100×, 1.4 NA oil immersion objective). Three images were recorded at a given SL and automatically superimposed by using a feature of the software (Global Lab Image, Data Translation). For measurements at another SL, a new preparation was used.
Measurement of Epitope Spacing from Immunofluorescence Images
The intensity profile along the myofibril axis was plotted, and the distances between intensity peaks were determined by two independent methods. First, a commercial peak detection program (AIDA; Raytest) was used to measure distances between the highest points on the plots. Second, to exclude the possibility that a distorted shape of some maxima influences the results, custom-written software (Borland Turbo Pascal) was used to determine the peak distances by a center-of-mass analysis (see Fig. 2 a, bottom). The program calculates the area under an intensity maximum, above a baseline drawn parallel to the abscissa (horizontal lines in the plot in Fig. 2 a, bottom). This baseline was either set to pass through the relative minimum between peaks reflecting the unlabeled Z-line region or was positioned by eye to above that minimum. The latter case (shown in Fig. 2 a, bottom) was applicable when peak spacings were increased and peak amplitudes were relatively large in highly stretched sarcomeres. Next, the program calculates those points on the abscissa at which a vertical straight line can be constructed that divides the area under each maximum into two equal parts (vertical lines in Fig. 2 a, bottom). These points were then used to measure the epitope spacings across the Z-line. Results obtained with the two analysis methods were similar within experimental error; in this study, we exclusively show data from the center-of-mass analysis. Because the method could be applied successfully only when peaks did not overlap, reliable detection required epitope spacings of at least ∼200 nm. Finally, the calculated peak distances were divided by two to obtain the Z-disk center to epitope distance in a half-sarcomere.
Figure 2.

Immunofluorescence images of stretched isolated cardiac myofibrils labeled with anti-titin antibodies. (a) A panel of antibodies (see Fig. 1) was used to stain single myofibrils at different SLs, here 2.3 and 2.7 μm. For comparison, phase-contrast images (PC) are also shown. Note that placed below the phase-contrast images are immunofluorescence images obtained with the only antibody specific to the N2-A titin isoform. The bottom trace shows the intensity profile of a fluorescence image to demonstrate how the spacing between intensity peaks was determined by performing a center-of-mass analysis (for detailed explanation, see Materials and Methods). (b) Irregular staining of a myofibril doublet labeled with I17 antibody. Fluorescence and phase-contrast (PC) images are shown, as well as the respective intensity profiles. Bars, 5 μm.
Immuno-EM
Immuno-EM was essentially carried out as follows: rapidly excised rabbit left ventricular tissue (mostly trabeculae, sometimes papillary muscles) was stretched to different degrees in relaxing buffer. Then, a solution containing 3.5% paraformaldehyde was added as fixative for no more than 2 h. As described elsewhere, specimens were processed for cryosectioning and immunolabeling with the titin antibodies and secondary antibody conjugated to 10-nm gold particles (Tokuyasu 1989; Mundel et al. 1991). Titin antiserum dilution was as follows: T12, 1:50; N2B, 1:5; I18, undiluted; I20/22, 1:40. For double staining experiments with the titin antibodies, N2B and I18, differently sized gold particles were used: N2B was visualized with 10-nm particles, I18 with 15-nm particles. Micrographs were taken with a Philips EM 301 at 80 kV. The center of the nanogold particles was used to measure the distance of a given antibody epitope from the center of the Z-disk.
Tension Recordings
Myofibrils were suspended between a micromotor and a sensitive force transducer, and passive length-tension curves were measured as described previously (Linke et al. 1996). Solutions contained 20 mM 2,3-butanedione monoxime to suppress force-generating actin-myosin interactions possibly remaining even under relaxing conditions. In a typical protocol, preparations were stretched in stages from slack length to a series of desired SLs. Stretch duration was ∼20 s; the hold period (to wait for stress relaxation) was 2–3 min. After stretching to a maximum SL, the specimen was released in stages to slack length. Then, the cycle was repeated two more times, extending the myofibril to a higher maximum SL in each cycle. To obtain passive tension, the cross-sectional area of a preparation was inferred from the diameter of the specimens as described (Linke et al. 1997).
Construct Preparations
GFP (green fluorescence protein)–N2-B fusion constructs: For the expression of N2-B sequences in cardiac myocytes, PCR fragments were amplified from a human cardiac cDNA library (Stratagene no. 936208) that contained either the complete cardiac-specific N2-B sequence in the center of I-band titin (EMBL accession number X90568), or fragments of it. For the amplification, the following primers were used (capital letters match X90568, whereas small letters denote cloning site tags): X218, tttagatct-GAA GGC ACT GGC CCA ATT TTC ATC AAA GAA; X219, tttgtcgac-ta-GTC TGT GTC TTC CAG AAG CAC AAG CAG CTC; X220, tttagatct-GAG GAT GGC CCC ATG ATA CAT ACA CCT TTA; X221, tttgtcgac-ta-CAC TGT CAC AGT TAG TGT GGC TGT ACA GCT; X222, tttagatct-ATG ACT GAT ACC CCC TGC AAA GCA AAG TCC; X223, tttgtcgac-ta-GCC ATC CTC TTT GAT TAA GCC ACC CTC AGC.
The NH2-terminal fragment X218-X219 encompasses the Ig repeats N2-B I16/17 and the COOH-terminal fragment X220-X221 contains the Ig repeats N2-B I18/I19 (Fig. 1). The intervening, cardiac-specific sequence (middle N2-B region) is contained in the construct X222-X223. The N2-B fragments amplified by the X218-X223 primers were inserted into the pEGFP-C1 vector (Clontech) that was linearized by digestion with BglII and Sal I restriction endonucleases (MBI Fermentas). The recombinant plasmid inserts were transformed into competent DH5-alpha E. coli cells, and recombinant plasmid DNAs were purified using Qiagen columns (Qiagen). Plasmids were verified by sequencing.
Cell Culture and Transfection Procedures
Cardiac myocytes were prepared from day 6 embryonic chick hearts and cultured as described previously (Gregorio and Fowler 1995). Isolated cells were plated in 35-mm tissue culture dishes containing 12-mm round coverslips (1 × 106 cells/dish). Approximately 15% of the cells in most of our primary cultures are fibroblasts. 24 h after plating, cultured myocytes were washed two times in OptiMEM, placed in 800 μl fresh OptiMEM, and returned to the incubator while DNA liposome complexes were prepared. Such complexes were prepared by combining 1 μg plasmid with 4 μl Lipofectamine and 6 μl PLUS Reagent in 200 μl serum-free OptiMEM. After 15 min, the complexes were added dropwise to the culture dish. 3 h later, 1 ml of MEM 10% FBS (Hyclone Laboratories, Inc.) was added to the dish. One to six days later, cells were gently washed with PBS and fixed with 2% formaldehyde in PBS for 10 min. Coverslips were washed and stored in PBS at 4°C until staining. Over 300 transfected cells per construct were analyzed. All tissue culture reagents (except where noted) were purchased from Life Technologies.
