

Abstract
We previously demonstrated contrasting roles for integrin α subunits and their cytoplasmic domains in controlling cell cycle withdrawal and the onset of terminal differentiation (Sastry, S., M. Lakonishok, D. Thomas, J. Muschler, and A.F. Horwitz. 1996. J. Cell Biol. 133:169–184). Ectopic expression of the integrin α5 or α6A subunit in primary quail myoblasts either decreases or enhances the probability of cell cycle withdrawal, respectively. In this study, we addressed the mechanisms by which changes in integrin α subunit ratios regulate this decision. Ectopic expression of truncated α5 or α6A indicate that the α5 cytoplasmic domain is permissive for the proliferative pathway whereas the COOH-terminal 11 amino acids of α6A cytoplasmic domain inhibit proliferation and promote differentiation. The α5 and α6A cytoplasmic domains do not appear to initiate these signals directly, but instead regulate β1 signaling. Ectopically expressed IL2R-α5 or IL2R-α6A have no detectable effect on the myoblast phenotype. However, ectopic expression of the β1A integrin subunit or IL2R-β1A, autonomously inhibits differentiation and maintains a proliferative state. Perturbing α5 or α6A ratios also significantly affects activation of β1 integrin signaling pathways. Ectopic α5 expression enhances expression and activation of paxillin as well as mitogen-activated protein (MAP) kinase with little effect on focal adhesion kinase (FAK). In contrast, ectopic α6A expression suppresses FAK and MAP kinase activation with a lesser effect on paxillin. Ectopic expression of wild-type and mutant forms of FAK, paxillin, and MAP/erk kinase (MEK) confirm these correlations. These data demonstrate that (a) proliferative signaling (i.e., inhibition of cell cycle withdrawal and the onset of terminal differentiation) occurs through the β1A subunit and is modulated by the α subunit cytoplasmic domains; (b) perturbing α subunit ratios alters paxillin expression and phosphorylation and FAK and MAP kinase activation; (c) quantitative changes in the level of adhesive signaling through integrins and focal adhesion components regulate the decision of myoblasts to withdraw from the cell cycle, in part via MAP kinase.
Keywords: MAP kinase, integrins, proliferation, FAK, paxillin
Cell proliferation and differentiation are governed by multiple stimuli including soluble growth factors, the extracellular matrix (Juliano and Haskill, 1993; Adams and Watt, 1993; Roskelly et al., 1995), and direct cell to cell interactions (Gumbiner, 1996). Whereas each of these signals uniquely regulates mitogenic responses and gene activity, the decision of a cell to proliferate, differentiate, or undergo apoptosis, for example, is an integrated response to its adhesive and growth factor environment (Schwartz and Ingber, 1994; Sastry and Horwitz, 1996). While the mechanisms by which growth factors produce mitogenic responses and regulate gene expression are becoming clearer, the pathways through which adhesive interactions modulate these responses are only beginning to emerge (reviewed in Howe et al., 1998).
Several studies demonstrate that integrins control proliferation and differentiation in numerous cell types (Varner et al., 1995; Watt et al., 1993). Clustering of integrins on the cell surface with ligand-coated microbeads induces focal adhesion-like structures that recruit numerous mitogenic signaling proteins to integrin receptors which include growth factor receptors (Plopper et al., 1995; Miyamoto et al., 1996), mitogen-activated protein (MAP)1 kinase, lipid second messengers, protein phosphatases, and small GTP-binding proteins (Miyamoto et al., 1995). Thus integrin-associated focal adhesions serve as signaling centers where adhesive and mitogenic pathways can integrate. Numerous physical interactions between integrins or focal adhesion components and mitogenic signaling proteins have been demonstrated. For example, integrins can interact with growth factor receptors through adaptor proteins like IRS-1 (Vuori and Ruoslahti, 1994) and shc (Mainiero et al., 1995; Wary et al., 1996). Focal adhesion kinase (FAK) can interact with PI 3-kinase (Chen and Guan, 1994) and with GRB2 (Schlaepfer et al., 1994). Through its interaction with GRB2, FAK potentially links integrin signaling to the ras/MAP kinase pathway.
Whereas these studies show a biochemical coupling between integrin and growth factor signaling pathways, the functional significance of these interactions in the context of the regulation of proliferation and differentiation is not well understood. MAP kinase stands out as a key point of convergence between integrin and growth factor pathways (Chen et al., 1994; Zhu and Assoian, 1995; Miyamoto et al., 1996; Renshaw et al., 1997) and is required for proliferation of most cells. However, mitogenic responses can be controlled by pathways that do not use MAP kinase (Olson et al., 1995; Klippel et al., 1998). In addition, MAP kinase can modulate other integrin-dependent cell responses including motility (Klemke et al., 1997) and integrin activation (Hughes et al., 1997) suggesting that its activation produces pleiotropic effects. Further, the focal adhesion proteins FAK and paxillin, which are phosphorylated in response to many soluble mitogenic stimuli (reviewed in Sastry and Horwitz, 1996) as well as in response to integrin engagement are likely to play an important role in integrin-growth factor synergy. Although recent studies indicate FAK plays a role in cell survival (Frisch et al., 1996; Hungerford et al., 1996; Ilic et al., 1998) and motility (Ilic et al., 1995; Cary et al., 1996; Gilmore and Romer, 1996), the role of FAK and paxillin, and variations in the level of their activation, in mitogenic signaling is not well understood.
We previously reported contrasting roles for integrin α subunits in proliferative signaling using myogenic differentiation as a model system (Sastry et al., 1996). Using ectopic expression of integrins in primary quail myoblasts we provided clear biological evidence that integrin α subunits uniquely alter the response of myoblasts to growth factors. We attributed this effect in part to perturbation of integrin α subunit ratios (on the order of a three- to fivefold increase in relative expression) which strikingly shifted the probability that a myoblast would either proliferate or withdraw from the cell cycle and initiate terminal differentiation. Ectopic expression of the α5 integrin enhanced the mitogenic response to favor a much increased probability of proliferation. In contrast, ectopic expression of the α6A integrin decreased the probability of continued proliferation and promoted differentiation. In addition, we also implicated the α subunit cytoplasmic domains in controlling proliferative versus differentiative signals through integrins.
In this study, we used ectopic expression of these two α subunits in primary skeletal muscle myoblasts as a convenient tool to drive either the proliferative or differentiative pathway through integrins. We used this approach to (a) assess the relative contribution of the individual α subunit cytoplasmic domains and (b) identify intracellular targets of integrins that modulate the probability of a myoblast to proliferate or withdraw from the cell cycle and initiate terminal differentiation. First, we demonstrate that the α subunit cytoplasmic domains indirectly regulate proliferative versus differentiative signals through the β1A cytoplasmic domain. The α5 cytoplasmic domain is permissive for proliferative signaling while a discrete region of the α6A cytoplasmic domain promotes cell cycle withdrawal. Furthermore, the β1A cytoplasmic domain is sufficient to initiate proliferative signals and inhibit differentiation. Second, we show that the ectopic α subunits differentially alter the expression and/or activation of FAK, paxillin, and MAP kinase. Ectopic expression of paxillin or CD2-FAK and their mutants recapitulate the effects of ectopic integrins on myoblast proliferation and differentiation. The effect of ectopic α5 or α6A on proliferation and differentiation can be reversed by altering the relative activity of MAP/erk kinase (MEK), an upstream activator of MAP kinase. These results suggest a model in which proliferative signaling occurs through the integrin β1A subunit which is modulated by the α subunit cytoplasmic domains. The level of signaling emanating from the β1A subunits controls the level of FAK, paxillin, or MAP kinase activation. Thus, in addition to changes in integrin ratios, quantitative changes in the level of focal adhesion signaling or MAP kinase activation shift the probability that a myoblast will proliferate or differentiate.
Materials and Methods
Primary Cell Culture
Primary myoblasts were isolated from pectoralis muscle of nine day Japanese quail embryos as previously described (Konigsberg, 1979). In brief, the breast muscle was dissected from the embryo and myoblasts were dissociated from muscle tissue with 0.1% dispase (Sigma Chemical Co.) in PBS. The cell suspension was filtered through a Sweeney filter; cells were seeded onto gelatin-coated tissue culture plates (0.1% gelatin in PBS). Myoblast cultures were maintained in complete myoblast medium (DMEM [Sigma Chemical Co.] containing 15% horse serum, 5% chick embryo extract, 1% pen/strep, and 1.25 mg/ml fungizone [GIBCO BRL]). Myoblasts were subcultured in trypsin-EDTA (0.06% trypsin, 0.02% EDTA) and used between passages 1 and 10.
Antibodies and Extracellular Matrix Ligands
The muscle α-actinin–specific mAb, 9A2B8, was kindly provided by D. Fishman (Cornell University, New York, NY) as a hybridoma supernatant. mAb VIF4, which recognizes the human α5 integrin extracellular domain was a gift of R. Isberg (Tufts University, Boston, MA). The chicken α6-specific polyclonal antibody, α6ex (de Curtis et al., 1991), was provided by L. Reichardt (University of California, San Francisco, CA). mAb 2B7 directed against the extracellular domain of the human α6 integrin (Shaw et al., 1993), was a gift of A. Mercurio (Harvard Medical School, Boston, MA). The mAb 165 is directed against paxillin (Turner et al., 1990). mAb 2A7 directed against FAK (Kanner et al., 1990) and the polyclonal Ab BC3 directed against FAK (Shaller et al., 1992) were gifts of T. Parsons (University of Virginia, Charlottesville, VA). The anti-FAK polyclonal antibody C-20, was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The anti-active MAPK polyclonal antibody, which specifically recognizes dually phosphorylated, activated MAPK, was purchased from Promega. The anti-MAPK mAb that recognizes erk-1, as well as the anti-phosphotyrosine antibodies, RC20H and PY20, were purchased from Transduction Laboratories. The anti-hemagglutinin (HA) mAb, 12CA5, was purchased from Boehringer Mannheim. The anti-human CD2 mAb, TS2/18.1.1 was purchased from the Developmental Studies Hybridoma Bank. TS2/16, a mAb against human β1 integrin (Hemler et al., 1984) was from M. Hemler (Dana-Farber Cancer Institute, Boston, MA). Antibody against the human IL2 receptor was purchased from Boehringer Mannheim. Poly-l-lysine was purchased from Sigma Chemical Co. Fibronectin was purified from human plasma by affinity chromatography as previously described (Ruoslahti et al., 1982). Laminin was isolated from murine Englebreth-Holm-Swarm sarcoma as previously described (Kleinman et al., 1982).
Expression Vectors
The human α5 cDNA in pRSVneo and the chicken α6 cDNA in pRSVneo were described previously (Sastry et al., 1996). The chicken α61044t truncation was constructed by first subcloning a 1.6-kb HindIII-SalI fragment of the chicken α6A DNA into M13 and then introducing an in-frame BclI site at amino acid position 1044 (de Curtis et al, 1991). Mutants were confirmed by restriction digestion of M13 clones with BclI and by single stranded DNA sequencing using the dideoxy-chain termination method according to the Sequenase™ protocol (United States Biochemical Corp.). An 800-bp BstXI-SalI fragment containing the mutation was subcloned into pRSVneoα6 partially digested with SalI and completely with BstXI. The human α6A and α6B cDNAs, in the expression plasmid pRc/ CMV (Shaw et al., 1993), were a generous gift of A. Mercurio (Harvard Medical School, Boston, MA). The pRSVneo-CH8β1 plasmid was constructed by subcloning a 1-kb HindIII fragment, containing the CH8 epitope tag, from the CH8β1 pBJ-1 construct received from Y. Takada (Scripps Research Institute, La Jolla, CA) (Takada and Puzon, 1993) into pRSVneoβ1 expression vector (Reszka et al., 1992). pRSVIL2R-α5 and pRSVIL2R-β1A were constructed by cloning an Nhe1-Xba1 fragment from pCMVIL2R-α5cyto or pCMVIL2R-β1A plasmids received from Susan LaFlamme (Albany Medical College, Albany, NY; Tahiliani et al., 1997) into the Xba1 site of the pRSVneo vector. Clones were screened for orientation by restriction digests. HA-tagged rat MEK1 and HA-tagged rat constitutively active (CA) MEK S218/220D in pCMVneo vector i.e., sodium-deoxycholate, sodium-pyrophosphate, sodium-orthorandate (Catling et al., 1995) were received from M. Weber (University of Virginia, Charlottesville, VA). The chicken paxillin cDNA, the Y118F, and the S188/ 190A mutants in the pcDNA3.0neo vector (Brown et al., 1997) were received from C. Turner. CD2FAK, CD2FAK(Y397F), and CD2FAK(K454R) in CDM8 vector (Chan et al., 1994) were received from A. Aruffo.
