

Abstract
Flightin is a multiply phosphorylated, 20-kD myofibrillar protein found in Drosophila indirect flight muscles (IFM). Previous work suggests that flightin plays an essential, as yet undefined, role in normal sarcomere structure and contractile activity. Here we show that flightin is associated with thick filaments where it is likely to interact with the myosin rod. We have created a null mutation for flightin, fln0, that results in loss of flight ability but has no effect on fecundity or viability. Electron microscopy comparing pupa and adult fln0 IFM shows that sarcomeres, and thick and thin filaments in pupal IFM, are 25–30% longer than in wild type. fln0 fibers are abnormally wavy, but sarcomere and myotendon structure in pupa are otherwise normal. Within the first 5 h of adult life and beginning of contractile activity, IFM fibers become disrupted as thick filaments and sarcomeres are variably shortened, and myofibrils are ruptured at the myotendon junction. Unusual empty pockets and granular material interrupt the filament lattice of adult fln0 sarcomeres. Site-specific cleavage of myosin heavy chain occurs during this period. That myosin is cleaved in the absence of flightin is consistent with the immunolocalization of flightin on the thick filament and biochemical and genetic evidence suggesting it is associated with the myosin rod. Our results indicate that flightin is required for the establishment of normal thick filament length during late pupal development and thick filament stability in adult after initiation of contractile activity.
Keywords: flightin, myosin, thick filaments, insect flight muscle, muscle mutant
Introduction
The force-generating properties of striated muscle are the result of the cyclical interaction between myosin crossbridges and actin in thin filaments. The organized arrangement of interdigitating thick and thin filaments translates the molecular movements of myosin motors into macroscopic movements and allows transduction of forces along the muscle into the adjoining appendage. A requirement for proper muscle function is the assembly of myosin molecules into bipolar filaments of specific and uniform length.
The mechanism by which thick filaments attain precise regularity in striated muscle remains unresolved. In vitro studies have demonstrated that myosin possesses self-assembly properties; however, the resulting synthetic myosin filaments (or paracrystals) lack important features of in vivo thick filaments, particularly uniform length (Huxley 1963; Squire 1986). Although myosin may contain all the information necessary for self-assembly, accessory proteins are likely to be required for dictating a particular and uniform thick filament length in vivo. The gigantic protein titin, which extends from the Z to the M line, has been proposed to act as a molecular ruler for thick filament assembly (Trinick 1996; Gregorio et al. 1999). The vertebrate myosin-binding protein MyPB-C affects the length, diameter, and uniformity of synthetic myosin filaments (Davis 1988; Winegrad 1999) and is required for the formation of normal thick filaments in striated muscle (Gilbert et al. 1996; for reviews, see Bennett et al. 1999; Winegrad 1999). Thick filaments in the body wall of the nematode Caenorhabditis elegans contain several nonmyosin core (coupling) proteins as well as possible assemblases that may act as cofactors or chaperones for assembly and/or stability (Liu et al. 1997; Ao and Pilgrim 2000). Drosophila and C. elegans thick filaments assemble around a core of varying amounts of paramyosin. Drosophila indirect flight muscles (IFM) thick filaments have the least amount of paramyosin (1%, compared with 6% in Lethocerus and 10% in Apis) and the most hollow appearance among insect flight muscles.
The indirect flight muscles of Drosophila melanogaster are an excellent experimental system in which to combine studies of muscle development, ultrastructure, and function with genetic manipulation (for reviews, see Bernstein et al. 1993; Maughan and Vigoreaux 1999). Drosophila IFM is an ideal system in which to examine filament and sarcomere assembly processes because of the synchrony of filament and sarcomere assembly over a short period of time (3 d). Furthermore, the high degree of order in the IFM makes it a sensitive indicator of alterations in structure and function of myofibrillar proteins. Thick and thin filaments increase in length in regulated unison. Initial IFM thick filaments are 1.6-μm long in early pupal stages, and grow uniformly longer throughout pupal development to reach the adult length of 3.0 μm (Reedy and Beall 1993). Actin filament length appears closely coregulated with thick filament length; both filaments increase by the same increment of length throughout normal development. Because the I band is very short and IFM changes length very little during contraction, sarcomere length closely reflects thick filament length. Even small changes in filament length or regularity can impair flight ability and give a clear signal of alterations caused by mutations.
It is important to learn how the structure and function of different muscles are modified to obtain different contractile properties, such as contraction speeds and power outputs. One factor may be modified kinetics of actin and myosin isoforms (Bernstein et al. 1993). Another is likely to be accessory proteins that modify the function or structure of the motor proteins or myofilaments. Drosophila IFM offers the opportunity to study the effects of accessory proteins on thick filament assembly and identify general principles of filament assembly in vivo.