Indirect Immunofluorescence Microscopy on Cardiac Cells
Cardiac myocytes were essentially stained as described (Gregorio et al. 1998). In brief, coverslips were permeabilized in 0.1% Triton X-100 and PBS for 15 min. To minimize nonspecific binding of antibodies, the coverslips were preincubated in 2% donkey serum, 2% BSA, and PBS for 30 min. Antibodies specific to various sarcomeric components (i.e., α-actinin, tropomyosin [CH1: Lin et al. 1985], myosin, MyBP-C, and titin) and phalloidin (to stain for actin filaments) were used to analyze the transfected cells. Details on the staining protocols used in the micrographs presented are described. To analyze the distribution of α-actinin, rabbit anti–α-actinin antibodies (1:1,000) (generously provided by Dr. S. Craig, Johns Hopkins University, Baltimore, MD) were used, followed by Texas red–conjugated F(ab) fragments of donkey anti–rabbit antibodies (1:200). To analyze the distribution of MyBP-C, polyclonal anti–MyBP-C antibodies (1:50) (see below) were used followed by Texas red–conjugated F(ab) fragments of donkey anti–rabbit antibodies (1:200). For triple labeling, rabbit anti-GFP antibodies (1:100) (Clontech) were used, to visualize transfected cells before the appearance of GFP fluorescence, followed by Cascade blue–conjugated goat anti–rabbit IgG antibodies (1:100; Molecular Probes Inc.). Monoclonal anti-striated muscle myosin antibodies (1:10) (F59, generously provided by Dr. F. Stockdale, Stanford University, Stanford, CA) were used to visualize thick filaments, followed by Texas red–conjugated F(ab′)2 fragments of donkey anti–mouse antibodies (1:600; when costained with titin antibodies) or FITC-conjugated donkey anti–mouse antibodies (1:200; when costained with phalloidin). Chicken anti-titin N2A antibodies were used to visualize titin (for sequence assignment, see Linke et al. 1996), followed by FITC-conjugated donkey anti–chicken antibodies. Texas red–conjugated phalloidin (Molecular Probes Inc.) was used to visualize actin. Stained cells were analyzed on a Zeiss Axiovert microscope using 63× (NA 1.4) and 100× (NA 1.3) objectives and micrographs were recorded as digital images on a SenSys cooled HCCD camera (Photometrics). Images were processed for presentation using Adobe Photoshop and printed using a Codonics NP1600 dye sublimation printer. All secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc., unless otherwise noted.
For the cardiac MyBP-C antibodies, residues 268–375 of mouse cardiac cDNA (Kasahara et al. 1994) were amplified from total mouse heart cDNAs by PCR (Saiki et al. 1985), and fragments were expressed as described above (see Antibodies to Cardiac Titin). The antibodies were raised in rabbits by Eurogentec and their specific IgG fraction was isolated by affinity chromatography. Antibody specificity was confirmed by Western blot analysis of extracts of rat cardiac muscle (data not shown).
Results
Extensibility of Cardiac Titins
By using a panel of sequence-assigned antibodies we aimed to establish the extension behavior of structurally distinct segments in both N2-A and N2-B cardiac titins. The epitope locations of the titin antisera are indicated in Fig. 1.
The mobility of titin antibody epitopes in each half-sarcomere was investigated in stretched single myofibrils isolated from rabbit left ventricle by immunofluorescence microscopy. Fig. 2 a shows typical images of myofibrils extended to two different SLs, 2.3 and 2.7 μm, and stained with the respective primary antibody and Cy-3–conjugated secondary antibody. Of the antibodies used, I17, N2B, and I18 are specific to the cardiac N2-B–titin region and did not stain rabbit soleus myofibrils (data not shown), whereas S54/56 is directed to Ig-repeats from the N2-A region. T12 (see Fig. 4 a), I20/22 and MIR recognize epitopes outside the differentially expressed I-band titin and should stain all titin isoforms. Both N2-A and N2-B–titin were found to be highly expressed in rabbit cardiac myofibrils, as judged from the strong staining intensity obtained with all antibodies except I17 (see below). At least at high stretch it was obvious that I17/N2B, I18, I20/22, and MIR stained different positions on N2-B–titin. I18 colocalized with the N2-A–specific S54/56 epitope.
Figure 4.
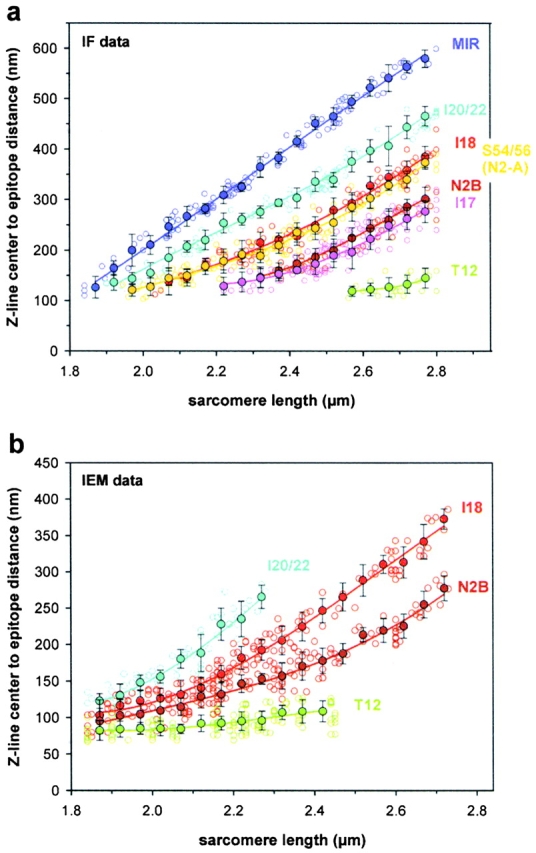
Antibody epitope mobility. (a) Summary of results of immunofluorescence (IF) experiments at SLs ranging from 1.84 to 2.8 μm. Color coding for the antibodies is as in Fig. 1. Small open circles indicate individual data points, larger filled circles show mean values calculated in 50-nm-wide SL bins; standard deviations are also shown. Data sets for each antibody type were fitted by third-order regressions. (b) Summary of results of immuno-EM (IEM) experiments. Presentation of data is as in a.
To more precisely map the epitope positions in a half-sarcomere, we plotted the intensity profiles along the myofibril axis and calculated the center-of-mass pixel position for each intensity peak with custom-written software (example in Fig. 2 a, bottom). The epitope spacing across the Z-disc could now be measured with satisfactory resolution at modest to long SLs. At short SLs, it was more difficult to obtain reasonable results with this analysis method, since the two epitopes around the Z-disk frequently (but not always) were seen to merge into one broader strip. Whereas the antibodies, T12, N2B, I18, and I20/22 were subsequently used for immunoelectron microscopy (immuno-EM), I17 appeared to be unsuitable for this technique. Possibly, the immunoreactive sequence in the Ig domain I17 becomes accessible only at very high sarcomere stretch. Although a tendency for stronger labeling at long SLs was observed sometimes by immunofluorescence microscopy, we regularly found both stained and unstained sarcomeres of comparable length side by side in a myofibril (Fig. 2 b), a finding never observed with any other antibody. Hence, we speculate that the native I17 domain of N2-B–titin may be partially inaccessible for the antibody, because its immunoreactive site is covered by some ligand (see Effects of Titin N2B Overexpression in Cardiac Myocytes).
The distance of a given epitope from the center of the Z-disk was determined at shorter SLs by immuno-EM. Fig. 3 a shows images of rabbit cardiac sarcomeres stretched to 1.9 and 2.3 μm, respectively, and labeled with one of four different antibodies. Using these antibodies, which flank the structurally distinct regions in N2-B–titin (proximal Ig-segment, middle N2-B region, PEVK domain), we confirmed that each region contributes to titin extensibility already at short to modest SLs. Whereas extension of the Ig-chains and the PEVK segment has been recognized previously (Linke et al. 1996; Granzier et al. 1997), the observation of considerable extensibility of the middle N2-B region is novel. To determine the SL at which this region begins to extend, we double-stained cardiac sarcomeres with both N2B and I18 and used secondary antibodies conjugated to different sized nanogold particles. As shown in Fig. 3 b, the epitopes could not yet be separated at 2.1 μm SL. However, separation was apparent in sarcomeres stretched to 2.35 μm. At 2.6 μm SL, the N2-B segment flanked by the antibody epitopes contributed substantially to I-band titin extensibility.
Figure 3.