Transfection and Flow Cytometry
Cells were transiently or stably transfected using a liposome-DNA solution as previously described previously (Sastry et al., 1996). In brief, replicating myoblasts (passages 1–3) were plated on 60-mm tissue culture plates coated with 0.1% gelatin in complete myoblast medium for 16–20 h. Cells were incubated for 8–16 h in a solution of 8 μg of plasmid DNA and 50 μg of Lipofectamine (GIBCO BRL) in complete myoblast medium. Transfected myoblasts were either washed with DMEM, refed with myoblast medium, and analyzed for transient expression or were trypsinized and plated into selection medium on gelatin-coated tissue culture plates (myoblast medium containing 0.4 mg/ml G418; GIBCO BRL) for 7–12 d and then into myoblast medium containing 0.2 mg/ml G418 (maintenance medium). For transfections with CD2FAK constructs, in order to generate stable populations, myoblasts were cotransfected with a pRSVneo or a pEGFP-C1neo plasmid (Clontech, Palo Alto, CA) at 1:7 ratio (neo resistance gene:CD2FAKcDNA) and selected in G418 as previously described. The chicken α6, human α6A or α6B, and α61044t transfections were selected and maintained on laminin-coated (20 μg/ml) tissue culture plates. For coexpression of hα6A integrin and CD2-FAK cells were cotransfected with pRc/CMVhα6A and CDM8CD2FAK vectors at a ratio of 1:7, respectively. Transiently transfected cells were sorted by flow cytometry (see below) for hα6A expression and the positive cells were grown in G418 containing medium. CDM8CD2FAK vector does not carry a neo resistance gene, therefore, only cells carrying both neo resistance (hα6A positive) and able to replicate (CD2FAK positive) will survive. Stable populations were analyzed both for hα6 and CD2 expression as described below.
Both transiently and stably transfected (the α5 phenotype was seen in transient as well as stable transfectants) myoblasts were analyzed for surface expression by flow cytometry as previously described (Sastry et al., 1996). Chicken α6A and α61044t transfected cells were stained with a chick α6-specific polyclonal antibody, α6ex, at 20 μg/ml in blocking buffer (Hepes-Hanks PBS-CMF with 2% BSA) and FITC-labeled goat anti–rabbit IgG (Cappel). Human α6A or B transfected myoblasts were analyzed with the human α6-specific mAb, 2B7, at 10 μg/ml in blocking buffer. Human α5 transfected cells were stained with VIF4 mAb. Cells transfected with CD2FAK or its mutants were stained with anti-CD2 mAb TS2/ 18.1.1. Chicken β1 transfected cells were stained with TS2/16 mAb against the human β1 epitope. IL2R-α5 or IL2R-β1A transfected cells were analyzed with an anti-IL2R antibody. The FACS profiles of the IL2R-α5 and IL2R-cyto-transfected cells were not stable, and we were unable to obtain populations greater than 40% positive, which were used for analysis. Flow cytometry was performed on a EPICS cell sorter (Coulter Electronics, Inc.) equipped with Cicero software for data analysis. As shown in Fig. 1, expression levels were assayed by fluorescence activated cell sorting. These profiles reflect enriched surface expression levels of ectopic integrin subunits in transfected myoblasts used in experiments.
Figure 1.

Surface expression of ectopic integrins in primary quail myoblasts. Myoblasts were transfected with (A) the human α5 (hα5) subunit, (B) the IL2Rα5 subunit, (C) a chimeric, chicken β1A subunit, CH8, in which an extracellular region of the chick sequence is replaced by the corresponding human β1 sequence to create an epitope-tag, (D) the IL2R-β1A subunit, (E) the human α6A (hα6A) subunit, (F) the IL2R-α6A subunit, or (G) the α61044t deletion mutant and analyzed for cell surface expression by flow cytometry. The hα5 (A) and hα6A (E) subunits were detected with hα5 subunit-specific mAb, VIF4, or hα6A-specific mAb, 2B7, respectively. (C) The CH8-β1A subunit was detected with TS2/16, an anti–human β1 integrin mAb. An anti-IL2R mAb was used to detect IL2R-α5 (B), IL2R-β1A (D), and IL2R-α6A (F). These mAbs do not cross react with UT myoblasts. (G) The α61044t deletion was detected with a polyclonal antibody, α6ex, which recognizes both the endogenous quail α6 subunit as well as the transfected chicken α6 subunit. qα6 is total α6 expression in untransfected cells and chα6 is total expression in transfected cells. The shift in the expression profile reflects a two- to threefold increase in total α6 integrin expression.
Cell Extracts, Western Blotting, and Immunoprecipitation
For Western blotting and immunoprecipitation experiments, untransfected and transfected myoblasts were plated on FN (UT and hα5 transfected cells), on LM (UT and chicken α6 and chicken α61044t or human α6A transfected cells) or on gelatin (UT, PAX, and CD2FAK transfected cells) for 24 h in complete myoblast medium. Since myoblasts will differentiate in the absence of serum and also secrete their own matrix, we were unable to test the effects of specific matrix ligands on the myoblast response. Therefore, all assays were conducted in the presence of serum and results are presented for steady state conditions. Cells were washed with ice-cold PBS containing 1 mM Na-orthovanadate and lysed in ice-cold modified RIPA extraction buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% NP-40, 1.0% Triton X-100, 0.25% sodium-deoxycholate, 2 mM EDTA, and 2 mM EGTA) with protease inhibitors (20 mg/ml leupeptin, 0.7 mg/ml pepstatin, 1 mM phenanthroline, 2 mM phenyl-methyl-sulfonyl-chloride, and 0.05 units aprotinin) and phosphatase inhibitors (30 mM sodium-pyrophosphate, 40 mM NaF, 1 mM sodium-orthovanadate). Protein content of the clarified lysates was determined using the Pierce bicinchoninic acid (BCA) method with bovine serum albumin as the standard.
For phosphotyrosine Western blots, 10–15 μg of lysates were separated on 10% SDS-PAGE gels (Laemmli, 1970) under reducing conditions and transferred to nitrocellulose membranes (Towbin et al., 1979). Membranes were blocked in 1% heat denatured BSA in TST buffer (10 mM Tris-HCl, pH 7.5, 100 mM NaCl, 0.1% Tween-20) overnight at 4°C. Phosphotyrosine containing proteins were detected by incubating the membranes with the anti-phosphotyrosine mAb, PY20, and a secondary horse radish peroxidase (HRP) conjugated anti–mouse antibody (Jackson ImmunoResearch Labs) or with RC20H, a directly conjugated HRP anti-phosphotyrosine Ab. Blots were visualized by chemiluminescence (Pierce Chemical Co.). Membranes were exposed to X-ray film (Kodak, X-OMAT AR) and developed in an automatic film processor. When indicated, membranes were stripped in stripping buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, and 100 mM β-mercaptoethanol) for 30 min at 60°C and reprobed with a different antibody.
For anti-MAPK Western blots, cells were trypsinized, washed once with soybean trypsin inhibitor (0.5 mg/ml), washed twice in Puck's Saline G (GIBCO BRL) and resuspended in serum-free medium containing 2% BSA. Cells were held in suspension for 1 h prior to plating on FN or LM in complete myoblast medium for 24 h. Cell extracts were prepared in RIPA buffer as described. 5 μg of cell lysates were separated on 12% SDS-PAGE gels under reducing conditions and the proteins transferred to nitrocellulose membranes. The membranes were blocked in 3% nonfat dry milk in TST overnight at 4°C. Active MAPK was detected by an anti-active MAPK pAb (Promega). Membranes were stripped and reprobed for total MAPK with an anti-erk1 mAb (Transduction Labs) or SC-94 anti-erk1 pAb (Santa Cruz Biotechnologies).
For paxillin, FAK, CD2-FAK, and HA immunoblot analysis, 5–20 μg cell lysates were resolved on 7.5% SDS-PAGE gels under reducing conditions and proteins transferred to nitrocellulose membranes. Membranes were blocked in TST buffer containing 3% nonfat milk and the proteins were detected with 165 mAb (anti-paxillin), BC3 pAb (anti-FAK), TS2/ 18.1.1 mAb (anti-CD2), or 12CA5 mAb (anti-HA).
For FAK immunoprecipitations, 100 μg of RIPA lysate was mixed with 1 μl of anti-FAK mAb, 2A7, 50 μl of packed agarose anti–mouse beads (blocked in 5% BSA; Sigma) in a final volume of 500 μl. The bead-antibody-antigen complex was incubated at 4°C for 2 h with continuous agitation. For paxillin immunoprecipitations, 100 μg of cell lysate and 1 μl of anti-paxillin mAb, 165 were incubated at 4°C with continuous agitation for 1 h. In a separate tube, 50 μl of packed protein A–agarose beads and 30 μg/ml rabbit anti–mouse IgG were incubated in lysis buffer for 1 h. The antigen-antibody mixture was then added to rabbit anti–mouse–protein A beads and incubated at 4°C an additional 2 h. The beads were pelleted gently and washed twice with lysis buffer. Bound protein was released from the beads by boiling in 100 μl Laemmli sample buffer containing 5% β-mercaptoethanol for 5 min. Equal aliquots of the precipitated protein for each antibody were loaded onto 7% SDS-PAGE gels. The FAK IP was blotted for FAK with C-20 and for phosphoFAK with RC20H. The paxillin IP was blotted for paxillin with the 165 mAb or for phosphopaxillin with RC20H. All immunoprecipitations and Western blots were detected by chemiluminescence.
Immunofluorescence Staining
Cells were grown on FN- or LM-coated coverslips. Immunostaining was done at room temperature. Cells were rinsed in PBS and fixed with 3% formaldehyde in PBS for 15 min then permeabilized with 0.4% Triton X-100 in PBS for 10 min, washed and blocked in 5% goat serum in PBS (BB) for 30 min. Cells were incubated with primary Ab in BB for 30 min, washed and incubated with FITC- or rhodamine-conjugated secondary Ab (Cappel) and DAPI (Sigma Chemical Co.) for additional 30 min. Coverslips were washed extensively and mounted in medium containing elvanol and p-phenylenediamine. Fluorescence was observed on a Zeiss Axioplan microscope.
Alteration of MAPK activity and differentiation
MAP kinase activity was manipulated in hα6A transfected myoblasts by coexpression of constitutively activate (CA) MEK1. Myoblasts were cotransfected with pRc/CMVhα6A and the HA-tagged pCMVneoMEK S218/220D vectors at a ratio of 1:7, respectively. Cells were selected in G418 and stable populations were sorted by flow cytometry for human α6A expression as described. Cell lysates were analyzed for HA expression by Western blotting as described. Stably cotransfected cells were plated on LM-coated plates and observed for 96 h.
To alter MAP kinase activity in hα5 transfected myoblasts, hα5-expressing cells were grown in the presence of the specific MEK inhibitor PD98059 (New England Biolabs) (Alessi et al., 1995). Transfected myoblasts were plated on FN-coated coverslips and on FN-coated TC plates. After 8 h in complete myoblast medium, the first dose of the inhibitor was added to the cells at 1, 10, 25, 50, or 100 μM final concentration. Cells were grown for an additional 24 h and a second dose of the inhibitor was added. After 24 and 48 h in presence of the inhibitor, coverslips were fixed and immunostained for DAPI and muscle α-actinin. At the same time, cells were extracted in RIPA buffer as described and lysates were analyzed by Western blotting for active MAPK and total erk1 expression as described above.
