

Abstract
Oriented cell growth requires the specification of a site for polarized growth and subsequent orientation of the cytoskeleton towards this site. During mating, haploid Saccharomyces cerevisiae cells orient their growth in response to a pheromone gradient overriding an internal landmark for polarized growth, the bud site. This response requires Cdc24p, Far1p, and a heterotrimeric G-protein. Here we show that a two- hybrid interaction between Cdc24p and Gβ requires Far1p but not pheromone-dependent MAP-kinase signaling, indicating Far1p has a role in regulating the association of Cdc24p and Gβ. Binding experiments demonstrate that Cdc24p, Far1p, and Gβ form a complex in which pairwise interactions can occur in the absence of the third protein. Cdc24p localizes to sites of polarized growth suggesting that this complex is localized. In the absence of CDC24-FAR1-mediated chemotropism, a bud site selection protein, Bud1p/Rsr1p, is essential for morphological changes in response to pheromone. These results suggest that formation of a Cdc24p-Far1p-Gβγ complex functions as a landmark for orientation of the cytoskeleton during growth towards an external signal.
Keywords: chemotropism, landmark, oriented growth, Ste4p Ste18p, yeast mating
Eukaryotic cells are able to polarize their growth in response to both external and internal signals. Polarization to external signals plays a crucial role in development and tissue formation. During yeast mating, cells of opposite mating type secrete peptide pheromones and respond to pheromone from their mating partner (for review see Sprague and Thorner, 1992; Chenevert, 1994; Leberer et al., 1997a). Mating pheromone binds to specific G-protein–coupled receptors on cells of opposite mating type (Bender and Sprague, 1989; Blumer et al., 1988). Receptor activation results in cell cycle arrest, transcriptional activation, morphological changes, and polarized growth towards a partner cell (Sprague and Thorner, 1992; Chenevert, 1994; Leberer et al., 1997a).
Cells respond to a gradient of mating pheromone by oriented growth along this gradient (Segall, 1993). Such chemotropic growth is essential for efficient mating (Dorer et al., 1995; Valtz et al., 1995; Nern and Arkowitz, 1998). During oriented growth, the actin cytoskeleton and secretory apparatus polarize towards the tip of the mating projection (Baba et al., 1989; Read et al., 1992). As a result cell wall and plasma membrane material is deposited at the tip of this pear-shaped cell known as a shmoo (Lipke et al., 1976; Tkacz and MacKay, 1979). Pheromone receptors and the heterotrimeric G-protein composed of Gα (GPA1), Gβ (STE4), and Gγ (STE18) are required for chemotropic growth (Jackson et al., 1991; Schrick et al., 1997; Xu and Kurjan, 1997). Certain alleles of the cyclin-dependent kinase inhibitor FAR1, such as far1-H7 (Valtz et al., 1995), and of the GDP-GTP exchange factor for the small GTPase Cdc42p CDC24, such as cdc24-m1 (Nern and Arkowitz, 1998), are specifically defective in chemotropic growth. These mutants are unable to orient in a pheromone gradient and select a site for mating projection growth adjacent to their previous bud site. Similarly, in the presence of saturating uniform concentrations of mating pheromone, shmoo formation occurs next to the previous bud site (Madden and Snyder, 1992; Dorer et al., 1995). The latter process has been referred to as default mating (Dorer et al., 1995).
During vegetative growth, haploid cells bud at a specific site next to their previous bud site, resulting in a characteristic axial budding pattern (Chant and Pringle, 1995). The BUD genes are required for this budding pattern (Chant and Herskowitz, 1991; Drubin and Nelson, 1996). During budding, cells polarize their actin cytoskeleton (Adams and Pringle, 1984; Kilmartin and Adams, 1984) and secretory apparatus towards the bud site (Tkacz and Lampen, 1972; Field and Schekman, 1980). However, this internal signal generated during budding is overridden upon exposure to a mating pheromone gradient, allowing cells to orient growth towards their mating partner (Madden and Snyder, 1992). How cells switch from an internally programmed polarized growth process to a process dictated by an external cue is unknown.
The pheromone receptors and the heterotrimeric G-protein are also required for cell cycle arrest, mitogen-activated protein (MAP)1-kinase–mediated gene induction, and cell morphological changes during mating (Sprague and Thorner, 1992; Chenevert, 1994; Leberer et al., 1997a). Genetic studies indicate that Gβγ activates all these processes (Whiteway et al., 1990) with Gα having a negative regulatory role (Dietzel and Kurjan, 1987; Miyajima et al., 1987). By analogy to other G-protein coupled receptors, receptor activation results in dissociation of Gα from Gβγ. Gβγ is found as a complex at the plasma membrane (Hirschman et al., 1997).
Previously we have shown that an association between Gβγ and Cdc24p is involved in oriented growth during mating (Nern and Arkowitz, 1998). We now show this Cdc24p–Gβ complex also contains Far1p. Genetic studies are consistent with the involvement of Far1p in this complex. Cdc24p localizes to sites of polarized growth in shmooing cells, suggesting that the complex is localized. In the absence of growth orientation mediated by this complex, cells form a mating projection adjacent to the bud site in a manner that is dependent on BUD1, suggesting Bud1p can regulate Cdc24p when chemotropic signaling is blocked. Together our results suggest that Cdc24p-Far1p-Gβγ acts as a landmark for cytoskeleton orientation in response to a pheromone gradient.
Materials and Methods
General Techniques
Standard techniques and media were used for growth and genetic manipulation of yeast (Rose et al., 1991). Unless otherwise indicated, yeast cells were grown at 30°C.
Strains and Plasmids
The yeast strains used in this study are described in Table I. In general, deletion mutants were constructed by PCR-based gene disruption as described (Arkowitz and Lowe, 1997; Nern and Arkowitz, 1998). ΔFar1 strains were constructed either by PCR-based gene disruption (Δ-1) or with a knockout cassette (Δ-2). This cassette contained the FAR1 ORF followed by 100 bp 3′ sequence with URA3Kl replacing all but the first 109 codons. Far1-H7, a far1 allele with a truncated COOH terminus (Valtz et al., 1995), was constructed by replacing codons 757–830 of FAR1 with a stop codon followed either by HIS5Sp or URA3Kl. Gene disruptions were confirmed by PCR and phenotype including mating defects with wild-type and enfeebled testers, mating pheromone growth arrest, and budding pattern. The Δbud1 cdc24-m1 double mutant was crossed with appropriate wild-type haploid and random spore analyses demonstrated that the inability to shmoo in the presence of pheromone specifically segregated with Δbud1 cdc24-m1 and this mating defect was observed in both mating types.
Table I.
Yeast Strains Used in This Study
| Strain | Genotype | Source | ||
|---|---|---|---|---|
| K699 | Mata, ura3, leu2-3,-112, trp1-1, ade2-1, can1-100, his3-11,-15, ssd1-Δ2, GAL | K. Nasmyth (IMP, Vienna) | ||
| PJ69-4A | Mata, trp1-901, leu2-3,-112, ura3-52, his3-200, gal4Δ, gal80Δ, GAL2-ADE2, | James et al., 1996 | ||
| LYS2::GAL1-HIS3, met2::GAL7-lacZ | ||||
| SEY6210 | Matα, leu2-3,-112, ura3-52, his3-Δ200, trp1-Δ901, lys2-801, suc2-Δ9 | S. Emr (University of | ||
| California, San Diego) | ||||
| SEY6211 | Mata, leu2-3,-112, ura3-52, his3-Δ200, trp1-Δ901, ade2, suc2-Δ9 | S. Emr | ||
| SFY526 | Mata, gal4, gal80, his3-200, trp1-901 ade2-101, lys2-801, ura3-52, | Clontech | ||
| leu2-3,-112, URA3::pGal1-lacZ, canr | ||||
| Y187 | MATα, gal4, gal80, his3, trp1-901, ade2-101, ura3-52, leu2-3,-112, URA3::pGal-lacZ | Clontech | ||
| RAY719 | Same as SEY6210 with bem1Δ-1::HIS3 | Arkowitz and Lowe, 1997 | ||
| RAY899 | Same as SEY6210 with ste20::LoxP HIS5Sp LoxP | This study | ||
| RAY910 | Same as SEY6211 with ste4::HASTE4 | This study | ||
| RAY912 | Same as SEY6211 with ste4::HASTE4 cdc24::TRP1 3xmyc CDC24 | This study | ||
| RAY931 | Same as SEY6211 with cdc24Δ-1::LoxP HIS5Sp LoxP and pEG(KT)CDC24 | Nern and Arkowitz, 1998 | ||
| RAY1034 | Same as SEY6211 with cdc24::TRP1 CDC24 | Nern and Arkowitz, 1998 | ||
| RAY1035 | Same as SEY6211 with cdc24::TRP1 cdc24-m1 | Nern and Arkowitz, 1998 | ||
| RAY1036 | Same as PJ69-4A with ste7Δ-1::URA3Kl | This study | ||
| RAY1041 | Same as SEY6210 with cdc24::TRP1 CDC24 | Nern and Arkowitz, 1998 | ||
| RAY1072 | Same as PJ69-4A with ste4Δ-1::URA3Kl | This study | ||
| RAY1074 | Same as PJ69-4A with ste5Δ-1::URA3Kl | This study | ||
| RAY1086 | Same as PJ69-4A with akr1Δ-1::URA3Kl | This study | ||
| RAY1109 | Same as RAY1034 with far1Δ-1::URA3Kl | This study | ||
| RAY1111 | Same as RAY1035 with far1Δ-1::URA3Kl | This study | ||
| RAY1113 | Same as PJ69-4A with far1Δ-2::URA3Kl | This study | ||
| RAY1114 | Same as PJ69-4A with ste20Δ-1::URA3Kl | This study | ||
| RAY1121 | Same as PJ69-4A with kss1Δ-1::URA3Kl | This study | ||
| RAY1123 | Same as PJ69-4A with ste11Δ-1::URA3Kl | This study | ||
| RAY1135* | Same as SEY6210 except LEU2 | This study | ||
| RAY1139‡ | Same as RAY1034 with bud1Δ-1::LoxP HIS5Sp LoxP | This study | ||
| RAY1142‡ | Same as RAY1035 with bud1Δ-1::LoxP HIS5Sp LoxP | This study | ||
| RAY1160§ | Same as RAY1035 with ste20Δ-1::LoxP HIS5Sp LoxP | This study | ||
| RAY1168‖ | Same as RAY1035 with bem1Δ-1::HIS3 | This study | ||
| RAY1173 ¶ | Same as RAY1034 with bem1Δ-1::HIS3 | This study | ||
| RAY1179 | Same as PJ69-4A with fus3Δ-1::URA3Kl | This study | ||
| RAY1182 | Same as Y187 with far1-H7::LoxP HIS5Sp LoxP | This study | ||
| RAY1183 | Same as Y187 with far1Δ-1::LoxP HIS5Sp LoxP | This study | ||
| RAY1246 | Same as RAY1034 with spa2Δ-1::LoxP HIS5Sp LoxP | This study | ||
| RAY1248 | Same as RAY1035 with spa2Δ-1::LoxP HIS5Sp LoxP | This study | ||
| RAY1249 | Same as RAY1034 with far1Δ-1::URA3Kl, bud1Δ-1::LoxP HIS5Sp LoxP | This study | ||
| RAY1254 | Same as K699 with cdc24::TRP1 3xmyc CDC24 | This study | ||
| RAY1258 | Same as K699 with cdc24::TRP1 3xmyc CDC24, far1::FAR1 ProtA HIS5Sp | This study | ||
| RAY1260 | Same as K699 with cdc24::TRP1 3xmyc CDC24-m1, far1::FAR1 ProtA HIS5Sp | This study | ||
| RAY1271** | Same as RAY1034 with ste20Δ-1::LoxP HIS5Sp LoxP | This study | ||
| RAY1276 | Same as SEY6211 with ste4::HASTE4 ProtA HIS5Sp, cdc24::TRP1 3xmyc CDC24 | This study | ||
| RAY1336 | Same as K699 with cdc24::TRP1 3xmyc cdc24, far1::far1-H7 ProtA HIS5Sp | This study | ||
| RAY1342‡ | Same as RAY1034 with sst2Δ-1::URA3Kl | This study | ||
| RAY1350‡ | Same as RAY1034 with sst2Δ-1::URA3Kl, bud1Δ-1::LoxP HIS5Sp LoxP | This study | ||
| RAY1360 | Same as RAY931 with p414Cdc24HAGFP instead of pEG(KT)CDC24 | This study | ||
| RAY1449 | Same as PJ69-4A with cdc24::TRP1 cdc24-m1 | This study |
HIS5Sp refers to HIS5 from S. pombe and URA3Kl refers to URA3 from K. lactis.
