

Abstract
Calpain 3 is known as the skeletal muscle–specific member of the calpains, a family of intracellular nonlysosomal cysteine proteases. It was previously shown that defects in the human calpain 3 gene are responsible for limb girdle muscular dystrophy type 2A (LGMD2A), an inherited disease affecting predominantly the proximal limb muscles. To better understand the function of calpain 3 and the pathophysiological mechanisms of LGMD2A and also to develop an adequate model for therapy research, we generated capn3-deficient mice by gene targeting. capn3-deficient mice are fully fertile and viable. Allele transmission in intercross progeny demonstrated a statistically significant departure from Mendel's law. capn3-deficient mice show a mild progressive muscular dystrophy that affects a specific group of muscles. The age of appearance of myopathic features varies with the genetic background, suggesting the involvement of modifier genes. Affected muscles manifest a similar apoptosis-associated perturbation of the IκBα/nuclear factor κB pathway as seen in LGMD2A patients. In addition, Evans blue staining of muscle fibers reveals that the pathological process due to calpain 3 deficiency is associated with membrane alterations.
Keywords: calpain, apoptosis, muscular dystrophies, IκBα/NF-κB pathway, knockout mice
Introduction
Calpains are calcium-dependent intracellular nonlysosomal proteases. The first characterized vertebrate calpains were the ubiquitous μ and m-calpains (Yoshimura et al. 1983). Identification of new members of the calpain family, expressed at various levels in different tissues, revealed the complexity of this family. In mammals, at least 11 different calpain genes have now been identified (Sorimachi et al. 1997; Dear and Boehm 1999). Among them, CAPN3, the calpain 3 gene, is expressed predominantly as a 94-kD protein in adult skeletal muscles (Sorimachi et al. 1989). It is, at present, the only member of the human calpain family whose deficiency is associated with a defined phenotype, as it was shown that mutations in CAPN3 lead to limb girdle muscular dystrophy type 2A (LGMD2A) (Richard et al. 1995). Atrophy and weakness of the proximal muscles of the limbs, especially those of posterior compartments, characterize this pathology (Fardeau et al. 1996).
Physiological functions of the calpains are still broadly unknown but, as proteases, they may regulate important cellular functions. Sorimachi et al. 1993 showed that calpain 3 undergoes an autolytic degradation immediately after translation, at least when expressed in COS-7 cells. It was also shown that when wild-type calpain 3 is expressed in COS-7 cells, the 230-kD intrinsic fodrin subunit is proteolyzed, yielding a 150-kD fragment (Ono et al. 1998). Calpain 3 may exert a selective action in the nucleus as it contains within its amino acid sequence a nuclear translocation signal (Sorimachi et al. 1989) and could also be detected in myonuclei (Baghdiguian et al. 1999). Increase in apoptosis and change in the localization profile of nuclear factor (NF)-κB, a transcription factor driving the transcription of several survival genes and of its inhibitor, IκBα, were found in biopsies from LGMD2A patients (Baghdiguian et al. 1999). In spite of these data, it is still unclear how a deficiency in this proteolytic enzyme could lead to a dystrophic process. It is worth mentioning that the majority of the other muscular dystrophies are caused by mutations in genes encoding structural proteins. This stands in contrast to the proteolytic deficiency in LGMD2A.
To better understand the function of calpain 3 and the pathophysiological mechanisms of LGMD2A and also to develop an adequate model for therapy research, we generated capn3-deficient mice by gene targeting. Although these mice seem indistinguishable from normal mice, examination of their skeletal muscles demonstrates clearly that they suffer from muscular dystrophy. The data obtained in these mice suggest that presence of the proteolytic activity of calpain 3 is necessary to protect myocytes from a death signal and show that its absence is associated with membrane alterations.
