

Abstract
The assembly of signaling molecules surrounding the integrin family of adhesion receptors remains poorly understood. Recently, the membrane protein caveolin was found in complexes with β1 integrins. Caveolin binds cholesterol and several signaling molecules potentially linked to integrin function, e.g., Src family kinases, although caveolin has not been directly implicated in integrin-dependent adhesion. Here we report that depletion of caveolin by antisense methodology in kidney 293 cells disrupts the association of Src kinases with β1 integrins resulting in loss of focal adhesion sites, ligand-induced focal adhesion kinase (FAK) phosphorylation, and adhesion. The nonintegrin urokinase receptor (uPAR) associates with and stabilizes β1 integrin/caveolin complexes. Depletion of caveolin in uPAR-expressing 293 cells also disrupts uPAR/integrin complexes and uPAR-dependent adhesion. Further, β1 integrin/caveolin complexes could be disassociated by uPAR-binding peptides in both uPAR-transfected 293 cells and human vascular smooth muscle cells. Disruption of complexes by peptides in intact smooth muscle cells blocks the association of Src family kinases with β1 integrins and markedly impairs their migration on fibronectin. We conclude that ligand-induced signaling necessary for normal β1 integrin function requires caveolin and is regulated by uPAR. Caveolin and uPAR may operate within adhesion sites to organize kinase-rich lipid domains in proximity to integrins, promoting efficient signal transduction.
Keywords: urokinase receptor, caveolin, integrins, adhesion, cell signaling
Integrins are heterodimeric adhesion receptors implicated in many biological processes characterized by cell movement, including inflammation, tissue remodeling, growth, and tumorigenesis (Hynes, 1992; Ruoslahti and Reed, 1994; Schwartz, 1997). Integrin-dependent cellular adhesiveness depends not only on specific α/β integrin chains expressed by cells but also on ligand-induced integrin redistribution and signal transduction leading to cytoskeletal reorganization (Schwartz et al., 1995; Burridge and Chrzanowska-Wodnicka, 1996; Parsons, 1996). Signaling through the integrin family of adhesion receptors requires their association with cytoplasmic elements capable of signal transduction because integrins lack intrinsic signaling capacity. In part, these dynamic aspects of integrin function are regulated by the physical association of integrins with nonintegrin membrane proteins. These include the tetraspan family of membrane proteins, integrin-associated protein, and CD98 (Lindberg et al., 1993; Berditchevski et al., 1996; Fenczik et al., 1997). In addition we have previously reported that β1 integrin function is regulated by its association with the GPI-linked nonintegrin receptor, the urokinase receptor (uPAR)1 (Wei et al., 1996). When human embryonic kidney cells are engineered by transfection to express uPAR, uPAR forms complexes with β1 integrins and promotes adhesion of these cells to vitronectin via a vitronectin binding site on uPAR. Incorporation of α5/β1 integrins into complexes with uPAR also inhibits the ability of these integrins to bind fibronectin. Disruption of the uPAR/integrin complexes by uPAR-binding peptides blocks adhesion to vitronectin and restores the normal adhesiveness of 293 cells for fibronectin. Thus, the ligand specificity and adhesive function of this class of integrins is altered by the presence of uPAR.
A second nonintegrin membrane protein, caveolin, was also found to coimmunoprecipitate with β1 integrins and uPAR in uPAR-transfected 293 cells (Wei et al., 1996). Independently, Wary et al. (1996) observed caveolin to coprecipitate with β1 integrins in detergent-lysed A431 cells, which express high levels of uPAR and caveolin. Caveolin, discovered as an Src kinase substrate, is an ∼21-kD membrane protein which binds cholesterol and a number of signaling molecules potentially linked with integrin function: Src family kinases, heterotrimeric G proteins, and H-ras (Li et al., 1995, 1996a). Caveolin has an intrinsic propensity to oligomerize and it is currently thought caveolin oligomers contribute to formation of cholesterol-sphingolipid-rich microdomains within membranes and the distinct membrane structures termed caveolae (Parton, 1996; Harder and Simons, 1997). Caveolae are enriched in a number of nonreceptor signaling proteins and growth factor receptor tyrosine kinases, but are not known to contain integrins (Lisanti et al., 1994; Mineo et al., 1996). However, while this work was in progress it was reported that a fraction of cellular caveolin associates with β1 integrins and promotes Fyn-dependent Shc phosphorylation in response to integrin ligation, leading to mitogen-activated protein kinase (MAPK) activation and cellular growth (Wary et al., 1998). In this report we provide direct evidence that caveolin is also important to β1 integrin–dependent fibronectin adhesion and focal adhesion kinase (FAK) activation and propose that this operates, at least in part, by virtue of the ability of caveolin to regulate Src family kinase activity surrounding integrins. Thus, caveolin appears to be a general regulator of β1 integrin function.
Materials and Methods
Generation and Transfection of Caveolin cDNA and Antisense Constructs
Human caveolin-1 cDNA was generated by reverse transcription and polymerase chain reaction, subcloned into TA cloning vector (Invitrogen), verified by nucleotide sequencing (Sequenase; United States Biochemical), digested with BamHI and Xhol, and then subcloned into the inducible expression vector pML1 (Lukashev et al., 1994) in an inverted order. Cells were transfected by electroporation, clones selected in hygromycin, and levels of caveolin-1 assessed by immunoblotting after overnight culture with 3 μM CdCl2. Caveolin cDNA was also subcloned into the CMV promoter-driven expression vector, pCEP9 (Invitrogen), and clones selected by growth in G418. Transfected cells were then screened for protein expression via immunoblot with antibody specific for caveolin-1 (Transduction Labs).
Adhesion and Binding Assays
Cells (5 × 104) were seeded in fibronectin (5 μg/ml; Sigma Chemical Co.) or vitronectin (1 μg/ml; Becton Dickinson) coated 96-well tissue culture plates, incubated at 37°C for 1 h, and adherent cells quantified as described previously (Wei et al., 1996). Before the adhesion assay, antisense caveolin 293 or uPAR/293 cells were induced with 3 μM CdCl2 at 37°C for 8 h.
Fibronectin binding was assessed on cells seeded in polylysine-coated 96-well tissue culture plates, cultured overnight, induced with 3 μM CdCl2 at 37°C for 8 h, and then washed with binding buffer (DMEM, 1 mg/ml BSA). Half of the cells were treated with β1 integrin activating antibody TS2/16 (3 μg/ml; Endogen) at 4°C for 30 min, and then all cells were incubated with buffer containing 125I-fibronectin (1, 5, and 10 nM) at 4°C for 1.5 h. After washing, cells were lysed in 0.2% SDS, 0.2% Triton X-100, 10% glycerol in PBS for 30 min, and radioactivity was measured. Nonspecific binding was determined by inclusion of 0.5 mM RGDS (American Peptide Co.) in the reaction mixture.