Differentiation was scored using the fusion index, which is the percentage of total nuclei in myotubes as described in Sastry et al. (1996).
Results
Integrin α Subunit Cytoplasmic Domains Modulate Proliferative Signals through the β1 Subunit
We recently reported a specificity for integrin α subunits and their cytoplasmic domains in controlling the proliferative to differentiative transition in primary quail myoblasts (Sastry et al., 1996). Ectopic expression of the human α5 integrin subunit (hα5) enhanced the fraction of myoblasts remaining in the proliferative phase and inhibited the initiation of terminal differentiation. In contrast, ectopic expression of the human α6A subunit of integrin (hα6A) inhibited myoblast proliferation and promoted differentiation. These effects resulted from a three- to fivefold increased surface expression of the α5β1 or the α6Aβ1 integrin (and a two- to threefold increase in total β1 integrin, see below) with little change in the relative expression of other integrin α subunits. These findings suggested that the α5 cytoplasmic domain promotes proliferative signals whereas the α6A cytoplasmic domain inhibits proliferation and enhances the fraction of cells initiating terminal differentiation.
To assess the contribution of these two cytoplasmic domains, we first examined the effect of ectopic α5 and α6A truncation mutants on myoblast proliferation and differentiation. (Fig. 2 and Table I). As we reported previously, ectopic expression of the hα5 truncation, α5GFFKR, which retains only the conserved GFFKR sequence, promoted proliferation and inhibited differentiation similar to the wild-type hα5 subunit (Sastry et al., 1996; Table I). These findings suggest that the majority of the α5 cytoplasmic domain is not required for proliferative signals. On the other hand, ectopic expression of an α6A truncation, α61044t, which deletes the COOH-terminal 11 amino acid residues, restores proliferative signaling and produces a phenotype similar to that of the ectopic α5 subunit. Myoblasts expressing α61044t remain in the proliferative phase and do not differentiate even in high density cultures (Fig. 2). Like hα5 transfected myoblasts (Fig. 2 A), myoblasts expressing α61044t do not express muscle α-actinin (Fig. 2), a myogenic differentiation marker, and exhibit a fusion index of 5% (Fig. 2 B) after 72 h of culture in a rich medium. This contrasts UT controls and hα6A transfected cells (Fig. 2) where a significant fraction of cells express muscle α-actinin and fuse into multinucleated myotubes (Fig. 2 B). Preliminary mapping of the COOH-terminal 11 amino acids points to S1071 (in hα6A) as a key residue, since its mutation to alanine produces a phenotype with enhanced proliferation (data not shown). Furthermore, the proliferation inhibiting effect of hα6 integrin is specific for the α6A cytoplasmic domain isoform. Ectopic expression of the hα6B subunit in myoblasts promotes proliferation and inhibits differentiation (data not shown, Table I). Consistent with these results, the α6A isoform is the predominant α6 integrin expressed in striated muscle (Hogervorst et al., 1993; and our own unpublished observations) and in embryonic cells of determined lineage (Cooper et al., 1991) whereas α6B is highly expressed in proliferating, totipotent or undifferentiated ES cells. Taken together, these observations suggest that the α5 and α6A cytoplasmic domains function differently: the α5 cytoplasmic domains appears permissive whereas a discrete region of the α6A cytoplasmic domain is inhibitory with respect to proliferation.
Figure 2.
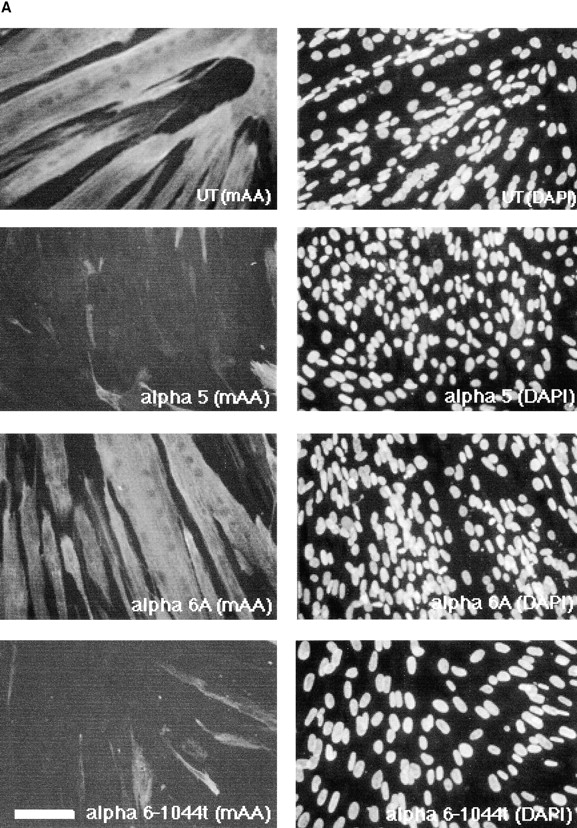

Effect of ectopic integrin subunits on muscle differentiation. (A) Control untransfected (UT) myoblasts, α5 transfected myoblasts, α6A transfected myoblasts, or α61044t transfected myoblasts were plated on coverslips in complete myo medium for 72 h and then double immunostained for a myogenic marker, muscle α-actinin or DAPI to visualize nuclei. The UT and α6A transfected cells show extensive fusion into multinucleated myotubes, alignment of nuclei, and expression of muscle α-actinin. Myoblasts expressing the α61044t subunit do not fuse into myotubes and only a small percentage of cells express muscle α-actinin. This phenotype resembles that of myoblasts expressing the ectopic α5 subunit. (B) Fusion index, the percentage of total nuclei in myotubes. Ectopic expression of α61044t, β1A, or IL2R-β1A inhibit myoblast fusion to a similar extent as the ectopic α5 subunit after 72 h of culture in serum-containing medium. Fusion of IL2R-α5 or IL2R-α6A transfected cells resembles that of the UT controls. Bar, 50 μm.
Table I.
Integrin Subunit Phenotypes
| Transfected subunit | Proliferation | Differentiation | ||
|---|---|---|---|---|
| Control | + | + | ||
| α5 | + | − | ||
| α6A | − | + | ||
| α5GFFKR | + | − | ||
| α6-1044t | + | − | ||
| α6B | + | − | ||
| β1 | + | − | ||
| IL2α5 | + | + | ||
| IL2β1 | + | − | ||
| α6A + IL2β1 | + | + |
To determine whether the α5 or α6A cytoplasmic domains act directly or indirectly, we assayed the effects of single-subunit cytoplasmic domain chimeras (LaFlamme et al., 1992), IL2R-α5 or IL2R-α6A, on the ability of myoblasts to proliferate or differentiate. Ectopic expression of either IL2R-α5 or IL2R-α6A had little detectable effect on myoblast proliferation or differentiation (Fig. 2 B). These cells behaved much like control, untransfected (UT) myoblasts. Thus the α subunit cytoplasmic domains do not directly initiate signals for myoblast proliferation or differentiation.
How then do these two integrin α subunits regulate proliferation and differentiation? Our observation that different integrins, e.g., α5, α6B, and α61044t, all produce a similar phenotype suggests an hypothesis in which these α subunits influence the proliferative signaling through the β1 subunit. In this view the α5 cytoplasmic domain (as well as that of the α6B, α61044t, and perhaps others) would permit signaling through the β1 subunit whereas the α6A cytoplasmic domain would inhibit it. Previous studies have shown that the α subunit cytoplasmic domain can regulate β1 integrin localization to focal adhesions (Briesewitz et al., 1993; Ylanne et al., 1993) and integrin activation (O'Toole et al., 1991); both localization and activation, however, are mediated by the β subunit cytoplasmic domain. Furthermore, the β1 cytoplasmic domain alone, when expressed as a single subunit chimera, IL2R-β1, can activate intracellular signals (Akiyama et al., 1994). To test this hypothesis, we first determined whether overexpression of the chicken β1A subunit of integrin would increase the fraction of proliferative myoblasts. We chose the β1A isoform since it is predominant in replicating myoblasts (Belkin et al., 1996). As reported in Sastry et al. (1996) ectopic expression of integrin α subunits also produces a two- to threefold increase in total β1 expression with little change in relative expression of the other endogenous α subunit levels. The increase in total β1 expression maintains myoblasts in the proliferative phase and inhibits terminal differentiation. Myoblasts with enhanced β1A expression grow to confluency but exhibit a fusion index of only ∼10% compared with 60–70% for untransfected cells (Fig. 2 E). Thus, increased β1A expression produces a phenotype resembling that of increased hα5 subunit expression. This finding agrees with a similar result reported previously for the β3 integrin (Blaschuk et al., 1997).
Next, we addressed whether the β1A cytoplasmic domain could independently affect myoblast proliferation or differentiation through ectopic expression of the single subunit chimera, IL2R-β1A. Previous studies show that this chimera localizes in focal adhesions (LaFlamme et al., 1992) and mediates enhanced integrin signaling (Akiyama et al., 1994). Myoblasts expressing IL2R-β1A remain replicative and proliferate until confluent with little detectable fusion into myotubes (Fig. 2 B). Like myoblasts expressing ectopic α5 subunit, they also only exhibit significant cell cycle withdrawal and differentiation if cultured under serum-free conditions (Sastry et al., 1996 and data not shown). These results demonstrate that the β1A cytoplasmic domain is sufficient to transmit proliferative signals and inhibit differentiation and thus modulate the growth factor response. Further, ectopic expression of IL2R-β1A can rescue the hα6A phenotype. Myoblasts that coexpress IL2R-β1A and hα6A integrins proliferate and differentiate like untransfected cells (data not shown; Table I). Taken together, these findings suggest that proliferative signaling through integrins occurs via the β1 subunit and that different α subunit cytoplasmic domains can modulate these signals. The effects of ectopic integrin subunits on myoblast proliferation and differentiation are summarized in Table I.
Ectopic Integrins Regulate FAK and Paxillin
We next sought to determine the effect of ectopic α5 or α6A expression on β1A integrin signaling pathways. Since integrins stimulate increased tyrosine phosphorylation of several intracellular proteins (Burridge et al., 1992; Kornberg et al., 1992; Bockholdt and Burridge, 1993; Petch et al., 1995; Vuori and Ruoslahti, 1995), we assayed the phosphotyrosine profile of myoblasts expressing different ectopic α subunits. Immunoblotting with an anti-phosphotyrosine antibody shows that myoblasts ectopically expressing the hα5 integrin contain elevated tyrosine phosphorylation of proteins migrating in the molecular mass ranges of 120–130 and 65–70 kD whereas cells transfected with hα6A show a marked, general decrease in tyrosine phosphorylation with no additional bands when compared with UT controls (data not shown). These observations are consistent with the phenotypic effects of the ectopic integrins presented above; i.e., the α subunits do not initiate separate pathways. Thus, ectopic α5 expression permits enhanced signaling through the β1A subunit, whereas the α6A integrin suppresses these signals.
Next, we pursued the identities of the phosphoproteins migrating at 120–130 and 65–70 kD. Focal adhesion kinase (pp125FAK) and paxillin (pp68) are downstream targets of integrin signaling pathways that migrate in these molecular mass ranges (Burridge et al, 1992). Therefore, we assayed the level of FAK and paxillin tyrosine phosphorylation in UT, hα5, and hα6A transfected myoblasts. A Western blot of a FAK immunoprecipitation shows that comparable levels of FAK were precipitated (Fig. 3 A, lanes 1–3). The level of tyrosine phosphorylated FAK decreased in hα6A (Fig. 3 A, lane 6) versus hα5 transfected (Fig. 3 A, lane 5) or UT myoblasts (Fig. 3 A, lane 4). However, the level of FAK tyrosine phosphorylation in hα5 and UT myoblasts does not differ (Fig. 3 A, lanes 4 and 5). Immunodepletion of FAK from the lysate, followed by Western blotting the supernatant for phosphotyrosine reveals an additional 120-kD band in the hα5 transfected cells that could account for the observed increase in phosphotyrosine (data not shown). Thus, whereas FAK phosphorylation decreases in myoblasts expressing ectopic α6A integrin, it is unaffected by ectopic α5 expression.