Flightin is a 20-kD myofibrillar protein that in Drosophila is expressed exclusively in IFM (Vigoreaux et al. 1993). Flightin undergoes extensive phosphorylation in an orderly temporal pattern in the hours preceding initial flight in newly eclosed adults, such that ∼50% of flightin is phosphorylated in mature adults (Vigoreaux and Perry 1994). Several results suggest that flightin is a component of the thick filaments: (a) light immunofluorescence microscopy localized flightin to the A band (overlap region) of the sarcomere (Vigoreaux et al. 1993); (b) IFM mutants null for myosin heavy chain [Ifm(2)2, renamed Mhc7; Bernstein et al., 1993], fail to assemble thick filaments and show loss of ∼12 other myofibrillar proteins, including flightin (Chun and Falkenthal 1988; Vigoreaux 1994); (c) biochemical fractionation of IFM fibers of the actin88F null mutant KM88, which lacks thin filaments, show flightin cosedimenting with the myosin-rich cytoskeletal fraction (Vigoreaux 1994).
We have initiated a genetic approach to elucidate the function of Drosophila flightin. Deletion of a region that included the flightin gene was homozygous lethal (Vigoreaux et al. 1998). However, flies heterozygous for that deficiency [Df(3L)fln] exhibited flight impairment and disruption of the periphery of the myofibrils (Vigoreaux et al. 1998), supporting an essential role for flightin in IFM structure or function. In this study, we have created fln0, a flightless strain that is null for flightin. We report the unexpected result that thick filaments and sarcomeres are ∼25% longer than normal in fln0 late pupa, while sarcomere structure is otherwise normal. Within hours after eclosion and the beginning of contractile activity, thick filaments shorten and sarcomere structure becomes increasingly disrupted as sarcomere length decreases, M lines disappear, actin filaments bend and buckle, and Z bands fragment. Cleavage of myosin heavy chain is also detected during this period. The immunolocalization, biochemical, and genetic results presented here establish that flightin codistributes with the thick filament, where it is probably associated with the myosin rod, and that flightin is essential for thick filament and sarcomere stability.
Materials and Methods
Fractionation of Myofibrillar Proteins
IFM fibers were dissected in a 4°C cooling chamber in solution YMG (50% glycerol, 20 mM phosphate buffer, pH 7.0, 2 mM MgCl2, 1 mM EGTA, and 8 mM DTT, 1 mM PMSF, 0.2 mM leupeptin). The fibers were centrifuged at 4°C for 5 min at ∼10,000 g, the supernatant was removed and discarded and the fiber pellet was resuspended in YMG containing 0.5% Triton X-100. After skinning for 1 h on ice, fibers were spun for 5 min (10,000 g at 4°C), the supernatant was discarded and the fiber pellet resuspended in washing solution (YMG without glycerol and Triton X-100) and centrifuged again. The wash was repeated twice, after which the fiber pellet was resuspended in myosin extraction buffer (1.0 M KCl, 50 mM K phosphate, pH 6.6, 10 mM NaPPi, 5 mM MgCl2, 8 mM DTT, and 0.5 mM EGTA, 1 mM PMSF, 0.2 mM leupeptin), incubated on ice for 10 min and centrifuged as above. The supernatant was transferred to an ultracentrifuge microtube, where it was diluted 10-fold with ice-cold water. Myosin was allowed to precipitate overnight in the cold room and then centrifuged at 100,000 g in a Beckman T100 ultracentrifuge for 10 min. The myosin pellet was resuspended in a small volume of 3 M KCl, and then diluted fourfold in myosin storage buffer (500 mM KCl, 20 mM MOPS, pH 7.0, 2 mM MgCl2, 8 mM DTT, 1 mM PMSF, 0.2 mM leupeptin).
Myosin Solubility
IFM fibers from 10 flies (<1-h old) were dissected in MgATP relaxing solution (16 mM K phosphate, 1 mM free Mg2+, 5 mM EGTA, 5 mM MgATP, 0.11 mM CaCl2, 20 mM BES buffer [N, N-bis(2-hydroxyethyl)-2-aminoethanesulphonic acid, titrated to pH 7], 10 mM DTT, 1 mM PMSF, 2 mM leupeptin; ionic strength was adjusted to 175 mM with added potassium methane sulfonate) without detergent (Dickinson et al. 1997), transferred to 25 μl of YMG with 0.5% Triton X-100, and skinned overnight at −20°C. The next morning, the samples were centrifuged for 10 min (∼14,000 g) at 4°C. The supernatant was removed, diluted with 25 μl of 2× Laemmli sample buffer and stored at −20°C until the rest of the samples were ready for gel electrophoresis. The fiber pellet was resuspended in wash buffer (see above), microcentrifuged for 5 min (4°C, 14,000 g), the supernatant was discarded, and the fiber pellet resuspended in 25 μl of relaxing solution. Fibers in relaxing solution were incubated for 1 h on ice, centrifuged, and the soluble fraction diluted with 2× Laemmli sample buffer. The pellet was resuspended in myosin extraction buffer with 0.1 instead of 1 M KCl, and incubated at room temperature for 10 min. After a 10-min centrifugation at 14,000 g at 4°C, the supernatant was removed and diluted with 2× Laemmli sample buffer and the pellet was dissolved in 1× Laemmli sample buffer. Duplicates of all samples were separated by SDS-PAGE in a 10% gel; one half of the gel was stained with coomassie blue and the other half electroblotted as described below.