Flow Cytometry
Cells transfected with caveolin antisense constructs were induced with 3 μM CdCl2 at 37°C for 8–18 h. Cells were then detached and incubated with PBS containing 0.1% BSA and primary antibodies to uPAR (R2) or integrins β1 (JB1A) (Wilkins et al., 1996), α5β1 (Chemicon), and αv (L230) (American Type Culture Collection) on ice for 30 min. After washing, cells were incubated with FITC-conjugated goat anti–mouse IgG (Sigma Chemical Co.) and analyzed on a flow cytometer (FACScan®; Becton Dickinson).
Immunofluorescence and Confocal Fluorescence Microscopy
To assess the distribution of caveolin and β1 integrins, human saphenous vein smooth muscle cells (SMC) and 293 or uPAR/293 cells coexpressing caveolin were cultured overnight on glass coverslips in 10% FBS. After washing, cells were incubated for 30 min with JB1A and/or K20 (Immunotec) antibodies to β1 or isotype controls at 4°C and fixed for 20 min in 3.7% paraformaldehyde. Fixed cells were permeabilized and incubated with rabbit polyclonal antibody to caveolin (Transduction Labs) for 30 min at room temperature, then incubated with FITC- or Rhodamine red– conjugated secondary antibodies and coverslips mounted in Prolong (Molecular Probes). Fluorescence staining was analyzed by confocal laser (model MRC1024; Bio-Rad Laboratories) attached to a Zeiss microscope (model Axiovert S100) using separate filters for each fluorochrome. Optical planes were imported into Adobe Photoshop and processed. To visualize integrin and caveolin clustering, SMC in suspension were incubated with FITC-K20 β1 antibodies without or with goat anti–mouse secondary antibodies for 1 h at 37°C, immobilized on 50 μg/ml polylysine-coated coverslips for 10 min, and then fixed and permeabilized as above. Cells were stained for caveolin and analyzed by confocal laser microscopy.
Immunoprecipitation and Blotting
Control or antisense caveolin clones were cultured for 8 h in serum-free DMEM containing 3 μM CdCl2 on either fibronectin (5 μg/ml), vitronectin (1 μg/ml), or polylysine (50 μg/ml) and lysed on ice for 30 min in buffer containing 50 mM Hepes (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM sodium orthovanadate, 1 mM phenylmethyl-sulfonylfluoride, and leupeptin (10 μg/ml). After preclearing with protein A–agarose, lysates were incubated with antibodies to β1 integrin (JB1A) or cortactin (Upstate Biotechnology Inc.) at 4°C overnight. In some experiments, the lysates were made 10 μM in the caveolin-related peptide, cav-1 (DGIWKASFTTFTVTKYWFYR), or the control cav-x peptide (WGIDKAFFTTSTVTYKWFRY), in 0.25% DMSO (Michel et al., 1997). The cav-x peptide has an identical amino acid composition and 60% identical sequence to the cav-1 peptide. The immunoprecipitates were blotted with caveolin antibody (Transduction Labs), stripped, and reblotted for Src family kinases (Santa Cruz Biotech), cortactin, or 4G10 (Upstate Biotechnology Inc.). To analyze FAK and EGF receptor activation, clones were induced with CdCl2 and plated on fibronectin- or polylysine-coated 100-mm dishes or their β1 integrins clustered with antibodies. Cells were lysed or stimulated with EGF (10 ng/ml; Upstate Biotechnology Inc.) for 5 min before lysing in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 1% deoxycholate, 0.1% SDS, 1% Triton X-100) supplemented with protease inhibitors. Lysates were immunoblotted for ErbB-2, -3, and -4 (Santa Cruz Biotech) and phosphotyrosine (Upstate Biotechnology Inc.). β1 immunoprecipitates from m1–m5 clones were immunoblotted with uPAR antibody 399R (American Diagnostica).
Kinase Assay
To measure total enzymatic activity of β1-associated Src kinases, induced cells were lysed in Triton X-100 buffer and immunoprecipitated with β1 antibody (JB1A). The immunocomplexes were suspended in 24 μl of reaction solution containing 50 mM Pipes, pH 7.0, 10 mM MnCl2, 5 μg of acid-denatured enolase, 10 μM ATP, 10 μCi of [γ-32P]ATP, and reacted at room temperature for 10 min. The kinase reaction was stopped by sample buffer, and samples were resolved on 10% SDS-PAGE and visualized by autoradiography.
For determination of basal activity of Src, induced cells were lysed in RIPA buffer and ∼500 μg of lysate immunoprecipitated with Src mAb (Calbiochem) and procedures described above followed.
Phospho-MAPK Assay
SMC were plated onto fibronectin- (5 μg/ml) or polylysine-coated (50 μg/ ml) dishes at 37°C for 20 min in serum-free DMEM/BSA with or without peptide 25 or 36 (75 μM). Cells were lysed in RIPA buffer and immunoblotted with phospho-specific MAPK antibody which detects p42 and p44 MAPK catalytically activated by phosphorylation at Tyr204/Thr202 (New England Biolabs). The membrane was stripped and reprobed for total MAPK protein as control.
Migration Assay
Passage 1–3 SMC (105) were seeded into Transwell (Costar; Becton Dickinson) inserts containing 8-μm polycarbonate filters precoated on the bottom with fibronectin, and then cultured overnight in serum-free DMEM containing BSA (5 mg/ml) with or without peptide 25 or 36 (50–100 μM). Cells on both sides of the filter were detached by trypsin-EDTA and counted. All assays were performed in triplicate and the data expressed as percent inhibition by the peptides.
Results
β1 Integrins and Caveolin Partially Colocalize
Because caveolin oligomers are relatively insoluble in nonionic detergents, the reported association between β1 integrins and caveolin could possibly be an artifact of cellular disruption by detergents. Therefore, the distribution of β1 integrins and caveolin in cells in situ was assessed by immunostaining. Primary cultures of human vascular SMC (Fig. 1, A and B) were examined because these cells express relatively high amounts of β1 integrins (red) and caveolin-1 (green). Confocal microscopic analysis indicated these two proteins had marked spotty or array-like colocalizations over the cell body of SMC grown on glass coverslips (Fig. 1 C). Substitution of isotype Ig controls for primary β1 and caveolin antibodies resulted in much weaker or no staining (Fig. 1 D) verifying the specificity of the immunostaining. Because β1 integrins and caveolin-1 only partially colocalize, we determined if β1 integrins and caveolin would cocluster. Clustering of SMC β1 integrins in suspension at 37°C resulted in the emergence of bright fluorescent signals of integrins (green) (Fig. 1 F) and caveolin (red) (Fig. 1 E) indicating clusters. Superimposition of the signals (Fig. 1 G) confirmed coclustering. No caveolin clustering was observed when the integrins were nonclustered (Fig. 1 H) or nonimmune Ig was substituted for the primary caveolin antibodies (Fig. 1 I). These images indicate that fractions of β1 integrins and caveolin colocalize and are physically linked in cells before cellular disruption with detergents and are consistent with the hypothesis that caveolin has a role in integrin function.