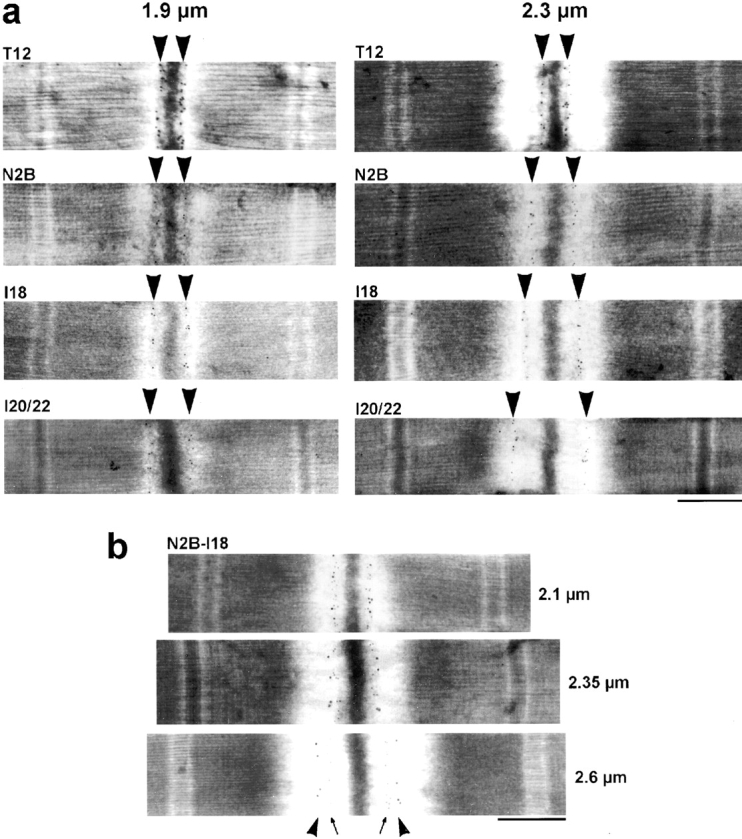
Immunoelectron micrographs of rabbit cardiac-muscle sarcomeres stained with titin antibodies. (a) Four different antibodies were used to label sarcomeres at different lengths, here 1.9 and 2.3 μm. This allowed us to follow the extension behavior of the three structurally distinct I-band titin segments of the N2-B isoform: proximal tandem-Ig segment (T12–N2B), middle N2-B region (N2B–I18), and PEVK domain (I18–I20/22). The nanogold particles indicate the respective epitope positions (arrowheads). (b) Stretched sarcomeres were double-stained with both N2B and I18 antibodies. The secondary antibody for N2B was conjugated to 10-nm gold particles (arrows), that for I18 to 15 nm particles (arrowheads). SLs are indicated on the right. Bars, 0.5 μm.
The results of the immunolabeling experiments are summarized in Fig. 4; separate graphs are shown for immunofluorescence (Fig. 4 a) and immuno-EM data (Fig. 4 b). For each antibody type, individual data points were pooled in 50-nm-wide SL bins, plotted as mean ± SD, and fitted by a third-order regression. When we compared results obtained with the two different techniques, epitope mobility was found to be similar within experimental error. The I17 and N2B antibodies stained almost the same position on N2-B–titin, as expected from their epitope location. I18, which comigrated with the N2-A–specific S54/56, labeled a position clearly different from that stained by I17/N2B or I20/22. The unique N2-B sequence flanked by I18 and N2B was confirmed by statistical analysis (unpaired Student's t test) of immuno-EM data to begin to lengthen significantly at ∼2.15 μm SL (P < 0.001; n = 17 for the I18 data set and n = 19 for the N2B data set at SL bin 2.15–2.19 μm).
By calculating the distances between fit curves at a given SL, we plotted the extensions of individual cardiac titin segments versus SL (Fig. 5 a). Data from both immuno-EM and immunofluorescence measurements were compiled because of the similarity of results obtained with the two techniques. Curves in the main Fig. 5 a indicate extensibility of the four structurally distinct regions that make up the entire elastic N2-B–titin. The proximal poly-Ig region elongated continuously up to the longest SL investigated, whereas the other three elements approached a plateau in their extension curves: the N2-B–PEVK domain at a SL of 2.3–2.4 μm, the distal poly-Ig region and the unique N2-B insertion at ∼2.6 μm. Substantial lengthening of the unique N2-B insertion began near 2.15 μm SL, and at 2.3–2.4 μm SL, this region reached 60–70 nm extension. When sarcomeres became highly stretched, the segment elongated to >100 nm. The inset in Fig. 5 a shows extension of the N2-A–PEVK domain and that of the Ig-segment flanked by T12 and S54/56. Whereas the Ig segment extended steadily at all SLs, the extension curve for N2-A–PEVK eventually reached a plateau at ∼2.6 μm SL.
Figure 5.
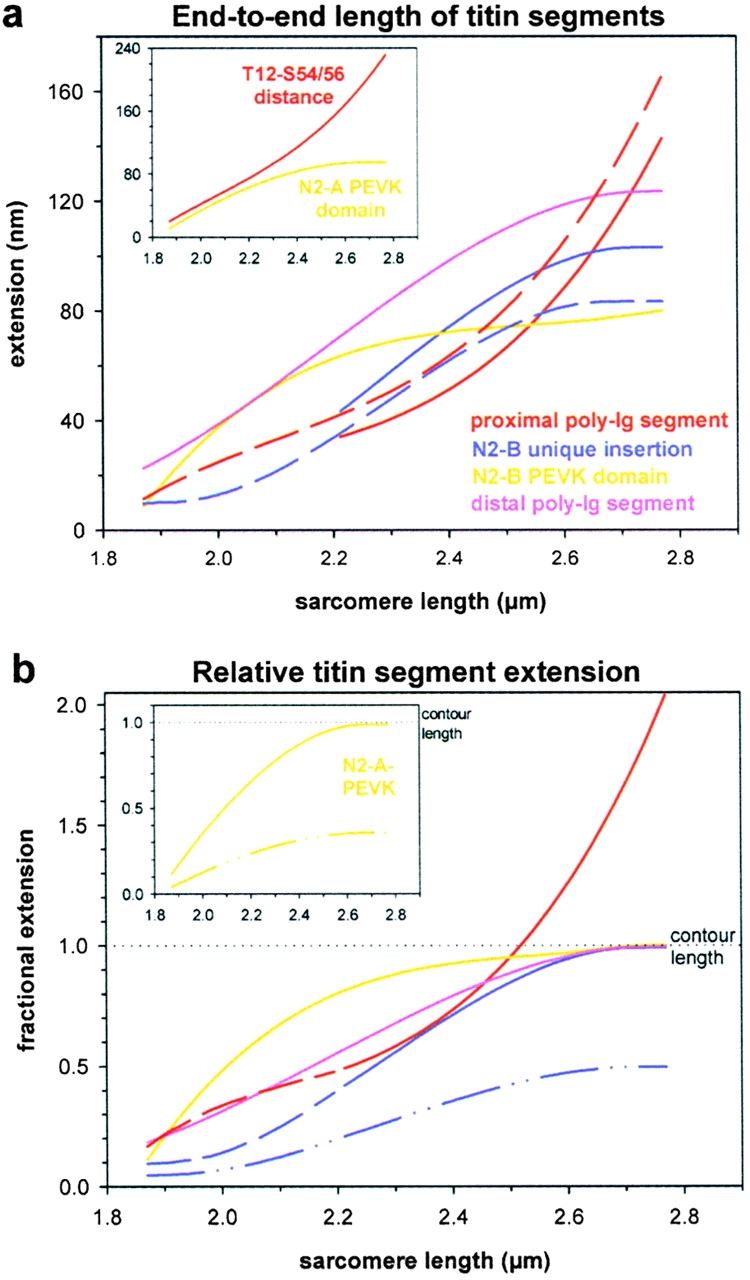
Extension of cardiac titin segments. (a) End-to-end lengths of titin segments plotted against SL. Curves were calculated from the regression curves in Fig. 4, a and b, as follows: proximal poly-Ig segment, T12 to I17 and T12 to N2B (red curves, solid and dashed, respectively); N2-B unique insertion, I17 to I18 and N2B to I18 (blue curves, solid and dashed, respectively); N2-B–PEVK domain, I18 to I20/22 (yellow curve); distal poly-Ig segment, I20/22 to MIR (pink curve). The inset shows the T12 to S54/56 distance and the extension of the N2-A–PEVK domain (S54/56 to I20/22). (b) Extension of titin segments relative to each segment's predicted contour length (see Table ). Solid curves shown in the main figure indicate relative extension of all those segments that together, make up the whole elastic I-band titin of the N2-B isoform. At short SLs, curves demonstrating extension of the proximal Ig-segment and of the full-length N2-B unique sequence were extrapolated (dashed lines). The inset shows results for the N2-A–specific PEVK domain. Color coding of curves is as in a. For explanation of differences between solid and dashed-dotted curves, see text.