Transformed with LEU2 fragment to make LEU2 +.
Strains made by deletion in a haploid, crossing with appropriate haploid followed by sporulation.
Made by crossing RAY1035 with RAY899 followed by sporulation.
Made by crossing RAY1035 with RAY719 followed by sporulation.
Made by crossing RAY1034 with RAY719 followed by sporulation.
Made by crossing RAY1034 with RAY899 followed by sporulation.
A single HA epitope (amino acids YPYDVPDYA) was added to the NH2 terminus of STE4 by PCR. HASTE4 (including 394 bp 5′ upstream of the ATG) was cloned into pRS406 and two-step gene replacement (Scherer and Davis, 1979) of STE4 was used to construct RAY910. Protein A–tagged Far1p (RAY1258 and RAY1336) and Ste4p (RAY1276) strains were constructed by PCR-based gene replacement using pZZ-His5 (Rayner and Munro, 1998) as template and oligonucleotides with 60 nucleotides 5′ and 3′ of the termination codon. Myc epitope-tagged Cdc24p and cdc24-m1p strains were constructed by PCR-mediated gene replacement as described (Nern and Arkowitz, 1998) except the sequence encoding a triple myc tag (MEQKLISEEDL MEQKLISEEDL MEQKLISEEDL) was directly fused to the Cdc24p NH2 terminus. Gene replacements were confirmed by PCR and expression of epitope-tagged proteins of the correct size by immunoblotting using either 12CA5 (anti-HA) mAB tissue culture supernatant at 1:40 dilution, anti-protein A mAb (Sigma) at 1:2,000 dilution, or anti-myc polyclonal serum (Santa Cruz) at 1:500 dilution followed by ECL (Amersham). Strains with tagged proteins mated with wild-type mating efficiencies and arrested growth normally in response to α-factor.
Cdc24HAGFP was constructed by fusing an HA epitope followed by PacI, SphI, NotI, and SacII restriction sites to the COOH terminus of Cdc24p using PCR and p414Cdc24 (Nern and Arkowitz, 1998) as a template. This resulted in p414Cdc24HA which had the amino acids YPYDVPDYAGLIKHARPPPRG fused to the COOH terminus. Yeast enhanced green fluorescent protein (GFP; Cormack et al., 1997) followed by the ADH terminator was PCR amplified from pMK199 (a gift from E. Schiebel) with an oligonucleotide that added a PacI site at the 5′ end and a NotI site at the 3′ end. This PCR product was cloned into p414Cdc24HA using PacI and NotI sites resulting in Cdc24p followed by YPYDVPDYAGLIKGSGAGAGAGAGA fused to GFP followed by the ADH terminator (p414Cdc24HAGFP). p416GalHASte4 was constructed by cloning HASTE4 into pRS416 containing the Gal1/10 promoter.
The ADE2 gene from pSP73Ade2 (cloned by PCR from genomic DNA with oligonucleotides that added an EcoRI site at the 3′ end and a XhoI site at the 5′ end) was released by digestion with EcoRI and BsrGI followed by blunting. This fragment was cloned into pRS425 in which the LEU2 gene had been removed by digestion with Tth111I and NaeI followed by blunting resulting in p2μA. TPI-STE18 (triose phosphate isomerase promoter) from p416TSte18 (pRS416 with TPI cloned into the SacII EagI sites and STE18 cloned into BamHI EcoRI sites) was cloned into the SacI EcoRI sites of p2μA resulting in p2μATSte18. An oligonucleotide encoding the GAL4 nuclear localization signal (NLS) MDKAELIPEPPKKKRKVEL followed by a NcoI restriction site was cloned into EagI BamHI sites of p2μATSte18 yielding p2μATNLSSte18. Subsequently, an oligonucleotide encoding an HA epitope tag was cloned into the NcoI BamHI sites resulting in the following NLS-HA sequence,
MDKAELIPEPPKKKRKVELPWMYPYDVPDYA fused to the NH2 terminus of Ste18p yielding p2μATNLSHASte18. An EcoRI SacI fragment of p2μATNLSHASte18 containing TPI-NLSHA-STE18 was then cloned into pRS413 resulting in p413TNLSHASte18. STE18 was removed from this vector by digestion with BamHI and EcoRI and replaced with the coding sequence of FAR1 from pGAD424Far1 (see below) yielding p413TNLSHAFar1.
The coding sequences of the entire FAR1 ORF and far1-H7 were amplified by PCR from genomic DNA and cloned into pGAD424. SpeI PstI fragments of FAR1 and far1-H7 from pGAD424 plasmids were cloned into pMal-c2 (New England Biolabs) resulting in pMFar1 (amino acid residues 133–831) and pMFar1H7 (amino acid residues 133–756). pMFar1ΔN (amino acid residues 638–831) and pMFar1ΔC (amino acid residues 133– 297) were derived from pMFar1 by removal of a BamHI or HindIII fragment, respectively. pMFar1H7ΔN (amino acid residues 638–756) was derived from pMFar1H7 by removal of a BamHI fragment. GSTCdc24 is comprised of the NH2-terminal 472 amino acids of Cdc24p fused to GST as described (Nern and Arkowitz, 1998).
Two-Hybrid
Two-hybrid interactions were tested by growth on SC-leu-trp-his as described (Nern and Arkowitz, 1998). Identical results were obtained with at least three transformants.
Expression of a LacZ reporter from Y187 derived two-hybrid strains was quantified by β-galactosidase assays (Miller, 1972). An EcoRI site was inserted by oligonucleotide-directed mutagenesis after amino acid 153 of Spa2p (Arkowitz and Lowe, 1997). This 153–amino acid Spa2p fragment was then cloned into pGAD424. PJ69-4A cdc24-m1 (RAY1449) was constructed by PCR-mediated gene replacement as described (Nern and Arkowitz, 1998) and confirmed by PCR and mating defect phenotype. Three independent PJ69-4A cdc24-m1 strains were used for two-hybrid analyses. Because TRP1 is used to replace CDC24 with cdc24-m1, STE4 cloned into the 2μ URA3 GAL4 DBD vector pGBDU-C1 (James et al., 1996) was used in this strain. Diploid two-hybrid strains were constructed by transformation of either DBD fusions or AD fusions along with p2μAT and p413T plasmids into SFY526 or Y187 and crossing these strains. After two-hybrid assays, phenotypes (diploid state and sterility) of diploid and haploid deletion two-hybrid strains were confirmed. Expression of NLSHAFar1p and NLSHASte18p in two-hybrid strains were confirmed by analysis of yeast extracts using SDS-PAGE, immunoblotting, probing with 12CA5 mAb, and ECL visualization.
Immunoprecipitation
RAY1254, RAY1258, RAY1260, and RAY1336 cells carrying p416GalHASte4 were grown to an OD600 of 0.5 in SC-ura with 2% (wt/vol) raffinose, galactose was added to a final concentration of 2% (wt/vol) and the cultures grown for 4 h. All subsequent steps were carried out at 4°C. Cells were harvested by centrifugation, and lysed by agitation with glass beads in buffer A (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM PMSF, 40 μg/ml each of leupeptin, chymostatin, pepstatin A, aprotinin, and antipain) containing 0.1% Triton X-100. Before use IgG–Sepharose was cross-linked with dimethylpimelimidate (Sigma; Harlow and Lane, 1988). Cell extracts were clarified by two centrifugations (10,000 g for 10 min). Supernatants, which contained the majority of the tagged proteins, were incubated with 20 μl of IgG-Sepharose (Pharmacia) equilibrated in buffer A containing 0.1% Triton X-100 for 1 h. Resin was then washed four times with buffer A containing 0.1% Triton X-100 and Far1-protein A fusions were specifically eluted by incubation with 20 U of TEV-protease (Boehringer Mannheim) for 4 h at 16°C in the same buffer. Eluates were analyzed by SDS-PAGE and immunoblotting using polyclonal sera against myc and Far1p (a gift from M. Peter) at 1:1,000 dilution followed visualization with ECL.
Protein Purification
All purification steps were carried out at 4°C. MBP and GST fusion proteins were expressed in E. coli with MBPFar1 and MBPFar1-H7 bacteria grown at 30°C. Cells were resuspended in buffer B (PBS, 1 mM DTT, 0.1% Triton X-100), frozen in liquid N2 and stored at −70°C. Cells were lysed by sonication in buffer B with 1 mM PMSF. Extracts were clarified by centrifugation (10,000 g for 10 min) and fusion proteins were isolated using glutathione-agarose (Sigma) or amylose resin (New England Biolabs). MBP fusion proteins were eluted with 10 mM maltose in buffer B and dialyzed against buffer C (50 mM Tris-HCl pH 7.4, 10 mM MgCl2, 1 mM DTT, 10% [vol/vol] glycerol). Protein concentrations were determined by the Bradford method or by comparing intensities of bands on Coomassie stained SDS-PAGE gels with BSA (Sigma) as a standard. For both MBPFar1 and MBPFar1-H7, concentrations used refer to the full-length protein and not proteolytic breakdown products.
HASte4-(TEV)-protein A was purified from RAY1276 cells using IgG-Sepharose under conditions similar to those described in (Song et al., 1996). Cells were grown in YEPD to an OD600 of ∼3, harvested by centrifugation, resuspended in 20 mM Tris-HCl pH 7.4 with 50 mM NaCl at ∼300 OD600/ml, snap frozen in liquid N2, and stored at −70°C. Typically 2,500 OD600 of cells were broken in buffer D (buffer A containing 2 mM EDTA and 3 mM MgCl2) by agitation with glass beads. Triton X-100 was added to cell extracts at a final concentration of 1%. After 1 h incubation the extract was centrifuged at 10,000 g for 20 min. The supernatant was incubated overnight with 250 μl IgG-Sepharose equilibrated in buffer D containing 1% Triton X-100. The resin was collected by centrifugation, washed once with buffer D containing 1% Triton X-100 and twice with buffer D containing 0.1% Triton X-100. HASte4p was specifically eluted by incubation with 20 U of TEV-protease in 400 μl buffer D containing 0.1% Triton X-100 for 5 h. Comparison of the amounts of total protein and HASte4p in yeast extracts (treated with TEV-protease) and eluted HASte4p preparations indicated that HASte4p was enriched over 1,000-fold in comparison to cell extracts. By immunoblotting both 3xmycCdc24p and Far1p were undetectable in HASte4p preparations (<0.01% of the starting level).
Binding Studies
Binding experiments were all carried out at 4°C. For binding of GSTCdc24 and MBP fusion proteins ∼10 μg of GSTCdc24 bound to glutathione-agarose was incubated with respective MBP fusion proteins in 100 μl of buffer C overnight. Glutathione-agarose samples were washed twice with 1 ml of buffer C and once with 1 ml of buffer B. Proteins were eluted with SDS-PAGE sample buffer and analyzed by SDS-PAGE followed by Coomassie blue staining or transfer to nitrocellulose, probing with anti-MBP mAb (Sigma) at 1:4,000 dilution and visualized by ECL. For binding experiments with yeast HASte4p, the HASte4p preparation was diluted 10-fold into buffer C and 100 μl was incubated with either resin bound GST or MBP fusions. MBP fusions were bound to amylose resin by incubation of ∼5 μg of each protein with 20 μl of amylose resin for 1 h. GSTCdc24–MBPFar1 was prepared by passing a bacterial extract (from 100 ml of cells) containing GSTCdc24 over a column with ∼500 μg of MBPFar1 bound to amylose resin. The column was washed with buffer B and then GSTCdc24–MBPFar1 was eluted with buffer B containing 10 mM maltose. The eluate was incubated with glutathione-agarose for 30 min which was then washed three times with buffer B. Proteins bound to the resin were analyzed by SDS-PAGE and Coomassie blue staining or used for HASte4p binding.