Materials and Methods
Gene Targeting
Screening of a mouse embryonic stem (ES) cell (strain 129Sv) library using two radiolabeled cDNA probes corresponding to exons 2–4 and exons 15–24, respectively, of the calpain 3 gene led to the identification of 13 recombinant λ phage clones. One of them (B1) was characterized by restriction mapping and limited sequencing. It was then used to construct a gene-targeting vector in a neomycin resistance (neoR) gene–containing plasmid (pD383; a gift of P. Kastner, IGBMG, Illkirch, France). A 6-kb HindIII-EcoRI fragment containing the distal part of intron 1 was inserted between the HindIII and EcoRI sites of a plasmid (p572GTI-2) carrying the herpes virus thymidine kinase (tk) gene and a 2.1-kb KpnI fragment containing exons 4 and 5 was inserted into the KpnI site of pD383. In a second step, the SalI fragment of the latter plasmid covering the KpnI fragment and tk gene was inserted at the SalI site of the p572GTI-2. Thereby, the targeting vector deletes the genomic sequence corresponding to exons 2 and 3 of capn3. This 12-kb vector was linearized at the NotI site of the multiple cloning site, and 20 μg of DNA was electroporated into 107 ES cells of 129Sv origin. 358 stable transfected cells were isolated after selection in medium supplemented first with 350 μg/ml of G418 and then with 2 μM gancyclovir. DNA was isolated from the various cell lines by phenol-chloroform extraction. To detect homologous recombination events at the capn3 locus, 100 ng of this DNA was used as template in a long-range PCR assay, using neoR-specific primers and external primers located 5′ and 3′ of the construct (see Fig. 1 a). Sequences of the primers are as follows: in 5′ upper primer (5′.a, GACCCACTTCTCCTCAATCCATGCA) and lower primer (Neo.1, CTTCACATACACCTTGTCTCCGACG); in 3′ upper primer (Neo.2 TACTTCCATTTGTCACGTCCTGCAC) lower primer (3′.m, ACCCCAGACTACTATTCCCAAAGCA). ES cells from one of the three correctly targeted clones (clone 32, 64, and 137) were injected into blastocysts derived from the C57BL/6 mice. Blastocysts were transferred to pseudopregnant foster mothers, and chimeric offsprings were identified by the presence of agouti hair. Chimeric males were mated to C57BL/6 females to obtain ES-derived offsprings that were analyzed by PCR on tail DNA to identify the heterozygous mice. The combination of gene-specific primers used for this purpose was for the neoR gene, Neo.3 (AGACTGCCTTGGGAAAAGCG)/ex4.m (ACCACATCTACCCAGTCTCC) and for capn3, ex2.a (GAGCCAACAGGACTCACATCT)/in2.m (CGCCTCTGCCCAAAAGACT) or ex3.a (CCTGCCTGACCCTGAATGAG)/ex3.m (TGGAAGTGGAAGATCCCTGC). DNA amplification was carried out for 35 cycles at 53°C for ex3.a/ex3.m or at 61°C for Neo.3/ex4.m and ex2.a/in2.m. Heterozygous mice were mated inter se to generate homozygous mutant mice.
Figure 1.

Gene targeting of mice. (a) Restriction map of the targeting construct, the wild-type capn3 locus, and the targeted locus. A fragment containing exons 2 and 3 encompassing C129, the cysteine participating to the catalytic site, was deleted and replaced by a neoR cassette. The herpes simplex virus tk (TK) cassette was added 5′ to the targeting vector for positive–negative selection. H, E, and K indicate HindIII, EcoRI, and KpnI, respectively. (b) Double-resistant ES cell clones were genotyped by long-range PCR using two primer pairs (5′.a/Neo.1 and Neo.2/3′.m). Primer sites are shown in panel a. The 6- and 2.1-kb PCR products are indicative of correct homologous recombination events in the 5′ and 3′ neoR flanking regions, respectively. (c) Genotype analysis of progeny from a heterozygote intercross. PCR analysis of tail DNA isolated from 3-wk-old mice was performed using the PCR primer pairs Neo.3/ex4.m (top) and ex3.a/ex3.m (bottom). Normal mice DNA (+/+) gave an amplification product only with exon 3 primer pairs; capn3 −/− mice DNA (−/−), only with neo primer pairs; and capn3 +/− mice DNA gave amplification products with both sets of primers. (d) Quantitative RT-PCR analysis of calpain 3 messenger level, expressed relative to wild-type mice, in RNA extracted from muscle. The analysis was performed according to TaqMan chemistry using primer pairs located in exon 1. Level of TFIID mRNA was used to normalize the results across different samples. The bars indicate the mean value ± the minimum or maximum. (e) Western blot analysis. A calpain 3–specific polyclonal antibody directed against the IS2 region of calpain 3 was used to detect calpain 3 expression in the muscle of wild-type (+/+) and capn3 −/− (−/−) mice. A Western blot using a calpain 2–specific polyclonal antibody was used as a control. Zinc staining was used to ascertain equivalent loading. (f) Western blot analyses were performed on lysates of transfected COS-7 cells to visualize proteolytic calpain 3 fragments using an antibody against the IS2-specific region of p94. The constructs used were the vector alone (pSRD) or derived constructs containing, respectively, the wild-type calpain 3 (p94), the cDNA corresponding to the calpain 3–deficient mice (KO), and a defective cDNA carrying the mutation C129S. White and black arrowheads indicate the full-length and proteolyzed products, respectively. (g) Proteolysis of fodrin was assessed using antibodies specific to the 150-kD α-fodrin fragment. The proteolized fragment was only detected for the wild-type calpain 3. (h) The titin-binding capacities of the cDNA was monitored in a yeast two-hybrid system, using as bait two different titin clones corresponding to the COOH-terminal (C-ter) region (pCNT-52) and to the region located in the N2A line of the sarcomere (pCNT-N2). Wild-type (p94), mutant calpain 3 (KO) cDNA, or vector alone (T-[pAS2C-1]) was cotransfected with the titin clones into Saccharomyces cerevisiae CG-1945 strain cells. Binding was visualized as growth on plates without leucine and without tryptophane. Deletion of exons 2 and 3 does not impair binding to the N2A region of titin but weakens binding to the COOH-terminal region.