Unfolding of Cardiac Titin Domains?
An important unresolved question is whether individual Ig domains are able to unfold at the stretch forces acting on titin filaments in normally functioning cardiac muscle (see Erickson 1997; Linke and Granzier 1998). That is, can a segment of Ig-modules be stretched to beyond the contour length predicted from its sequence structure assuming folded domains? To answer this question, we used the available sequence data for cardiac titin to predict the contour length of both the proximal and the distal tandem-Ig segments of the N2-B isoform (Table ). A value of 4.6 nm was taken as the maximum spacing between folded domains (Linke et al. 1998b; Trombitas et al. 1998). Because only incomplete sequence information is available for cardiac N2-A titin, we could not predict the contour length of the Ig segment flanked by T12 and S54/56 (see Fig. 1). However, contour length predictions could be made for all other I-band segments in N2-B–titin and for the N2-A–PEVK domain (Table ). By relating extension of a given titin segment to predicted contour length, the fractional extension of each segment was calculated (Fig. 5 b). We found that the distal poly-Ig region stretched up to its predicted contour length but not beyond. The proximal Ig domain region of N2-B–titin exceeded the contour length value at a SL of ∼2.5 μm, perhaps as a result of domain unfolding. Thus, proximal N2-B Ig domains might potentially be able to unfold in highly stretched sarcomeres, but are unlikely to do so in normally functioning heart where maximum SLs are presumably below 2.4 μm.
Table 1.
Predicted Contour Length of Cardiac I-Band Titin Segments
| Contour length predicted for | |||||
|---|---|---|---|---|---|
| Isoform | I-Band segment | No. of domains/residues | Maximum average domain/ residue spacing | Folded/partially folded region | Completely unfolded region |
| nm | nm | nm | |||
| Both | Distal Ig | 22 domains (+5 domains at the A-band edge) | 4.6 | 124 | |
| N2-A | Proximal Ig | ? | |||
| PEVK | ∼700 residues | 0.38 | ∼265 | ||
| 0.14 | 97 | ||||
| N2-B | Proximal Ig | 15 domains | 4.6 | ∼70 | |
| Unique N2-B sequence | 572 residues | 0.38 | ∼215 | ||
| 0.19 | ∼108 | ||||
| PEVK | 163 residues | 0.38 | 62 (incl. flanking sequences: 78) | ||
Sequence data are from Labeit and Kolmerer 1995 and from our own preliminary work (unpublished results) on the primary structure of rabbit cardiac titin.
For all other titin segments, the end-to-end lengths were predicted to either reach or stay below the assumed contour lengths (Fig. 5 b). Interestingly, when we assumed that the PEVK segments and the unique N2-B insertion are capable of complete unfolding (maximum residue spacing, 0.38 nm), only the curve for N2-B–PEVK was predicted to reach the contour length value (main Fig. 5 b, yellow curve). The unique N2-B insertion (Fig. 5 b, blue dashed-dotted curve) and the N2-A–PEVK domain (Fig. 5 b, inset, dashed-dotted curve) at best extended to half the predicted maximum value. On the other hand, because both segments did approach a plateau in their extension curves, we argue that these regions may not unfold entirely. Rather, they may adopt some permanent structural fold, which would lead to a shorter contour length; the resulting curves are shown in Fig. 5 b (solid blue curve) and Fig. 5 b, inset (solid yellow curve), respectively.
Reversible Extension of Titin
We aimed to determine the SL range within which cardiac titins can be extended and released in a completely reversible manner. Therefore, nonactivated single cardiac myofibrils were repeatedly stretched and released in steps while passive force was recorded (Fig. 6). Between steps, the specimens were rested for 2–3 min, to allow force to drop to a quasi steady state level. In each cycle, the maximum SL was increased and between cycles the slack SL was monitored. Thus, we could study the maximum length from which cardiac sarcomeres can be released to a completely recoverable slack SL.
Figure 6.
Passive length-tension curves during three consecutive stretch-release cycles, performed on two different single cardiac myofibrils (a and b). The stretch/release-hold protocols are indicated above the panels. Maximum SL in each cycle was progressively increased. Note that the myofibrils shortened down to their initial slack length after the first cycle, in which they had been stretched to SLs of 2.4 μm (a) and 2.45 μm (b), respectively. However, slack SL was increased and hysteresis was much larger after the second and third cycles, when specimens had been stretched to beyond 2.5 μm SL.


Results obtained with four different myofibrils (Fig. 6 shows two representative examples) demonstrated that sarcomeres could be extended to ∼2.5 μm while still returning to their initial slack length after release (cycle 1). However, when the maximum stretch clearly exceeded 2.5 μm, the force-stretch/release curves exhibited large hysteresis and the specimens' slack SL was increased (cycles 2 and 3). This finding implies that cardiac titin filaments can be stretched reversibly to well above physiological length. Further stretch likely results in irreversibly increased contour lengths of titin. This, in turn, leads to dramatic changes in the passive length–tension relation of cardiac myofibrils (in Fig. 6, a and b, compare the stretch curve of cycle 1 and cycle 2 to that of cycle 3).
Effects of Titin N2B Overexpression in Cardiac Myocytes
To gain further insight into the functional significance of the N2-B region of cardiac titin in cells, we overexpressed this region in primary cultures of chick cardiac myocytes in the hope that the expressed fragment would compete with the endogenous titin domain to generate dominant-negative effects on sarcomere structure. Initially, we transfected cells with a plasmid encoding the entire region of titin N2-B. A GFP tag was used to distinguish the recombinant protein from endogenous protein. Although no significant disruption of actin filaments was observed in cells overexpressing GFP alone, substantial disruption of thin filament structure was observed in ∼70% of all transfected cells where N2-B–titin was overexpressed, as seen by a marked loss in actin filaments (Fig. 7, compare cells transfected with GFP alone, which demonstrated a strong striated staining pattern of actin [b], with those transfected with N2-B [f]). Moreover, when the transfected cells overexpressing titin N2-B were stained for tropomyosin (CH1; Lin et al. 1985), a similar disruption of thin filament structure was observed (data not shown). In contrast, preliminary data (not shown) suggested that overexpression of the entire N2-A region of cardiac titin has no effect on sarcomere structure.
Figure 7.

Overexpression of the NH2-terminal segment of titin N2-B in cardiac myocytes results in marked disruption of actin (thin) filaments; also, Z-disk integrity is lost, but to a lesser degree. Cardiac myocytes expressing GFP alone (a and c), GFP-titin N2-B (e and g), or GFP-tagged NH2-terminal (N; i and k), middle (M; m and o) and COOH-terminal (C; q and s) domains of titin N2-B were fixed 3–5 d after transfection and stained with Texas red–conjugated phalloidin to visualize F-actin (b, f, j, n, and r). To study α-actinin distribution, cells were also labeled with antibodies to this protein followed by staining with Texas red–conjugated F(ab) fragments of donkey anti–rabbit antibodies (d, h, l, p, and t). Arrows point to the typical striated staining pattern observed in cardiac myocytes with Texas red phalloidin (b, n, and r) and with anti–α-actinin antibodies (d, p, and t); arrowheads point to disrupted staining patterns in cardiac myocytes; short arrows point to slightly misaligned Z-lines. Note the strongly stained remnant of a myofibril in the top left corner of the micrograph in f. Bar, 10 μm.
To identify the specific domain(s) within titin N2-B responsible for the disruption of thin filaments, we also transfected cells with plasmids encoding GFP-tagged subdomains of N2-B specific to the 25-kD NH2 terminus (N-N2-B), a 70-kD middle region (M-N2-B), and the 25-kD COOH terminus (C-N2-B) (see Fig. 1). Cells overexpressing N-N2-B revealed severe disruption of actin filament structure in all transfected cells (Fig. 7 j). In fact, the number of live myocytes that were transfected with N-N2-B declined drastically as the levels of this fusion protein increased; the cells were observed to round up and detach from the coverslips. This observation was unique to this construct.