Figure 1.
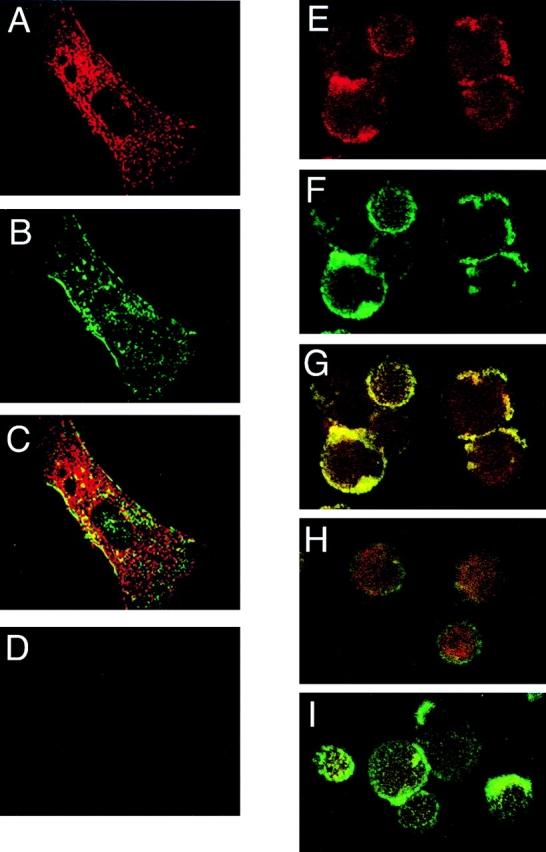
Caveolin and β1 integrins colocalize and cocluster. Early passage human SMC (A–C) were cultured in 10% FBS, stained with JB1A/K20 mAb to localize β1 integrins (A–C), and then fixed/permeabilized and stained with polyclonal caveolin antibodies. Secondary antibodies were then used to visualize optical planes of the cells by confocal microscopy: integrins (red), caveolin (green), and sites of colocalization (yellow). Substitution of isotype mouse Ig and rabbit IgG for primary antibodies resulted in much less or no immunostaining of SMC (D). SMC were also examined for integrin-induced coclustering with caveolin (E–G). SMC were stained in suspension with FITC-K20 antibodies to β1 integrins followed by clustering with secondary antibodies and then stained for caveolin. In this case integrin (green) and caveolin (red) fluorescent signals emerge and colocalize (E–G) with clustering in comparison with that of nonclustered cells (H) or clustered cells stained without primary caveolin antibodies (I).
Caveolin Depletion in 293 Cells Blocks β1 Integrin Function
To explore more directly the importance of caveolin to β1 integrin function, parental 293 cells were transfected with antisense full-length mRNA for caveolin-1 under the influence of an inducible murine metallothionein promoter. Several clones with inducible suppression of caveolin protein expression were identified and examined. After 12–18 h of induction in the presence of 3 μM cadmium, two clones with normal caveolin levels, 6 and 9 (Fig. 2 A), displayed strong adhesion to fibronectin (Fig. 2 B). By contrast, two representative antisense clones, 31 and 32, which had 15– 50% the level of caveolin in parental 293 cells (Fig. 2 A), displayed markedly impaired adhesion to fibronectin (Fig. 2 B). Although 293 cells have only weak vitronectin adhesion, mediated by αvβ5 and αvβ1, this adhesion was also abrogated in caveolin-deficient cells (not shown). As is also indicated in Fig. 2 A, there was substantially less caveolin in these clones even without induction, indicating leakage of antisense expression, which has been reported for this expression vector in previous studies (Lukashev et al., 1994). Adhesion assays mirrored the extent of caveolin depletion: although they were capable of spreading on fibronectin, clones 31 and 32 were less adhesive even in the absence of induction (Fig. 2 B). Nonetheless, clones 31 and 32 grew nearly as well as clones 6 and 9 over many months of passage. Flow cytometric analyses revealed no differences in surface level of β1 or αv or α5β1 heterodimers among these clones (Fig. 2 C), indicating that depletion of caveolin expression affected the function rather than expression of β1 or its dimerization with α5 or other integrins.
Figure 2.


Caveolin is required for β1 integrin–dependent adhesion to fibronectin. (A) Lysates of 293 cells (clones 6, 9, 31, and 32) expressing antisense caveolin before (−) and after (+) cadmium (CdCl2) induction were immunoblotted for caveolin-1 and reprobed for cortactin as a control (left). Relative caveolin and cortactin levels among the clones were quantified by densitometry (right) and confirmed in three separate experiments. (B) Clones 31 and 32 with marked depletion of caveolin (A) did not adhere to fibronectin, whereas clones 6 and 9 and control 293 cells adhered avidly. (C) FACS® analysis of β1, αv, and α5β1 integrin expression on control clone 6, and antisense clone 31 which has a markedly reduced caveolin level. B and C were performed three times with similar results. (D) Photographs of phase images (a and b) and paxillin immunostaining (c and d) of clones 6 and 31 after plating on fibronectin and subsequent induction with CdCl2.
Caveolin antisense clones also exhibited time-dependent changes in cell shape with decreasing caveolin expression. Clones 6 and 31 were allowed to spread onto fibronectin for 1 h at 37°C, and then antisense caveolin mRNA induced. Within 8 h, clone 31 cells, but not clone 6 cells, became less spread. Within 18 h, clone 31 cells became round (Fig. 2 D) and numerous cells actually detached from the matrix. The shape changes paralleled loss of focal adhesion sites as indicated by loss of focal paxillin accumulation on ventral surfaces (Fig. 2 D). In spite of the marked changes in adhesiveness and shape, most cells were found to respread and adhere better within hours of removing cadmium.
Caveolin Depletion in 293 Cells Does Not Impair Integrin Ligand Binding or Lateral Movement but Blocks Signaling through β1 Integrins
We next considered mechanisms by which caveolin could influence integrin function. Because integrin expression per se was not changed in caveolin-depleted cells (Fig. 2 C), the capacity of the various clones to bind 125I-fibronectin in the absence and presence of a stimulating antibody (TS2/16) known to promote fibronectin binding by the major fibronectin receptor, α5β1, was determined (Masumoto and Hemler, 1993). After induction, clones 6 and 31 were found to bind fibronectin equally at 4°C and exposure of the cells to TS2/16 enhanced fibronectin binding to a similar degree (Fig. 3 A), indicating that the affinity states of α5β1 in control and caveolin-depleted clones are similar. Further, when surface β1 integrins were cross-linked by antibodies, the β1 integrins of normal (clone 6) and caveolin-depleted cells (clones 31 and 32) clustered equally well as judged by confocal microscopic analysis (not shown). In spite of the capacity of β1 integrins on caveolin-depleted clones to engage ligand, the changes in cell shape and adhesiveness provoked by caveolin depletion (Fig. 2) could not be reversed by the stimulatory β1 integrin antibody (TS2/16). These data imply that a postreceptor binding event(s) important to adhesion, rather than binding per se, is most likely impaired in caveolin-depleted cells.