Statistical Analysis of Transmission Ratio Distortion
The number of animals of each genotype expected according to Mendel's law was calculated. The significant departure of observed values compared with expected values was estimated using a χ2 test.
RNA and Protein Analysis
Expression of the calpain 3 gene was investigated by a real-time quantitative reverse transcriptase (RT)-PCR method using TaqMan probes (PerkinElmer) (Heid et al. 1996) as described (Herasse et al. 1999). Investigation of autolysis, proteolysis of in vitro substrates, and titin-binding capacity was performed as described (Herasse et al. 1999). The following primary antibodies were used: anti-p94 (Baghdiguian et al. 1999) and antifodrin (Ono et al. 1998).
Determination of Serum Level of Creatine Kinase
Blood was collected from the retroorbital sinus of mice, and the serum was stored at −20°C until measurements were taken. Quantitative kinetic determination of creatine kinase (CK) activity in serum of control and capn3 −/− mice was measured by Vetfrance.
Analysis of Muscle Specimen
Four or five affected animals were examined at each age for each of the two genetic backgrounds. Striated muscles were removed from the animals, frozen in isopentane cooled in liquid nitrogen, and kept at −80°C until use. Longitudinal and transverse 7-μm sections were made and processed for hematoxilin and eosin (H&E) histological staining or fluorescence labeling. Nuclear DNA fragmentation visualization and indirect immunofluorescence confocal microscopy were performed as described (Baghdiguian et al. 1999). The following primary antibodies were used: anti-IκBα (FL, sc-847; Santa Cruz Biotechnology, Inc.) and anti–NF-κB (C20, sc-372; Santa Cruz Biotechnology, Inc.). The number of centronucleated fibers was counted by examination of the H&E-stained sections (three different sections for each animal) under a light microscope and the total number of fibers present in the section was counted on digitized images.
Evans Blue Dye Staining
Intraperitoneal injection of Evans blue (EB) dye (0.5 mg/10 g of body weight; Sigma-Aldrich) was performed on 6-mo-old mice of 129Sv background (n = 4 for each genotype). Mice were killed 12 h after injection. Muscles were examined for blue coloration indicating dye uptake. Muscle cryosections were fixed in acetone cooled at −20°C, rinsed in PBS, and mounted with Vectashield medium (Vector Laboratories). Sections were examined under a Leica fluorescence or a ZEISS confocal microscope. Sections were also stained for terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) positivity and propidium iodide.
Results and Discussion
Generation of capn3−/− Mice
In a previous study, Ono et al. 1998 showed in vitro that impairment of calpain 3–mediated proteolysis was a common feature of several LGMD2A mutations. Therefore, we targeted the proteolytic function for gene disruption using homologous recombination to produce calpain 3–deficient mice. Since exon 3 encodes the critical cysteine participating in the proteolytic site (Sorimachi et al. 1989), we constructed a replacement vector in which exons 2 and 3 are substituted by a neoR cassette (Fig. 1 a). After electroporation and selection of G418-resistant ES cell clones carrying the recombinant allele (Fig. 1 b), chimeric animals were generated and subsequently mated to obtain capn3 +/− progeny. Mating of heterozygous animals enabled the production of capn3 −/− mice (Fig. 1 c). As the phenotypic consequences of a natural or introduced mutation could depend on genetic background, appropriate crosses were undertaken to obtain congenic strains of 129Sv and C57BL/6 backgrounds. The genetic backgrounds of the examined mice were either of pure 129Sv or a mixture of C57BL/6 and 129Svter (with a ratio of 3:1 or 7:1), the latter being referenced here as the mix line.
Skeletal muscles of the capn3 −/− mice showed a dramatic decrease of capn3 mRNA and a complete absence of calpain 3 protein as verified, respectively, by quantitative RT-PCR (Fig. 1 d) and Western blot analyses (Fig. 1 e). Cloning and sequencing of the recombinant mRNA produced by capn3 −/− mice demonstrated that the corresponding protein would carry an in-frame deletion of amino acids 104–166 as the neoR cassette was spliced out. In vitro investigation of known biochemical characteristics of the corresponding recombinant calpain 3 protein showed that autolysis (Fig. 1 f) and proteolysis of fodrin (Fig. 1 g), an in vitro substrate, were abolished, whereas titin-binding ability in the N2A region, assayed in a yeast two-hybrid system, was preserved (Fig. 1 h). Thus, the effect of the disruption of the mouse capn3 gene can be related to the effects of the human mutations, at least with respect to proteolysis.