Remarkably, in cardiac myocytes in which M-N2-B was overexpressed, no obvious disruption of thin filaments was observed (Fig. 7 n). Similarly, thin filaments showed no detectable disruption in cells expressing C-N2-B (Fig. 7 r). In this regard, it is important to note that it is well documented that the process of myofibril assembly in primary cultures of cardiac myocytes is temporarily irregular; thus, numerous structures representing different stages of assembly are observed within the same cell, as well as within different cells in a culture dish (Wang et al. 1988; Lin et al. 1989; Rhee et al. 1994; Gregorio and Fowler 1995). Thus, ∼20% of nontransfected cells, cells transfected with GFP alone, M-N2-B, or C-N2-B demonstrate a disruption-like distribution of actin filaments (data not shown).
The striking thin filament disruption observed in cardiac myocytes as a result of the overexpression of titin N2-B, and specifically, N-N2-B, suggested that other sarcomeric components might also be disrupted. To study this, the distribution of the major Z-line component, α-actinin, and the thick filament–associated proteins, myosin and MyBP-C, were analyzed in the transfected myocytes. As demonstrated in Fig. 7 d, mature myofibrils in cardiac myocytes probed with α-actinin antibodies exhibited a sharp repeating (∼2 μm apart) staining pattern. As found for actin filaments, overexpression of N2-B or N-N2-B resulted in disruption of Z-lines also (Fig. 7h and Fig. l, respectively), but to a somewhat lesser degree. That is, Z-disks could still be detected in the transfected cells, although they were frequently distributed in a less ordered fashion (Fig. 7h and Fig. l, short arrows). Again, consistent with the observations on thin filament disruption, overexpression of M-N2-B, C-N2-B or GFP alone did not significantly disrupt α-actinin staining patterns (Fig. 7p, Fig. t, and Fig. d, respectively). On the other hand, when cells overexpressing N2-B or N-N2-B were stained for thick-filament proteins, including MyBP-C, no altered distribution was detected (Fig. 8, compare similar MyBP-C staining patterns in cells overexpressing GFP alone [b], N2-B [d], or N-N2-B [f]). Since it was difficult to imagine such well-formed sarcomeres visible by MyBP-C staining in the absence of thin filaments, we performed triple labeling experiments to visualize this phenomenon within identical myofibrils. Costaining cells overexpressing N2-B for actin and myosin confirmed our results: thin filament structure was disrupted in the same myofibrils that demonstrated intact thick filaments (Fig. 9, compare actin and myosin staining patterns in N2-B–transfected cells [e and f] with staining in control cells overexpressing GFP alone [b and c]). Furthermore, analysis of identical cells overexpressing titin N2-B stained for myosin and several epitopes of titin along the length of the molecule demonstrated that the titin filaments (data shown using anti–N2-A antibodies) also remain intact (Fig. 9, compare the similarity in myosin and titin N2-A staining in N2-B overexpressing cells [k and l] with staining in control cells overexpressing GFP alone [h and i]). Similarly, when cells overexpressing GFP-N2-B were triple stained for GFP, actin, and N2A, the distribution patterns of titin looked unperturbed in the identical myofibrils that demonstrated a disrupted staining pattern for actin (data not shown). Finally, we note that overexpression of GFP alone, titin N2-B, or any of the three N2-B fragments resulted in no observable alteration to the actin-containing stress fibers in the fibroblasts contaminating our cultures, as determined by phalloidin staining (data not shown).
Figure 8.

Thick filament structure is unaffected in cardiomyocytes overexpressing titin N2-B. Cells expressing GFP alone (a), GFP-titin N2-B (c), or GFP-tagged NH2-terminal N2-B (e) were fixed 3–5 d after transfection and stained with antibodies to MyBP-C (C-protein), as a marker for thick filaments, followed by Texas red–conjugated F(ab) fragments of donkey anti–rabbit antibodies (b, d, and f). Arrows point to the typical MyBP-C striated staining pattern observed in cardiac myocytes. Bar, 10 μm.
Figure 9.

Triple staining of cells overexpressing GFP-tagged titin N2-B reveals a marked disruption of actin (thin) filaments while myosin and titin filaments remain intact within identical myofibrils. Cardiac myocytes expressing GFP alone (a–c) or GFP-tagged titin N2-B (d–f) were fixed 1–3 d after transfection and triple stained with rabbit anti-GFP antibodies followed by Cascade blue–conjugated goat anti–rabbit antibodies (to identify transfected cells before the appearance of GFP fluorescence; a and d), Texas red–conjugated phalloidin (b and e), and monoclonal anti-striated muscle myosin antibodies followed by FITC-conjugated donkey anti–mouse antibodies (c and f). In addition, cardiac myocytes expressing GFP alone (g–i) or GFP-tagged titin N2-B (j–l) were fixed 1-3 days after transfection and triple stained with rabbit anti-GFP antibodies followed by Cascade blue–conjugated goat anti–rabbit antibodies (g and j), avian anti-titin N2-A antibodies followed by FITC-conjugated donkey anti–chicken antibodies (h and k) and monoclonal anti-striated muscle myosin antibodies followed by Texas red–conjugated donkey anti–mouse antibodies (i and l). Arrows point to the typical striated staining pattern observed in cardiac myocytes for actin (b), titin N2-A (h and k), and muscle myosin (c, f, i, and l); arrowheads point to disrupted phalloidin staining patterns in cardiac myocytes (e). Note, staining of nuclei using Cascade blue in d and j, and enhanced Z-line staining commonly observed using phalloidin in cardiac myocytes (e.g., Gregorio and Fowler 1995). Bar, 10 μm.
We conclude that the NH2-terminal region of chick cardiac N2-B–titin possesses structural properties necessary to maintain the integrity of thin filaments. We would also like to stress that similar results were obtained in transfection experiments with primary cultures of neonatal rat cardiac myocytes.
Discussion
Recently, progress has been made in understanding the nature of titin elasticity in skeletal muscle sarcomeres (Linke et al. 1998a,Linke et al. 1998b; Trombitas et al. 1998). In contrast, cardiac titin is expressed in different length isoforms, N2-A and N2-B, which makes an investigation of the protein's mechanical properties less straightforward. Insights into the function of cardiac I-band titin have been obtained in mechanical and immunolabeling studies on isolated cells (e.g., Granzier et al. 1996, Granzier et al. 1997) and single myofibrils (e.g., Linke et al. 1996, Linke et al. 1997). It was proposed that in unstretched cardiac sarcomeres, titin's elastic section is in a contracted state (Granzier et al. 1996). The small passive forces developing upon low stretch to ∼2.0 μm SL were suggested to be entropic in nature and to arise from straightening of I-band titin. With further stretch and exponential tension rise, the molecular domains within titin were thought to unfold. In a later report, in light of new experimental evidence, the SL for the onset of unfolding (of titin's Ig domains) was proposed to be ∼2.2 μm (Granzier et al. 1997). Similarly, it was concluded in an immunofluorescence microscopical study of stretched single myofibrils stained with sequence-assigned antibodies (Linke et al. 1996), that cardiac titin's Ig domains may begin to unfold above 2.2–2.3 μm SL. (For comparison, the maximum SL reached in situ is probably 2.3–2.4 μm [Allen and Kentish 1985; Rodriguez et al. 1992]). These predictions were made based on the hypothesis that titin contains two structurally distinct extensible elements, poly-Ig chains and the PEVK domain (Labeit and Kolmerer 1995). We now demonstrate in this study that this property is indeed a characteristic of the N2-A isoform, but not of the N2-B isoform.
The extension properties of cardiac titin isoforms were investigated by immunolabeling techniques, using isoform-specific antibodies. Perhaps most striking was our observation on the extensibility of I-band segments in N2-B–titin. We found that this isoform contains, besides poly-Ig chains and the PEVK segment, a third extensible element, the middle N2-B sequence. The results led us to propose a novel model for titin isoform extension in cardiac muscle (Fig. 10). We suggest that in both isoforms, low sarcomere extension is brought about by elongation of tandem-Ig-segments and the PEVK domain (Fig. 10, stretch from stage 1 to stage 2). Above 2.15 μm SL, also the 572-residue N2-B region in the middle of I-band titin begins to elongate substantially (Fig. 10, stretch from stage 2 to stage 3) and may compensate for the relatively short length of the N2-B–PEVK segment (163 residues), whose extensibility would soon be exhausted. Elasticity of the unique N2-B sequence may help the shorter N2-B–titin isoform to adjust its range of extension to that of the longer N2-A isoform. The model in Fig. 10 also predicts that elongation of poly-Ig segments is brought about by straightening, rather than by domain unfolding. Moreover, cardiac titin extension may still be fully reversible after stretch to 2.5 μm SL (see Fig. 6). Then, complete reversibility of stretch appears to be a characteristic of all three elastic elements, the middle N2-B region, the PEVK segments, and the poly-Ig chains (with folded domains).