In Vitro Proteolysis
IFM fibers from <15-min-old adults were dissected and skinned in YMG containing 0.5% Triton X-100. After two washes to remove YMG, the fibers were resuspended in 100 mM Tris, pH 8.0, with or without endoproteinase Arg-C (Boehringer). The amount of enzyme was adjusted to 1/50 the amount of fiber protein and the sample incubated at 37°C. After 15 min, the samples were diluted with an equal volume of 2× Laemmli sample buffer containing protease inhibitors and subjected to electrophoresis on 10% SDS-PAGE.
Protein Gel Electrophoresis and Western Blotting
A description of our SDS-PAGE and Western blot protocols has been published (Vigoreaux et al. 1993). All antibodies to muscle proteins are described in Saide et al. 1989 and Vigoreaux et al. 1993. mAb 3E8 recognizes an epitope in the S2 region of myosin heavy chain (MHC, our unpublished observation). Antibodies to proteosome subunits α and C8 were provided by Donald Mykles (Department of Biology, Colorado State University, Fort Collins, CO).
The relative amounts of flightin and myosin essential light chain (ELC) in skinned fibers were determined by scanning densitometry of SDS-PAGE.
Fly Stocks
KM88 is a null mutation of the IFM-specific actin88F gene (Okamoto et al. 1986). Mhc7[Ifm(2)2] is a mutation in the myosin heavy chain gene that prevents expression in IFM (Chun and Falkenthal 1988). KM88 and Mhc7flies were obtained from John Sparrow. Df(3L)fln is a lethal genetic deficiency in the 76BE region of the third chromosome that deletes the gene for flightin (Vigoreaux et al. 1998). Y57, a transgenic line expressing an IFM headless myosin in an Mhc7background (Cripps et al. 1999), and Mhc13, a myosin rod point mutation (Kronert et al. 1995), were obtained from Sandy Bernstein. The troponin I point mutant hdp2 was obtained from Ted Homyk.
Genetic Screen to Isolate a Flightin Null Mutant
To isolate new alleles of flightin, three batches of ∼100 males each from an isogenized w e line were aged 1–4 d before feeding overnight with a 1% sucrose solution containing 3 mM ethyl methanesulphonate (Sigma-Aldrich). Males were then mated to w; TM3 e Sb/TM6B e Tb virgin females. Individual w; e Sb F1 males were then mated to virgin Df(3L)fln/TM6C e Sb females and non-Sb progeny tested for the presence of flightin by immunoblotting as follows. Single flies were placed on individual wells of 96 microtiter plates containing 40 μl of Laemmli SDS sample buffer and homogenized with a multiprong replicator. A small amount of each sample was transferred with the replicator and imprinted onto a nitrocellulose membrane. The membrane was allowed to dry at room temperature for 1 h before proceeding with the immunoblotting procedure. The membranes were blocked, incubated with anti–flightin mAb 7f8 followed by incubation with secondary anti–mouse alkaline phosphatase antibody colorimetric assay (GIBCO BRL; for details, see Vigoreaux et al. 1993). Homozygous fly lines were established from siblings of flies in which no flightin was detected. The absence of flightin was then verified by Western blots of SDS-PAGE of IFM proteins.
Flight Test
A full description of the method can be found in Vigoreaux et al. 1998 and references therein.
DNA Cloning and Sequence Analysis
Total RNA was extracted from late stage pupa and very young adults with Trizol reagent (GIBCO BRL) following the manufacturer's protocol. 2 μg total RNA was reverse transcribed with 1 U AMV reverse transcriptase and 250 ng of oligo dT for 1 h at 42°C. The cDNA product was then amplified by PCR using two primers specific for flightin: forward primer: AGGATTCGGGGTACCCCGATGGCAGACGAAGA; reverse primer: CTTGGTATTTCCCGGGCCACTCC. The following conditions were used: one 5-min cycle at 94°C; 25 cycles of 30 s at 94°C, 45 s at 55°C, and 45 s at 72°C; one 5-min cycle at 72°C. The PCR product was ligated into the pGemT vector for 12 h at 12°C. DNA was purified from transformed colonies using Wizard prep (GIBCO BRL) and sequenced. DNA sequence was obtained from both strands of two independent clones for each fly strain (wild type and mutant).