Figure 3.


Effect of ectopic α5 and α6A expression on tyrosine phosphorylation of FAK and paxillin. (A) FAK was immunoprecipitated from UT, hα5, or hα6A transfected myoblasts with an anti-FAK antibody, mAb 2A7. Cell lysates were normalized for protein content. Immunoprecipitated proteins were detected by Western blot for FAK with an anti-FAK polyclonal antibody, C-20 (left panel, lanes 1–3) and for phosphoFAK with RC20H (right panel, lanes 4–6). The FAK Western blot shows that equivalent amounts of FAK were isolated from the different cell types. An RC20H, phosphotyrosine, blot of the FAK immunoprecipitation (IP) shows that FAK phosphorylation decreases in hα6A transfected myoblasts (lane 6) compared with UT (lane 4) or hα5 (lane 5) transfected cells. (B) Paxillin is upregulated in hα5 transfected myoblasts. Paxillin was immunoprecipitated with an anti-paxillin antibody, mAb 165, from equal protein quantities of UT, hα5, or hα6A transfected myoblast lysates. Immunoprecipitated proteins were resolved by SDS-PAGE, transferred to nitrocellulose and Western blotted for paxillin with mAb 165 (lanes 1–3) or phosphopaxillin with RC20H (lanes 4–6). Hα5 transfected myoblasts (lane 2) contain significantly elevated levels of paxillin compared to UT myoblasts (lane 1). UT (lane 1) and hα6A transfected cells (lane 3) contain comparable amounts of paxillin. The paxillin band at 68 kD is indicated by an arrow. The 55-kD IgG band is similar in all three lanes and serves as a loading control. On the right, phosphopaxillin increases in hα5 transfected myoblasts (lane 5) or slightly decreases in hα6A transfected cells (lane 6) compared to UT (lane 4).
To determine if paxillin phosphorylation is differentially regulated in UT, hα5, and hα6A transfected myoblasts, paxillin was immunoprecipitated with an anti-paxillin mAb. In contrast to observations with FAK, we observed a major difference in the level of paxillin expression between UT and hα5 transfected myoblasts. As seen in Fig. 3 B, paxillin is significantly upregulated in hα5 transfected myoblasts (Fig. 3 B, lane 2) when compared to UT (lane 1) or hα6A transfected (lane 3) myoblasts. The comparable intensity of a 55-kD band in the paxillin immunoprecipitation corresponds to reduced IgG and serves as a loading control. In addition to elevated levels of paxillin, a phosphotyrosine Western blot of the paxillin immunoprecipitation shows a concomitant increase in tyrosine phosphorylation of paxillin in hα5 transfected myoblasts (Fig. 3 B, lane 5) compared to UT myoblasts (lane 4). Tyrosine phosphorylation of paxillin in hα6A transfected cells (lane 6) is somewhat decreased relative to untransfected cells. The enhanced paxillin expression observed in hα5 transfected myoblasts does not arise as a direct effect of the hα5 integrin. Myoblasts expressing IL2R-β1A also show increased paxillin expression whereas myoblasts expressing IL2R-α5 do not (data not shown). Taken together, these results indicate that enhanced paxillin expression accompanies increased α5 (or β1A) levels, whereas decreased FAK phosphorylation coincides with increased α6A levels. These data also suggest an uncoupling of FAK and paxillin signaling.
Paxillin and FAK Regulate the Proliferative to Differentiative Transition
The altered expression and activation of paxillin and FAK presented above prompted us to examine the effects of ectopic paxillin or FAK expression on myoblast proliferation and differentiation. As shown in Fig. 4 A (lane 2), the level of paxillin expression increases when compared with controls (Fig. 4 A, lane 1) after transfection of a paxillin cDNA. Ectopic expression of wild-type paxillin inhibits differentiation and results in a proliferative phenotype (Fig. 5, Table II). Myoblasts expressing ectopic paxillin proliferate until confluent but neither fuse into multinucleated myotubes (Fig. 5) nor express muscle α-actinin (Fig. 5). This phenotype is similar to that of hα5 transfected myoblasts (Fig. 2). Thus ectopic paxillin expression alone can recapitulate the effects of the hα5 or IL2R-β1A integrin subunits. Paxillin expression levels in control cells do not differ in replicating myoblasts versus differentiated cultures (data not shown).
Figure 4.

Ectopic expression of adhesive signaling molecules. Myoblasts were transfected with expression plasmids encoding a wild-type chicken paxillin, paxillin mutants Y118F and S188/190A, or a membrane-targeted, chimeric FAK construct, CD2-FAK. (A) Western blot analysis of myoblasts expressing ectopic paxillin (lane 2) or paxillin mutants Y118F (lane 3) or S188/190A (lane 4) shows significant overexpression of these proteins compared with endogenous levels in untransfected myoblasts (lane 1). Paxillin was detected with mAb 165. (B) Expression of CD2-FAK on the cell surface was confirmed by flow cytometry using an anti-CD2 antibody.
Figure 5.
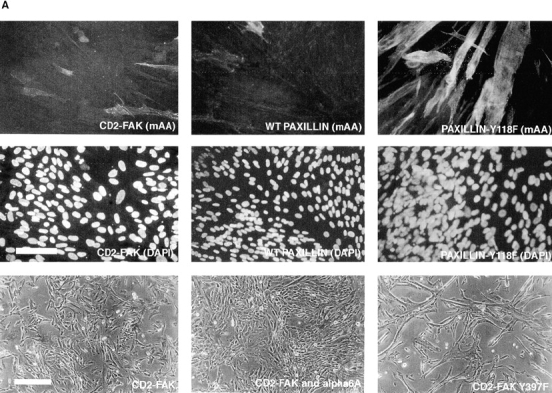

Effect of ectopic adhesive signaling molecules on muscle differentiation. (A) Myoblasts expressing CD2-FAK, wild-type paxillin, or paxillin Y118F were grown in serum-containing medium for 72 h and then double immunostained for muscle α-actinin (mAA) and DAPI. Both the ectopic CD2-FAK or wild-type paxillin inhibit expression of muscle α-actinin in the majority of cells. In contrast, paxillin Y118F does not inhibit differentiation. These cells fuse to form multinucleated myotubes and express muscle α-actinin. Phase contrast pictures show that myoblasts expressing CD2-FAK-Y397 exhibit extensive fusion into myotubes much like control, UT myoblasts, whereas myoblasts that express CD2-FAK or that coexpress hα6A integrin and CD2-FAK proliferate until confluent with minimal fusion. (B) Fusion index, the percentage of total nuclei in myotubes: CD2-FAK, CD2-FAK K454R, or paxillin expression inhibits myoblast fusion when compared with UT controls. The fusion of paxillin Y118F, paxillin S188/190A, or CD2-FAK-Y397F transfected myoblasts is similar to UT controls. Bars: (fluorescence micrographs) 50 μm; (phase micrographs) 35 μm.
Table II.
Focal Adhesion Molecules and Differentiation
| Transfected molecule | Proliferation | Differentiation | ||
|---|---|---|---|---|
| Untransfected | + | + | ||
| FAK | + | + | ||
| FAK(Y397F) | + | + | ||
| CD2-FAK | + | − | ||
| CD2-FAK(K454R) | + | − | ||
| CD2-FAK(Y397F) | + | + | ||
| PAX WT | + | − | ||
| PAX(S188/190A) | + | + | ||
| PAX(Y118F) | + | + |
Two major sites of phosphorylation in paxillin in response to adhesion to fibronectin are Y118 and S188/190 (Bellis et al., 1997). The tyrosine phosphorylation site, Y118, is also the site phosphorylated by FAK (Bellis et al., 1995). Therefore, we next tested the effect of a Y118F mutation (Fig. 5) or the double mutation S188/190A on the myoblast phenotype. When expressed in myoblasts (Fig. 4 A, lanes 3 and 4), neither of these mutants showed the enhanced proliferation seen when wild-type paxillin was expressed. Instead the paxillin mutants (Fig. 5) exhibited a phenotype characteristic of UT myoblasts (Fig. 5, Table II). This result suggests that these tyrosine and serine phosphorylation sites in paxillin participate in proliferative signaling that regulates myoblast cell cycle withdrawal.
Since FAK phosphorylation decreased in parallel with the inhibition of proliferation in myoblasts expressing hα6A integrin, we next tested the effect of an ectopic FAK mutant, Y397F, which lacks the autophosphorylation site and cannot bind src-family kinases (Schaller and Parsons, 1994), on proliferation and differentiation of myoblasts. Since ectopic expression of soluble FAK often produces short-lived or weak phenotypes (Richardson and Parsons, 1996), we used CD2-FAK, a membrane bound, chimeric FAK construct, which is constitutively active (Chan et al., 1994). Presumably, this results from increased adhesive signaling that arises from its constitutive membrane association and consequent localization in focal adhesions. Ectopic expression of CD2-FAKY397F, inhibits myoblast proliferation while promoting differentiation (Fig. 5). These cells are reminiscent of myoblasts transfected with the hα6A integrin subunit except that their proliferation is not inhibited as completely. They also show extensive fusion into multinucleated myotubes (Fig. 5). Interestingly, myoblasts transfected with wild-type CD2-FAK (Fig. 4 B), remain proliferative and do not initiate terminal differentiation (Fig. 5) when compared to UT controls. Fewer than 5% of CD2-FAK–expressing myoblasts fuse into multinucleated myotubes (Fig. 5) or express muscle α-actinin (Fig. 5). Using FACS analysis of propidium iodide labeled cells to measure G1/S progression, CD2-FAK transfected myoblasts show an increased ratio of G2 to G1 cells compared to untransfected cells (data not shown). Similarly, ectopic expression of CD2-FAK454, which is kinase defective, also inhibits differentiation and promotes proliferation (Fig. 5). Ectopic expression of wild-type CD2-FAK in hα6A transfected myoblasts results in a proliferative phenotype. Myoblasts that coexpress CD2-FAK and hα6A integrin grow to confluency and do not differentiate (Fig. 5). In sum, the hα6A phenotype can be recapitulated by expression of a FAK mutant expressed in control myoblasts or rescued by coexpression of an activated form of FAK. These data suggest that one potential mechanism by which hα6A integrin inhibits myoblast proliferation is through altering FAK phosphorylation. Thus, changes in the level of focal adhesion signaling, through FAK or paxillin, significantly affect the likelihood of myoblasts to proliferate or withdraw from the cell cycle and differentiate. Table II summarizes the effects of ectopic focal adhesion molecules.
MAP Kinase Activity Modulates Integrin-mediated Proliferation and Differentiation
Several reports implicate MAP kinase in adhesion dependent regulation of proliferation (Chen et al., 1994; Zhu and Assoian, 1995; Wary et al., 1996; Miyamoto et al., 1996; Lin et al., 1997). In addition, MAP kinase activation plays an important role in muscle differentiation (Bennet and Tonks, 1997). Therefore, we investigated if the ectopic integrins altered MAP kinase activation to control myoblast proliferation and differentiation. MAP kinase activation was assessed by immunoblotting cell lysates with an antibody that specifically recognizes phosphorylated, or active, forms of the 42- and 44-kD MAP kinases. Quail myoblasts express the 44-kD MAP kinase, erk-1, which was detected using an anti–erk-1 mAb (data not shown). Fig. 6 shows Western blots of active MAP kinase in myoblasts expressing ectopic integrins. In all cases, control or transfected cells were cultured for 24 h in complete serum-containing medium before extraction. The results shown reflect differences in steady state levels of MAP kinase activity. Compared with UT myoblasts (Fig. 6, bottom, lane 1), hα5 (lane 2), or IL2R-β1A (lane 3) transfected myoblasts contain elevated levels of active MAP kinase. In contrast, the level of active MAP kinase in hα6A transfected cells is significantly decreased (Fig. 6, top, lane 3) compared with controls (top, lane 1). The different intensities for control levels of active MAP kinase (top versus bottom) is due to different exposure times of the Western blots to film. As presented earlier, the α6A cytoplasmic domain truncation, α61044t, did not produce the proliferation inhibiting effects of hα6A integrin, and instead promoted proliferation and inhibited cell cycle withdrawal. This also altered the level of MAP kinase activation, as assayed by Western blotting. Compared with myoblasts expressing hα6A, those expressing α61044t display enhanced levels of active MAP kinase (Fig. 6, top, lane 2). Stripping and reprobing these membranes for total erk1 levels showed similar expression in all cells tested (data not shown). Therefore, the level of MAP kinase activity depends on the expression of specific integrins and their cytoplasmic domains.