Figure 10.

Extension model for I-band titin isoforms in rabbit cardiac muscle. Shown is the functionally elastic section of titin at three different stages of extension corresponding to SLs reached under physiological conditions: stage 1, 1.95 μm SL; stage 2, 2.15 μm SL; stage 3, 2.35 μm SL. Upon low stretch (stage 1–stage 2), proximal and distal Ig-segments, as well as PEVK domains, begin to extend. Up to ∼2.15 μm SL, exclusively these segments confer elasticity to cardiac sarcomeres. However, on further stretch, the middle N2-B region (unique sequence insertion) also begins to elongate (stage 2–stage 3). This may help prevent potentially catastrophic events such as irreversible unfolding of Ig domains at high physiological stretch. Extensions are fully reversible for stretches up to ∼2.5 μm SL. The elastic behavior of cardiac titin might be constrained by protein/protein interactions involving the region bounded by the domains I16/17 of the N2-B isoform (indicated by the red oval). The nature of this putative association remains to be discovered, but it is clear that the I16/17 position on N2-B–titin is required to maintain the integrity of thin filaments. Dashed curves indicate missing sequence information.
The model suggests that titin's tandem-Ig modules, earlier thought to unfold above 2.2 μm SL, may not do so within the normal working range of cardiac muscle. Rather, they are likely to remain in a folded state even at high physiological stretch, just as recently suggested also for skeletal muscle titin (Linke et al. 1998b; Trombitas et al. 1998). These findings, which confirm and extend previous conclusions on the extensibility of the distal Ig domain segment of cardiac titin (Gautel et al. 1996), address an intriguing issue arising from the results of earlier single-molecule mechanical experiments on titin (Kellermayer et al. 1997; Rief et al. 1997; Tskhovrebova et al. 1997). In these studies, Ig domains were found to unravel at high stretch forces of 20–300 pN (the exact value depends on stretch speed). However, refolding of the modules occurred relatively slowly and only when the external force was lowered to a few piconewtons, resulting in enormous hysteresis; thus, the Ig domain behaves like a spring that loses considerable energy in each stretch (unfold)–release cycle. It seems unlikely that such an ineffective spring exists in the heart, which undergoes millions of stretch-release cycles during a lifetime. Our results provide strong evidence that the experimentally inducible mechanical denaturation/renaturation of Ig repeats does not take place during normal heart function. If at all, unfolding may be found perhaps in the proximal Ig domain region and only in pathologically overstretched cardiac muscle.
Although our understanding of the elastic properties of cardiac N2-A titin is still incomplete, mainly due to limited availability of sequence data, this study nevertheless provides interesting insights. For example, our findings indicated that the ∼700-residue-long N2-A–PEVK domain does not unravel completely even at the highest sarcomere stretch applied (2.8 μm SL). This hints at the fact that this domain may well be able to adopt some structural fold (see Witt et al. 1998), the nature of which remains to be identified. Also, since our analysis of N2-B–titin extensibility excluded that the proximal and distal Ig domains unfold at physiological stretch (Fig. 5 b), we find it also unlikely that the titin segment NH2-terminal to the N2-A–PEVK domain elongates by unfolding of Ig domains. Support for this conclusion comes from earlier observations that in different regions of the titin molecule, the thermodynamic and mechanical stabilities of Ig domains are comparable (Politou et al. 1995; Rief et al. 1998). Furthermore, our results clearly show that the segment flanked by the T12 antibody epitope and the I17 epitope extends much less than that located between T12 and S54/56 (Fig. 5 a). Then, all evidence taken together, we reason that cardiac N2-A titin should contain more (folded) Ig modules than the N2-B isoform in the proximal tandem-Ig region (see Fig. 10). It will be interesting to see whether sequence data will eventually confirm this prediction.
The elastic properties of cardiac titin have previously been modeled according to entropic polymer elasticity theory, by assuming that two different elastic elements, the Ig domain segments and the PEVK domain, act as worm-like chains (WLCs) in series (Granzier et al. 1997). With the knowledge that a third type of elastic element exists in the cardiac N2-B isoform, it is obvious that the WLC modeling for cardiac titin needs modification. A complication is that our data on N2-B expression in myocytes (Fig. 7 Fig. 8 Fig. 9) raise the possibility that part of the N2-B–titin region might be involved in protein–protein interactions (Fig. 10). Thus, titin may not be allowed to move freely in the sarcomere, which could affect the elastic properties of the protein chain. Because it is unsettled how this possible association will change the (hypothesized) WLC behavior of cardiac titin, we did not perform a detailed WLC analysis at this stage.
A fascinating observation that emerges from our results is that some portions of cardiac N2-B–titin appear to have properties necessary to maintain an ordered thin filament structure. Although this is, to our knowledge, the first direct evidence for a structural role of I-band titin in cardiac muscle, preliminary, indirect evidence existed before. From the study of deep-etch replica electron micrographs prepared to analyze the filament network present in cardiac I-bands before and after selective removal of actin, it was proposed that the elastic titin could associate laterally with thin filaments (Funatsu et al. 1993). However, despite several reports that titin interacts with actin in vitro (e.g., Soteriou et al. 1993; Jin 1995; Kellermayer and Granzier 1996), potential actin–titin interaction sites could not be identified in intact sarcomeres, except within a region near the Z-disk (Linke et al. 1997; Trombitas and Granzier 1997). Thus, the nature of the interaction of I-band titin with the thin filaments suggested by Funatsu et al. 1993 remained elusive. In this study, we reasoned that a most characteristic segment of cardiac titin is the central I-band region of the N2-B isoform. Therefore, we investigated the functional significance of this region of titin in the context of cardiac myocytes.
The central N2-B–titin region, as well as distinct subfragments of it, were overexpressed in primary cultures of cardiac myocytes. Using transfection techniques, overexpression of the entire N2-B region or its NH2-terminal segment encompassing the Ig domains I16 and I17 resulted in severe thin filament disruption (Fig. 7 and Fig. 9). In contrast, overexpression of the middle N2-B region or the COOH domains I18 and I19 resulted in more or less normal phenotypes. Interestingly, whereas the Z-lines were also affected in cells overexpressing the full N2-B region or the NH2-terminal segment of this region, the disruption appeared less severe. This observation suggests that the disruption of the Z-lines was a secondary effect of the thin filaments being disrupted. The picture emerging from these results is one in which the region bounded by the N2-B–specific Ig domains I16 and I17 is important for the stability of thin filaments. Also, the integrity of N2-B–titin appears to be a prerequisite specifically for thin filament structure, because overexpression of N2-B–titin segments did not affect the thick filaments (Fig. 8 and Fig. 9). The latter finding lends further support to the hypothesis that nucleation and assembly of I-Z-I bodies (containing several Z-line and thin filament components and titin) occurs independently of the formation of sarcomeric thick filaments, in primary cultures of heart and skeletal muscle cells, as well as in heart development in vivo (Manasek 1968; Antin et al. 1981; Schultheiss et al. 1990; Holtzer et al. 1997). Moreover, the observation that the titin filaments (Fig. 9) were unaffected in cells overexpressing N2-B, is consistent with the idea that titin may be the structural element keeping the A-bands aligned, in the absence of thin filaments.