Figure 3.

Caveolin depletion does not block integrin clustering but abrogates integrin signaling through FAK. (A) Fibronectin binding to clones 6 (▵, ▴) and 31 (□, ▪) in the absence and presence of the stimulatory TS2/16 antibody. Data are shown as specific binding (mean ± SD of triplicate determinations) as a function of 125I-fibronectin concentration. Similar results were seen in three separate experiments. (B) Clones 6 and 31 after induction and clustering were immunoprecipitated with FAK antibody. The immune complexes were analyzed for tyrosine phosphorylation by antiphosphotyrosine antibody (4G10) (top). The same membrane was stripped and blotted for FAK (bottom). (C) Clones 6 and 31 were induced and allowed to adhere on either polylysine (PL) or fibronectin (Fn) for 40 min before being analyzed for FAK phosphorylation. (D) Clones 6 and 31 were induced and allowed to adhere on fibronectin. Cells were stimulated with EGF for 5 min and lysed and immunoblotted for tyrosine-phosphorylated ErbB-2 (top). The membrane was then blotted for total ErbB-2. EGF binding to ErbB-2/ErbB-3 heterodimers in epithelial cells was reported recently (Pinkas-Kramarski et al., 1998). All of the experiments involving cell lysates (Figs. 3–6) were performed three to four times with similar results.
Src family tyrosine kinases are required for fibronectin receptor α5β1 and vitronectin receptor αvβ5-mediated cell adhesion (Bergman et al., 1995; Kaplan et al., 1995). Moreover, tyrosine phosphorylation of FAK and other signaling molecules important to cytoskeletal reorganization and cell spreading after ligand engagement of β1 integrins may depend on interactions between FAK and Src family kinases (Parsons, 1996; Schlaepfer and Hunter, 1997). Because caveolin has been shown previously to interact with c-Src (Li et al., 1996 a,b), it is possible that integrin-mediated tyrosine phosphorylation depends on caveolin. To test this hypothesis, we initially treated 293 cells with the tyrosine kinase inhibitor genestein (100 μM) and noted that 293 cells failed to attach and spread on fibronectin (data not shown). When β1 integrins were clustered with antibodies or 293 cells were allowed to adhere to fibronectin, increased FAK phosphorylation was apparent (Fig. 3, B and C). In contrast, 293 cells with suppressed caveolin expression failed to exhibit FAK phosphorylation whether in response to antibody-induced clustering or ligand binding. This defect was not a general defect in tyrosine phosphorylation as autophosphorylation of ErbB-2, an EGF receptor–like tyrosine kinase, after binding of EGF, was not different between control and caveolin-deficient clones (Fig. 3 D).
The β1 integrins were next examined for interaction with nonreceptor tyrosine kinases by coprecipitation. Caveolin depletion of 293 cells (clones 31 and 32) resulted in loss of caveolin and Src family kinases in the β1 immunoprecipitates (Fig. 4 A). The antibody used in this experiment was a pan-Src antibody recognizing multiple members of the src gene family. In additional experiments using specific antibodies we found c-Src, Fyn, and Yes in the β1 immunoprecipitates, all of which were almost completely lost in the β1 immunoprecipitates of caveolin-depleted cells (not shown). A major substrate for Src kinase, cortactin, implicated in adhesion and migration (Vuori and Ruoslahti, 1995), was also markedly depleted in these precipitates. In situ kinase assays of β1 immunoprecipitates confirmed markedly reduced activity in cells (clones 31 and 32) depleted of caveolin (Fig. 4 B).
Figure 4.

Caveolin depletion leads to loss of integrin-associated Src family kinases. (A) The top panel shows immunoprecipitates of β1 integrins of antisense caveolin 293 cells immunoblotted sequentially for caveolin, cortactin, Src family kinases, and β1 integrin. The bottom panel shows immunoprecipitates of cortactin in Cd-induced 293 cells sequentially immunoblotted for phosphotyrosine, Src family kinases (pan-Src Ab), and cortactin. (B) β1 integrin immunoprecipitates were assayed in situ for total Src family tyrosine kinase activity (top). The bottom panel shows the relative amounts of integrin precipitated in each sample.
Although little or no Src family kinase or cortactin associates with β1 integrins in caveolin-depleted cells (Fig. 4 A), this was not because tyrosine kinases could not be activated in these cells. Rather, kinase assays of immunoprecipitated c-Src confirmed a two- to fourfold increase in total c-Src activity in the caveolin-deficient clones (not shown). Previously, caveolin levels have been linked inversely to cellular Src kinase activity (Li et al., 1996a). Enhanced total kinase activity correlated with a striking hyperphosphorylation of cortactin in the caveolin-depleted clones (31 and 32) compared with the control clone 6 (Fig. 4 A). Immunoblots of the cortactin immunoprecipitates revealed increased Src family kinases associated with tyrosine-phosphorylated cortactin, possibly through Src SH2 domain (Thomas and Brugge, 1997). These data indicate that caveolin-deficient 293 cells have dysregulated Src family kinase activity and little or no tyrosine kinase activity associated with their β1 integrins.
Caveolin Depletion Disrupts uPAR/Integrin Complexes and Abrogates uPAR-mediated Adhesion to Vitronectin
uPAR forms complexes with β1 and β2 integrins and such complexes are important to uPAR-dependent adhesion to matrix vitronectin via a vitronectin binding site on uPAR (Wei et al., 1996). Caveolin is also found in uPAR/β1 integrin complexes. To explore the functional importance of caveolin to these complexes, we created stable cotransfectants of 293 cells expressing both uPAR and the antisense full-length mRNA for caveolin-1. Several clones with inducible suppression of caveolin expression were again identified. Control clones expressing uPAR but without reduction in caveolin levels adhered avidly to vitronectin (Fig. 5 A). Clones with <50% the normal level of caveolin protein (clones m2–m5) adhered weakly or not at all to vitronectin (Fig. 5 A) even though uPAR and β1 integrin surface expression was not changed as judged by FACS® analysis (data not shown). Immunoprecipitation of β1 integrins coprecipitated uPAR in proportion to caveolin levels and the ability of the cells to adhere to vitronectin (Fig. 5 A), indicating that caveolin is required for stable uPAR/ integrin complexes and vitronectin adhesion. As with nontransfected 293 cells (Fig. 4), depletion of caveolin in uPAR/293 cells resulted in almost complete loss of Src family kinases from the β1 integrin immunoprecipitates (Fig. 5 B). As expected, the caveolin-depleted uPAR/293 cells also failed to adhere to fibronectin (not shown).
Figure 5.