Figure 6.
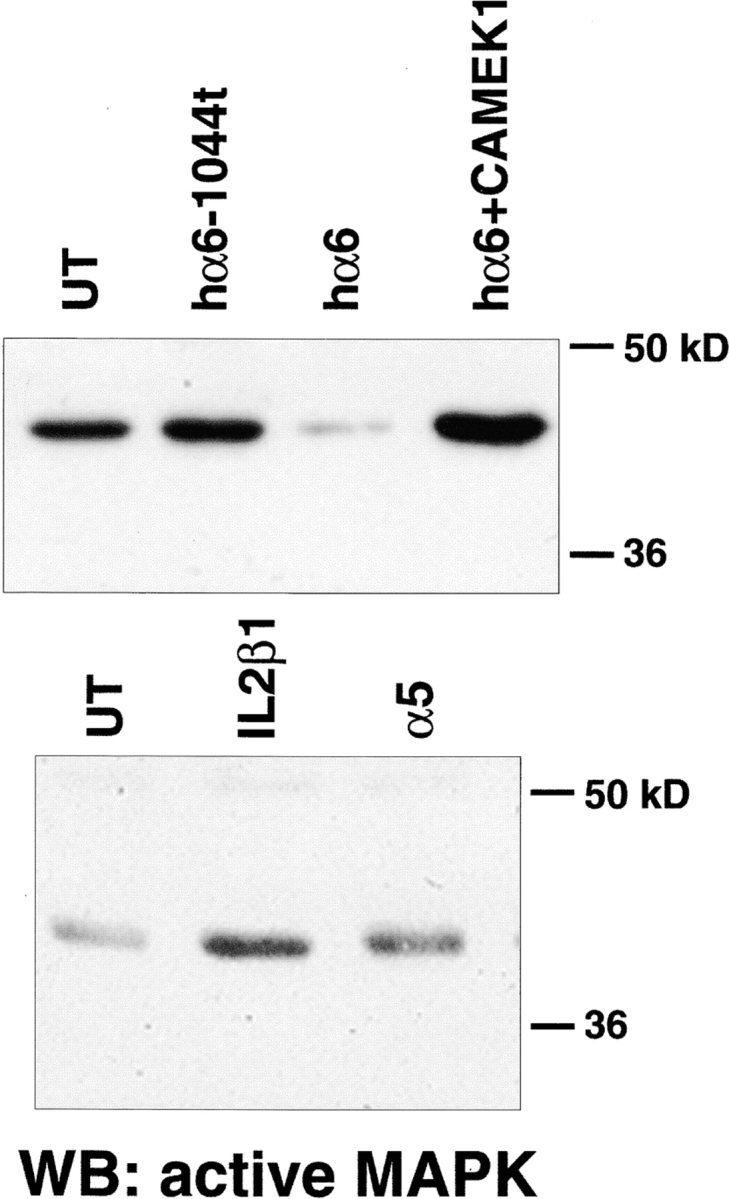
Ectopic integrins regulate MAP kinase activity. Different ectopic integrins alter activation of the level of erk-1 MAP kinase in 24-h cultures grown in serum-containing medium. MAP kinase activity was determined by Western blotting with an antibody that recognizes dually phosphorylated, active MAP kinase. Top panel: ectopic hα6A expression (lane 3) suppresses MAP kinase activation compared to untransfected controls (lane 1). Ectopic expression of the α6A truncation mutant, a61044t (lane 2), does not inhibit MAP kinase activation. Coexpression of hα6A and constitutive MEK-1 reverses the hα6A inhibition of MAP kinase activity (lane 4). (Bottom) Ectopic hα5 (lane 3) or IL2R-β1A (lane 2) expression enhance MAP kinase activation compared to untransfected controls (lane 1). The differing intensities for untransfected controls between the top and lower panels reflect different exposure times of the membranes to film.
We next investigated whether altering the activation state of MAP kinase could reverse the hα5 or hα6A induced phenotypes. We manipulated the level of active MAP kinase through overexpression of MEK-1, an upstream activator of MAP kinase, or by addition of PD-98059, a specific MEK inhibitor (Alessi et al., 1995). Cotransfection of myoblasts with constitutively active MEK (CA-MEK; Catling et al., 1995) and hα6A integrin restores a proliferative phenotype to myoblasts expressing hα6A. These cells stably express both the hα6A subunit (Fig. 7 B) and CA-MEK (Fig. 7 A) after drug selection and continue to proliferate for the lifetime of the cells in culture. FACS analysis of propidium iodide labeled cells shows an increased ratio of G2 to G1 cells in the hα6A/ CA-MEK cotransfectants (data not shown). As reported previously (Sastry et al., 1996), we were unable to isolate cells stably overexpressing only the α6A integrin. The hα6A-CA-MEK transfected cells are similar to the hα5 transfected myoblasts; i.e., they remain proliferative and do not differentiate appreciably (Fig. 7 E) compared with hα6A transfected (Fig. 7 D) or UT cells (Fig. 7 C). The level of active MAP kinase is enhanced in hα6A-CA-MEK cells (Fig. 6, top, lane 4) when compared to hα6A (Fig. 6, top, lane 3) or UT (Fig. 6, top, lane 1) myoblasts. Interestingly, we were unable to obtain stable expression of CA-MEK in untransfected, control myoblasts. Presumably, excessive levels of activated MEK, or MAP kinase, leads to increased cell death or decreased cell growth.
Figure 7.
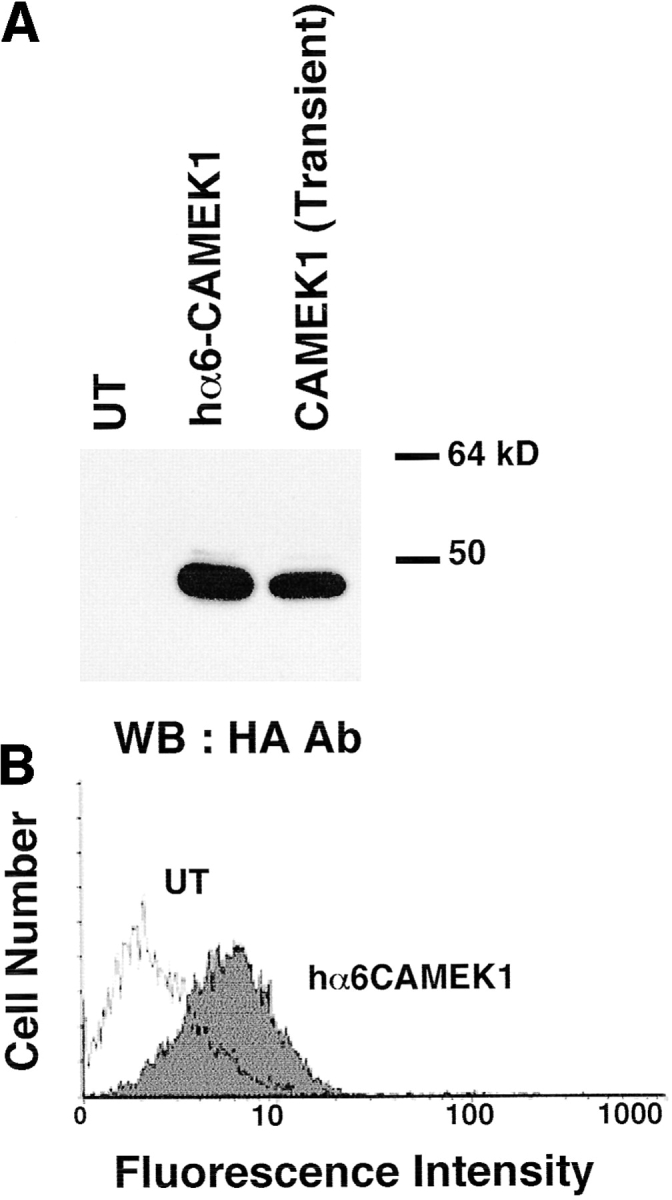



The effect of ectopic α6A integrin on proliferation is reversed by MAP kinase activation. (A) Constitutive HA-tagged MEK-1 (CA-MEK1) is efficiently expressed in control (lane 3) and hα6A transfected (lane 2) myoblasts in a transient transfection. MEK-1 expression is detected by a Western blot with an antibody specific for the HA epitope. (B) FACS analysis of hα6A integrin expression in myoblasts cotransfected with CA-MEK1. The hα6A subunit was detected with mAb 2B7, which is specific for hα6A. Both the hα6A integrin and CA-MEK are stably expressed in the double transfected cells after G418 selection. After 72 h of culture in serum-containing medium, hα6A/CA-MEK1 transfected myoblasts (E) grow to confluency with little detectable fusion into myotubes while both UT (C) and hα6A transfected (D) myoblasts differentiate and form myotubes. Bar, 40 μm.
We next determined whether decreasing the level of active MAP kinase, using the specific MEK inhibitor PD-98059, would reverse the hα5 phenotype. hα5 transfected myoblasts were plated onto FN-coated plates in serum-containing medium and allowed to attach for 8–12 h. Increasing concentrations of the MEK inhibitor, PD-98059 were then added for an additional 24–48 h. With increasing inhibitor concentration, the fraction of differentiated cells increased, whereas at high inhibitor concentrations, the total number of cells decreased, presumably due to inhibited proliferation (Fig. 8 and data not shown). After 48 h, hα5 transfected myoblasts treated with a 25 μM or greater concentrations (Fig. 8 C) of the MEK inhibitor displayed marked differentiation into myotubes compared with untreated hα5 transfected cells (Fig. 8 B) resembling UT controls (Fig. 8 A). A Western blot for the level of active MAP kinase shows that increasing concentrations of PD-98059 reduces MAP kinase activity in hα5 transfected cells (Fig. 8 E). Taken together, our observations demonstrate that quantitative changes in integrins closely parallels changes in MAP kinase activation. Moreover, the level of active MAP kinase appears to be a critical determinant of myoblast proliferation versus differentiation.
Figure 8.


The effect of ectopic α5 integrin on proliferation and differentiation requires active MAP kinase. MEK-1 inhibitor, PD98059, was added to hα5 transfected myoblasts. After 72 h in serum-containing medium, hα5 transfected cells treated with PD98059 exhibit extensive fusion into myotubes (C) and resemble control, UT myoblasts (A). (B) Untreated hα5 transfected myoblasts grow to confluency without significant fusion. (E) Increasing concentrations of PD98059 reduce the level of active MAP kinase. At greater than 25–50 μM inhibitor, the level of active MAP kinase in hα5 transfected cells is reduced to that observed in UT controls (D). Bar, 30 μm.
Discussion
In this study we addressed the mechanisms by which integrin α subunits modulate intracellular signal transduction events that lead to phenotypic changes in cell proliferation and differentiation. Skeletal myoblasts are well suited for this kind of study. The probability that a myoblast withdraws from the cell cycle and initiates terminal differentiation is highly sensitive to environmental cues including growth factors and ECM components (Sastry et al., 1996). The sharply contrasting effects of ectopic α5 or α6A integrin on myoblast proliferation and differentiation therefore provide a useful system to address α subunit specific signaling and the mechanisms by which integrins regulate the myoblast decision to withdraw from the cell cycle and initiate terminal differentiation. The data presented here demonstrate that the ectopic α subunits differentially regulate proliferative signaling through the β1 subunit. They also demonstrate that paxillin, FAK, and MAP kinase serve as important regulators of cell cycle withdrawal and the onset of terminal differentiation. Finally, our results show that the decision to withdraw from the cell cycle and initiate terminal differentiation is highly poised and regulated by quantitative changes in the level of MAP kinase activation, which in turn is regulated by quantitative changes in levels of integrin signaling. These observations provide a rationale for: (a) the apparently disparate requirements of different muscle cell lines and primary cultures for proliferation and cell cycle withdrawal, (b) the modulation of integrin subunits during muscle differentiation (Menko and Boettiger, 1987; Muschler and Horwitz, 1991; Bronner-Fraser et al., 1992; Lakonishok et al., 1992; Blaschuk and Holland, 1994; Boettiger et al., 1995) and (c) the hyperproliferative phenotypes observed when integrins are modulated in some other cell types (Carroll et al., 1995).