A possible explanation for the severe disruption of the thin filaments is that a dominant-negative phenotype occurred. That is, overexpressed titin N2-B (or the NH2-terminal domains of this region) competed for the interaction of endogenous N2-B–titin with an intracellular ligand, thus preventing native titin from taking on its usual intracellular role. A speculative idea is that titin N2-B may interact directly or indirectly with tropomyosin, a protein that assembles head-to-tail, forming two polymers along the sides of the actin filaments and stabilizes them (e.g., Wegner and Walsh 1981; Broschat 1990). Alternatively, based on its sarcomeric location, titin N2-B may interact directly or indirectly with nebulette in cardiac muscle. Nebulette is an ∼100-kD protein of unknown function that is related to the giant protein nebulin in skeletal muscle, the predicted ruler of thin filament assembly (Moncman and Wang 1995; Millevoi et al. 1998). Taken together, the interaction between N2-B–titin domains and their ligand could be functionally relevant in several ways: (a) it may be critical for the maintenance of sarcomeric structure and (b) may participate in the sequential assembly of I-Z-I bodies; (c) it may constrain the elastic behavior of cardiac titin, thereby affecting myofibril stiffness; and (d) it could also affect the relative sliding of thick and thin filaments past one another when the sarcomere changes length. An intriguing possibility is that the binding properties of cardiac N2-B–titin might be different in pathologically altered myocardium, resulting in dramatic changes in the heart's mechanical performance. It shall therefore be interesting to investigate how the I-band titin interaction affects the mechanical properties of cardiac myofibrils, both under physiological and pathophysiological conditions. Future studies will also focus on elucidating whether the NH2-terminal region of N2-B–titin directly binds to tropomyosin, nebulette, actin, to other thin filament components, and/or to some still unidentified sarcomeric protein(s).
In summary, two main conclusions seem especially noteworthy. First, the N2-B region in the center of I-band titin contains, at its NH2 terminus, a cardiac-specific segment that is directly or indirectly critical for the stability of thin filament structure. Second, the N2-B region also contains an elastic element, the unique sequence insertion, which extends toward the high end of the physiological sarcomere-length range. Cardiac titin thus represents a molecular spring consisting of three elements: the middle N2-B region, the PEVK domain, and poly-Ig regions with folded modules. N2-B–titin must be viewed as a unique isoform critical for both reversible extensibility and structural maintenance of cardiac myofibrils.
Acknowledgments
We thank Hiltraud Hosser for help with immuno-EM, Thilo Welsch for writing a computer program, Mary Martin for performing the initial transfection experiments, Abigail McElhinny and Catherine McLellan for helpful comments on the transfection experiments, Bernhard Kolmerer for making an N2-B construct, and Christel Bletz and Reinhold Wojciechowski for expert technical assistance.
This work is supported by the Deutsche Forschungsgemeinschaft (Li 690/2-2 to W.A. Linke, La 668/5-1 to S. Labeit, and SFB 320 to W.A. Linke and S. Labeit), the Forschungsfond für Klinische Medizin Mannheim (to S. Labeit), National Institutes of Health HL57461 (to C.C. Gregorio), and the Human Frontier Science Program (to S. Labeit and C.C. Gregorio). The Leica confocal microscope used in this study is supported by the NIEHS Southwest Environmental Health Science Grant ES-06694.
Footnotes
1.used in this paper: GFP, green fluorescence protein; MyBP-C, myosin-binding protein C; SL, sarcomere length; WLC, worm-like chain
References
- Allen D.G., Kentish J.C. The cellular basis of the length-tension relation in cardiac muscle. J. Mol. Cell. Cardiol. 1985;17:821–840. doi: 10.1016/s0022-2828(85)80097-3. [DOI] [PubMed] [Google Scholar]
- Antin P.B., Forry-Schaudies S.L., Friedman T.M., Tapscott S.J., Holtzer H. Taxol induces postmitotic myoblasts to assemble interdigitating microtubule-myosin arrays which exclude actin filaments. J. Cell Biol. 1981;90:300–308. doi: 10.1083/jcb.90.2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broschat K.O. Tropomyosin prevents depolymerization of actin filaments from the pointed end. J. Biol. Chem. 1990;265:21323–21329. [PubMed] [Google Scholar]
- Erickson H.P. Stretching single protein moleculestitin is a weird spring. Science. 1997;276:1090–1092. doi: 10.1126/science.276.5315.1090. [DOI] [PubMed] [Google Scholar]
- Funatsu T., Kono E., Higuchi H., Kimura S., Ishiwata S., Yoshioka T., Maruyama K., Tsukita S. Elastic filaments in situ in cardiac muscledeep-etch replica analysis in combination with selective removal of actin and myosin filaments. J. Cell Biol. 1993;120:711–724. doi: 10.1083/jcb.120.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürst D.O., Osborn M., Nave R., Weber K. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopya map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J. Cell Biol. 1988;106:1563–1572. doi: 10.1083/jcb.106.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautel M., Lehtonen E., Pietruschka F. Assembly of the cardiac I-band region of titin/connectinexpression of the cardiac-specific regions and their relation to the elastic segments. J. Muscle Res. Cell Motil. 1996;17:449–461. doi: 10.1007/BF00123361. [DOI] [PubMed] [Google Scholar]
- Granzier H., Helmes M., Trombitás K. Nonuniform elasticity of titin in cardiac myocytesa study using immunoelectron microscopy and cellular mechanics. Biophys. J. 1996;70:430–442. doi: 10.1016/S0006-3495(96)79586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier H., Kellermayer M., Helmes M., Trombitas K. Titin elasticity and mechanism of passive force development in rat cardiac myocytes probed by thin-filament extraction. Biophys. J. 1997;73:2043–2053. doi: 10.1016/S0006-3495(97)78234-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio C.C., Fowler V.M. Mechanisms of thin filament assembly in embryonic chick cardiac myocytestropomodulin requires tropomyosin for assembly. J. Cell Biol. 1995;129:683–695. doi: 10.1083/jcb.129.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio C.C., Granzier H., Sorimachi H., Labeit S. Muscle assemblya titanic achievement? Curr. Opin. Cell Biol. 1999;11:18–25. doi: 10.1016/s0955-0674(99)80003-9. [DOI] [PubMed] [Google Scholar]
- Gregorio C.C., Trombitás K., Centner T., Kolmerer B., Stier G., Kunke K., Suzuki H., Obermayr F., Herrmann B., Granzier H., Sorimachi H., Labeit S. The NH2 terminus of titin spans the Z disc; its interaction with a novel 19 kD ligand (T-cap) is required for sarcomeric integrity. J. Cell Biol. 1998;143:1013–1027. doi: 10.1083/jcb.143.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzer H., Hijikata T., Lin Z.X., Zhang Z.Q., Holtzer S., Protasi F., Franzini-Armstrong C., Sweeney H.L. Independent assembly of 1.6 microns long bipolar MHC filaments and I-Z-I bodies. Cell Struct. Funct. 1997;22:83–93. doi: 10.1247/csf.22.83. [DOI] [PubMed] [Google Scholar]
- Jin J.-P. Cloned rat cardiac titin class I and class II motifs. J. Biol. Chem. 1995;270:6908–6916. [PubMed] [Google Scholar]
- Kasahara H., Itoh M., Sugiyama T., Kido N., Hayashi H., Saito H., Tsukita S., Kato N. Autoimmune myocarditis induced in mice by cardiac C-protein. Cloning of complementary DNA encoding murine cardiac C-protein and partial characterization of the antigenic peptides. J. Clin. Invest. 1994;94:1026–1036. doi: 10.1172/JCI117416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellermayer M.S., Granzier H.L. Calcium-dependent inhibition of in vitro thin-filament motility by native titin. FEBS Lett. 1996;380:281–286. doi: 10.1016/0014-5793(96)00055-5. [DOI] [PubMed] [Google Scholar]