Mating and Pheromone Response Assays
Quantitative matings were carried out as described in (Arkowitz and Lowe, 1997; Nern and Arkowitz, 1998) with Mata cells as indicated and Matα RAY1135 cells. Pheromone induced cell cycle arrest and induction of a Fus1LacZ reporter were assayed as described (Nern and Arkowitz, 1998). For pheromone treatment ∼0.2 OD600 of log-phase cells were collected by centrifugation, resuspended in 2 ml YEPD containing 12 μM α-factor (synthesized by David Owen, MRC LMB) and incubated for 3 h. Cells were fixed with formaldehyde and actin was visualized as described (Nern and Arkowitz, 1998) using rhodamine phalloidin (Molecular Probes). To examine cell morphologies in mating mixtures, Mata cells were stained with 10 μg/ml Calcofluor white (Pringle, 1991) (Sigma) in YEPD for 5 min at rt and subsequently washed extensively with YEPD. Approximately 5 × 106 stained cells were then mixed with unstained Mata (RAY1135) cells and incubated on filters. After 2 h cells were washed from the filters, briefly sonicated, resuspended in PBS and fixed with formaldehyde. Images of cells were taken using a Zeiss Axioskop microscope with either a NA 1.4 ×63 or NA 1.3 ×100 objective and recorded with a Princeton Instrument Micromax CCD camera. Fluorescence and differential interference-contrast (DIC) images were merged to permit identification of Mata cells.
Localization of Cdc24p
Cdc24HAGFP (p414Cdc24HAGFP) was transformed into RAY931 which is deleted for CDC24 and kept alive by the rescuing plasmid pEG(KT)Cdc24 (Nern and Arkowitz, 1998). This strain was able to lose the rescuing plasmid (both by extensive growth in SC-trp media or counter-selection on 5-FOA) as determined by markers and PCR, resulting in RAY1360, indicating that Cdc24HAGFP was functional. RAY1360 grew normally at 22°C, 30°C, and 37°C on YEPD plates. Budding patterns were determined as described (Arkowitz and Lowe, 1997) and mating efficiency was determined as described above. Cdc24HAGFP expression and size was verified by SDS-PAGE, immunoblotting, probing with 12CA5 mAb and ECL visualization. Confocal microscopy was carried out as described (Arkowitz and Lowe, 1997) except cells were grown in SC supplemented with 55 μg/ml adenine to reduce fluorescence due to ade2. Pheromone treatment was with 140 μM α-factor. Cells were imaged after 1 h in order to observe early localization. For latrunculin A treatment of budding cells 2 μl of either 10 mM latrunculin A (Molecular Probes) in DMSO or DMSO was added to 200 μl of log-phase cells (final concentration latrunculin A 0.1 mM) and cells were incubated for 3 h (Ayscough et al., 1997). For latrunculin A treatment of shmoos, cells were incubated with 140 μM α-factor for 1 h and then 0.1 mM latrunculin A or DMSO was added to cells which were incubated for 2 h. After observation by confocal microscopy, actin depolymerization was confirmed by staining fixed cells with rhodamine phalloidin as described above.
Results
The Cdc24p–Gβγ Interaction Requires FAR1 but Not Pheromone-dependent Signaling
Cdc24-m alleles are defective in growth orientation along a pheromone gradient, yet do not affect pheromone-dependent MAP-kinase pathway signaling (Nern and Arkowitz, 1998). These mutants are also unable to interact with Gβ (Ste4p) in two-hybrid assays. In Cdc24p–Gβ two-hybrid assays Gβ was overexpressed, which has been shown to activate the MAP-kinase pathway (Whiteway et al., 1990). Hence it was possible that MAP-kinase signaling is required for this interaction. Two-hybrid experiments revealed that while Cdc24p and Gβ interact in a haploid strain, no detectable interaction was observed in a diploid (compare Fig. 1, A and B), in which several mating specific proteins, including Gγ (Ste18p), are not expressed (Sprague and Thorner, 1992). Gγ is required for this interaction in a haploid (Nern and Arkowitz, 1998). Surprisingly, overexpression of Gγ in a diploid did not restore the Cdc24p–Gβ interaction, whereas overexpression of Gγ in a Δste18 haploid restored this interaction (Table II). This result is consistent with the notion that either a haploid specific component and/or pheromone-dependent signaling is required for the Cdc24p–Gβγ interaction.
Figure 1.

Two-hybrid interaction between Cdc24p and Gβ requires Gγ and Far1p. (A) Overexpression of Gγ and Far1p enhances the Cdc24p–Gβ interaction. Assays were carried out in a Y187 strain grown in SC-leu-trp-ade or SC-leu-trp-ade-his. LacZ values are the average of three to five determinations with bars showing standard deviation. As indicated, SPA2 (NH2-terminal 153 amino acids), CDC24, STE4, STE18, or FAR1 (entire ORF) were fused either to GAL4 activation domain (AD) or GAL4 DNA binding domain (DBD). Spa2 serves as a DBDCdc24 negative control and Cdc24-m1 serves as an ADSte4 negative control. For overexpression, NLSHA-STE18 and NLSHA-FAR1 in plasmids p2μATPI and p413TPI were used. (B) Optimal interaction between Cdc24p and Gβ in a diploid requires Gγ and Far1p. Assays were carried out in a diploid strain made by crossing SFY526 and Y187 as described above. (C) Cdc24p–Gβ interaction requires FAR1 orientation function. Assays were carried out in a Y187 strain in which FAR1 was either deleted (RAY1183) or replaced by far1-H7 (RAY1182).
Table II.
Requirements of Cdc24p Ste4p Interaction
| Strain | Cdc24p Ste4p interaction | +Ste18p | ||
|---|---|---|---|---|
| Δste4 | ++ | ND | ||
| Δste18 | - | ++ | ||
| Δste5 | - | + | ||
| Δste20 | - | ± | ||
| Δste11 | - | + | ||
| Δste7 | - | + | ||
| Δkss1 | ++ | ND | ||
| Δfus3 | ++ | ND | ||
| Δste12 | - | ± | ||
| Δfar1 | - | - | ||
| Δbem1* | ++ | ND | ||
| Δakr1 | ++ | ND | ||
| Δbud6 | ++ | ND | ||
| Δskm1 | ++ | ND |
Two-hybrid assays were carried out in strain PJ69-4A with the indicated gene deletions. Identical results were obtained with at least three transformants. ++ Denotes clear growth on selective plates lacking histidine.
Plates incubated at 25°C.
To examine the role of the pheromone-dependent MAP- kinase pathway in the Cdc24p–Gβγ interaction, two-hybrid strains were constructed in which each component of this pathway was deleted. The MAP-kinase scaffolding protein Ste5p, the PAK kinase Ste20p which phosphorylates Ste11p, the MAPKKK Ste11p, the MAPKK Ste7p, the MAPK Fus3p or Kss1p, and the transcription factor Ste12p were each individually disrupted in a two-hybrid strain. In addition, the Ste20p homologue Skm1p, the cyclin-dependent kinase inhibitor required for α-factor cell cycle arrest Far1p, the polarity establishment protein Bem1p, the Gβγ effector Akr1p, and the bipolar bud site selection protein Bud6p were deleted from this strain. Several of these proteins including Ste5p, Fus3p, and Far1p are only expressed in haploids (Sprague and Thorner, 1992) and thus are candidates for haploid specific components required for the Cdc24p–Gβγ interaction. Deletion of SKM1, BEM1, AKR1, or BUD6 had no effect on the Cdc24p–Gβ interaction. In contrast, removal of any component of the pheromone-dependent MAP-kinase pathway (with the exception of Fus3p and Kss1p which are functionally redundant for mating) resulted in the loss of the Cdc24p–Gβ two-hybrid interaction. Because Gγ appeared to be required for the Cdc24p–Gβ interaction (Nern and Arkowitz, 1998), we examined whether overexpression of Gγ was able to restore the Cdc24p–Gβ interaction in these strains. Table II shows that overexpression of Gγ partially restored the Cdc24p–Gβ interaction in Δste5, Δste11, and Δste7 strains and to a lesser extent in Δste20 and Δste12 strains. These results indicate that signaling through the pheromone-dependent MAP-kinase cascade per se is not required for the Cdc24p–Gβγ interaction. However, deletion of FAR1 resulted in a loss of the Cdc24p–Gβ interaction which was not restored upon overexpression of Gγ (Table II and Fig. 1 C), suggesting that Far1p may be essential for this interaction.
The requirement for Gγ and Far1p in the Cdc24p–Gβ two-hybrid interaction suggested an explanation for the low level of LacZ reporter activity observed in the haploid Y187 two-hybrid strain and the absence of an interaction in the diploid two-hybrid strain, namely that these two proteins were limiting in haploids and absent in diploids. To test this possibility, we overexpressed Gγ and Far1p individually and together in the Y187 haploid two-hybrid strain. Fig. 1 A shows that overexpression of Gγ in the presence of pAS1Cdc24 and pGAD424Ste4 resulted in an approximately twofold increase in LacZ activity, whereas the additional overexpression of Far1p resulted in a further increase in LacZ activity by ∼3.5-fold. In diploids, overexpression of Gγ did not result in a Cdc24p–Gβ interaction. However overexpression of Far1p resulted in LacZ reporter activity (Fig. 1 B) and this was further increased by additional overexpression of Gγ, suggesting that in the absence of pheromone-dependent signaling Far1p is sufficient for restoring the Cdc24p–Gβγ interaction.
FAR1 is necessary for both pheromone-dependent growth arrest and oriented growth towards a pheromone gradient (Chang and Herskowitz, 1990; Valtz et al., 1995). These two functions of FAR1 can be separated, with the Far1p NH2 terminus necessary for cell cycle arrest and the COOH terminus necessary for growth orientation. Our two-hybrid results indicate FAR1 is necessary for the Cdc24p–Gβ interaction, yet it is unclear which function of FAR1 this corresponds to. Because cdc24-m and far1-s appear phenotypically identical and both exhibit orientation defects (Valtz et al., 1995; Nern and Arkowitz, 1998), we examined the effect of the far1-s allele far1-H7 on this interaction. This far1 mutation results in a COOH-terminal 75– amino acid deletion and despite its orientation defect is normal for cell cycle arrest. Fig. 1 C shows that a far1-H7 mutation prevents the Cdc24p–Gβ interaction. These results suggest that the FAR1 orientation function is required for the Cdc24p–Gβ association, consistent with the role of this interaction in growth orientation.
We next investigated whether Far1p interacted with Cdc24p and Gβ. Fig. 1 and Table III show that in two-hybrid assays Far1p can interact with both Cdc24p and Gβ. The Cdc24p–Far1p interaction was observed in strains deleted for STE4, STE18, FUS3, or STE12, indicating that it does not require Gβγ nor pheromone-dependent MAP-kinase signaling (including Fus3p-dependent phosphorylation of Far1p). Similarly, the Far1p–Gβ interaction did not require STE18, FUS3, or STE12. The Far1p–Gβ interaction also did not require the CDC24 orientation function as we observed this interaction in a cdc24-m1 two-hybrid strain. In addition we examined Cdc24-m1p, which we had previously shown does not interact with Gβ, and found that Cdc24-m1p also did not interact with Far1p (data not shown). Together these results suggest that Far1p and Cdc24p can associate and this association is independent of pheromone signaling.
Table III.
Far1p Interacts with Cdc24p and Ste4p
| Strain | Far1 Cdc24 interaction | Far1 Ste4 interaction | ||
|---|---|---|---|---|
| Wild-type | ++ | ++ | ||
| Δste4 | ++ | ++ | ||
| Δste18 | ++ | ++ | ||
| Δfus3 | ++ | ++ | ||
| Δste12 | ++ | ++ | ||
| cdc24-m1 | NΔ | ++ |
Two-hybrid assays were carried out in strain PJ69-4A with the indicated gene replacements. Identical results were obtained with at least three transformants. ++ Denotes growth on SC-leu-trp-his or in the case of cdc24-m1 SC-leu-ura-his.
Far1p Binds Cdc24p
To further investigate these interactions epitope-tagged versions of Far1p and Cdc24p were constructed. Myc and protein A domains were fused to Cdc24p and Far1p, respectively and these fusions were used to replace wild-type genes. Far1-protein A fusions had a tobacco etch virus (TEV) protease cleavage site between Far1p and protein A to allow specific elution. These strains grew normally and exhibited normal vegetative morphology. Furthermore, both fusions mated with similar efficiencies as a wild-type strain when crossed to a wild-type tester or to an enfeebled tester. Far1-protein A fusions promoted normal cell cycle arrest and cells carrying this fusion formed shmoos that appeared normal upon exposure to mating pheromone. Together these results indicated that the fusion proteins were functional. Fig. 2 shows that when Far1-protein A was isolated with IgG-Sepharose, myc-tagged Cdc24p was bound (compare lanes 1 and 2 with 3 and 4). When Cdc24p or Far1p orientation mutants were used, a substantial decrease in the amount of Cdc24p bound to Far1p was observed in both cases (lanes 5–8). These results reveal the molecular basis for the similar phenotypes of cdc24-m1 and far1-H7 mutants. Although two-hybrid results indicated a Far1p–Gβ association, this was apparently not stable enough to observe by immunoprecipitation.