How does the α5β1 integrin potentiate signaling while the α6β1 integrin attenuates it? Our data show that the β1A subunit (i.e., cytoplasmic domain) is sufficient to transmit proliferative signals. The α5 subunit is permissive and allows β1 signaling while the α6A inhibits it. Previous studies indicate that signaling through integrins requires receptor ligation or clustering into focal adhesions (Kornberg et al., 1991; Miyamoto et al., 1996). In the myoblast system, both the ectopic α5 and α6A subunits form functional receptors with the endogenous β1 subunit for FN or LM, respectively (data not shown, and Sastry et al., 1996). Therefore, the differences in signaling we observe are most likely not due to an inability to bind ligand. Furthermore, since IL2R-β1A alone stimulates proliferation, receptor-ligation does not appear necessary except perhaps to form focal adhesions. Our observation used cells cultured on a complex ECM containing serum and secreted FN, also indicating that receptor ligation may not be a critical factor, for the inhibitory effects of ectopic α6. Interestingly, the α5β1 and the α6β1 integrins exhibit distinct subcellular distributions in muscle when cultured on the appropriate matrix ligand (Sastry, S., J. Muschler, and M. Lakonishok, unpublished observations). The α5β1 integrin localizes in focal adhesions on a FN substrate. In contrast, the α6β1 integrin displays a diffuse cell surface distribution on a laminin substrate. Since the initiating event for signaling through integrins is receptor clustering, the contrasting localizations of α5β1 and α6β1 could reflect a difference in their ability to signal. The clustering of integrins recruits signaling proteins into the focal adhesion signaling complex (Miyamoto et al., 1995). In this view, the α5β1 integrin (or any other β1 integrin with a permissive α subunit) recruits and activates proteins like FAK, paxillin, and MAP kinase to stimulate signaling. The α6β1 integrin, though able to bind to LM, is unable to recruit and/or activate the requisite signaling complex.
In the context of a model in which integrin signaling occurs primarily through the β1 subunit, how might the α subunits regulate signaling? A possible mechanism is that the α5 and α6A cytoplasmic domains differentially regulate the accessibility of binding sites on the β1 cytoplasmic domain. The α5 subunit (and likely several other α subunits) would permit exposure of critical binding sites, whereas the α6A subunit would mask them. A similar mode of regulation is proposed for the α subunit cytoplasmic domain in the ligand-dependent localization of integrins in focal adhesions (Briesewitz et al., 1993; Ylanne et al., 1993). Other models of regulation are also possible. They include a steric masking of the β1 cytoplasmic domain by the α6 cytoplasmic domain through interaction with an inhibitory binding protein, or through an α6-mediated signaling event that results in the activation of a phosphatase. The observation that IL2R-β1A expression rescues the hα6A phenotype, suggests that α6A masks some critical site on β1A cytoplasmic domain. Our observation that ectopic IL2R-α6A does not inhibit proliferation argues against an α6β1 binding protein but does not rule it out.
The critical regulatory site(s) within the α6 cytoplasmic domain appears to reside in the eleven COOH-terminal amino acids of the α6A sequence (Table I). This COOH-terminal region of the α6A cytoplasmic domain contains two putative serine phosphorylation sites (Shaw et al., 1990; Delwel et al., 1993), which could potentially participate in negative regulation of signaling by the α6A integrin. Preliminary mapping of the α6A cytoplasmic domain implicates one of these phosphorylation sites in α6A (S1071) in inhibition of proliferation (unpublished observations). However, the physiological relevance of α6 phosphorylation in this system is not yet clear.
Our observations also identify important roles for intracellular components of integrin signaling pathways in regulating cell cycle withdrawal and the onset of terminal differentiation. FAK and paxillin, stand out as potential integrin proximal mediators of adhesive signaling. These proteins are phosphorylated on tyrosine in response to cell adhesion to ECM in many cell types (Guan et al., 1991; Burridge et al., 1992; Hanks 1992; Kornberg et al., 1992; Schaller et al., 1992; Schwartz et al., 1995). In addition, both FAK and paxillin bind to synthetic peptides derived from the β1A cytoplasmic domain (Schaller et al., 1995). However, their role in cell cycle withdrawal and the onset of terminal differentiation is not well understood. Some evidence implicates FAK in proliferation and cell survival. Displacement of endogenous FAK from focal adhesions in endothelial cells, through microinjection of the focal adhesion targeting domain, interferes with cell cycle progression (Gilmore and Romer, 1996) resulting in apoptosis. Similarly, microinjection of β1A peptides corresponding to the FAK binding site, or anti-FAK antibodies induces apoptosis in cultured fibroblasts (Hungerford et al., 1996). Expression of constitutively active FAK, CD2-FAK, in epithelial cells protects them from apoptosis (Frisch et al., 1996).
Our data support a requirement for FAK activation in basal myoblast proliferation; but it does not contribute to the enhanced proliferation, i.e., the inhibited cell cycle withdrawal, observed in cells expressing ectopic α5. Phosphorylation of FAK on tyrosine is not significantly altered in myoblasts expressing ectopic α5 integrin. In most cell types, FAK phosphorylation peaks within 1 h after cell attachment to the ECM (Guan et al., 1991; Burridge et al., 1992; Hanks et al., 1992; Schaller et al., 1992). A transient increase in FAK phosphorylation is observed in α5 transfected myoblasts after initial attachment on a FN substrate (data not shown). However, we do not observe a sustained increase in FAK activation that coincides with the sustained proliferative phenotype induced by ectopic α5 integrin. Additionally, overexpression of FAK, as well as several FAK mutants and FRNK (Richardson and Parsons, 1996), had no detectable effect on myoblast proliferation or differentiation (unpublished observations). However, ectopic expression of CD2-FAK, a membrane-bound, activated form of FAK, does promote proliferation and inhibit differentiation, thus resembling the effect of ectopic α5 or β1A integrin. Presumably this arises because CD2-FAK is targeted to the membrane and therefore available to recruit additional adhesive signaling complexes. In contrast, the level of FAK phosphorylation is reduced in myoblasts expressing ectopic α6A integrin, correlating with the increased cell cycle withdrawal. Ectopic expression of CD2-FAKY397F, which lacks an autophosphorylation site, acts as a dominant negative and inhibits proliferation and promotes differentiation, thus revealing its role in basal proliferation. This phenotype is similar to but less dramatic than that of ectopic α6A integrin expression. It is interesting to note that Y397 is a binding site for src family kinases (Schaller and Parsons, 1994). Finally, coexpression of CD2-FAK and hα6A results in proliferation. Taken together, these observations are consistent with a role for FAK phosphorylation in the basal level of proliferation seen in control myoblasts and show that it lies on the pathway downstream of the α6A-mediated inhibition of the β1 integrin signaling.
In contrast to FAK, both the expression and subsequent tyrosine phosphorylation of paxillin are significantly enhanced in α5 transfected myoblasts when compared to untransfected or α6A transfected cells. A role for paxillin in proliferation or differentiation has not been demonstrated previously. Paxillin is a multidomain adapter protein that mediates numerous interactions with different signaling proteins, including FAK (Schaller and Parsons, 1994). However, paxillin has no known enzymatic activity. Paxillin does contain multiple LIM domains, which are involved in protein-protein interactions and targeting of paxillin to focal adhesions (Turner and Miller, 1994; Brown et al., 1996). Finally, paxillin has been implicated in growth factor dependent differentiation where an increase in its tyrosine phosphorylation correlates with neuronal differentiation (Khan et al., 1995; Leventhal and Feldman, 1996). In myoblasts, the upregulation of paxillin in response to ectopic α5 integrin coincides with a proliferative phenotype, which can be recapitulated by ectopic expression of paxillin. Our results with paxillin mutants implicate both the Y118F and S188/190A phosphorylation sites as key. Whereas the mechanism by which paxillin inhibits cell cycle withdrawal, i.e., stimulates proliferation, is not known, increased paxillin expression and phosphorylation enhances the formation of adhesive signaling complexes as do CD2-FAK and ectopic α5 expression (unpublished observation). In contrast to the significant effects of ectopic α5 expression on paxillin expression and phosphorylation, ectopic α6A expression had only a very modest effect on paxillin phosphorylation and none on expression. Coexpression of paxillin and hα6A did not relieve the α6A phenotype (data not shown). Thus the level of paxillin expression and phosphorylation appear to contribute the myoblast decision to proliferate or differentiate. However, it may not play a role in the inhibition of proliferation by ectopic α6A expression. It is noteworthy that our results also indicate an uncoupling of FAK and paxillin signaling, since they are not activated in parallel as reported for fibroblasts (Burridge et al., 1992).
Although our results demonstrate an inhibitory role for the α6A integrin in signaling, this integrin is capable of activating intracellular signals in other cell systems. In macrophages, for example, α6Aβ1-mediated attachment to laminin leads to increased tyrosine phosphorylation of paxillin (Shaw et al., 1995). In addition, antibody clustering of the α6 integrin in endothelial cells leads to a distinct profile of tyrosine phosphorylated proteins, which do not include FAK or paxillin (Jewell et al., 1995).
The contrasting effects of ectopic α5 or α6A integrin expression on MAP kinase activation suggests quantitative changes in MAP kinase activation plays a major role in regulating myoblast cell cycle withdrawal. Perturbing the ratio of different integrins in the cell alters the level of MAP kinase activation. In contrast to our findings, the α6A integrin, but not the α6B subunit, activates MAP kinase in macrophages (Wei et al., 1998). Cell type differences likely explain these conflicting observations. In the muscle system, the effect of ectopic α5 or α6A integrins and their mutants on MAP kinase activation parallels their phenotypic effects on myoblast proliferation and differentiation. Likewise, perturbing the activation state of MAP kinase, using a MEK inhibitor or CA-MEK, can overcome the integrin induced effect on both MAP kinase activity and the myoblast phenotype. Consistent with these results, the activation of MAP kinase is apparently an important determinant of myogenic differentiation. Inactivation of MAP kinase by overexpression of MAP kinase phosphatase renders C2C12 myoblasts insensitive to mitogenic stimuli, favoring expression of muscle-specific genes like myoD (Bennet and Tonks, 1997). Finally, our data show that MAP kinase activation in myoblasts requires both adhesive and growth factor signals. We do not observe active MAP kinase in the absence of serum or on a non-specific adhesive substrate like poly-l-lysine (unpublished observations). Taken together, these findings reveal a role for integrins in controlling myoblast proliferation and differentiation through a pathway that regulates the level of MAP kinase activation.
The ability of different integrins to activate MAP kinase is proposed to occur via an association of integrins with the adaptor protein, shc. In fibroblasts or endothelial cells, α5β1 and several other integrins interact with shc and thus can activate MAP kinase whereas the α6 integrin cannot bind shc or activate MAP kinase (Wary et al., 1996). However, the interaction with shc does not require integrin cytoplasmic domains. Our data do not preclude an involvement of shc; the increase in MAP kinase activation seen in response to the α61044t truncation may reflect a difference in the binding of shc, or an unidentified adapter or regulatory protein.