Inactivation of Calpain 3 Leads to Transmission Ratio Distortion
The distribution of wild-type, heterozygous, and homozygous calpain 3 mutant mice among 250 offsprings of heterozygous crosses revealed a statistically significant departure from Mendelian transmission with, surprisingly, a ratio in favor of capn3 −/− pups in both the mix and 129Sv lines (Table ; P < 0.01). Although this observation demonstrates that inactivation of calpain 3 does not lead to lethality in utero or shortly after birth, it also suggests that calpain 3–null alleles tend to be transmitted preferentially. Litter sizes are normal, suggesting that the underlying phenomenon acts before development, probably during gametogenesis or fertilization. This transmission ratio distortion is likely to be due to the impairment of calpain 3, as the genomes of the +/+ and −/− mice in the 129Sv line differ only by the introduced mutation. Existence of transmission ratio distortion, if validated on human populations, may have direct implications for the study of LGMD2A, especially for genetic counseling. Furthermore, understanding the mechanism underlying this phenomenon may lead to new insights on the function of calpain 3, as it suggests a role in the germ line. Analyzing mRNA from testis and ovary, transcripts for differnent calpain 3 isoforms were detected, although at a low level, compared with muscle (data not shown). These observations further increase the complexity of calpain 3 expression, as it is now known that alternative promoter or splicing gives rise to several calpain 3 isoforms in a large variety of tissues such as smooth and cardiac muscles, brain, thymus, leukocyte, and eye tissues (Ma et al. 1998; Herasse et al. 1999).
Table 1.
The Distribution of Wild-Type, Heterozygous, and Homozygous Offsprings of Heterozygous Crosses
| Genotypes | |||
|---|---|---|---|
| +/+ | +/− | −/− | |
| Observed | 44 | 128 | 84 |
| Expected | 62.5 | 125 | 62.5 |
| χ2 | 5.48 (P value <0.02) | 0.07 | 7.40 (P value <0.01) |
Data show observed and expected numbers of animals according to their genotypes at the capn3 locus.
capn3−/− Mutant Mice Develop a Progressive Mild Muscular Dystrophy
capn3 −/− mice exhibit a postnatal appearance similar to that of wild-type littermates and develop normal behavioral activity. They are fertile and have been kept alive for >1.5 yr with no increase in spontaneous deaths.
Increase in serum CK level is a criterion used to ascertain muscle fiber degeneration. Hence, it was evaluated in animals from both genotypes (−/− and +/+) of 129Sv genetic background. Animals were grouped into age classes (2–3, 3–4, and 5–7 mo of age). Fig. 2 shows that the difference in mean CK levels between the two genotypes increases with age. It should be noted, however, that this parameter cannot be used as a test to distinguish between −/− and +/+ due to the high variability of individual levels.
Figure 2.
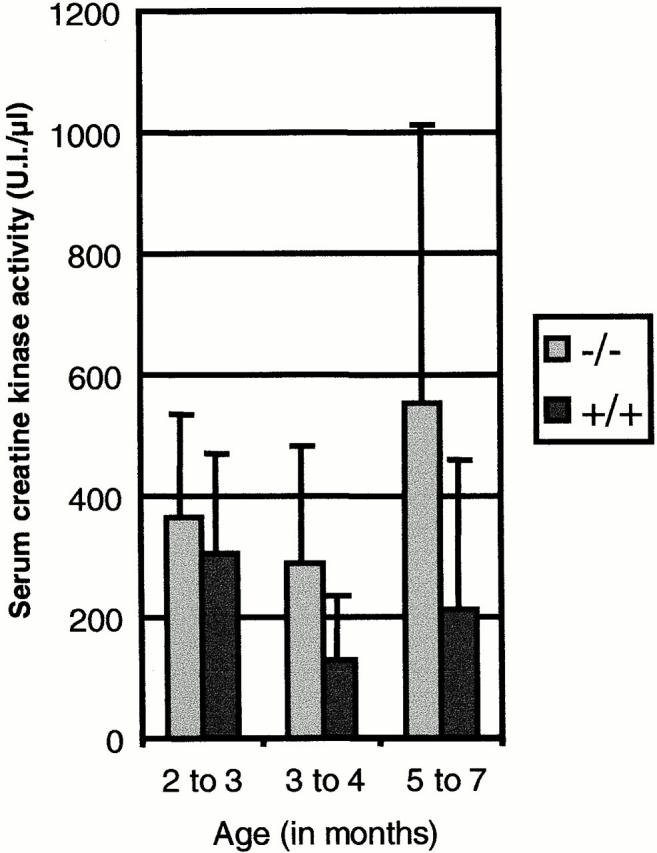
Serum CK activity. This parameter was evaluated on the 129Sv background. Animals were grouped into three classes according to their age. For the 2–3-mo-old animals, n = 38 for −/−, n = 25 for +/+; for the 3–4-mo-old animals, n = 68 for −/−, n = 6 for the +/+; and for the 5–7-mo-old animals, n = 37 for −/− and n = 21 for +/+. The bars indicate the mean values ± SD. P < 0.09 at 2–3 mo of age, P < 0.03 at 3–5 mo of age, and P < 0.002 at 6 mo of age.