Figure 2.

Cdc24p specifically binds Far1p in vivo. Extracts from strains (lanes 1 and 2 RAY1254; lanes 3 and 4 RAY1258; lanes 5 and 6 RAY1260; lanes 7 and 8 RAY1336) with an activated pheromone response carrying Far1-protein A (lanes 3–6), Far1-H7-protein A (lanes 7 and 8), 3xmycCdc24 (lanes 1–4 and 7 and 8), and 3xmycCdc24-m1 (lanes 5 and 6) were incubated with IgG-Sepharose and bound proteins analyzed by 10% SDS-PAGE. Upper panel was probed with anti-myc, lower panel with anti-Far1p serum. Bacterially expressed MBPFar1 was added to the sample in lane 1.
To address whether these protein interactions were direct, binding experiments were carried out using purified proteins. Far1p and the NH2-terminal half of Cdc24p (amino acids 1–472; Nern and Arkowitz, 1998) were purified from bacteria as fusions to maltose binding protein (MBP) and glutathione-S-transferase (GST), respectively. Fig. 3 A shows that MBPFar1 bound GSTCdc24 but not GST alone. MBPFar1-H7 (Fig. 4 A) does not significantly bind GSTCdc24p, consistent with immunoprecipitation results above (Fig. 2). In these binding experiments an excess of GSTCdc24 is used and an increase in MBPFar1 binding occurs as its concentration in the binding reaction is increased (Fig. 3 B). These results demonstrate that Far1p can bind Cdc24p directly in the absence of other proteins.
Figure 3.
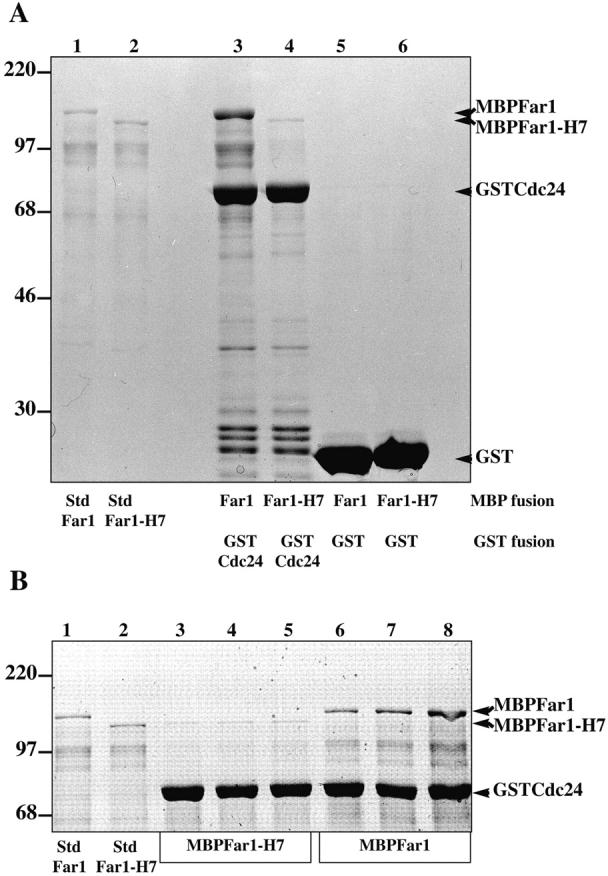
Cdc24p directly binds Far1p. (A) MBPFar1 (0.3 μM, lanes 3 and 5) or MBPFar1-H7 (0.3 μM, lanes 4 and 6) was incubated the NH2-terminal half of Cdc24 fused to GST denoted GSTCdc24 (1 μM, lanes 3 and 4) or GST (3 μM, lanes 5 and 6) bound to glutathione-agarose. Bound proteins were analyzed by 10% SDS-PAGE, and Coomassie blue staining. Lanes 1 and 2 show standards representing 5% of added MBPFar1 or MBPFar1-H7. Bands at ∼97 kD are breakdown products of MBPFar1 and MBPFar1-H7 that reacted with anti-MBP mAb hence lack the COOH termini. While full-length MBPFar1 was substantially enriched over this MBPFar1 fragment in GSTCdc24 pulldowns, these breakdown products were still observed in resin eluates consistent with MBPFar1 oligomerization. (B) Concentration dependence of MBPFar1 binding to GSTCdc24. MBPFar1-H7 (lanes 3, 4, and 5) and MBPFar1 (lanes 6, 7, and 8) at concentrations 75 nM (lanes 3 and 6), 150 nM (lanes 4 and 7), and 300 nM (lanes 5 and 8) was added to 1 μM GSTCdc24 bound to glutathione-agarose. Samples were analyzed as described above. Lanes 1 and 2 show standards representing 20% (for lanes 3 and 6), 10% (for lanes 4 and 7), and 5% (for lanes 5 and 8) of added MBPFar1 or MBPFar1-H7.
Figure 4.


Delineation of Far1p region necessary for Cdc24p and Gβ binding. (A) Far1p fragments. Far1 fragments contained amino acid residues 133–831 for Far1, residues 133–756 for Far1-H7, residues 133–297 for Far1ΔC, residues 638–831 for Far1ΔN, and residues 638–756 for Far1-H7ΔN fused to MBP. (B) The Far1p COOH terminus is necessary for Cdc24p binding. GSTCdc24 (0.9 μM) bound to glutathione-agarose was incubated with MBP fusions (∼0.5 μM) as indicated. Samples were analyzed by 10% SDS-PAGE, followed by immunoblotting with anti-MBP mAb and ECL. Lanes 1–6 show standards representing 5% of added MBP fusions and lanes 7–12 show resin eluates. GST alone did not bind any MBP fusions. (C) The NH2 terminus of Far1p is necessary for binding yeast Gβ. Indicated MBP fusions bound to amylose resin were incubated with yeast HASte4p purified from RAY1276. Samples were prepared and analyzed as described above by 10% SDS-PAGE using anti-HA mAb. Anti-MBP immunoblots revealed lanes 4–7 had similar amounts of MBP fusions whereas lanes 2 and 3 had approximately two- to threefold less MBP fusion. Lanes 1 and 8 show standard representing 12.5% and 6.2% of added HASte4p, respectively.
These binding studies demonstrated that the COOH terminus of Far1p is necessary for GSTCdc24 binding, hence we examined if this region was also sufficient for binding. Fig. 4 B shows that a 200–amino acid Far1p COOH-terminal fragment (lane 10) is not sufficient for GSTCdc24 binding and furthermore an NH2-terminal Far1p fragment did not bind GSTCdc24 (lane 9). MBPFar1-H7 and MBPFar1ΔC, which do not bind GSTCdc24 are unlikely to be grossly misfolded as they retain the ability to bind Gβ (see below). These results indicate that although the COOH terminus of Far1p is necessary for binding Cdc24p it is not sufficient. An immunoblot of the MBPFar1 bound to GSTCdc24 (Fig. 4 B, lane 7) revealed that proteolytic fragments of Far1 with as little as 25 kD of the NH2-terminus (approximately residues 133–350) are coprecipitated with MBPFar1 and GSTCdc24. This region of Far1p, which includes a Lim domain (Sanchezgarcia and Rabbitts, 1994), does not bind Cdc24p directly (Fig. 4 B, lane 9), suggesting that this region may mediate Far1p multimerization.
Far1p Binds Gβ
Hemagglutinin (HA)-tagged Gβ (Ste4p) was purified from yeast in order to examine its binding to MBPFar1 (Fig. 4 C). For this purpose a strain in which the wild-type copy of STE4 was replaced with HASte4-(TEV)-protein A was used. This fusion was functional for mating and cell cycle arrest. The Ste4p fusion was isolated with IgG-Sepharose and eluted by specific cleavage between Ste4p and the protein A domains using TEV protease, yielding HASte4p which was over 1,000-fold enriched compared with cell extracts. This HASte4p preparation had undetectable levels of Far1p and Cdc24p (<0.01% of the starting level, data not shown). Aliquots of HASte4p were incubated with various MBPFar1 fragments immobilized on amylose resin and bound HASte4p was analyzed by SDS-PAGE and immunoblotting. Fig. 4 C shows that HASte4p bound equally well to MBPFar1 and MBPFar1-H7 (lanes 2 and 3), whereas both NH2- and COOH-terminal Far1p fragments bound substantially less HASte4p (lanes 4–6). Of these smaller Far1p fragments, only Far1ΔC (amino acid residues 133–297) bound substantial amounts of HASte4p, suggesting that this region which includes the Lim domain is involved in Gβ binding. These experiments show that Gβ can bind Far1p in the absence of Cdc24p and suggest that perhaps Gβ and Cdc24p bind to different regions of Far1p.
A Cdc24p-Far1p-Gβ Complex
Since both Cdc24p and Far1p bind Gβ, we addressed whether the addition of MBPFar1 to GSTCdc24 bound to glutathione-agarose could compete for HASte4p (Gβ) binding. Fig. 5 A shows that addition of MBPFar1 did not prevent HASte4p binding to GSTCdc24 (compare lanes 1 and 2 with 3 and 4), but rather increased binding by about twofold. Coomassie blue staining of glutathione-agarose eluates revealed that MBPFar1 bound GSTCdc24. These results indicate that Far1p binding to Cdc24p does not displace Gβ, and are consistent with the formation of a complex of all three proteins.
Figure 5.

Cdc24p, Far1p, and Gβ form a trimeric complex. (A) Cdc24p binds Gβ in the presence of MBPFar1. GSTCdc24 (0.7 μM) bound to glutathione-agarose was incubated with yeast HASte4p and 0.5 μM MBPFar1 or 7.4 μM MBP as indicated. Samples prepared and analyzed by 10% SDS-PAGE as described in Fig. 4 C (α-HA) or analyzed by Coomassie blue staining. For the α-HA blot 10% of added HASte4p was used as a standard. GST alone did not bind HASte4p. (B) A trimeric Cdc24p-Far1p-Gβ complex. GSTCdc24-MBPFar1 was prepared and ∼10% of this complex was analyzed by SDS-PAGE and Coomassie blue staining. Approximately 30% GSTCdc24-MBPFar1 or GSTCdc24 alone was incubated with yeast HASte4p. Samples were prepared and analyzed as described above using anti-HA mAb (α-HA) or anti-MBP mAb (α-MBP). For the α-HA blot 2.5% of added HASte4p was used as a standard.
To directly test whether a complex of all three proteins could form we determined whether a stoichiometric complex of Cdc24p–Far1p could bind Gβ (Ste4p). GSTCdc24– MBPFar1 was isolated by sequential purification using amylose and glutathione resin. Fig. 5 B shows that GSTCdc24–MBPFar1 contained roughly equal amounts of these two fusion proteins. Purified HASte4p was then incubated either with this complex or GSTCdc24 alone. Densitometric quantification showed that twofold more HASte4p bound to Cdc24p–Far1p (Fig. 5 B compare lanes 1 and 2) than to a similar amount of Cdc24p alone, demonstrating that trimeric Cdc24p-Far1p-Gβ can form. This increase in Gβ binding does not appear to be cooperative and is more likely to be the sum of contributions from Cdc24p and Far1p. Because both Far1p and Cdc24p can individually bind each other or Gβ it is likely that in a trimeric complex each protein contacts the other two proteins. These binding studies together with the two-hybrid results suggest that Cdc24p-Far1p-Gβ is necessary for mating projection orientation.