Caveolin is required for uPAR/β1 integrin complexes and uPAR-dependent adhesion to vitronectin. (A) The bottom panel shows uPAR/293 cells expressing varying levels of antisense caveolin (clones m1–m5) tested for adhesion to vitronectin as described in Materials and Methods. The corresponding decrease in caveolin levels for each of the clones, as judged by densitometry of immunoblots, was as follows: m1, 0% decrease; m2, 83%; m3, 89%; m4, 96%; and m5, 73%. The top panel shows β1 integrins of uPAR/293 cells cotransfected with antisense caveolin (clones m1–m5) immunoprecipitated and immunoblotted for relative amounts of uPAR as quantified by densitometry. (B) Immunoprecipitates of β1 integrins sequentially immunoblotted for caveolin, Src, and cortactin as in Fig. 4. (C) uPAR-expressing or uPAR and caveolin–coexpressing 293 cells were detached and allowed to adhere on vitronectin (Vn) fibronectin (Fn)-coated 96-well plates. After washing, the percentage of adherent cells was quantitated by spectrophotometric absorbance of stained cells. Adhesion assays were performed three times with identical results.
In prior work, we observed that incorporation of α5/β1 integrins into complexes with uPAR, while stabilizing caveolin/integrin interactions, inhibited the ability of these integrins to bind fibronectin. Indeed, uPAR-transfected 293 cells had impaired fibronectin adhesion (Wei et al., 1996). In light of the critical role for caveolin in 293 cell adhesion to fibronectin (Fig. 2) and the fact that 293 cells have relatively low levels of caveolin, we considered the possibility that uPAR expression also inhibited β1 integrin function indirectly by sequestering caveolin into uPAR/ integrin complexes. Therefore, we transfected uPAR/293 cells with wild-type caveolin-1 and assessed the capacity of these transfectants to adhere to vitronectin and fibronectin. Concurrent caveolin overexpression in uPAR/293 cells had no effect on uPAR-dependent adhesion to vitronectin but completely restored the capacity of these cells to adhere to fibronectin (Fig. 5 C). These data suggest competition for limited caveolin between unbound β1 integrin and uPAR/β1 integrin complexes may largely explain the impaired fibronectin adhesion of uPAR-transfected 293 cells.
Disruption of β1 Integrin/Caveolin Complexes with Caveolin and uPAR-Binding Peptides
To explore further the functional connection between caveolin and uPAR in assembly of integrin-associated signaling molecules, lysates of uPAR/293 cells were immunoprecipitated for β1 integrins in the presence of either a peptide (peptide 25) (Wei et al., 1996) which disrupts uPAR/integrin associations or a peptide derived from the “scaffolding domain” of caveolin-1 (cav-1). In experiments using purified proteins, cav-1 disrupts self-oligomerization of caveolin and blocks direct interactions between caveolin and other proteins, including c-Src (Li et al., 1996a; Michel et al., 1997). Both peptide 25 and cav-1 blocked coprecipitation of caveolin with β1 integrins in lysates prepared from either uPAR/293 cells or primary human vascular SMC (Fig. 6 A). Both peptides, but not inactive control peptides, also blocked coprecipitation of Src family kinases and cortactin with β1 integrins (Fig. 6 A), verifying that the stable association of these cytoplasmic proteins with integrins is dependent upon the membrane proteins caveolin and uPAR in these cells. Neither control peptide (peptide 36 and cav-x) had any effect on caveolin, cortactin, or Src coprecipitation with β1 integrins when compared with a no peptide control (Fig. 6 A). Further, peptide 25 had no effect on coprecipitation of caveolin (or Src kinases) with β1 integrins in nontransfected 293 cells (not shown), confirming the specificity of this peptide for uPAR.
Figure 6.

Disruption of β1 integrin/uPAR/caveolin complexes in SMC leads to loss of integrin-associated Src kinases. (A) β1 integrins of uPAR/293 cells and SMC were immunoprecipitated in the presence of 100 μM of either peptide 25 or 36, 10 μM cav-1, or the control cav-x peptide, then sequentially immunoblotted for caveolin, cortactin, Src family kinases, and β1 integrins. (B) Intact SMC were incubated with 75 μM peptide 25 or control 36 and then immunoprecipitated with β1 integrin antibodies and the precipitates immunoblotted sequentially for Src kinases and β1 integrins. SMC were also pretreated with peptides as above and plated onto polylysine (PL) or fibronectin (Fn) for 30 min at 37°C. Lysates were immunoblotted for total MAPK and phosphorylated MAPK as an indicator of the active kinase. (C) SMC were placed in the upper chambers of Transwell inserts undercoated with fibronectin in the presence of peptide 25 or 36 at concentrations indicated in the figure. SMC on the filter bottom were counted after 24 h. The assay was done three times and the data are expressed as mean (± SD) percent inhibition by either peptide.
Vascular SMC are known to express uPAR and β1 integrins (Clyman et al., 1992; Okada et al., 1995) and we confirmed uPAR expression by immunostaining. Clustering of β1 integrins resulted in uPAR coclustering, though no uPAR was detectable in these β1 immunoprecipitates by immunoblotting (data not shown). Although complex formation of β1 integrins with uPAR could be expected to inhibit fibronectin adhesion (Wei et al., 1996), SMC attach and spread well on fibronectin, mostly via α5β1. As discussed above, this may be explained by the relatively high levels of caveolin in SMC as compared with 293 cells. Nonetheless, peptide 25 but not a control peptide, was found to disrupt caveolin and Src kinase associations with β1 integrins almost completely in lysates of SMC (Fig. 6 A). Therefore, we used this peptide to explore the role of caveolin in the integrin function of intact SMC. Incubation of SMC adherent to fibronectin with peptide 25 before lysis resulted in loss of Src kinases (as well as caveolin) in the β1 immunoprecipitates (Fig. 6 B). Consistent with the loss of signaling molecules, integrin-dependent MAPK activation was also blocked by peptide 25 (Fig. 6 B). It should be noted that the pathway of integrin and uPAR– dependent MAPK activation in these cells is not yet defined. Although MAPK is reproducibly phosphorylated within 30 min of fibronectin attachment, we can detect no Shc or FAK phosphorylation (not shown). The inhibitory effects of the peptide on association of Src family kinases with integrins was correlated with loss of integrin function. Although neither peptide blocked adhesion, peptide 25 delayed SMC spreading on fibronectin and blocked migration of human SMC across microbore filters to fibronectin-coated surfaces (Fig. 6 C), consistent with its effect on integrin signaling.