Scoring IFM Fiber Morphology
Normal and mutant flies were aged for the indicated times and their thoraces were dissected in half along the midline (Kronert et al. 1995). Dorsolongitudinal (DLM) fibers in each hemithorax were scored under a dissecting microscope following the scale described in Kronert et al. 1995, where 0 is assigned to a normal length fiber and 3 to a fiber >50% shortened length. Values represent mean ± SD.
Electron Microscopy
Fly thoraces for scanning electron microscopy were prepared as described in Kronert et al. 1995. Transmission EM used glutaraldehyde/tannic acid fixation as described previously (Reedy and Beall 1993). Glycerination and rigor were done as described in Reedy et al. 1989.
For immunoelectron microscopy, thoraces from wild type or the actin null mutant KM88 were divided in half along the midline after removing the gut. Half thoraces in ice-cold rigor buffer (0.1 M NaCl, 20 mM Na phosphate, pH 6.8, 5 mM MgCl2, 5 mM EGTA, 5 mM NaN3) were infiltrated with 2.1 M sucrose in rigor buffer on ice for 15 min, with three changes of solution. The half thoraces were mounted on aluminum pins and, after removing excess sucrose, the pins were dropped into liquid ethane cooled with liquid N2. The pins were then transferred under liquid N2 into a Reichert CS Auto cryosubstitution apparatus and processed by a modification of the method of van Genderen et al. 1991. Samples were dehydrated for 48 h at −90°C, followed by an increase in temperature of 2°C/h for 24 h to a final temperature of −45°C. After washing in methanol, samples were infiltrated with mixtures of Lowicryl HM20 and methanol in ratios of 1:1, 2:1, and pure resin, each for 2 h with one change, and then overnight with pure resin. Half thoraces were embedded at −45°C in 0.5-ml Eppendorf tubes and the resin was polymerized with UV light for 48 h. 50–60-nm-thick sections were cut from the outer surface of the IFM on a Reichert OMU3 ultramicrotome with a diamond knife and mounted on 200-mesh gold grids coated with Formavar and carbon. Sections were rehydrated on a drop of rigor buffer and labeled with antiflightin rabbit serum diluted 1:30, followed by Protein A gold (10 nm) diluted 1:65, as described by Lakey et al. 1990. Sections were stained with 6% uranyl acetate and viewed in a Philips CM20 Bio-Twin microscope at 100 kV.
Electron micrographs of wild-type IFMs were digitized with a scanner (SCAI; Carl Zeiss, Inc.) at 28-μm pixel size. Images were displayed on a Macintosh computer and gold particle positions were measured using NIH Image software. Histograms showing the distribution of the position of gold particles were calculated using Kaleidagraph (Abelbeck Software).
Production of Antiflightin Polyclonal Antibodies
Recombinant flightin was expressed and purified from insect Sf9 cells (PharMingen) transfected with a recombinant baculovirus vector containing a flightin cDNA (Vigoreaux et al. 1993). Transfected Sf-9 cells were precipitated by microcentrifugation and the cell pellet resuspended in cell lysis buffer (334 mM NaCl, 10 mM phosphate buffer pH 7.4, 1.0% Triton X-100). The sample was spun in a microcentrifuge and the flightin containing pellet was resuspended in a denaturing buffer (8 M urea, 100 mM sodium phosphate, 10 mM Tris, pH 8.0). After a brief microcentrifugation, the supernatant was incubated with Ni-NTA agarose (Quiagen) and flightin was purified following manufacturer's instructions (Quiagen). The purified flightin fraction was subjected to electrophoresis on 12% SDS-PAGE, stained with Coomassie blue, and the band containing flightin was cut out from the gel. Approximately 2.5 mg of flightin was used to immunize rabbits. Antibody production was performed by Cocalico Biologicals.
Results
Flightin Is Associated with Thick Filaments
Four lines of evidence indicate that flightin is associated with thick filaments. (a) We used a modified high salt fractionation protocol commonly used to extract myosin from muscle preparations (Starr and Offer 1982). Skinned IFM fibers from wild-type flies were treated with a solution containing 1 M KCl and the solubilized proteins recovered by ultracentrifugation, as seen in Fig. 1. The fraction containing MHC and its associated light chains (ELC and RLC), also contains flightin. (b) We used the same high salt extraction on skinned fibers from IFM of Y57 transgenic flies that express a “headless” myosin in their IFM (Cripps et al. 1999). This headless myosin lacks all sequences NH2 terminal to the RLC binding site. As seen in Fig. 1, the KCl-soluble fraction contains the MHC rod polypeptide, RLC, and flightin. As expected, ELC is not detected in this fraction, suggesting that absence of its binding site prevents ELC from accumulating in the IFM. This result suggests that flightin is associated with the rod portion of the myosin molecule. (c) Immunoelectron microscopy provided additional evidence that flightin is associated with the myosin thick filaments. We used an antiflightin polyclonal antibody and gold label to locate flightin in wild-type IFM and in the IFM of the actin88F null KM88. The actin null mutant completely lacks thin filaments, but does assemble large numbers of thick filaments that form bundles. Electron microscopy of thin Lowicryl sections of wild-type IFM showed the distribution of antiflightin gold over the A band (Fig. 2 A). The histogram shows there is a gap of 800 nm in the label in the Z-band region and one of ∼170 nm in the M-line region. Therefore, flightin is in the A band, but is not uniformly distributed along its length. Antiflightin gold labeling of KM88 IFM showed extensive labeling localized over the thick filaments (Fig. 2 B), strongly suggesting that flightin is associated with the thick filaments. (d) Genetic studies provide another line of evidence that flightin is a thick filament protein (see below).