The pathological aspect of H&E-stained frozen cross sections of various muscles was evaluated in calpain 3–deficient mice from the ages of 8 wk to 19 mo and compared with those of age-matched normal mice. Pathological changes characteristic of muscular dystrophy such as centronucleation, area of necrosis/regeneration, splitting of fibers, and foci of mononuclear cell infiltrates can be seen in both genetic backgrounds (Fig. 3). It should be noted that, in most cases, fibers with internal nuclei are clustered. Alterations increase with age, and the level of abnormality is variable depending on the muscle examined. Considering the combined effect of all alterations, psoas, soleus, and deltoid are the most affected muscles; tibialis anterior and biceps are affected but to a lower extent, whereas the quadriceps, gastrocnemius, and triceps brachii do not seem to be substantially affected.
Figure 3.
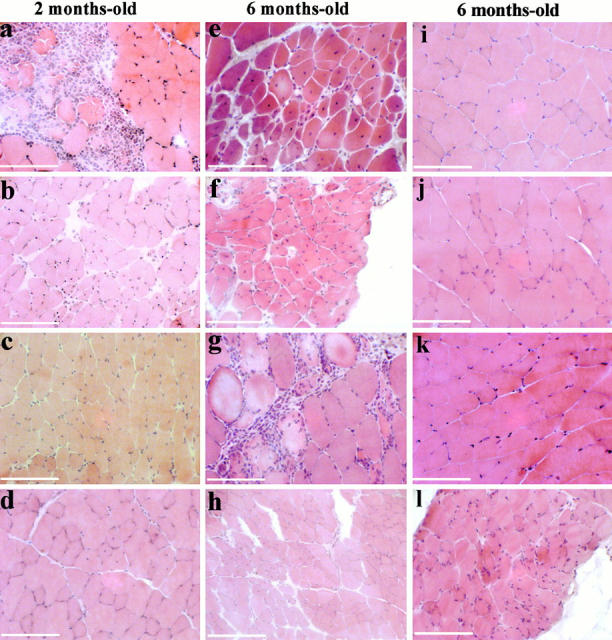
Histologic analysis of capn3-deficient mice. H&E-stained transverse muscle cryosections (7 μm) from mice of 129Sv genetic background. (a–d) 2-mo-old mice; (e–l) 6-mo-old mice. Dystrophic changes are evident in psoas (a and e), soleus (b and f), deltoid (c and g), and tibialis anterior (d and h), whereas quadriceps (i), gastrocnemius (j), and triceps (k) present a normal aspect. Diaphragm in l presents very slight abnormalities. The main features encountered are fibers with internal nuclei (b–f, and h) or area of infiltration of mononuclear cells (a and g). A cluster of small regenerating fibers can be seen in e. Bars, 50 μm.
The distribution of the impairment is the same on both genetic backgrounds. However, in the 129Sv congenic line, signs were already evident at 2 mo of age, whereas in 6-mo-old animals of mixed background, only discrete abnormalities are seen (Table ). The diaphragm is the only muscle examined that seems to be similarly affected in both lines. These observations suggest that the phenotypic consequences of a mutation in capn3 can be affected by the presence of a modifier gene(s). The same assumption can be made in humans, as phenotypic variations have been reported among siblings carrying identical CAPN3 mutations (Penisson-Besnier et al. 1998).
Table 2.
Distribution of Centronucleation with Respect to Genetic Background and Muscle Type in capn3−/− Mice
| 6-mo-old C57BL/6/129Sv (n =2) | 9-mo-old C57BL/6/129Sv (n =5) | 2-mo-old 129Sv (n =5) | 6-mo-old 129Sv (n =4) | |
|---|---|---|---|---|
| Psoas | <1.0 | 5.8 (3.0–10.0) | 3.8 (<1.0–6.5) | 7.3 (3.0–10) |
| Soleus | <1.0 | 8.2 (4.0–11.0) | 2.0 (<1.0–6.0) | 16.0 (2.0–28) |
| Deltoid | ≈3.0 | 4.2 (1.5–8.0) | 1.2 (<1–1.9) | 4.0 (3.0–5.5) |
| Extensor digitorum longus plus tibialis anterior | <1.0 | 3.9 (1.5–7.5) | 1.9 (<1.0–2.0) | 4.2 (4.0–6.0) |
| Biceps | ≈1.5 | ≈2.0 | 1 (<1.0–2.0) | 2.5 (2.0–5.5) |
| Quadriceps | <1.0 | 1.5 (1.0– 3.0) | <1.0 | <1.0 (<1.0–2.0) |
| Gastrocnemius | 2.0 | 1 (<1.0–2.5) | <1.0 | <1.0 (<1.0–2.0) |
| Triceps | <1.0 | <1.0 | <1.0 | <1.0 (<1.0–1.5) |
Mean ratio of fibers with internal nuclei among fibers of n mice on C57BL/6/129Sv background (at 6 and 9 mo of age) and on 129Sv background (at 2 and 6 mo of age). The lowest and highest levels are indicated in parentheses. The levels of centronucleation were examined in the same muscles of wild-type mice of the same genetic backgrounds as presented here and were never found to be >1%.