Discussion
Expression of the capacity of integrins to mediate cellular adhesion has several distinct facets: (a) the conformational state of integrins, affecting the affinity of ligand binding; (b) ligand-induced integrin clustering, enhancing the strength of attachment and promoting interactions between kinases and their substrates important to signal transduction; and (c) signaling itself, activating a cascade of events leading to organization of the cytoskeleton and cell spreading (Schwartz et al., 1995; Burridge and Chrzanowska-Wodnicka, 1996; Parsons, 1996). Numerous in vitro studies and recent evaluations of mice with targeted deletions in nonreceptor tyrosine kinases implicate FAK and Src family kinases as key mediators of integrin signaling (Furata et al., 1995; Ilic et al., 1997; Meng and Lowell, 1998). Thus, the molecular events leading to assembly and activation of these kinases surrounding integrins are important determinants of integrin function. This conclusion has focused much attention on nonintegrin membrane proteins which might regulate this process and our data identify one such protein as caveolin. While caveolin does not affect integrin expression or the intrinsic capacity of β1 integrins to bind fibronectin (Figs. 2 and 3), caveolin is required for the normal assembly of adhesion plaques which develop in response to ligand engagement and integrin clustering. In the absence of sufficient β1 integrin–associated caveolin, there is loss of integrin-associated Src kinase activity, little or no FAK activation after ligand binding, and impaired association of integrins with structural proteins such as tyrosine-phosphorylated cortactin important to β1 integrin– dependent adhesion (Fig. 4). As a result, the characteristic accumulation of enzymes and structural proteins which comprise integrin-dependent adhesion sites fails to develop (Fig. 2).
Our results confirm and extend the recent findings of Wary and colleagues (1998) that caveolin expression is required for the association of Fyn kinase with β1 integrins and ligand-dependent Shc phosphorylation. These investigators began with cells (Fisher rat thyroid cells) expressing little or no caveolin or α5 integrin and transfected both proteins. They found that physical association of the Src family kinase Fyn with α5/β1 required concurrent coexpression of caveolin-1. Fyn but not Src was required for α5/β1-dependent Shc phosphorylation. Beginning with cells having functional β1 integrins and expressing caveolin-1, we suppressed caveolin expression and found β1 integrin association with several Src family kinases to be completely dependent on caveolin. In both cases, deficient β1 integrin–associated Src family kinase activity led to correspondingly marked changes in integrin function. Together, these findings indicate caveolin is a general regulator of β1 integrin function.
How does caveolin promote integrin signaling? Although our understanding is incomplete, we favor a model (Fig. 7) in which caveolin functions primarily to sequester Src family kinases in an inactive configuration at sites proximate to integrins, promoting their presentation to integrins and activation during ligand-induced integrin clustering. Caveolin has been shown previously to interact predominantly with the inactive form of Src and to reduce Src activity when overexpressed in 293 cells, perhaps by sequestering Src kinases from activating phosphatases and substrates (Li et al., 1996b; Rodgers and Rose, 1996). Consistent with this model, caveolin-deficient 293 cells were found to have two- to fourfold higher levels of basal c-Src kinase activity (not shown) and to exhibit striking hyperphosphorylation of a major Src substrate, cortactin (Fig. 4). If one function of caveolin is to suppress basal Src kinase activity, the model in Fig. 7 also offers a possible molecular explanation of how caveolin contributes to Src family kinase activation as a consequence of integrin clustering. Caveolin interactions with Src family kinases are thought to occur through the same membrane proximal sites on caveolin as those involved in formation of caveolin homo-oligomers (Okamoto et al., 1998). Because coclustering of caveolin with integrins (Fig. 1) could be expected to favor caveolin homo-oligomer formation, integrin clustering is likely to modify the physical interaction of caveolin with Src kinases. This may lead to outright release of these kinases from caveolin (as shown in Fig. 7) and/or an altered, more accessible position of these kinases in the cluster favoring their activation through contact with substrate or through a phosphatase. In either case, these studies directly implicate caveolin function in the membrane proximate events of ligand-induced, β1 integrin–dependent tyrosine kinase activation. As caveolin levels are reportedly low in a number of transformed cells (Koleske et al., 1995; Okamoto et al., 1998), our data raise the possibility that low caveolin levels contribute to the defective fibronectin adhesion and matrix assembly seen in many cancer cells and thought to be important to their metastatic potential (Hynes, 1992; Schwartz, 1997).
Figure 7.

Model for regulation of β1 integrin signaling by caveolin. The model indicates that caveolin binds and maintains Src family kinases in an inactive configuration (dark shade) proximate to a fraction of β1 integrins (complex is encircled). Whether these complexes exists in caveolae as signaling units or separately from caveolae is currently unknown (see text). Ligand engagement of integrins and clustering of caveolin along with integrins results in homo-oligomerization of caveolin and release of Src kinases from caveolin. This in turn allows Src kinases to activate (light shade) through a phosphatase and/or through direct binding to substrates at the developing focal contact site. Available evidence suggests that Fyn kinase may primarily function in MAPK activation whereas Src/Yes may primarily function to activate FAK and phosphorylate other cytoskeletal proteins leading to the organization and development of an adhesion plaque. The model also proposes that as integrins activate in response to ligand-induced clustering, uPAR moves into the complexes enriching them with caveolin and its associated signaling molecules. The importance of uPAR, when present, to the complexes is illustrated by the ability of uPAR-binding peptides to disrupt signaling through β1 integrins in SMC (Fig. 6).
A further implication of the studies reported here and the recent studies of Wary and colleagues (1998) is that Fyn and Src may have overlapping but distinct roles in β1 integrin signaling: Fyn being primarily required for Shc phosphorylation and activation of an MAPK-dependent growth pathway and Src being primarily required for FAK and cytoskeletal phosphorylation and assembly of focal adhesion sites. This concept is supported by results of prior studies examining mice deficient in the negative regulator of Src family kinase activity, Csk (Thomas et al., 1995). Fibroblasts from Csk− embryos exhibit hyperphosphorylation of adhesion sites and striking hyperphosphorylation of cortactin, reminiscent of that seen in caveolin-deficient cells (Fig. 4). The hyperphosphorylation of adhesion sites and cortactin was largely corrected by crossing the Csk− mice with Src− mice but not by crossing with Fyn− mice, again implicating Src as primarily being involved in regulation of integrin-dependent adhesion. Clearly this is an oversimplification since we also observe Yes as well as Fyn and Src in β1 integrin/caveolin complexes and even kinase-inactive Src can promote focal contact assembly in Src− cells (Kaplan et al., 1995). Still, these recent studies support the possibility that different members of the src gene family have favored roles in integrin signaling even in cells expressing multiple family members.
The structural basis for the association between caveolin and β1 integrins is uncertain. First, most if not all cellular caveolin is reportedly found in purified caveolae (Lisanti et al., 1994; Schnitzer et al., 1995). Although integrins have not been observed in purified caveolae, when we subject 293 cells to mechanical lysis, sonicate, and centrifuge the postnuclear supernatant through a sucrose gradient, caveolin in 293 cells appears at the 5/35% sucrose interface, typical for that reported for caveolae preparations in 293 and other cells (Song et al., 1996; Ikezu et al., 1998). Almost all of the β1 integrins appear in the fractions containing caveolin (Yang, X., unpublished observation). These findings are consistent with the scenario that β1 integrin/ caveolin/Src family kinase complexes (encircled in Fig. 7) exist as signaling units in caveolae and separate from caveolae in response to ligand-induced clustering and cytoskeletal reorganization. However, the exact composition of caveolae remains controversial and appears strongly dependent on the method of purification (Lisanti et al., 1994; Schnitzer et al., 1995; Mineo et al., 1996; Stan et al., 1998). An alternative possibility is that some fraction of caveolin traffics outside caveolae, as has been argued by Wary and colleagues (1998). This will be an important issue for future experiments. In either case, the fraction of β1 integrins which colocalize and cocluster with caveolin (Fig. 1) appears to be critical to β1 integrin signaling.