Figure 1.

Flightin cofractionates with myosin after high salt extraction of skinned IFM fibers. A 10% SDS-PAGE of proteins from a high ionic strength extraction and fractionation of skinned IFM fibers from wild-type flies (MHC+) and from Y97 transgenic flies (MHCY97) expressing a truncated MHC lacking the motor domain (Cripps et al. 1999). F shows total myofibrillar proteins from a skinned fiber before extraction, and P shows the 1-M KCl insoluble pellet after ultracentrifugation at 100,000 g. The supernatant from the 1-M KCl extraction was then diluted 10-fold and subjected to a second ultracentrifugation at 100,000 g. S shows the supernatant and M shows the precipitated, myosin-enriched fraction, which includes MHC, RLC, phosphorylated RLC, ELC, and flightin (FLN). By immunoblot analysis, flightin is detected in the M fraction, but not in the P or S fractions (not shown). Note that flightin is present in the thick filament-enriched M fraction from IFM of Y97 and, as expected, the ELC is absent. The difference in flightin mobility is due to phosphorylation (Vigoreaux et al. 1993). These results suggest that flightin is associated with the thick filament shaft.
Figure 2.

The distribution of flightin in myofibrils of wild type and the actin null, KM88. Thin Lowicryl longitudinal sections of Drosophila IFM labeled with anti–flightin polyclonal antibody and Protein A gold. Both sections show tissue from adults >2 d old. (A) Wild type shows labeling on the A band with gaps each side of the Z band and at the H zone. The histogram shows the distribution of gold particles measured in five sarcomeres from different myofibrils. (B) KM88 actin null, where no thin filaments are present, has labeling on bundles of thick filaments. The resolution of the probe is not sufficient to distinguish between label on or between thick filaments, but the presence of flightin in the actin null suggests flightin is associated with the thick filaments. Scale bars: 0.5 μm.
A Null Mutation in Flightin Abolishes Flight
A genetic screen was conducted to identify mutations that prevent flightin expression in the IFM (Fig. 3 A). Male flies were fed EMS (a depurinating agent), mated to female Df(3L)fln flies, and the hemizygous progeny was tested for the presence of flightin via dot blots. Of 1,280 flies tested, one (2987) did not express flightin (Fig. 3 B). A homozygous line established from a sibling did not express flightin (Fig. 3 C).
Figure 3.
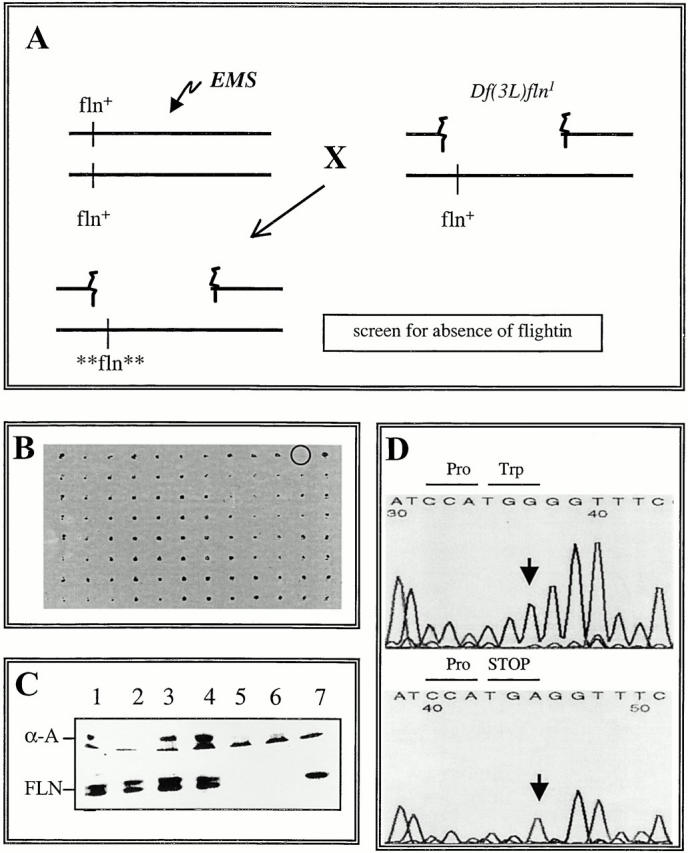
Generation and characterization of a null allele of flightin. (A) Summary of genetic scheme to isolate null mutation in the flightin gene. Male flies fed the mutagen EMS were mated to females heterozygous for Df(3L)fln and the hemizygous progeny were scored for the absence of flightin by immunodot blots. (B) A representative dot blot. The sample encircled, 2987, did not react with an antiflightin mAb. (C) Strain 2987 does not express flightin. Western blot showing the accumulation of α-actinin (top) and flightin (bottom) in IFM of the following strains: (1) Oregon R (wild-type); (2) Df(3L)kto1/+, a deficiency of the 76BD region that does not delete the flightin gene; (3) Df(3L)fln/+; (4) 2987/+; (5) 2987/Df(3L)fln; (6) 2987/2987; (7) +/+ pupa. Note that flightin is not detected in 2987 homozygotes nor 2987/Df(3L)fln transheterozygotes. (D) DNA sequence analysis of the flightin gene from wild-type flies (top) and 2987 (bottom). Note the G to A transition converts a Trp codon at the eighth amino acid position into a stop codon. Herein we will refer to line 2987 as fln0.