Our observations can be summarized in a working model for adhesive regulation of myoblast withdrawal from the cell cycle and the onset of terminal differentiation. In this model, the myoblast decision to proliferate or withdraw from the cell cycle is regulated, at least in part, by the level of activated MAP kinase. This decision appears highly poised and sensitive to quantitative fluctuations in the level MAP kinase activation: increased MAP kinase activation favoring proliferation and decreased MAP kinase activation favoring cell cycle withdrawal. MAP kinase activation, in turn, is regulated by the signaling emanating from both adhesive and growth factor pathways. Evidently, neither pathway alone is sufficient to sustain proliferation. The absence of either growth factor or adhesive signaling promotes cell cycle withdrawal and the onset of terminal differentiation, which appears to function as a default. At one level, the synergy is compatible with an additive model since increased signaling via either enhanced growth factor or adhesive signaling leads to a decreased probability of cell cycle withdrawal. However, the synergy between the growth factor and adhesive signaling systems likely has interactions that are more consistent with an additive, threshold model, in which signals are required from both the growth factor and adhesive pathways. MAP kinase is not activated and targetted to the nucleus in cells that are either adhering to a nonspecific adhesive substrate like poly-l-lysine or that lack serum growth factors. Recent studies point to RAF as an integration point for adhesive and growth factor signals (Chen et al., 1996; Renshaw et al., 1997; Howe et al., 1998).
This working model provides a rationale for diverse observations on the myoblast decision to proliferate or differentiate. Infection of myoblasts with viruses encoding src family kinases (Alemo and Tato, 1987) or erbB (Olson, 1992), a truncated form of the EGF receptor with intrinsic tyrosine kinase activity, or the ectopic expression of CD2-FAK or paxillin all inhibit cell cycle withdrawal and produce a proliferative phenotype similar to that of myoblasts ectopically expressing the α5 integrin. It is likely that this arises from increased adhesive and/or growth factor signaling resulting in increased MAP kinase activation. Similarly, the general requirement for high confluency and low serum for differentiation of diverse muscle cell lines likely also appears to reflect enhanced mitogenic signaling, i.e., increased MAP kinase activation, which in this case results from immortalization or adaptation to culture. Previous reports show alterations in the expression of integrin subunits during muscle differentiation, our results raises the possibility that they contribute to cell cycle withdrawal and the onset of terminal differentiation. Finally, our studies appear pertinent to keratinocyte proliferation in vivo as well as to myoblast differentiation in vitro. Transgenic mice expressing two- to threefold increased expression of α5β1 or α2β1 integrins exhibit epidermal hyper-proliferation, perturbed keratinocyte differentiation and other features of psoriasis, a skin disease (Carroll et al., 1995). This epidermal hyper-proliferation is similar to our observations on myoblasts expressing ectopic α5 integrin. Our working model provides a possible mechanism. Therefore, it is likely that quantitative changes in mitogenic signaling through alterations in adhesive signaling produces phenotypic alterations that operate through common mechanisms in diverse systems.
Acknowledgments
We thank J.T. Parsons, M. Weber, Y. Yakada, S. LaFlamme, L. Reichardt, R. Isberg, M. Hemler, D. Fishman, Y. Takada, and A. Mercurio for generous gifts of reagents. We also thank A. Richardson and J.T. Parsons for their advice and M. Berg for her comments on the manuscript.
This work was supported by National Institutes of Health grants GM23244 (A. Horwitz) and GM47607 (C. Turner). C. Turner is an Established Investigator of the American Heart Association.
Abbreviations used in this paper
- BCA
bicinchoninic acid
- CA-MEK
constitutively active MEK
- FAK
focal adhesion kinase
- HA
hemagglutinin
- MAP
mitogen-activated protein
- MEK
MAP/erk kinase
- UT
untransfected
Footnotes
Dr. Sastry's present address is Department of Cell Biology and Anatomy, University of North Carolina, Chapel Hill, NC 27599.
Dr. Horwitz's address after 1 July 1999 will be Department of Cell Biology, Box 439, Health Sciences Center, University of Virginia, Charlottesville, VA 22908.
References
- Adams J, Watt F. Regulation of development and differentiation by the extracellular matrix. Development. 1993;117:1183–1198. doi: 10.1242/dev.117.4.1183. [DOI] [PubMed] [Google Scholar]
- Alema S, Tato F. Interaction of retroviral oncogenes with the differentiation program of myogenic cells. Adv Canc Res. 1987;49:1–28. doi: 10.1016/s0065-230x(08)60792-7. [DOI] [PubMed] [Google Scholar]
- Alessi D, Cuenda A, Cohen P, Dudley D, Saltiel A. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase in vitro and in vivo. . J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Akiyama S, Yamada S, Yamada K, LaFlamme S. Transmembrane signal transduction by integrin cytoplasmic domains expressed in single-subunit chimeras. J Biol Chem. 1994;269:15691–15694. [PubMed] [Google Scholar]
- Belkin A, Zhidkova N, Balzac F, Altruda F, Tomatis D, Maier A, Tarone G, Koteliansky V, Burridge K. β1D integrin displaces the β1A isoform in striated muscles: localization at junctional structures and signaling potential in nonmuscle cells. J Cell Biol. 1996;132:211–226. doi: 10.1083/jcb.132.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellis S, Miller J, Turner C. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J Biol Chem. 1995;270:17437–17441. doi: 10.1074/jbc.270.29.17437. [DOI] [PubMed] [Google Scholar]
- Bellis S, Perrotta J, Curtis M, Turner C. Adhesion of fibroblasts to fibronectin stimulates both serine and tyrosine phosphorylation of paxillin. Biochem J. 1997;325:375–381. doi: 10.1042/bj3250375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennet A, Tonks N. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997;278:1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- Blaschuk K, Holland P. The regulation of α5β1 integrin expression in human muscle cells. Dev Biol. 1994;164:475–483. doi: 10.1006/dbio.1994.1217. [DOI] [PubMed] [Google Scholar]
- Blaschuk K, Guerin C, Holland P. Myoblast αvβ3 integrin levels are controlled by transcriptional regulation of expression of the β3 subunit and down-regulation of β3 subunit expression is required for skeletal muscle cell differentiation. Dev Biol. 1997;184:266–277. doi: 10.1006/dbio.1997.8527. [DOI] [PubMed] [Google Scholar]
- Bockholdt S, Burridge K. Cell spreading on extracellular matrix proteins induces tyrosine phosphorylation of tensin. J Biol Chem. 1993;268:14565–14567. [PubMed] [Google Scholar]
- Boettiger D, Enomoto-Iwamoto M, Yoon H, Hofer U, Menko A, Chiquet-Ehrismann R. Regulation of integrin α5β1 affinity during myogenic differentiation. Dev Biol. 1995;169:261–272. doi: 10.1006/dbio.1995.1142. [DOI] [PubMed] [Google Scholar]
- Briesewitz R, Kern A, Marcantonio E. Ligand-dependent and –independent integrin focal contact localization: the role of the alpha chain cytoplasmic domain. Mol Biol Cell. 1993;4:593–604. doi: 10.1091/mbc.4.6.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner-Fraser M, Artinger M, Muschler J, Horwitz A. Developmentally regulated expression of the α6 integrin in avian embryos. Development. 1992;115:197–211. doi: 10.1242/dev.115.1.197. [DOI] [PubMed] [Google Scholar]
- Brown M, Perrotta J, Turner C. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol. 1996;135:1109–1123. doi: 10.1083/jcb.135.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Turner C, Romer L. Tyrosine phosphorylation of paxillin and pp125(FAK) accompanies cell adhesion to extracellular matrix—a role in cytoskeletal assembly. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Romero M, Watt F. Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell. 1995;83:957–968. doi: 10.1016/0092-8674(95)90211-2. [DOI] [PubMed] [Google Scholar]
- Cary L, Chang J, Guan JL. Stimulation of cell migration by overexpression of focal adhesion kinase and its association with src and fyn. J Cell Sci. 1996;109:1787–1794. doi: 10.1242/jcs.109.7.1787. [DOI] [PubMed] [Google Scholar]
- Catling A, Schaeffer H, Reuter C, Reddy G, Weber M. A proline-rich sequence unique to MEK1 and MEK2 is required for raf binding and regulates MEK function. Mol Cell Biol. 1995;15:5214–5225. doi: 10.1128/mcb.15.10.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan P, Kanner B, Whitney G, Aruffo A. A transmembrane-anchored chimeric focal adhesion kinase is constitutively activated and phosphorylated at tyrosine residues identical to pp125FAK. J Biol Chem. 1994;269:20567–20574. [PubMed] [Google Scholar]
- Chen H, Guan JL. Stimulation of phosphatidylinositol 3′-kinase association with focal adhesion kinase by platelet-derived growth factor. J Biol Chem. 1994;269:31229–31233. [PubMed] [Google Scholar]
- Chen Q, Kinch M, Lin T, Burridge K, Juliano R. Integrin-mediated cell adhesion activates mitogen-activated protein kinases. J Biol Chem. 1994;269:26602–26605. [PubMed] [Google Scholar]
- Cooper H, Tamura R, Quaranta V. The major laminin receptor of mouse embryonic stem cells is a novel isoform of the α6β1 integrin. J Cell Biol. 1991;115:843–850. doi: 10.1083/jcb.115.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCurtis I, Quaranta V, Tamura R, Reichardt L. Laminin receptors in the retina: sequence analysis of the chick integrin α6 subunit. J Cell Biol. 1991;113:405–416. doi: 10.1083/jcb.113.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedhar S, Hannigan G. Integrin cytoplasmic interactions and bidirectional transmembrane signaling. Curr Opin Cell Biol. 1996;8:657–669. doi: 10.1016/s0955-0674(96)80107-4. [DOI] [PubMed] [Google Scholar]
- Delwel G, Hogervorst F, Kuikman I, Paulsson M, Timpl R, Sonnenberg A. Expression and function of the cytoplasmic variants of the integrin α6 subunit in transfected K562 cells: activation-dependent adhesion and interaction with isoforms of laminin. J Biol Chem. 1993;268:25865–25875. [PubMed] [Google Scholar]
- Frisch S, Vuori K, Ruoslahti E, Chan-Hui P. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore A, Romer L. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Trevithik J, Hynes R. Fibronectin/integrin interaction induces tyrosine phosphorylation of a 120-kDa protein. Cell Regul. 1991;2:951–964. doi: 10.1091/mbc.2.11.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner B. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Hanks S, Calalb M, Harper M, Patel S. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc Natl Acad Sci USA. 1992;89:8487–8491. doi: 10.1073/pnas.89.18.8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemler M, Sanchez-Madrid F, Flotte T, Krensky A, Burakoff S, Bhan A, Springer T, Strominger J. Glycoproteins of 210, 000 and 130,000 m.w. on activated T cells: cell distribution and antigenic relation to components on resting cells and T cell lines. J Immunol. 1984;132:3011–3018. [PubMed] [Google Scholar]
- Hogervorst F, Admiraal L, Niessen C, Kuikman I, Janssen H, Daams H, Sonnenberg A. Biochemical characterization and tissue distribution of the A and B variants of the integrin α6 subunit. J Cell Biol. 1993;121:179–191. doi: 10.1083/jcb.121.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe A, Aplin AE, Alahari SK, Juliano R. Integrin signaling and cell growth control. Curr Opin Cell Biol. 1998;10:220–231. doi: 10.1016/s0955-0674(98)80144-0. [DOI] [PubMed] [Google Scholar]
- Hughes P, Renshaw M, Pfaff M, Forsyth J, Keivens V, Schwartz M, Ginsberg M. Suppression of integrin activation: a novel function of a Ras/ Raf-initiated MAP kinase pathway. Cell. 1997;88:521–530. doi: 10.1016/s0092-8674(00)81892-9. [DOI] [PubMed] [Google Scholar]