Apoptosis and the IκBα/NF-κB Pathway
The analysis of muscle biopsies of LGMD2A patients revealed that calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation in the expression of the transcription factor NF-κB and of its inhibitor IκBα (Zabel et al. 1993; Baldwin 1996; Baghdiguian et al. 1999). Therefore, we examined whether the same events can be seen in calpain 3–deficient mice. A TUNEL assay and immunostaining using polyclonal antibodies against NF-κBp65 and IκBα were performed on histologically affected muscles. The results demonstrate the presence of myofibers presenting TUNEL-positive nuclei associated with nuclear localization of IκBα (Fig. 4 a). With respect to NF-κB staining, half of the calpain 3–deficient muscle sections examined present a subsarcolemmal localization (Fig. 4 b), whereas the other half present a normal pattern, that is labeling of some myonuclei. The coexistence of these two patterns suggests that the deficiency in calpain 3 is not sufficient to lead to an abnormal localization of NF-κB and that another element (e.g., a death signal) should intervene. In the absence of such a signal, the intervention of NF-κB may not be necessary and, therefore, a calpain 3–deficient muscle would not differ from a normal muscle with respect to the NF-κB/IκBα expression. The idea of the necessity of a signal is compatible with the observation that the pathological features present in the muscle sections of the calpain 3–deficient mice are often clustered in particular areas.
Figure 4.

Apoptosis and immunolocalization of IκBα and NF-κB in muscles of calpain 3–deficient mice. (a) A longitudinal muscle section of a calpain 3–deficient mouse was triple labeled for apoptosis (green myonuclei superimposed over the phase–contrast field), IκBα (red), and chromatin (DAPI, right). Note that TUNEL-positive myonuclei were always IκBα-positive (arrowheads). (b) Subsarcolemmal localization of NF-κB (blue fluorescence, left). Note that in this calpain 3–deficient tissue, NF-κB was not found concentrated in the myonuclei (stained with DAPI, right, arrowheads). (c) As a control, NF-κB (blue fluorescence) was normally found concentrated in some myonuclei of wild-type mice (arrowheads). The DAPI fields were recorded with a CCD camera mounted on an additional port of the confocal microscope. For a, 10 mm corresponds to 72.3 μm, and for b and c, 10 mm corresponds to 57 μm.
Sarcolemmal Integrity, Apoptosis, and Calpain 3 Deficiency
Loss of sarcolemmal integrity is a characteristic feature of several muscular dystrophies (Petrof et al. 1993; Matsuda et al. 1995; Straub et al. 1997). It was not clear if a deficiency in calpain 3 could also lead to sarcolemmal damage. No alterations of the gene products known to be involved in other progressive muscular dystrophies disclosing similar clinical signs and classified under the term LGMD2 (Roberds et al. 1994; Bönnemann et al. 1995; Lim et al. 1995; Noguchi et al. 1995; Nigro et al. 1996) were seen in capn3 −/− mice; immunohistochemical investigation of α-, β-, and γ- sarcoglycan and of dystrophin did not reveal any abnormality (data not shown). These data are consistent with observations made in humans. To test further if mutation in calpain 3 could result in impairment of membrane integrity, membrane permeability was assessed by intraperitoneal injection of EB dye, a normally impermeant molecule. Tracer uptake was detected by microscopic inspection in a limited number of muscle cells of calpain 3–deficient mice (Fig. 5 a). This observation is a clear demonstration that membrane alterations exist during the pathological process induced by calpain 3 deficiency. TUNEL assays performed on muscle sections revealed that only EB-stained fibers present apoptotic myonuclei (Fig. 5 b). A similar observation was reported for mdx mice (Matsuda et al. 1995). We have shown previously in LGMD2A patients that apoptotic levels parallel CK levels, i.e., they are highest when CK leaks are highest (Baghdiguian et al. 1999). The concordance of apoptotic nuclei and EB positive cells suggests that elevation of CK level is presumably associated with loss of the sarcolemmal integrity. It remains unclear whether activation of the apoptotic cascade is a cause or a consequence of membrane impairment. In support of the former, apoptosis was reported to be associated, at least in one occurrence, with changes in permeability of the cell membrane through the effect of perforin excreted by cytotoxic lymphocytes (Dourmashkin et al. 1980). Furthermore, in mdx mice, apoptosis occurs by perforin-mediated cytotoxicity (Spencer et al. 1997). This suggests that a similar mechanism could also happen in LGMD2A and that common pathological features could exist between dystrophin and calpain 3 deficiencies. On the other hand, if the latter hypothesis is correct, deficiency of calpain 3 could directly lead to membrane damage by an yet unknown mechanism. This could cause extracellular calcium influx into the cytoplasm eventually triggering apoptosis.