To determine whether 2987 flies have a genetic lesion in their flightin gene, we isolated total RNA from a mixed population of late stage pupa and very young adults (the stages at which flightin expression is maximal; Vigoreaux et al. 1993) and generated cDNA to serve as a template for PCR analysis using flightin-specific primers. PCR products from wild-type controls and 2987 flies were then subjected to DNA sequence analysis. As seen in Fig. 3 D, the flightin cDNA from 2987 flies has a G-to-T transition at the 8 amino acid codon converting Trp to a stop codon. Therefore, the absence of flightin immunoreactivity in dot and Western blots is explained by premature termination of the open reading frame. These results, together with the observation that no other myofibrillar protein appears to be missing or reduced in 2987 IFM (data not shown), strongly suggest that the observed mutant phenotype is due to a lesion in the flightin gene. Herein we will refer to line 2987 as fln0.
Homozygous fln0 flies, as well as hemizygous fln0/Df(3L)fln, are flightless (Table ) and show defects in their wing position, holding their wings ventrolaterally as opposed to dorsally. However, their fecundity and viability are not affected, suggesting that the mutation that impairs flight ability has little or no effect on muscles essential for viability. To establish that the flightless phenotype results from the mutation in the flightin gene, we mapped the genetic position of the flightless mutation by meiotic recombination. The mutation was found to map 23.1 map units from ebony and 0.05 map units from Df(3L)kto2 (Hernandez 1998). The flightin gene is located at ∼76E1, just outside the proximal breakpoint of Df(3L)kto2 (Vigoreaux et al. 1998). The genetic mapping results strongly suggest that the flightless mutation lies within or very close to the flightin gene. Given that no other muscle protein genes are known to exist within the region where the mutation maps, we conclude that the mutation in flightin is responsible for the flightless phenotype of fln0.
Table 1.
Flight Index of flightin, Mhc, and Actin Null Mutant Heterozygotes and Double Heterozygotes
| Genotype | Flight index | |
|---|---|---|
| n | mean ± SD | |
| +/+ | 10 | 5.6 ± 1.1 |
| fln0/fln0 | 25 | 0 |
| fln0/Df(3L)fln1 | 10 | 0 |
| Mhc7/+ | 8 | 0.1 ± 0.4 |
| KM88/+ | 15 | 0.1 ± 0.3 |
| Mhc7/+; KM88/+ | 27 | 3.3 ± 1.2 |
| fln0/+ | 10 | 5.0 ± 0.8 |
| Mhc7/+; fln0/+ | 24 | 0.3 ± 0.6 |
| fln0+/+KM88 | 19 | 3.4 ± 1.7 |
The availability of fln0allows genetic analysis to further test the hypothesis that flightin is a thick filament component. We conducted experiments to determine whether removing one functional flightin gene copy can restore the flightless phenotype of KM88 heterozygotes and/or Mhc7 heterozygotes. Previous studies have shown that the flight impairments and myofibrillar defects of KM88/+ and Mhc7/+ flies are due to thin-to-thick filament imbalances (Beall et al. 1989). This is supported by the observation that Mhc7/+; KM88/+ double heterozygotes are flight competent and have sarcomeres that are more normal than those of either KM88/+ or Mhc7/+ single heterozygotes (Beall et al. 1989). These results show that an actin to mhc gene dosage ratio of 1:1 restores thin to thick filament stoichiometry and overcomes the structural defects engendered by single heterozygotes.