Cdc24p and Far1p Function in the Same Shmoo Orientation Process
To examine if CDC24 and FAR1 function in the same process we compared the mating efficiencies of both single and double Δfar1 and cdc24-m1 mutants. Fig. 6 shows that the presence of a cdc24-m1 mutation in a Δfar1 background did not result in a further decrease in mating efficiency, suggesting that FAR1 and CDC24 function in the same orientation process. The mating defect of the double mutant is closer to that of the Δfar1 mutant that in addition to a chemotropism defect does not arrest growth in response to mating pheromone. If cdc24-m1 affected chemotropism similarly to Δfar1, then a double mutant with Δspa2, a gene required for the default mating pathway (Dorer et al., 1995), should have a mating defect greater than the product of the individual mating defects, a phenomenon known as synthetic sterility (Dorer et al., 1995). Fig. 6 shows that Δspa2 cdc24-m1 mutants exhibited synthetic sterility.
Figure 6.

CDC24 and FAR1 function in same shmoo orientation pathway. Quantitative matings were carried out with a wild-type tester (RAY1135) and mating efficiencies (number of diploid cells divided by total number of cells) are the average of three to five determinations with wild-type mating efficiency (42%) set to 100%. Strains RAY1034, RAY1035, RAY1109, RAY1111, RAY1246, RAY1248, RAY1271, RAY1160, RAY1173, and RAY1168 were used. Bars indicate standard deviation.
We also examined genetic interactions between cdc24- m1 and Δbem1 or Δste20, two genes involved in polarized growth and mating. Bem1p is associated with the cytoskeleton (Leeuw et al., 1995), binds Cdc24p (Peterson et al., 1994; Zheng et al., 1995), and Far1p (Lyons et al., 1996). Bem1 mutants are unable to form shmoos and instead form round cells in the presence of mating pheromone (Chenevert et al., 1992). ΔBem1 cdc24-m1 cells showed similar temperature sensitive growth and morphological defects (large round cells) as Δbem1 cells, providing further evidence that cdc24-m1 has no effect on vegetative growth. Even in cells lacking BEM1 which cannot form shmoos, cdc24-m1 resulted in a substantial decrease in mating efficiency (Fig. 6), i.e., synthetic sterility. Because deletion of the PAK kinase STE20 in our strain background did not result in complete sterility, we were able to examine the mating defect of Δste20 cells in the presence and absence cdc24-m1. In the absence of STE20, cdc24-m1 resulted in a further decrease in mating efficiency. Together these results suggest that in Δbem1 and Δste20 mutants, which are unable to form shmoos, polarization may still be necessary for mating perhaps for the localization of proteins necessary for cell fusion. Furthermore, because BEM1 and STE20 are not required for default mating (Dorer et al., 1997), such synthetic mating defects with cdc24-m1 are consistent with a genetic linkage between shmoo formation and orientation.
Cdc24p Localization
If Cdc24p transmits signals from bud site selection proteins or Gβγ, it might be localized to regions of polarized growth. We therefore examined the localization of a Cdc24p green fluorescent protein (GFP) fusion. Cdc24HAGFP expressed from its own promoter on a CEN plasmid complemented Δcdc24 as determined by growth at different temperatures, budding patterns, and mating efficiencies (data not shown). Fig. 7 A shows the localization of Cdc24HAGFP in living cells at different stages in the cell cycle. In unbudded cells Cdc24p localized as a tight patch at the membrane, and in cells with small buds at the growing end. In larger buds, this localization became more spread out. Finally, during cytokinesis Cdc24p generally localized to the mother-bud neck. Curiously, a preliminary report showed that an overexpressed GSTCdc24 fusion protein had a circumcellular distribution in budding cells (Pringle et al., 1995). Cdc24HAGFP also localized to sites of polarized growth after α-factor treatment. Fig. 7 B shows different shmoos in which Cdc24HAGFP is observed as a patch at the tip of the mating projection. Cdc24HAGFP was localized similarly in mating mixtures (data not shown). Furthermore, as the sole copy of Cdc24p in a Δcdc24 strain, Cdc24-m1HAGFP also localized to sites of polarized growth in budding and mating cells (data not shown), indicating that this mutant is not defective in its localization to sites of polarized growth. These data demonstrate that Cdc24p localizes to sites of polarized growth.
Figure 7.
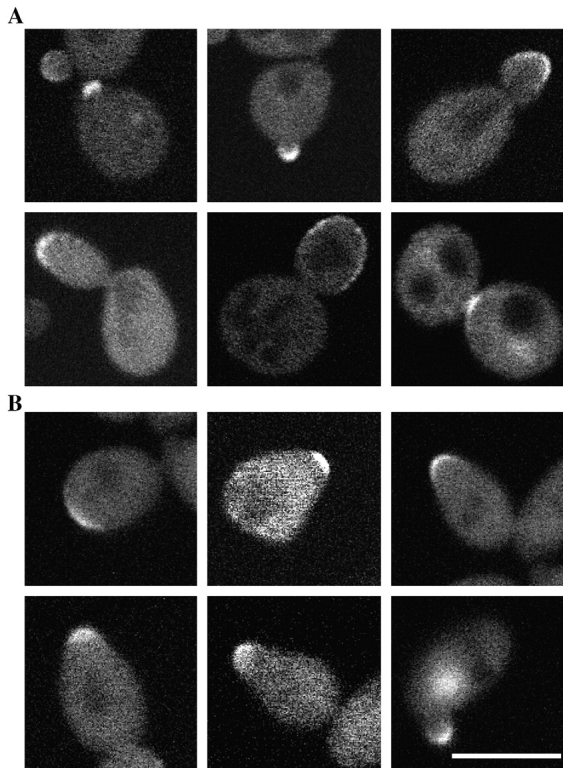
Cdc24p localizes to sites of polarized growth. (A) Cdc24p localizes to sites of polarized growth in budding cells. Confocal micrographs of live Δcdc24 p414Cdc24HAGFP cells (RAY1360) at different stages in the cell cycle. At each stage in the cell cycle background fluorescence was observed in the cytosol that varied from cell to cell. (B) Cdc24p localizes to the shmoo tip. Cells treated with 140 μM α-factor for 1 h were imaged as described above. Bar, 5 μm.
The early localization of Cdc24p in the cell cycle and its localization to the shmoo tip are consistent with its function in polarity establishment. The localization of Cdc24p is similar to that of its substrate Cdc42p (Ziman et al., 1993). To determine whether the actin cytoskeleton was necessary for polarized Cdc24p localization, budding and shmooing cells were treated with the actin depolymerizing drug latrunculin A (Ayscough et al., 1997). Fig. 8 A shows that even in the absence of actin polymerization, Cdc24p is localized to sites of polarized growth in budding cells. In contrast, latrunculin A treatment of shmoos resulted in a substantial decrease in Cdc24p localization (Fig. 8 B). Upon latrunculin A treatment the number of cells with Cdc24p localized to the shmoo tip decreased by fivefold (n = 100) and in cells that exhibited localized Cdc24p, there appeared to be a decrease in the amount of Cdc24HAGFP localized to the shmoo tip and an increase in fluorescence throughout the cell. These results are consistent with the effects of latrunculin A on Cdc42p localization in budding and shmooing cells (Ayscough et al., 1997; Ayscough and Drubin, 1998).
Figure 8.
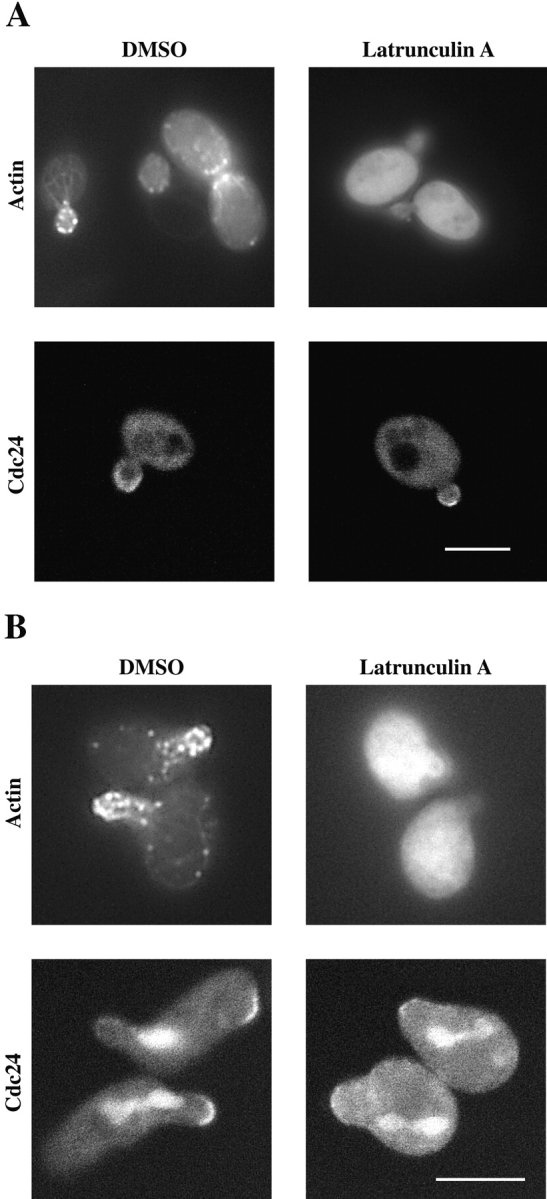
Cdc24p localization requires the actin cytoskeleton in shmoos but not budding cells. (A) Cdc24p localization in budding cells does not require the actin cytoskeleton. ΔCdc24 p414Cdc24HAGFP cells (RAY1360) were treated with either 0.1 mM latrunculin A or DMSO for 3 h. Cdc24HAGFP was detected as described in Fig. 7 A. Cells were also fixed and stained with rhodamine phalloidin to visualize actin cytoskeleton. (B) Cdc24p localization in shmoos requires the actin cytoskeleton. RAY1360 cells were treated with 140 μM α-factor for 1 h and then either 0.1 mM latrunculin A or DMSO was added for 2 h. Cdc24HAGFP and actin cytoskeleton were visualized as described above. Note the upper cell in Cdc24 panel treated with DMSO is initiating a second mating projection where Cdc24HAGFP is observed. Fluorescent material within cells in Cdc24 panels is attributed to fluorescence due to ade2. Bar, 5 μm.
In the Absence of CDC24- or FAR1-mediated Chemotropism the Bud Site Selection Machinery Is Essential for Shmoo Formation
During mating, a pheromone gradient serves as the external cue for growth orientation. This external signal allows haploid cells to orient growth in a pheromone gradient emanating from any direction (Madden and Snyder, 1992), whereas the site for bud formation in haploids is fixed adjacent to the previous bud site (Chant and Pringle, 1995). The selection of a site for the mating projection must override the fixed location of the bud. If Cdc24p acts as a switch between internal signals during budding and external signals during mating (Nern and Arkowitz, 1998), we would predict that bud site selection proteins become important for cell mating when the capacity for shmoo orientation is lost in mutants such as cdc24-m1.
The ras related small G-protein Bud1p/Rsr1p is essential for bud site selection, yet is not required for chemotropic or default mating in saturating pheromone (Roemer et al., 1996; Dorer et al., 1997). However Bud1p can directly associate with Cdc24p (Zheng et al., 1995; Park et al., 1997) and this association is likely to be functionally important (Bender and Pringle, 1989; Michelitch and Chant, 1996). We therefore examined the phenotype of Δbud1 cdc24-m1 double mutants to determine if the loss of CDC24-mediated chemotropism caused a BUD1-dependent mating defect. Both Δbud1 and Δbud1 cdc24-m1 cells grew normally, were not temperature sensitive for growth, and had the expected random budding pattern (data not shown). Strikingly, the Δbud1 cdc24-m1 double mutant showed a stronger mating defect (an eightfold further decrease in mating efficiency) than cdc24-m1 alone (Fig. 9 A). In contrast, Δbud1 alone had no effect on mating efficiency in agreement with previous studies (Chant and Herskowitz, 1991; Dorer et al., 1997). Microscopic observation of Δbud1 cdc24-m1 double mutants treated with a high concentration of mating pheromone (Fig. 9 B) or exposed to pheromone gradients in mating mixtures (Fig. 9 C) revealed that these cells were defective in shmoo formation. Instead of forming typical pear-shaped shmoos most cells were enlarged and round. On closer inspection a small protrusion was occasionally observed on these cells. Furthermore, the actin cytoskeleton in the double mutants was depolarized, with actin cortical patches and cables disorganized (Fig. 9 D). In contrast, both Δbud1 and cdc24-m1 single mutants formed shmoos. Otherwise Δbud1 cdc24-m1 double mutants responded normally to pheromone by undergoing cell cycle arrest and pheromone-dependent gene induction (data not shown). These results suggest that in the absence of chemotropism, BUD1 and perhaps the bud site selection machinery becomes essential for shmoo formation. Surprisingly, in saturating uniform concentrations of mating pheromone Δbud1 does not result in a mating defect (Dorer et al., 1997), raising the possibility that this novel role of BUD1 is revealed specifically when signaling from Gβγ to Cdc24p is blocked.