Figure 5.

EB staining of cryosections of calpain 3–deficient muscles reveals apoptosis-associated disruption of sarcolemmal integrity. Four 6-mo-old animals of each genotype were killed 12 h after being injected with EB dye and biopsied. Regions of EB uptake are seen as red cytoplasmic staining on fluorescence microscopy. A mean of three EB-positive cells was detected per muscle section of calpain 3–deficient mice. (a) Calpain 3–deficient biceps showing EB staining. No EB uptake is seen in wild-type biceps. (b) Triple labeling for EB, TUNEL, and propidium iodide (PI) of a muscle section of calpain 3–deficient mouse. From left to right, EB-positive cells (red pseudocolor), TUNEL-positive myonuclei, propidium iodide–stained nuclei (blue pseudocolor), and a superposition of all stainings (white arrows and/or green pseudocolor refer to apoptotic nuclei). Bars: (a) 50 μm; (b) 20 μm.
Conclusion
The selectivity of the muscular impairment, the compatibility of the biopsy appearance with a necrosis/regeneration pattern, and the preservation of the dystrophin-associated complex (data not shown) are elements showing similarity with the corresponding human disease. In addition, apoptosis in these mice myonuclei seems to be associated both with a perturbation of the NF-κB/IκBα pathway and with an impairment of membrane permeability. The calpain 3–deficient mice will thus provide a valuable resource for further dissection of the molecular events governing dystrophic changes and apoptosis in muscle as well as for the development of therapeutic rescue strategies.
Acknowledgments
We thank Philip Kastner and Jean-Pierre Chambon for their help in the homologous recombination process; Marianne Le Meur and Maryline LeMée for taking care of the colony; Christian Pastoret, Bernard Gjata, and Corinne Laplace for their help in the analysis of muscles; Catherine Astier for providing anti-calpain 3 antibody; Takaomi C. Saido for providing antifodrin antibody; and Xavier Montagutelli and Jean-Louis Guénet for helpful discussion.
This work was funded by the Association Française contre les Myopathies.
Footnotes
Abbreviations used in this paper: CK, creatine kinase; ES, embryonic stem; EB, Evans blue; H&E, hematoxilin and eosin; LGMD2A, limb girdle muscular dystrophy type 2A; neoR, neomycin resistance; NF, nuclear factor; RT, reverse transcriptase; tk, thymidine kinase; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
References
- Baghdiguian S., Martin M., Richard I., Pons F., Astier C., Bourg N., Hay R.T., Chemaly R., Halaby G., Loiselet J. Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IκBa/NF-kB pathway in limb-girdle muscular dystrophy type 2A. Nat. Med. 1999;5:503–511. doi: 10.1038/8385. [DOI] [PubMed] [Google Scholar]
- Baldwin A.S., Jr. The NFκB and IκB proteinsnew discoveries and insights. Annu. Rev. Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Bönnemann C.G., Modi R., Noguchi S., Mizuno Y., Yoshida M., Gussoni E., McNally E.M., Duggan D.J., Angelini C., Hoffman E.P., Ozawa E., Kunkel L.M. β-Sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat. Genet. 1995;11:266–273. doi: 10.1038/ng1195-266. [DOI] [PubMed] [Google Scholar]
- Dear T.N., Boehm T. Diverse mRNA expression patterns of the mouse calpain genes Capn5, Capn6 and Capn11 during development. Mech. Dev. 1999;89:201–209. doi: 10.1016/s0925-4773(99)00214-2. [DOI] [PubMed] [Google Scholar]
- Dourmashkin R.R., Deteix P., Simone C.B., Henkart P. Electron microscopic demonstration of lesions in target cell membranes associated with antibody-dependent cellular cytotoxicity. Clin. Exp. Immunol. 1980;42:554–560. [PMC free article] [PubMed] [Google Scholar]
- Fardeau M., Eymard B., Mignard C., Tomé F.M.S., Richard I., Beckmann J.S. Chromosome 15-linked limb-girdle muscular dystrophyclinical phenotypes in Réunion Island and French metropolitan communities. Neuromuscul. Disord. 1996;6:447–453. doi: 10.1016/s0960-8966(96)00387-2. [DOI] [PubMed] [Google Scholar]