We crossed fln0 flies to KM88 to generate fln0+/+KM88 double heterozygotes and fln0 flies to Mhc7 flies to generate Mhc7/+; fln0/+ double heterozygotes. Table summarizes the results of a flight test of single and double heterozygous lines. Removal of one flightin gene copy partially restores the flight impairment of KM88 heterozygotes (fln0+/+KM88) but not of Mhc7heterozygotes (Mhc7/+; fln0/+). Furthermore, the flight ability of fln0+/+KM88 double heterozygotes is comparable to that of Mhc7/+; KM88/+ double heterozygotes. One possibility is that the improvement in flight performance of fln0+/+KM88 over KM88/+ results from a restoration of thick to thin filament stoichiometry, analogous to the principle in the experiment described above for Mhc7/+; KM88/+. This result implies that flightin is present in large amounts relative to actin and myosin and supporting this are our unpublished results (Ayer, G., and J.O. Vigoreaux) indicating a 1–1.5 ratio of ELC to flightin. These results are consistent with the interpretation that flightin is a thick filament protein.
fln0 Fibers Have Abnormal Morphology by Light and Scanning Electron Microscopy
The absence of flightin has a dramatic effect on IFM cell morphology in pupa as well as in adults. In wild-type flies, the DLM IFM fibers are straight and parallel to the body axis, and extend fully from the anterior to the posterior end of the thorax (Fig. 4 A). In contrast, the DLM of many fln0 late-stage pupa and pharate adults are wavy, as if crimped to fit within the thoracic cavity (Fig. 4 B). Light microscopy of 1-μm sections show that some fibers are narrower than normal and/or misoriented with respect to the anterior–posterior body axis. Light microscopy of sections also revealed that not all DLM fibers are equally disordered during the first 12 h after eclosion. The two longest DLM fibers located nearer the ventral thorax appear more severely affected than the other four DLM fibers. Furthermore, the fibers are not uniformly affected along their length. The two most affected fibers are paler staining with toluidine blue than the others. Regions near the myotendon junction appear very disordered (results not shown).
Figure 4.

Absence of flightin results in large changes in IFM morphology. Scanning electron micrographs of entire half-thoraces from wild-type adult (A), newly eclosed (<30 min) fln0adult (B), and 3-d-old fln0adult (C). In wild-type flies, DLM fibers (*) extend the entire antero-posterior length of the thoracic cavity (A). In contrast, newly eclosed fln0adults occasionally show “wavy” fibers that are longer than normal (arrow). In C, all DLM fibers have detached from the posterior cuticle (arrow). For all figures, anterior is right and dorsal is top. Magnification 150× (A and B) or 200× (C). (d) Time course of fln0fiber degeneration. Flies aged for the indicated times were dissected and their DLM fibers were scored on an arbitrary scale of 0 (normal) to 3 (severely shortened). Each time point represents at least five flies (mean ± SEM). Note that by 8 h, all fibers appear completely degenerated. A similar abnormality has been described for Mhc13(Kronert et al. 1995).
During the first day of adult life, the morphology of DLM fibers changes dramatically from long and wavy to shortened and torn. Scanning EM of the DLM fibers from 1-d-old adults shows only small remnants remain attached to the cuticle (Fig. 4 C). We routinely observe fibers in which the bulk of the mass appears to be pulled towards one of the cuticle anchoring sites, most often toward the anterior end, resulting from rupture at the myotendon junction from the opposite end. In some flies, all six DLM appear to shorten to the same extent while, in other flies, some fibers are shortened and some appear normal or less affected. The appearance of the shortened fibers occurs over a time course of a few hours after eclosion, very similar to that described previously for the myosin rod mutant Mhc13 (Fig. 4 D; Kronert et al. 1995).
IFM Sarcomeres Are Abnormally Long in fln0 Pupa
EM examination of fln0 DLM of late stage pupa in thin longitudinal sections shows that myofibrils pass in and out of the section plane over short distances, only staying well oriented in the section plane for two or three sarcomere lengths. It is immediately apparent upon simple inspection that the fln0 pupal sarcomeres are narrower and longer than wild-type sarcomeres (Fig. 5). fln0 sarcomeres also vary over a wider range (wild type, 3.1–3.3 μm, average 3.1 μm, n = 24; fln0, 3.6–4.1 μm, average 3.81 μm, n = 36). The longer sarcomere length reflects the longer length of the thick filaments, but it is important to note that the thin filaments are also uniformly longer, since they extend to the M line.
Figure 5.