Figure 9.

In the absence of chemotropic signaling BUD1 functions in mating. (A) Combination of Δbud1 with either cdc24-m1 or Δfar1 results in a synthetic mating defect. Quantitative mating carried out as described in Fig. 6. Strains RAY1034, RAY1139, RAY1035, RAY1142, RAY1109, RAY1249, RAY1342, and RAY1350 were used. (B) BUD1 is necessary for shmoo formation in pheromone treated cdc24-m1 cells. DIC images of α-factor treated (12 μM for 3 h) cells (genotype indicated). (C) BUD1 is necessary for shmoo formation in mating mixtures. Cells with genotype indicated stained with Calcofluor white, mated with a wild-type. Fluorescence and DIC images were merged. (D) Actin cytoskeleton is depolarized in cdc24-m1 Δbud1 cells treated with α-factor. Cells with genotype indicated were treated with α-factor and actin cytoskeleton visualized as described in Fig. 7 D. Bars, 5 μm.
Our results indicate that Far1p is required for signaling from Gβγ to Cdc24p. If the shmoo formation defect of Δbud1 cdc24-m1 cells is due to a defect in this signaling, a Δbud1 Δfar1 double mutant should show an analogous decrease in mating efficiency. ΔBud1 Δfar1 cells had a stronger mating defect (an eightfold decrease in mating efficiency) than Δfar1 cells. As a control the effect of Δbud1 was examined in a Δsst2 strain. ΔSst2 cells are supersensitive to mating pheromone as SST2 negatively regulates the heterotrimeric G-protein (Dohlman et al., 1996). Therefore Δsst2 cells mate as though they are saturated with mating pheromone, mating by the default pathway (Dorer et al., 1997). ΔBud1 Δsst2 cells had a similar mating defect as Δsst2 alone, indicating that the absence of chemotropic mating by itself is not sufficient to reveal BUD1 function in mating. These synthetic mating defects of Δbud1 with cdc24-m1 or far1 show that Bud1p, which normally functions in bud site selection, can play a role in shmoo formation, presumably by regulating Cdc24p.
Discussion
During mating yeast cells grow in a polarized fashion towards their mating partner (Segall, 1993; Dorer et al., 1995; Valtz et al., 1995; Nern and Arkowitz, 1998). Yeast cells are able to sense pheromone gradients and orient their actin cytoskeleton and secretion towards such a gradient. Here we show that a complex comprised of Cdc24p, Far1p, and Gβγ can form and is likely to be required for orientation towards a mating partner. The formation of this complex does not directly require signaling via the pheromone-dependent MAP-kinase pathway. Analyses of mating defects of double mutants indicate that FAR1 and CDC24 both function in the same cell orientation process. Cdc24p localizes to sites of polarized growth suggesting that Cdc24p-Far1p-Gβγ is localized. Cdc24p localization does not depend on the actin cytoskeleton during budding but does depend on the actin cytoskeleton during shmooing. In the absence of signaling from Gβγ to Cdc24p, the bud site selection protein Bud1p is required for shmoo formation, demonstrating a molecular link between growth site selection in mating and budding. Together these results suggest that binding of Gβγ to Far1p and Cdc24p creates an internal landmark for growth towards an external signal.
A Complex Comprised of Cdc24p, Far1p, and Gβγ Links External Signals to Cytoskeleton Orientation
Detection of a pheromone gradient and orientation of growth in such a gradient is a process central to yeast mating and is analogous to Dictyostelium chemotaxis and nerve cell chemotropism (Arkowitz, 1999). Alleles of both far1 (Valtz et al., 1995) and cdc24 (Nern and Arkowitz, 1998) are specifically defective in orientation towards a pheromone gradient. Cells mutant for the α-factor pheromone receptor (Ste2p) or the heterotrimeric G-protein, discriminate poorly between pheromone signaling and nonsignaling mating partners suggesting that these components are also required for chemotropism (Jackson et al., 1991; Schrick et al., 1997). Cdc24-m mutants are unable to interact with the Gβ subunit of the heterotrimeric G-protein (Nern and Arkowitz, 1998). These results led to a model in which Gβγ locally activates or recruits Cdc24p, which could then activate Cdc42p and other downstream targets required for cytoskeleton orientation. We conclude from two-hybrid, binding, and genetic data that Far1p is involved in signaling from Gβγ to Cdc24p by forming a complex with these proteins.
Our two-hybrid results suggest that the Far1p–Gβ interaction does not require the CDC24 orientation function, yet Far1p is essential for the Cdc24p–Gβ interaction. In contrast, in vitro binding experiments show that Cdc24p is able to bind to Gβ purified from bacteria (Nern and Arkowitz, 1998) and yeast in the absence of Far1p. We attribute this difference between two-hybrid and in vitro binding results to the different methods used. For example, in the two-hybrid experiments interactions occur in the nucleus and on the other hand the in vitro binding studies are carried out with high concentrations of purified proteins. We suggest that although Far1p is important for the Cdc24p–Gβ interaction it is not absolutely essential, whereas Cdc24p is not necessary for the Far1p–Gβ interaction. We have demonstrated that a triple complex comprised of Cdc24p-Far1p-Gβγ can form using purified proteins and believe that in vivo this complex links receptor activation to cytoskeleton organization (Fig. 10). These results show at a molecular level the role of Far1p in growth orientation. Consistent with the specific phenotype of far1 and cdc24 orientation alleles, we find that pheromone-dependent MAP-kinase cascade signaling is not necessary for the association of this complex. This result indicates that the MAP-kinase cascade is not directly required for chemotropic growth, in agreement with recent mating partner discrimination studies (Schrick et al., 1997). Furthermore, the formation of this complex does not require FUS3, which normally phosphorylates Far1p in a pheromone-dependent fashion (Chang and Herskowitz, 1992; Elion et al., 1993). This phosphorylation of Far1p is necessary for cell cycle arrest, indicating that the cell cycle arrest function of FAR1 is not required for interactions between Cdc24p, Far1p, and Gβγ.
Figure 10.

Model of Cdc24p signaling in growth orientation. Empty rings represent bud scars. Cdc24p is localized to the bud site during vegetative growth and upon exposure to a mating pheromone gradient associates with Far1p and Gβγ that is released by receptor activation (not shown). The dashed line between Cdc24p at the bud site and the site for oriented growth during mating indicates that Cdc24p can switch between these two locations. Presumably Bud1p is involved in signaling to Cdc24p at the bud site. It is assumed that signaling to Cdc24p during mating and budding regulates Cdc42p which is necessary for appropriate orientation of the actin cytoskeleton.
How is the formation of this protein complex regulated by pheromone activation of the receptor? Pheromone binding to the receptor is believed to trigger dissociation of Gα from Gβγ. Recent studies suggest Gα binds the pheromone receptor (Kallal and Kurjan, 1997; Medici et al., 1997) and that Gβγ must be membrane associated in order to function (Pryciak and Huntress, 1998). GFP fused to Gγ is localized preferentially to the plasma membrane of the mating projection after pheromone treatment (Nern and Arkowitz, unpublished observation). Upon pheromone stimulation, Far1p levels increase (Chang and Herskowitz, 1992; Valtz et al., 1995) resulting in increased levels of Cdc24p–Far1p (Nern and Arkowitz, unpublished observation). As Far1p localizes to the nucleus during vegetative growth (Henchoz et al., 1997), it would appear likely that Far1p must exit the nucleus in order to carry out its mating orientation function. We envision that released Gβγ recruits Cdc24p–Far1p to the vicinity of activated receptors and Cdc24p-Far1p-Gβγ ultimately directs the cytoskeleton towards this internal landmark. Such a mechanism provides a means of translating local activation of pheromone receptors to cytoskeletal orientation. We would predict that Far1p, like Cdc24p, localizes to the tip of the mating projection in pheromone treated cells.
While this work was being reviewed a paper examining the role of Far1p in polarized growth during mating was published (Butty et al., 1998). In general, our results agree with the findings of this work. The authors postulate that Far1p functions as an adaptor or linker between Gβγ and polarity establishment proteins including Cdc24p. Our in vitro binding results indicate that even in the absence of Far1p, Gβγ can still bind Cdc24p, suggesting that perhaps Far1p is not simply a physical adaptor but may have more complex functions. Overexpressed GFPFar1 was shown to relocalize from the nucleus to the cytoplasm upon treatment with a saturating uniform concentration of pheromone for two hours. In these conditions GFPFar1 does not appear to accumulate at shmoo tips. It will be interesting to determine whether wild-type levels of Far1p localizes similarly in cells exposed to a pheromone gradient for various times.
How are these protein interactions involved in transmitting spatial information? Previous studies have indicated that in a pheromone gradient, shmoo orientation improves as a function of time (Segall, 1993). This appears to be due to reorientation of the shmoo tip as it grows (Segall, 1993; Nern and Arkowitz, unpublished observation), indicating that shmoo orientation is a continuous process unlike bud site selection. Perhaps Cdc24p-Far1p-Gβγ dissociates reasonably fast such that this complex is continually dissociating and forming. Such a dynamic process would provide a means for continuous reorientation during mating and could play a central role in translating initial small differences in receptor occupancy into oriented growth.
Another difference between bud and shmoo formation is that in budding the polarity establishment proteins Cdc42p and Bem1p localize independent of the actin cytoskeleton (Ayscough et al., 1997) whereas in the latter process the actin cytoskeleton is necessary for the efficient localization of these proteins (Ayscough and Drubin, 1998). During shmoo formation the actin cytoskeleton requirement for localization of Cdc24p and these other polarity establishment proteins appears to be similar. Why is the actin requirement for localization of this group of proteins different in budding and shmooing cells? Perhaps the continuous nature of the shmooing process compared with the committed directional growth required for budding underlies this different dependence on the actin cytoskeleton. It will be important to examine the role of the actin cytoskeleton in cells responding to a pheromone gradient.
Coordination of Different Pheromone Responses
Pheromone stimulation results in gene induction, cell cycle arrest, and morphological changes (Sprague and Thorner, 1992; Chenevert, 1994; Leberer et al., 1997a). The timing and coordination of these different responses is important for efficient mating. Our genetic studies are consistent with Cdc24p and Far1p being part of the same protein complex functioning in growth orientation and we examined two additional genes that might have a role in coordinating various pheromone responses.
The PAK kinase Ste20p is important for MAP-kinase signaling during mating. It interacts with Bem1p, Ste4p, Ste5p, and Cdc42p (Leeuw et al., 1995; Zhao et al., 1995; Peter et al., 1996; Leberer et al., 1997b; Leeuw et al., 1998). Recent mating partner discrimination studies (Schrick et al., 1997) suggest that STE20 may not be required for chemotropism. Our two-hybrid results suggest that STE20 has some effect on the Cdc24p–Gβ interaction, however cdc24- m1 results in a further mating defect in Δste20 cells. While Ste20p binds Gβ, it is unclear how this association relates to the Far1p, Cdc24p, Gβγ interaction. Further studies will be necessary to elucidate the roles of STE20 in various aspects of mating.
Bem1p is required for polarized growth both during mating and budding (Bender and Pringle, 1991; Chenevert et al., 1992). ΔBem1 cells are defective in shmoo formation, mating pheromone-dependent cell cycle arrest and efficient signaling via the MAP-kinase cascade (Chenevert et al., 1992; Lyons et al., 1996). At the molecular level Bem1p interacts with many components required for polarized growth such as the G-protein Bud1p (Zheng et al., 1995; Park et al., 1997), Cdc24p (Peterson et al., 1994; Zheng et al., 1995), Far1p (Lyons et al., 1996), actin (Leeuw et al., 1995), Ste5p (Leeuw et al., 1995; Lyons et al., 1996), and Ste20p (Leeuw et al., 1995). Although Bem1p binds both Cdc24p and Far1p, Bem1p is not required for the formation of Cdc24p-Far1p-Gβγ. Results from Δbem1 cdc24-m1 mutants suggest that even for cells unable to form shmoos, polarization is important. Perhaps this is because the molecules necessary for cell fusion must be correctly localized. What is the molecular function of Bem1p in mating? We favor the idea that Bem1p acts as a scaffolding component linking pheromone-dependent MAP-kinase signaling, shmoo formation, and shmoo orientation.