Whether β1 integrins directly bind caveolin is also uncertain. Wary and colleagues (1998) reported that the α5 transmembrane domain was required for coprecipitation of α5/β1 and caveolin. However, no direct binding has been demonstrated. Most if not all other proteins that are reported to bind caveolin bind sequences of caveolin that are membrane proximal but in the cytoplasmic compartment (Okamoto et al., 1998). Indeed, in SMC we observed a peptide comprised of a membrane proximal region of caveolin (amino acids 82–101) blocking coprecipitation of caveolin with β1 integrins (Fig. 5). Thus, it is uncertain whether β1 integrins and caveolin directly bind or associate indirectly through a common affinity for certain membrane lipids or a third protein such as uPAR which may directly interact both with integrins and cholesterol-rich lipid domains enriched in caveolin. The physical basis for the association of β1 integrins and caveolin also requires further investigation.
The marked inhibition of β1 integrin function by uPAR-binding peptides in human SMC (Fig. 6) is remarkable. Although we could detect uPAR in SMC by immunostaining and could cocluster uPAR along with β1 integrins, no uPAR was detected in the β1 immunoprecipitations of SMC. This suggests the association of uPAR with β1 integrins and caveolin in these cells is relatively weak. There also appears to be a stoichiometric excess of β1 integrins compared with uPAR in SMC. Thus, it is surprising that the coprecipitation of most caveolin- and integrin-associated Src kinases with β1 integrins would be blocked by peptide 25, raising the possibility of an in vitro artifact. This is an unlikely explanation for the findings because the uPAR-binding peptide (peptide 25), but not two control peptides (peptide 36 and a scrambled version of peptide 25), also had clear biochemical and functional effects on intact SMC. Prior treatment of SMC with the active peptide blocked Src kinase association with β1 integrins and inhibited both signaling and migration of the cells on fibronectin (Fig. 6). These results indicate that, although not intrinsically required for the function of integrin/caveolin (Fig. 2), the presence of uPAR organizes caveolin and its associated signaling molecules in an interdependent manner such that integrins, caveolin, and uPAR form a unit promoting integrin function. Perhaps this is possible because only a fraction of β1 integrins associates with caveolin (Fig. 1) and uPAR associates mainly with activated integrins (Wei et al., 1996). As indicated in Fig. 7, integrin activation initiated by ligand-induced integrin clustering may promote formation of these signaling complexes. Similar mechanisms may account for observations that uPAR is required for normal function of the β2 integrins, CD11/ CD18 (Mac-1; Simon et al., 1996; Sitrin et al., 1996; May et al., 1998), and for migration on vitronectin mediated by αvβ5 (Yebra et al., 1996), suggesting a promoting effect of uPAR on integrin function may be a widespread phenomenon.
Acknowledgments
The authors thank Martin Hemler for helpful discussions and Thomas Michel for providing cav-1 and cav-x peptides.
This work was supported by a grant from the National Institutes of Health (HL44712) to H.A. Chapman.
Abbreviations used in this paper
- FAK
focal adhesion kinase
- MAPK
mitogen-activated protein kinase
- SMC
smooth muscle cells
- uPAR
urokinase receptor
Footnotes
Y. Wei and X. Yang contributed equally to this work.
References
- Berditchevski F, Zutter MM, Hemler ME. Characterization of novel complexes on the cell surface between integrins and proteins with 4 transmembrane domains (TM4 proteins) Mol Biol Cell. 1996;7:193–207. doi: 10.1091/mbc.7.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman M, Joukov V, Virtanen I, Alitalo K. Overexpressed Csk tyrosine kinase is localized in focal adhesions, causes reorganization of αvβ5 integrin, and interferes with HeLa cell spreading. Mol Cell Biol. 1995;15:711–722. doi: 10.1128/mcb.15.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–519. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Clyman RI, Mauray F, Kramer RH. Beta1 and beta3 integrins have different roles in the adhesion and migration of vascular smooth muscle cells on extracellular matrix. Exp Cell Res. 1992;200:272–284. doi: 10.1016/0014-4827(92)90173-6. [DOI] [PubMed] [Google Scholar]
- Fenczik CA, Sethi T, Ramos JW, Hughes PE, Ginsberg MH. Complementation of dominant suppression implicates CD98 in integrin activation. Nature. 1997;390:81–85. doi: 10.1038/36349. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Ilic D, Kanazawa S, Takeda N, Yamamoto T, Aizawa S. Mesodermal defect in late phase of gastrulation by a targeted mutation of focal adhesion kinase, FAK. Oncogene. 1995;11:1989–1995. [PubMed] [Google Scholar]
- Harder T, Simons K. Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Curr Opin Cell Biol. 1997;9:534–542. doi: 10.1016/s0955-0674(97)80030-0. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Ikezu T, Trapp BD, Song KS, Schlegel A, Lisanti MP, Okamoto T. Caveolae, plasma membrane microdomains for α-secretase-mediated processing of the amyloid precursor protein. J Biol Chem. 1998;273:10485–10495. doi: 10.1074/jbc.273.17.10485. [DOI] [PubMed] [Google Scholar]
- Ilic D, Damsky CH, Yamamoto T. Focal adhesion kinase: at the crossroads of signal transduction. J Cell Sci. 1997;110:401–407. doi: 10.1242/jcs.110.4.401. [DOI] [PubMed] [Google Scholar]
- Kaplan KB, Swedlow JR, Morgan DO, Varmus HE. c-Src enhances the spreading of Src−/− fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 1995;9:1505–1517. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- Koleske AJ, Baltimore D, Lisanti MP. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci USA. 1995;92:1381–1385. doi: 10.1073/pnas.92.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Okamoto T, Chun M, Sargiacomo M, Casanova JE, Hansen SH, Nishimoto I, Lisanti MP. Evidence for a regulated interaction between heterotrimeric G proteins and caveolin. J Biol Chem. 1995;270:15693–15701. doi: 10.1074/jbc.270.26.15693. [DOI] [PubMed] [Google Scholar]
- Li S, Couet J, Lisanti MP. Src tyrosine kinases, G alpha subunits, and H-ras share a common membrane-anchored scaffolding protein, caveolin. J Biol Chem. 1996a;271:29182–29190. doi: 10.1074/jbc.271.46.29182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Seitz R, Lisanti MP. Phosphorylation of caveolin by Src tyrosine kinases. J Biol Chem. 1996b;271:3863–3868. [PubMed] [Google Scholar]