- Hungerford J, Compton M, Matter M, Hoffstrom B, Otey C. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–1390. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanzawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Alzawa S. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Ilic D, Almeida E, Schlaepfer D, Dazin P, Aizawa S, Damsky C. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. 1998;143:547–560. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell K, Kapron-Bras C, Jeevaratnam P, Dedhar S. Stimulation of tyrosine phosphorylation of distinct proteins in response to antibody-mediated ligation and clustering of α3 and α6 integrins. J Cell Sci. 1995;108:1165–1174. doi: 10.1242/jcs.108.3.1165. [DOI] [PubMed] [Google Scholar]
- Juliano RL, Haskill S. Signal transduction from the extracellular matrix. J Cell Biol. 1993;120:577–585. doi: 10.1083/jcb.120.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner S, Reynolds A, Vines R, Parsons JT. Monoclonal antibodies to individual tyrosine-phosphorylated protein substrates of oncogene-encoded tyrosine kinases. Proc Natl Acad Sci USA. 1990;87:3328–3332. doi: 10.1073/pnas.87.9.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Okumura N, Okada M, Kobayashi S, Nakagawa H. Nerve growth factor stimulates tyrosine phosphorylation of paxillin in PC12h cells. FEBS Lett. 1995;362:201–204. doi: 10.1016/0014-5793(95)00250-d. [DOI] [PubMed] [Google Scholar]
- Kleinman H, McGarvey M, Liotta L, Robey P, Tryggvason K, Martin G. Isolation and characterization of type IV procollagen, laminin, and heparan sulfate proteoglycan from the EHS sarcoma. Biochemistry. 1982;21:6188–6193. doi: 10.1021/bi00267a025. [DOI] [PubMed] [Google Scholar]
- Klemke R, Cai S, Giannini A, Gallagher P, deLanerolle P, Cheresh D. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A, Escobedo M, Wachowicz M, Apell G, Brown T, Giedlin M, Kavanaugh W, Williams L. Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol Cell Biol. 1998;18:5699–5711. doi: 10.1128/mcb.18.10.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konigsberg I. Skeletal myoblasts in culture. Methods Enzymol. 1979;58:511–527. doi: 10.1016/s0076-6879(79)58166-x. [DOI] [PubMed] [Google Scholar]
- Kornberg L, Earp HS, Turner C, Prockop C, Juliano R. Signal transduction by integrins—increased protein tyrosine phosphorylation caused by clustering of beta 1 integrins. Proc Natl Acad Sci USA. 1991;88:8392–8396. doi: 10.1073/pnas.88.19.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg L, Earp HS, Parsons JT, Schaller M, Juliano R. Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion-associated tyrosine kinase. J Biol Chem. 1992;267:23439–23442. [PubMed] [Google Scholar]
- Laemmli U. Cleavage of the structural proteins during assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- LaFlamme S, Akiyama S, Yamada K. Regulation of fibronectin receptor distribution. J Cell Biol. 1992;117:437–447. doi: 10.1083/jcb.117.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakonishok M, Muschler J, Horwitz A. The α5β1 integrin associates with a dystrophin-containing lattice during muscle development. Dev Biol. 1992;152:209–220. doi: 10.1016/0012-1606(92)90129-5. [DOI] [PubMed] [Google Scholar]
- Leventhal P, Feldman E. Tyrosine phosphorylation and enhanced expression of paxillin during neuronal differentiation in vitro. . J Biol Chem. 1996;271:5957–5960. doi: 10.1074/jbc.271.11.5957. [DOI] [PubMed] [Google Scholar]
- Lin T, Aplin A, Shen Y, Chen Q, Schaller M, Romer L, Aukhil I, Juliano R. Integrin-mediated activation of MAP kinase is independent of FAK: evidence for dual integrin signaling pathways in fibroblasts. J Cell Biol. 1997;136:1385–1395. doi: 10.1083/jcb.136.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainiero F, Pepe A, Wary K, Spinardi L, Mohamaddi M, Schlessinger J, Giancotti F. Signal transduction by the α6β4 integrin: distinct β4 subunit sites mediate the recruitment of shc/GRB2 and association with the cytoskeleton of hemidesmosomes. EMBO (Eur Mol Biol Organ) J. 1995;14:4470–4481. doi: 10.1002/j.1460-2075.1995.tb00126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menko A, Boettiger D. Occupation of the extracellular matrix receptor, integrin, is a control point for myogenic differentiation. Cell. 1987;51:51–57. doi: 10.1016/0092-8674(87)90009-2. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Teratomo H, Coso O, Gutkind J, Burbelo P, Akiyama S, Yamada K. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol. 1995;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Teramoto H, Gutkind J, Yamada K. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: roles of integrin aggregation and occupancy of receptors. J Cell Biol. 1996;135:1633–1642. doi: 10.1083/jcb.135.6.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschler J, Horwitz A. Down-regulation of the chicken a5b1 integrin fibronectin receptor during development. Development. 1991;113:327–337. doi: 10.1242/dev.113.1.327. [DOI] [PubMed] [Google Scholar]
- Olson M, Ashworth A, Hall A. An essential role for rho, rac, and cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- O'Toole T, Mandelman D, Forsyth J, Shattil S, Plow E, Ginsberg M. Modulation of the affinity of integrin αIIbβ3 (GPIIb-IIIa) by the cytoplasmic domain of αIIb. Science. 1991;254:845–847. doi: 10.1126/science.1948065. [DOI] [PubMed] [Google Scholar]
- Petch L, Bockholdt S, Bouton A, Parsons JT, Burridge K. Adhesion-induced tyrosine phosphorylation of the p130 src substrate. J Cell Sci. 1995;108:1371–1379. doi: 10.1242/jcs.108.4.1371. [DOI] [PubMed] [Google Scholar]
- Plopper G, McNamee H, Dike L, Bojanowski K, Ingber D. Convergence of integrin and growth factor receptor signaling pathways within the focal adhesion complex. Mol Biol Cell. 1995;6:1349–1365. doi: 10.1091/mbc.6.10.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw M, Ren X, Schwartz M. Growth factor activation of MAP kinase requires cell adhesion. EMBO (Eur Mol Biol Organ) J. 1997;16:5592–5599. doi: 10.1093/emboj/16.18.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reszka A, Hayashi Y, Horwitz AF. Identification of amino acid sequences in the integrin β1 cytoplasmic domain implicated in cytoskeletal association. J Cell Biol. 1992;117:1321–1330. doi: 10.1083/jcb.117.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A, Parsons JT. A mechanism for regulation of the adhesion-associated protein tyrosine kinase pp125FAK. Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- Roskelly C, Srebrow A, Bissell M. A hierarchy of ECM-mediated signaling regulates tissue-specific gene expression. Curr Opin Cell Biol. 1995;7:736–747. doi: 10.1016/0955-0674(95)80117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E, Hayman E, Pierschbacher M, Engvall E. Fibronectin: purification, immunochemical properties, and biological activities. Methods Enzymol. 1982;82:803–831. doi: 10.1016/0076-6879(82)82103-4. [DOI] [PubMed] [Google Scholar]
- Sastry S, Horwitz AF. Integrin cytoplasmic domains: mediators of cytoskeletal linkages and intra- and extracellular initiated transmembrane signaling. Curr Opin Cell Biol. 1993;5:819–831. doi: 10.1016/0955-0674(93)90031-k. [DOI] [PubMed] [Google Scholar]
- Sastry S, Horwitz AF. Adhesion-growth factor interactions during differentiation: an integrated biological response. Dev Biol. 1996;180:455–467. doi: 10.1006/dbio.1996.0319. [DOI] [PubMed] [Google Scholar]
- Sastry S, Lakonishok M, Thomas D, Muschler J, Horwitz AF. Integrin α subunit ratios, cytoplasmic domains, and growth factor synergy regulate muscle proliferation and differentiation. J Cell Biol. 1996;133:169–184. doi: 10.1083/jcb.133.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M, Parsons JT. Focal adhesion kinase and associated proteins. Curr Opin Cell Biol. 1994;6:705–710. doi: 10.1016/0955-0674(94)90097-3. [DOI] [PubMed] [Google Scholar]
- Schaller M, Borgman B, Cobb B, Vines R, Reynolds A, Parsons JT. Pp125FAK, a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M, Otey C, Hildebrand J, Parsons JT. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D, Hanks S, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to the Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Ingber D. Integrating with integrins. Mol Biol Cell. 1994;5:389–393. doi: 10.1091/mbc.5.4.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Schaller M, Ginsberg M. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- Shaw L, Messier J, Mercurio A. The activation dependent adhesion of macrophages to laminin involves cytoskeletal anchoring and phosphorylation of α6β1 integrin. J Cell Biol. 1990;110:2167–2174. doi: 10.1083/jcb.110.6.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw L, Lotz M, Mercurio A. Inside-out integrin signaling in macrophages: analysis of the role of the α6Aβ1 and α6Bβ1 integrin variants in laminin adhesion by cDNA expression in an α6 integrin-deficient macrophage cell line. J Biol Chem. 1993;268:11401–11408. [PubMed] [Google Scholar]
- Shaw L, Turner C, Mercurio A. The α6Aβ1 and α6Bβ1 integrin variants signal differences in the tyrosine phosphorylation of paxillin and other proteins. J Biol Chem. 1995;270:23648–23652. doi: 10.1074/jbc.270.40.23648. [DOI] [PubMed] [Google Scholar]
- Tahiliani P, Singh L, Auer K, LaFlamme S. The role of conserved amino acid motifs within the integrin beta3 cytoplasmic domain in triggering focal adhesion kinase phosphorylation. J Biol Chem. 1997;272:7892–7898. doi: 10.1074/jbc.272.12.7892. [DOI] [PubMed] [Google Scholar]
- Takada Y, Puzon W. Identification of a regulatory region of integrin β1 subunit using activating and inhibiting antibodies. J Biol Chem. 1993;268:17597–17601. [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner C, Miller J. Primary sequence of paxillin contains putative SH2 and SH3 binding motifs and multiple LIM domains: identification of a vinculin and pp125FAK binding region. J Cell Sci. 1994;107:1583–1591. doi: 10.1242/jcs.107.6.1583. [DOI] [PubMed] [Google Scholar]
- Turner C, Glenney J, Burridge K. Paxillin: a new vinculin-binding protein present in focal adhesions. J Cell Biol. 1990;111:1059–1068. doi: 10.1083/jcb.111.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varner J, Emerson D, Juliano R. Integrin α5β1 expression negatively regulates cell growth: reversal by attachment to fibronectin. Mol Biol Cell. 1995;6:725–740. doi: 10.1091/mbc.6.6.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuori K, Ruoslahti E. Association of insulin receptor substrate-1 with integrins. Science. 1994;266:1576–1578. doi: 10.1126/science.7527156. [DOI] [PubMed] [Google Scholar]
- Vuori K, Ruoslahti E. Tyrosine phosphorylation of p130cas and cortactin accompanies integrin-mediated cell adhesion to extracellular matrix. J Biol Chem. 1995;270:22259–22262. doi: 10.1074/jbc.270.38.22259. [DOI] [PubMed] [Google Scholar]
- Wary K, Mainiero F, Isakoff S, Marcantonio E, Giancotti F. The adaptor protein shc couples a class of integrins to the control of cell cycle progression. Cell. 1996;87:733–743. doi: 10.1016/s0092-8674(00)81392-6. [DOI] [PubMed] [Google Scholar]
- Watt F, Kubler M, Hotchin N, Nicholson L, Adams J. Regulation of keratinocyte terminal differentiation by integrin-extracellular matrix interactions. J Cell Sci. 1993;106:175–182. doi: 10.1242/jcs.106.1.175. [DOI] [PubMed] [Google Scholar]
- Wei J, Shaw L, Mercurio A. Regulation of mitogen-activated protein kinase activation by the cytoplasmic domain of the α6 integrin subunit. J Biol Chem. 1998;273:5903–5907. doi: 10.1074/jbc.273.10.5903. [DOI] [PubMed] [Google Scholar]
- Williams E, Hughes P, O'Toole T, Ginsberg M. The inner world of cell adhesion: integrin cytoplasmic domains. Trends Cell Biol. 1994;4:109–112. doi: 10.1016/0962-8924(94)90059-0. [DOI] [PubMed] [Google Scholar]
- Ylanne J, Chen Y, O'Toole T, Loftus J, Takada Y, Ginsberg M. Distinct functions of integrin α and β subunit cytoplasmic domains in cell spreading and formation of focal adhesions. J Cell Biol. 1993;122:223–233. doi: 10.1083/jcb.122.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Assoian R. Integrin-dependent activation of MAP kinase: a link to shape-dependent cell proliferation. Mol Biol Cell. 1995;6:273–282. doi: 10.1091/mbc.6.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]