Cdc24p as a Switch between Growth Site Selection in Mating and Budding
An attractive model (Fig. 10) is that Cdc24p acts as a selector switch that responds to input signals from bud site selection (Sloat et al., 1981) and mating projection orientation (Nern and Arkowitz, 1998). We envision that the localization and activation of Cdc24p is essential for its function in both bud site selection and mating projection orientation. During budding it is likely that local activation of the G-protein Bud1p marks the site for bud formation (Chant et al., 1991; Michelitch and Chant, 1996). The GTP bound form of Bud1p binds Cdc24p (Zheng et al., 1995; Park et al., 1997) and this interaction may be required for Cdc24p localization to the bud site. Interactions of Cdc24p with the bud site selection machinery dictate the site of mating projection growth in the absence of local activation of Cdc24p by Gβγ, such as in the case of far1 (Valtz et al., 1995) or cdc24 (Nern and Arkowitz, 1998) mutations or in the presence of saturating mating pheromone (Madden and Snyder, 1992; Dorer et al., 1995), wherein the mating projection forms adjacent to the previous bud. We show Bud1p becomes essential for shmoo formation specifically in the absence of signaling from Gβγ to Cdc24p. This demonstrates that the bud site selection machinery can function in shmoo formation. It is surprising that under these conditions, BUD1 functions in shmoo formation, while during budding it appears only to function in bud site selection and not bud formation. Interestingly, a specific role for BUD2 in bud formation has been observed in triple mutant combinations with Δcln1 and Δcln2 (Benton et al., 1993; Cvrckova and Nasmyth, 1993). A possible explanation for these different functions of BUD1 is that mating projection orientation is a continuous process, in contrast to bud site selection in which once a site for growth is chosen, subsequent directed growth is fixed to this site and there may no longer be a requirement for BUD genes.
We attribute the role of BUD1 in shmoo formation to a synthetic effect with cdc24/far1 suggesting this function of BUD1 is normally redundant yet revealed in the absence of Gβγ-mediated chemotropism. Recently it has been proposed that BUD1 is involved in cell fusion (Elia and Marsh, 1998), yet the effects of Δbud1 we observe in cdc24-m1 mutants, i.e., the inability to form a shmoo, are unlikely to be a result of its role in fusion as we observe this morphological defect in response to mating pheromone without a mating partner. Furthermore, in contrast to the results of Elia and Marsh (1998) but in agreement with previous studies (Dorer et al., 1997), Δbud1 does not result in a mating defect in our strain background. In addition, Δbud1 does not affect mating in the presence of saturating uniform mating pheromone concentration (Dorer et al., 1997). Therefore while both mating in the presence of saturating pheromone or mating in a cdc24 or far1 mutant block chemotropic growth, at the molecular level these two situations are not equivalent and this difference is consistent with the suggestion that Cdc24p must be localized or locally activated to function properly (Fig. 10). We imagine that during mating in saturating uniform pheromone concentrations, the Cdc24p-Far1p-Gβγ linkage is intact, but the external spatial signal is absent. In contrast, in a cdc24-m1 or Δfar1 mutant while the external signal is present, signaling from Gβγ to Cdc24p is prevented. Furthermore, the early localization of Cdc24p during shmoo and bud formation supports the proposed role of Cdc24p in linking a spatial landmark to polarity establishment.
A simple mechanism for growth site selection during mating and budding is that a threshold level of locally activated Cdc24p is necessary to catalyze the GDP-GTP exchange of Cdc42p. This activation of Cdc24p is presumably generated in part by Bud1p during budding and switched to the region of the cell adjacent to the pheromone source by released Gβγ during mating. In such a mechanism, it would not be necessary to inhibit or erase the incipient bud site during mating as previously suggested (Dorer et al., 1995). It is, however, possible that the binding of Cdc24p to Far1p results not only in an increased level of interaction with Gβγ but also a decrease in the amount of Cdc24p at the bud site, perhaps by decreasing its affinity for Bud1-GTP. We favor the notion of a balance between Cdc24p activation at the new bud site and at the region of the plasma membrane adjacent to pheromone source. We propose that Far1p serves to bias this equilibrium, i.e., shift the balance, towards the site for shmoo formation.
Cells from a variety of organisms undergo polarized growth in response to external signals. For example, in C. elegans embryonic development it is the sperm entry site that determines antero-posterior axis (Goldstein et al., 1993). In Dictyostelium, cell aggregation occurs via cAMP-mediated chemotaxis (Parent and Devreotes, 1996) and local activation of G-protein signaling events occurs in the absence of cell movement (Parent et al., 1998). Chemotaxis is necessary for cell migration responses for example of lymphocytes (Arkowitz, 1999). Chemotropism is also essential for axonal guidance and neuronal growth cone remodeling and extension (Tessier-Lavigne and Goodman, 1996). Such processes are crucial for tissue and organ development. Many of these chemotactic and chemotropic processes appear similar to chemotropism during yeast mating, in that they depend on chemoattractant gradients that are recognized and transmitted by a molecular machinery including G-protein coupled receptors, rho-family GTPases, and their exchange factors. Chemotropic growth in yeast is therefore a suitable model for understanding the molecular basis of many different chemotropic and chemotactic processes.
Acknowledgments
We thank S. Munro, M. Peter, and E. Schiebel for plasmids and antibodies. We are grateful to M. Bretscher, A. Gonzalez-Reyes, S. Munro, and H. Pelham for their critical reading of the manuscript.
This work was supported by the Medical Research Council. A. Nern was supported by a Marie Curie fellowship of the European Commission.
Abbreviations used in this paper
- GFP
green fluorescent protein
- GST
glutathione-S-transferase
- HA
hemagglutinin
- MAP
mitogen-activated protein
- MBP
maltose binding protein
- TPI
triose phosphate isomerase
- TEV
tobacco etch virus
Note Added in Proof:
We have now demonstrated that in the absence of CDC24-mediated chemotropism, in addition to Bud1p, other components of the general bud site selection machinery are important for shmoo formation (Nern, A., and R.A. Arkowitz, manuscript submitted for publication).
References
- Adams AE, Pringle JR. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic mutant Saccharomyces cerevisiae. . J Cell Biol. 1984;98:934–945. doi: 10.1083/jcb.98.3.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkowitz RA. Responding to attraction: chemotaxis and chemotropism in Dictyosteliumand yeast. Trends Cell Biol. 1999;9:20–27. doi: 10.1016/s0962-8924(98)01412-3. [DOI] [PubMed] [Google Scholar]
- Arkowitz RA, Lowe N. A small conserved domain in the yeast Spa2p is necessary and sufficient for its polarized localization. J Cell Biol. 1997;138:17–36. doi: 10.1083/jcb.138.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough KR, Drubin DG. A role for the yeast actin cytoskeleton in pheromone receptor clustering and signalling. Curr Biol. 1998;8:927–930. doi: 10.1016/s0960-9822(07)00374-0. [DOI] [PubMed] [Google Scholar]
- Ayscough KR, Stryker J, Pokala N, Sanders M, Crews P, Drubin DG. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin A. J Cell Biol. 1997;137:399–416. doi: 10.1083/jcb.137.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Baba N, Ohsumi Y, Kanaya K, Osumi M. Three-dimensional analysis of morphogenesis induced by mating pheromone alpha factor in Saccharomyces cerevisiae. . J Cell Sci. 1989;94:207–216. doi: 10.1242/jcs.94.2.207. [DOI] [PubMed] [Google Scholar]
- Bender A, Pringle JR. Multicopy suppression of the cdc24 budding defect in yeast by CDC42 and three newly identified genes including the ras-related gene RSR1. . Proc Natl Acad Sci USA. 1989;86:9976–9980. doi: 10.1073/pnas.86.24.9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Pringle JR. Use of a screen for synthetic lethal and multicopy suppressor mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. . Mol Cell Biol. 1991;11:1295–1305. doi: 10.1128/mcb.11.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Sprague GF., Jr Pheromones and pheromone receptors are the primary determinants of mating specificity in the yeast Saccharomyces cerevisiae. . Genetics. 1989;121:463–476. doi: 10.1093/genetics/121.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton BK, Tinkelenberg AH, Jean D, Plump SD, Cross FR. Genetic analysis of Cln Cdc28 regulation of cell morphogenesis in budding yeast. EMBO (Eur Mol Biol Organ) J. 1993;12:5267–5275. doi: 10.1002/j.1460-2075.1993.tb06222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumer KJ, Reneke JE, Thorner J. The STE2 gene product is the ligand binding component of the alpha-factor receptor of Saccharomyces cerevisiae. . J Biol Chem. 1988;263:10836–10842. [PubMed] [Google Scholar]
- Butty AC, Pryciak PM, Huang LS, Herskowitz I, Peter M. The role of Far1p in linking the heterotrimeric G protein to polarity establishment proteins during yeast mating. Science. 1998;282:1511–1516. doi: 10.1126/science.282.5393.1511. [DOI] [PubMed] [Google Scholar]
- Chang F, Herskowitz I. Identification of a gene necessary for cell cycle arrest by a negative growth factor of yeast: FAR1 is an inhibitor of a G1 cyclin, CLN2. . Cell. 1990;63:999–1011. doi: 10.1016/0092-8674(90)90503-7. [DOI] [PubMed] [Google Scholar]
- Chang F, Herskowitz I. Phosphorylation of FAR1in response to alpha-factor: a possible requirement for cell-cycle arrest. Mol Biol Cell. 1992;3:445–450. doi: 10.1091/mbc.3.4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J, Herskowitz I. Genetic control of bud site selection in yeast by a set of gene products that constitute a morphogenetic pathway. Cell. 1991;65:1203–1212. doi: 10.1016/0092-8674(91)90015-q. [DOI] [PubMed] [Google Scholar]
- Chant J, Pringle JR. Patterns of bud-site selection in the yeast Saccharomyces cerevisiae. . J Cell Biol. 1995;129:751–765. doi: 10.1083/jcb.129.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chant J, Corrado K, Pringle JR, Herskowitz I. Yeast BUD5, encoding a putative GDP-GTP exchange factor, is necessary for bud site selection and interacts with bud formation gene BEM1. . Cell. 1991;65:1213–1224. doi: 10.1016/0092-8674(91)90016-r. [DOI] [PubMed] [Google Scholar]
- Chenevert J. Cell polarization directed by extracellular cues in yeast. Mol Biol Cell. 1994;5:1169–1175. doi: 10.1091/mbc.5.11.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenevert J, Corrado K, Bender A, Pringle J, Herskowitz I. A yeast gene (BEM1)necessary for cell polarization whose product contains two SH3 domains. Nature. 1992;356:77–79. doi: 10.1038/356077a0. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Bertram G, Egerton M, Gow N, Falkow S, Brown A. Yeast-enhanced green fluorescent protein yEGFP: A reporter of gene expression in Candida albicans. . Microbiology UK. 1997;143:303–311. doi: 10.1099/00221287-143-2-303. [DOI] [PubMed] [Google Scholar]
- Cvrckova F, Nasmyth K. Yeast G(1) cyclins Cln1 and Cln2 and a gap-like protein have a role in bud formation. EMBO (Eur Mol Biol Organ) J. 1993;12:5277–5286. doi: 10.1002/j.1460-2075.1993.tb06223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzel C, Kurjan J. The yeast SCG1gene a Gα-like protein implicated the a- and α-factor response pathway. Cell. 1987;50:1001–1010. doi: 10.1016/0092-8674(87)90166-8. [DOI] [PubMed] [Google Scholar]
- Dohlman HG, Song JP, Ma DR, Courchesne WE, Thorner J. Sst2, a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: expression, localization, and genetic interaction and physical association with Gpa1 (the G-protein alpha subunit) Mol Cell Biol. 1996;16:5194–5209. doi: 10.1128/mcb.16.9.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorer R, Boone C, Kimbrough T, Kim J, Hartwell LH. Genetic analysis of default mating behavior in Saccharomyces cerevisiae. . Genetics. 1997;146:39–55. doi: 10.1093/genetics/146.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorer R, Pryciak PM, Hartwell LH. Saccharomyces cerevisiaecells execute a default pathway to select a mate in the absence of pheromone gradients. J Cell Biol. 1995;131:845–861. doi: 10.1083/jcb.131.4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Elia L, Marsh L. A role for a protease in morphogenic responses during yeast cell fusion. J Cell Biol. 1998;142:1473–1485. doi: 10.1083/jcb.142.6.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion EA, Satterberg B, Kranz JE. Fus3 phosphorylates multiple components of the mating signal transduction cascade–evidence for Ste12 and Far1. Mol Biol Cell. 1993;4:495–510. doi: 10.1091/mbc.4.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field C, Schekman R. Localized secretion of acid phosphatase reflects the pattern of cell surface growth in Saccharomyces cerevisiae. . J Cell Biol. 1980;86:123–128. doi: 10.1083/jcb.86.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, B., S.N. Hird, and J.G. White. 1993. Cell polarity in early C. elegans development. Dev. Suppl. 279–287. [PubMed]
- Harlow, E., and D. Lane. 1988. Antibodies: A Laboratory Manual. Cold Spring Harbor Press, Cold Spring Harbor, NY. 726 pp.