- Heid C.A., Steven J., Livak K.J., Williams P.M. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Herasse M., Ono Y., Fougerousse F., Kimura E., Beley C., Montarras D., Pinset C., Sorimachi H., Suzuki K., Beckmann J.S., Richard I. Expression and functional characteristics of calpain 3 isoforms generated through tissue-specific transcriptional and post-transcriptional events. Mol. Cell. Biol. 1999;19:4047–4055. doi: 10.1128/mcb.19.6.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L.E., Duclos F., Broux O., Bourg N., Sunada Y., Allamand V., Meyer J., Richard I., Moomaw C., Slaughter C. β-Sarcoglycan (43DAG)characterization and role in limb-girdle muscular dystrophy linked to chromosome 4q12. Nat. Genet. 1995;11:257–265. doi: 10.1038/ng1195-257. [DOI] [PubMed] [Google Scholar]
- Ma H., Fukiage C., Azuma M., Shearer T.R. Cloning and expression of mRNA for calpain Lp82 from rat lenssplice variant of p94 Invest Ophthalmol.Vis. Sci. 391998. 454 461 [PubMed] [Google Scholar]
- Matsuda R., Nishikawa A., Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blueevidence of apoptosis in dystrophin-deficient muscle. J. Biochem. 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- Nigro V., de Sà Moreira E., Piluso G., Vainzof M., Belsito A., Politano L., Puca A.A., Passos-Bueno M.R., Zatz M. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the δ-sarcoglycan gene. Nat. Genet. 1996;14:195–198. doi: 10.1038/ng1096-195. [DOI] [PubMed] [Google Scholar]
- Noguchi S., McNally E.M., Ben Othmane K., Hagiwara Y., Mizuno Y., Yoshida M., Yamamoto H., Bönnemann C.G., Gussoni E., Denton P.H. Mutations in the dystrophin-associated protein γ-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270:819–822. doi: 10.1126/science.270.5237.819. [DOI] [PubMed] [Google Scholar]
- Ono Y., Shimada H., Sorimachi H., Richard I., Saido T.C., Beckmann J.S., Ishiura S., Suzuki K. Functional defects of a muscle-specific calpain, p94, caused by mutations associated with limb-girdle muscular dystrophy type 2A. J. Biol. Chem. 1998;273:17073–17078. doi: 10.1074/jbc.273.27.17073. [DOI] [PubMed] [Google Scholar]
- Penisson-Besnier I., Richard I., Beckmann J.S., Fardeau M. Phenotypic variations of calpain deficiency in two siblings. Muscle Nerve. 1998;21:1078–1080. doi: 10.1002/(sici)1097-4598(199808)21:8<1078::aid-mus15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Petrof B.J., Shreger J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscular contraction. Proc. Natl. Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard I., Broux O., Allamand V., Fougerousse F., Chiannilkulchai N., Bourg N., Brenguier L., Devaud C., Pasturaud P., Roudaut C. Mutations in the proteolytic enzyme, calpain 3, cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- Roberds S.L., Leturcq F., Allamand V., Piccolo F., Jeanpierre M., Anderson R.D., Lim L.E., Lee J.C., Tome F.M.S., Romero N.B. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78:625–633. doi: 10.1016/0092-8674(94)90527-4. [DOI] [PubMed] [Google Scholar]
- Sorimachi H., Imajoh-Ohmi S., Emori Y., Kawasaki H., Ohno S., Minami Y., Suzuki K. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-types. Specific expression of the mRNA in skeletal muscle. J. Biol. Chem. 1989;264:20106–20111. [PubMed] [Google Scholar]
- Sorimachi H., Toyama-Sorimachi N., Saido T.C., Kawasaki H., Sugita H., Miyasaka M., Arahata K., Ishiura S., Suzuki K. Muscle-specific calpain, p94, is degraded by autolysis immediately after translation, resulting in disappearance from muscle. J. Biol. Chem. 1993;268:10593–10605. [PubMed] [Google Scholar]
- Sorimachi H., Ishiura S., Suzuki K. Structure and physiological function of calpains. Biochem. J. 1997;328:721–732. doi: 10.1042/bj3280721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer M.J., CM. Walsh K.A., Dorshkind E.M., Rodriguez, Tidball J.G. Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J. Clin. Invest. 1997;99:2745–2751. doi: 10.1172/JCI119464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V., Rafael J.A., Chamberlain J.S., Campbell K.P. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura N., Kikuchi T., Sasaki T., Kitahara A., Hatanaka M., Murachi T. Two distinct Ca2+ proteases (calpain I and calpain II) purified concurrently by the same method from rat kidney. J. Biol. Chem. 1983;258:8883–8889. [PubMed] [Google Scholar]
- Zabel U., Henkel T., Silva M.S., Baueuerle P.A. Nuclear uptake control of NF-κB by MAD-3, an IκB protein present in the nucleus. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:201–211. doi: 10.1002/j.1460-2075.1993.tb05646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]