- Lindberg FP, Gresham HD, Schwarz E, Brown EJ. Molecular cloning of integrin-associated protein: an immunoglobulin family member with multiple membrane-spanning domains implicated in αvβ3-dependent ligand binding. J Cell Biol. 1993;123:485–496. doi: 10.1083/jcb.123.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisanti MP, Scherer PE, Vidugiriene J, Tang Z, Hermanowski-Vosatka A, Tu YH, Cook RF, Sargiacomo M. Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol. 1994;126:111–126. doi: 10.1083/jcb.126.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashev ME, Sheppard D, Pytela R. Disruption of integrin function and induction of tyrosine phosphorylation by the autonomously expressed β1 integrin cytoplasmic domain. J Biol Chem. 1994;269:18311–18314. [PubMed] [Google Scholar]
- Masumoto A, Hemler ME. Multiple activation states of VLA-4. Mechanistic differences between adhesion to CS1/fibronectin and to vascular cell adhesion molecule-1. J Biol Chem. 1993;268:228–234. [PubMed] [Google Scholar]
- May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT. Urokinase receptor (CD87) regulates leukocyte recruitment via β2 integrins in vivo. J Exp Med. 1998;188:1029–1037. doi: 10.1084/jem.188.6.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Lowell CA. A β1 integrin signaling pathway involving Src-family kinases, Cbl and PI-3 kinase is required for macrophage spreading and migration. EMBO (Eur Mol Biol Organ) J. 1998;17:4391–4403. doi: 10.1093/emboj/17.15.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel JB, Feron O, Sase K, Prabhakar P, Michel T. Caveolin versus calmodulin: counterbalancing allosteric modulators of endothelial nitric oxide synthase. J Biol Chem. 1997;272:25907–25912. doi: 10.1074/jbc.272.41.25907. [DOI] [PubMed] [Google Scholar]
- Mineo C, James GL, Smart EJ, Anderson RGW. Localization of epidermal growth factor-stimulated ras/raf1 interaction to caveolae membrane. J Biol Chem. 1996;271:11930–11935. doi: 10.1074/jbc.271.20.11930. [DOI] [PubMed] [Google Scholar]
- Okada SS, Tomaszewski JE, Barnathan ES. Migrating vascular smooth muscle cells polarize cell surface urokinase receptors after injury in vitro. . Exp Cell Res. 1995;217:180–187. doi: 10.1006/excr.1995.1077. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- Parsons JT. Integrin-mediated signalling: regulation by protein tyrosine kinases and small GTP-binding proteins. Curr Opin Cell Biol. 1996;8:146–152. doi: 10.1016/s0955-0674(96)80059-7. [DOI] [PubMed] [Google Scholar]
- Parton RG. Caveolae and caveolins. Curr Opin Cell Biol. 1996;8:542–548. doi: 10.1016/s0955-0674(96)80033-0. [DOI] [PubMed] [Google Scholar]
- Pinkas-Kramarksi R, Lenferink AE, Bacus SS, Lyass L, van de Poll ML, Klapper LN, Tzahar E, Sela M, van Zoelen EJ, Yarden Y. The oncogenic ErbB-2/ErbB-3 heterodimer is a surrogate receptor of the epidermal growth factor and betacellulin. Oncogene. 1998;16:1249–1258. doi: 10.1038/sj.onc.1201642. [DOI] [PubMed] [Google Scholar]
- Rodgers W, Rose JK. Exclusion of CD45 inhibits activity of p56lck associated with glycolipid-enriched membrane domains. J Cell Biol. 1996;135:1515–1523. doi: 10.1083/jcb.135.6.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E, Reed JC. Anchorage dependence, integrins, and apoptosis. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hunter T. Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to Erk2/mitogen-activated protein kinase through interactions with and activation of c-src. J Biol Chem. 1997;272:13189–13195. doi: 10.1074/jbc.272.20.13189. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE, McIntosh DP, Dvorak AM, Liu J, Oh P. Separation of caveolae from associated microdomains of GPI-anchored proteins. Science. 1995;269:1435–1439. doi: 10.1126/science.7660128. [DOI] [PubMed] [Google Scholar]
- Schwartz MA. Integrins, oncogenes, and anchorage independence. J Cell Biol. 1997;139:575–578. doi: 10.1083/jcb.139.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- Simon DI, Rao NK, Xu H, Wei Y, Majdic O, Ronne E, Kobzik L, Chapman HA. Mac-1 (CD11b/CD18) and the urokinase receptor (CD87) form a functional unit on monocytic cells. Blood. 1996;88:3185–3194. [PubMed] [Google Scholar]
- Sitrin RG, Todd RF, III, Petty HR, Brock TG, Shollenberger SB, Albrecht E, Gyetko MR. The urokinase receptor (CD87) facilitates CD11b/CD18-mediated adhesion of human monocytes. J Clin Invest. 1996;97:1942–1951. doi: 10.1172/JCI118626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SK, Li SW, Okamoto T, Quilliam LA, Sargiacomo M, Lasanti MP. Co-purification and direction interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. J Biol Chem. 1996;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- Stan RV, Roberts WG, Predescu D, Ihida K, Saucan L, Ghitescu L, Palade GE. Immunoisolation and partial characterization of endothelial plasmalemmal vesicles (caveolae) Mol Biol Cell. 1998;8:595–605. doi: 10.1091/mbc.8.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Soriano P, Imamoto A. Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature. 1995;376:267–271. doi: 10.1038/376267a0. [DOI] [PubMed] [Google Scholar]
- Vuori K, Ruoslahti E. Tyrosine phosphorylation of p130cas and cortactin accompanies integrin-mediated cell adhesion to extracellular matrix. J Biol Chem. 1995;270:22259–22262. doi: 10.1074/jbc.270.38.22259. [DOI] [PubMed] [Google Scholar]
- Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Wary KK, Mariotti A, Zurzolo C, Giancotti FG. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 1998;94:625–634. doi: 10.1016/s0092-8674(00)81604-9. [DOI] [PubMed] [Google Scholar]
- Wei Y, Lukashev ME, Simon DI, Bodary SC, Rosenberg S, Doyle MV, Chapman HA. Regulation of integrin function by the urokinase receptor. Science. 1996;273:1551–1555. doi: 10.1126/science.273.5281.1551. [DOI] [PubMed] [Google Scholar]
- Wilkins JA, Li A, Ni H, Stupack DG, Shen C. Control of β1 integrin function. J Biol Chem. 1996;271:3046–3051. [PubMed] [Google Scholar]
- Yebra M, Parry GCN, Stromblad S, Mackman N, Rosenberg S, Mueller BM, Cheresh DA. Requirement of receptor-bound urokinase-type plasminogen activator for integrin alphav beta5-directed cell migration. J Biol Chem. 1996;271:29393–29399. doi: 10.1074/jbc.271.46.29393. [DOI] [PubMed] [Google Scholar]