- Henchoz S, Chi Y, Catarin B, Herskowitz I, Deshaies RJ, Peter M. Phosphorylation- and ubiquitin-dependent degradation of the cyclin-dependent kinase inhibitor Far1p in budding yeast. Genes Dev. 1997;11:3046–3060. doi: 10.1101/gad.11.22.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschman JE, DeZutter GS, Simonds WF, Jenness DD. The G beta gamma complex of the yeast pheromone response pathway—subcellular fractionation and protein-protein interactions. J Biol Chem. 1997;272:240–248. doi: 10.1074/jbc.272.1.240. [DOI] [PubMed] [Google Scholar]
- Jackson CL, Konopka JB, Hartwell LH. S. cerevisiaealpha pheromone receptors activate a novel signal transduction pathway for mating partner discrimination. Cell. 1991;67:389–402. doi: 10.1016/0092-8674(91)90190-a. [DOI] [PubMed] [Google Scholar]
- James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallal L, Kurjan J. Analysis of the receptor binding domain of Gpa1p, the Gα subunit involved in the yeast pheromone response pathway. Mol Cell Biol. 1997;17:2897–2907. doi: 10.1128/mcb.17.5.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV, Adams AE. Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. . J Cell Biol. 1984;98:922–933. doi: 10.1083/jcb.98.3.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberer E, Thomas DY, Whiteway M. Pheromone signalling and polarized morphogenesis in yeast. Curr Opin Genet Dev. 1997a;7:59–66. doi: 10.1016/s0959-437x(97)80110-4. [DOI] [PubMed] [Google Scholar]
- Leberer E, Wu CL, Leeuw T, Fourest-Lieuvin A, Segall JE, Thomas DY. Functional characterization of the Cdc42p binding domain of yeast Ste20p protein kinase. EMBO (Eur Mol Biol Organ) J. 1997b;16:83–97. doi: 10.1093/emboj/16.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuw T, Fourest-Lieuvin A, Wu C, Chenevert J, Clark K, Whiteway M, Thomas DY, Leberer E. Pheromone response in yeast: association of Bem1p with proteins of the MAP kinase cascade and actin. Science. 1995;270:1210–1213. doi: 10.1126/science.270.5239.1210. [DOI] [PubMed] [Google Scholar]
- Leeuw T, Wu CL, Schrag JD, Whiteway M, Thomas DY, Leberer E. Interaction of a G-protein beta-subunit with a conserved sequence in Ste20/PAK family protein kinases. Nature. 1998;391:191–195. doi: 10.1038/34448. [DOI] [PubMed] [Google Scholar]
- Lipke PN, Taylor A, Ballou CE. Morphogenic effects of α-factor on the yeast Saccharomyces cerevisiae. . J Bacteriol. 1976;127:610–618. doi: 10.1128/jb.127.1.610-618.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DM, Mahanty SK, Choi KY, Manandhar M, Elion EA. The SH3-domain protein bem1 coordinates mitogen-activated protein-kinase cascade activation with cell-cycle control in Saccharomyces cerevisiae. . Mol Cell Biol. 1996;16:4095–4106. doi: 10.1128/mcb.16.8.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden K, Snyder M. Specification of sites for polarized growth in Saccharomyces cerevisiaeand the influence of external factors on site selection. Mol Biol Cell. 1992;3:1025–1035. doi: 10.1091/mbc.3.9.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici R, Bianchi E, DiSegni G, Tocchini-Valentini GP. Efficient signal transduction by a chimeric yeast-mammalian G protein alpha subunit Gpa1-Gs alpha covalently fused to the yeast receptor Ste2. EMBO (Eur Mol Biol Organ) J. 1997;16:7241–7249. doi: 10.1093/emboj/16.24.7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelitch M, Chant J. A mechanism of Bud1p GTPase action suggested by mutational analysis and immunolocalization. Curr Biol. 1996;6:446–454. doi: 10.1016/s0960-9822(02)00512-2. [DOI] [PubMed] [Google Scholar]
- Miller, J.H. 1972. Experiments in Molecular Genetics. Cold Spring Harbor Press, Cold Spring Harbor, NY. 466 pp.
- Miyajima I, Nakafuku M, Nakayama N, Brenner C, Miyajima A, Kaibuchi K, Arai K, Kaziro Y, Matsumoto K. GPA1, a haploid-specific essential gene, encodes a yeast homolog of mammalian G protein which may be involved in mating factor signal transduction. Cell. 1987;50:1011–1019. doi: 10.1016/0092-8674(87)90167-x. [DOI] [PubMed] [Google Scholar]
- Nern A, Arkowitz RA. A GTP-exchange factor required for cell orientation. Nature. 1998;391:195–198. doi: 10.1038/34458. [DOI] [PubMed] [Google Scholar]
- Parent CA, Blacklock BJ, Froehlich WM, Murphy DB, Devreotes PN. G protein signaling events are activated at the leading edge of chemotactic cells. Cell. 1998;94:81–91. doi: 10.1016/s0092-8674(00)81784-5. [DOI] [PubMed] [Google Scholar]
- Parent CA, Devreotes PN. Molecular genetics of signal transduction in Dictyostelium. . Annu Rev Biochem. 1996;65:411–440. doi: 10.1146/annurev.bi.65.070196.002211. [DOI] [PubMed] [Google Scholar]
- Park HO, Bi EF, Pringle JR, Herskowitz I. Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc Natl Acad Sci USA. 1997;94:4463–4468. doi: 10.1073/pnas.94.9.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M, Neiman AM, Park HO, Van Lohuizen M, Herskowitz I. Functional analysis of the interaction between the small GTP binding protein Cdc42 and the Ste20 protein kinase in yeast. EMBO (Eur Mol Biol Organ) J. 1996;15:7046–7059. [PMC free article] [PubMed] [Google Scholar]
- Peterson J, Zheng Y, Bender L, Myers A, Cerione R, Bender A. Interactions between the bud emergence proteins Bem1p and Bem2p and rho-type GTPases in yeast. J Cell Biol. 1994;127:1395–1406. doi: 10.1083/jcb.127.5.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle JR. Staining of bud scars and other cell wall chitin with calcofluor. Methods Enzymol. 1991;194:732–735. doi: 10.1016/0076-6879(91)94055-h. [DOI] [PubMed] [Google Scholar]
- Pringle JR, Bi E, Harkins HA, Zahner JE, Devirgilio C, Chant J, Corrado K, Fares H. Establishment of cell polarity in yeast. Cold Spring Harbor Symp Quant Biol. 1995;60:729–744. doi: 10.1101/sqb.1995.060.01.079. [DOI] [PubMed] [Google Scholar]
- Pryciak PM, Huntress FA. Membrane recruitment of the kinase cascade scaffold protein Ste5 by the G beta gamma complex underlies activation of the yeast pheromone response pathway. Genes Dev. 1998;12:2684–2697. doi: 10.1101/gad.12.17.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner JC, Munro S. Identification of the MNN2 and MNN5 mannosyltransferases required for forming and extending mannose branches of the outer chain mannan of Saccharomyces cerevisiae. . J Biol Chem. 1998;273:26836–26843. doi: 10.1074/jbc.273.41.26836. [DOI] [PubMed] [Google Scholar]
- Read EB, Okamura HH, Drubin DG. Actin- and tubulin-dependent functions during Saccharomyces cerevisiaemating projection formation. Mol Biol Cell. 1992;3:429–444. doi: 10.1091/mbc.3.4.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemer T, Vallier LG, Snyder M. Selection of polarized growth sites in yeast. Trends Cell Biol. 1996;6:434–441. doi: 10.1016/s0962-8924(96)10039-8. [DOI] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1991. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Press, Cold Spring Harbor, NY. 198 pp.
- Sanchezgarcia I, Rabbitts TH. The Lim domain—a new structural motif found in zinc-finger like proteins. Trends Genetics. 1994;10:315–320. doi: 10.1016/0168-9525(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Scherer S, Davis RW. Replacement of chromosomal segments with altered DNA sequences constructed in vitro. . Proc Natl Acad Sci USA. 1979;76:4951–4955. doi: 10.1073/pnas.76.10.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrick K, Garvik B, Hartwell LH. Mating in Saccharomyces cerevisiae: The role of the pheromone signal transduction pathway in the chemotropic response to pheromone. Genetics. 1997;147:19–32. doi: 10.1093/genetics/147.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall JE. Polarization of yeast cells in spatial gradients of alpha mating factor. Proc Natl Acad Sci USA. 1993;90:8332–8336. doi: 10.1073/pnas.90.18.8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloat BF, Adams A, Pringle JR. Roles of the CDC24 gene product in cellular morphogenesis during the Saccharomyces cerevisiaecell cycle. J Cell Biol. 1981;89:395–405. doi: 10.1083/jcb.89.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JP, Hirschman J, Gunn G, Dohlman HG. Regulation of membrane and subunit interactions by N-myristoylation of a G-protein alpha-subunit in yeast. J Biol Chem. 1996;271:20273–20283. doi: 10.1074/jbc.271.34.20273. [DOI] [PubMed] [Google Scholar]
- Sprague, G.F.J., and J.W. Thorner. 1992. In Pheromone Response and Signal Transduction during the Mating Process of Saccharomyces cerevisiae. Vol. 2. E.W. Jones, J.R. Pringle, and J.R. Broach, editors. Cold Spring Harbor Press, Cold Spring Harbor, NY. 657–744.
- Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- Tkacz JS, Lampen JO. Wall replication in Saccharomycesspecies: use of fluorescein-conjugated concanavalin A to reveal the site of mannan insertion. J Gen Microbiol. 1972;72:243–247. doi: 10.1099/00221287-72-2-243. [DOI] [PubMed] [Google Scholar]
- Tkacz JS, MacKay VL. Sexual conjugation in yeast. J Cell Biol. 1979;80:326–333. doi: 10.1083/jcb.80.2.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtz N, Peter M, Herskowitz I. FAR1is required for oriented polarization of yeast cells in response to mating pheromones. J Cell Biol. 1995;131:863–873. doi: 10.1083/jcb.131.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteway M, Hougan L, Thomas DY. Overexpression of the STE4 gene leads to mating response in haploid Saccharomyces cerevisiae. . Mol Cell Biol. 1990;10:217–222. doi: 10.1128/mcb.10.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B-E, Kurjan J. Evidence that mating by the Saccharomyces cerevisiae gpa1 Val50mutant occurs through the default mating pathway and a suggestion of a role for ubiquitin mediated proteolysis. Mol Biol Cell. 1997;8:1649–1664. doi: 10.1091/mbc.8.9.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZS, Leung T, Manser E, Lim L. Pheromone signalling in Saccharomyces cerevisiaerequires the small GTP-binding protein Cdc42p and its activator CDC24. Mol Cell Biol. 1995;15:5246–5257. doi: 10.1128/mcb.15.10.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Bender A, Cerione RA. Interactions among proteins involved in bud-site selection and bud-site assembly in Saccharomyces cerevisiae. . J Biol Chem. 1995;270:626–630. doi: 10.1074/jbc.270.2.626. [DOI] [PubMed] [Google Scholar]
- Ziman M, Preuss D, Mulholland J, O'Brien JM, Botstein D, Johnson DI. Subcellular localization of Cdc42p, a Saccharomyces cerevisiaeGTP-binding protein involved in the control of cell polarity. Mol Biol Cell. 1993;4:1307–1316. doi: 10.1091/mbc.4.12.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]