- Kellermayer M.S.Z., Smith S.B., Granzier H.L., Bustamante C. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science. 1997;276:1112–1116. doi: 10.1126/science.276.5315.1112. [DOI] [PubMed] [Google Scholar]
- Labeit S., Kolmerer B., Linke W.A. The giant protein titin. Emerging roles in physiology and pathophysiology. Circ. Res. 1997;80:290–294. doi: 10.1161/01.res.80.2.290. [DOI] [PubMed] [Google Scholar]
- Labeit S., Gautel M., Lakey A., Trinick J. Towards a molecular understanding of titin. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:1711–1716. doi: 10.1002/j.1460-2075.1992.tb05222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labeit S., Kolmerer B. Titins, giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- Lin J.J., Chou C.S., Lin J.L. Monoclonal antibodies against chicken tropomyosin isoformsproduction, characterization and application. Hybridoma. 1985;4:223–242. doi: 10.1089/hyb.1985.4.223. [DOI] [PubMed] [Google Scholar]
- Lin Z., Holtzer S., Schultheiss T., Murray J., Masaki T., Fischman D.A., Holtzer H. Polygons and adhesion plaques and the disassembly and assembly of myofibrils in cardiac myocytes. J. Cell Biol. 1989;10:2355–2367. doi: 10.1083/jcb.108.6.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke W.A., Granzier H. A spring talenew facts on titin elasticity. Biophys. J. 1998;75:2613–2614. doi: 10.1016/S0006-3495(98)77706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke W.A., Ivemeyer M., Olivieri N., Kolmerer B., Rüegg J.C., Labeit S. Towards a molecular understanding of the elasticity of titin. J. Mol. Biol. 1996;261:62–71. doi: 10.1006/jmbi.1996.0441. [DOI] [PubMed] [Google Scholar]
- Linke W.A., Ivemeyer M., Labeit S., Hinssen H., Rüegg J.C., Gautel M. Actin-titin interaction in cardiac myofibrilsprobing a physiological role. Biophys. J. 1997;73:905–919. doi: 10.1016/S0006-3495(97)78123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke W.A., Ivemeyer M., Mundel P., Stockmeier M.R., Kolmerer B. Nature of PEVK-titin elasticity in skeletal muscle Proc. Natl. Acad. Sci. USA. 95 1998. 8052 8057a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke W.A., Stockmeier M.R., Ivemeyer M., Hosser H., Mundel P. Characterizing titin's I-band Ig domain region as an entropic spring J. Cell Sci. 111 1998. 1567 1574b [DOI] [PubMed] [Google Scholar]
- Manasek F.J. Embryonic development of the heart. I. A light and electron microscopic study of myocardial development in the early chick embryo. J. Morph. 1968;125:329–366. doi: 10.1002/jmor.1051250306. [DOI] [PubMed] [Google Scholar]
- Maruyama K. Connectin/titin, giant elastic protein of muscle. FASEB J. 1997;11:341–345. doi: 10.1096/fasebj.11.5.9141500. [DOI] [PubMed] [Google Scholar]
- Mayans O., van der Ven P.F., Wilm M., Mues A., Young P., Fürst D.O., Wilmanns M., Gautel M. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature. 1998;395:863–869. doi: 10.1038/27603. [DOI] [PubMed] [Google Scholar]
- Millevoi S., Trombitas K., Kostin S., Schaper J., Pelin K., Kolmerer B., Granzier H., Labeit S. Characterization of nebulette and nebulin and emerging concepts of their roles for vertebrate Z-discs. J. Mol. Biol. 1998;282:111–123. doi: 10.1006/jmbi.1998.1999. [DOI] [PubMed] [Google Scholar]
- Moncman C.L., Wang K. Nebulettea 107 kD nebulin-like protein in cardiac muscle. Cell Motil. Cytoskelet. 1995;32:205–225. doi: 10.1002/cm.970320305. [DOI] [PubMed] [Google Scholar]
- Mundel P., Gilbert P., Kriz W. Podocytes in glomerulus of rat kidney express a characteristic 44 kD protein. J. Histochem. Cytochem. 1991;39:1047–1056. doi: 10.1177/39.8.1856454. [DOI] [PubMed] [Google Scholar]
- Politou A.S., Thomas D.J., Pastore A. The folding and stability of titin immunoglobulin-like modules, with implications for the mechanism of elasticity. Biophys. J. 1995;69:2601–2610. doi: 10.1016/S0006-3495(95)80131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politou A.S., Gautel M., Pfuhl M., Labeit S., Pastore A. Immunoglobulin-type domains of titinsame fold, different stability? Biochemistry. 1994;33:4730–4737. doi: 10.1021/bi00181a604. [DOI] [PubMed] [Google Scholar]
- Rhee D., Sanger J.M., Sanger J.W. The premyofibrilevidence for its role in myofibrillogenesis. Cell Motil. Cytoskel. 1994;28:1–24. doi: 10.1002/cm.970280102. [DOI] [PubMed] [Google Scholar]
- Rief M., Gautel M., Schemmel A., Gaub H.E. The mechanical stability of immunoglobulin and fibronectin III domains in the muscle protein titin measured by atomic force microscopy. Biophys. J. 1998;75:3008–3014. doi: 10.1016/S0006-3495(98)77741-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rief M., Gautel M., Oesterhelt F., Fernandez J.M., Gaub H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- Rodriguez E.K., Hunter W.C., Royce M.J., Leppo M.K., Doulas A.S., Weisman H.F. A method to reconstruct myocardial sarcomere lengths and orientations at transmural sites in beating canine hearts. Am. J. Physiol. 1992;263:H293–H306. doi: 10.1152/ajpheart.1992.263.1.H293. [DOI] [PubMed] [Google Scholar]
- Saiki R.K., Scharf S.J., Faloona F., Mullis G.T., Erlich H.A. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- Schultheiss T., Lin Z., Lu M.-H., Murray J., Fischman D.A., Weber K., Masaki M., Imamura M., Holtzer H. Differential distribution of subsets of myofibrillar proteins in cardiac nonstriated and striated myofibrils. J. Cell Biol. 1990;110:1159–1172. doi: 10.1083/jcb.110.4.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soteriou A., Gamage M., Trinick J. A survey of the interactions made by titin. J. Cell Sci. 1993;104:119–123. doi: 10.1242/jcs.104.1.119. [DOI] [PubMed] [Google Scholar]
- Studier F.W., Moffat B.A. RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1991;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- Tokuyasu K.T. Use of poly(vinylpyrrolidone) and poly(vinyl alcohol) for cryoultramicrotomy. Histochem. J. 1989;21:163–171. doi: 10.1007/BF01007491. [DOI] [PubMed] [Google Scholar]
- Trinick J. Cytoskeletontitin as a scaffold and spring. Curr. Biol. 1996;6:258–260. doi: 10.1016/s0960-9822(02)00472-4. [DOI] [PubMed] [Google Scholar]
- Trombitas K., Granzier H. Actin removal from cardiac myocytes shows that near the Z-line titin attaches to actin while under tension. Am. J. Physiol. 1997;273:C662–C670. doi: 10.1152/ajpcell.1997.273.2.C662. [DOI] [PubMed] [Google Scholar]
- Trombitas K., Greaser M., Labeit S., Jin J.-P., Kellermayer M., Helmes M., Granzier H. Titin extensibility in situentropic elasticity of permanently folded and permanently unfolded molecular segments. J. Cell Biol. 1998;140:853–859. doi: 10.1083/jcb.140.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tskhovrebova L., Trinick J., Sleep J.A., Simmons R.M. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature. 1997;387:308–312. doi: 10.1038/387308a0. [DOI] [PubMed] [Google Scholar]
- Wang K. Titin/connectin and nebulingiant protein rulers of muscle structure and function. Adv. Biophys. 1996;33:123–134. [PubMed] [Google Scholar]
- Wang S.M., Greaser M.L., Schultz E., Bulinski J.C., Lin J.J., Lessard J.L. Studies on cardiac myofibrillogenesis with antibodies to titin, actin, tropomyosin, and myosin. J. Cell Biol. 1988;107:1075–1083. doi: 10.1083/jcb.107.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegner A., Walsh T.P. Interaction of tropomyosin-troponin with actin filaments. Biochemistry. 1981;20:5633–5642. doi: 10.1021/bi00522a043. [DOI] [PubMed] [Google Scholar]
- Witt C.C., Olivieri N., Centner T., Kolmerer B., Millevoi S., Morell J., Labeit D., Labeit S., Jockusch H., Pastore A. A survey of the primary structure and the interspecies conservation of I-band titin's elastic elements in vertebrates. J. Struct. Biol. 1998;122:206–215. doi: 10.1006/jsbi.1998.3993. [DOI] [PubMed] [Google Scholar]