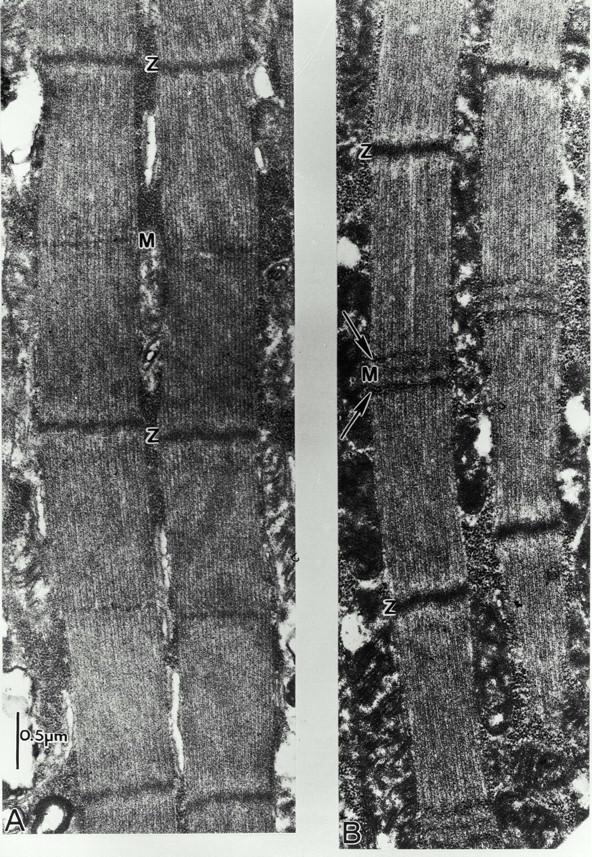
Flightin null assembles long myofilaments and sarcomeres during pupal development. EMs of thin longitudinal sections of IFM from wild-type (left) and fln0 (right) late stage pupa. At this developmental stage, wild-type sarcomeres show an average length of 3.1 μm, while fln0 sarcomeres show an average length of 3.8 μm and are narrower. Note the presence of two transverse electron dense stripes flanking the M line (arrows). These stripes are reminiscent of the transient, particle-dense stripes that have been observed in some late pupa of wild-type IFM (Reedy and Beall 1993). In normal flies, the double band is present for a few hours just before the end of pupation; in fln0 flies, the double band sometimes persists into adulthood.
Pupal IFM (wild-type as well as fln0 and other IFM mutants) is characterized by very dense packing of ribosome-like granules, mitochondria, and myofibrils (Fig. 5). The composition of the granules has not been determined, but they may represent ribosomes, glycogen, and/or proteosomes. Though abundant in pupa, the particles are reduced to almost undetectable amounts in wild-type IFM. A notable feature of fln0 pupal myofibrils is the presence of two transverse electron-dense stripes flanking the M line (Fig. 5). The peri-M line stripes seen in fln0 pupa resemble the transient, particle-dense stripes observed in late pupa of wild-type IFM (Reedy and Beall 1993). In wild-type pupa, the double band is apparently only present for a few hours just before the end of pupation, so even with 10-h sampling, the double band is not observed in all wild-type pupa. Because of the limited number of wild-type pupa (three) sectioned as controls for these experiments, examples of this double band were not seen. However, the prominence of the double bands in late fln0 pupa, together with the presence and distribution of similar particles persisting in fln0 adult IFM (see Fig. 8), suggests that this feature may be particularly significant, although its function remains unknown.
Figure 8.
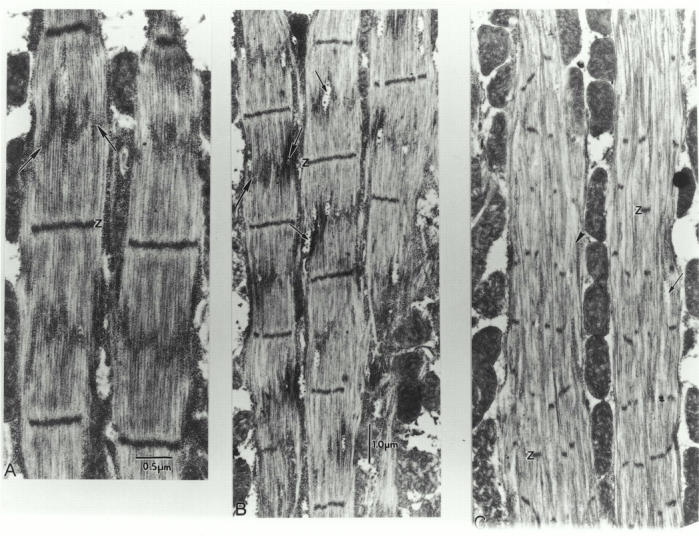
Unusual accumulation of particles in degenerating fln0adult IFM. Longitudinal 60-nm sections of adult IFM. (A) Note abundance of dense particles, similar to those observed in pupa but never seen in wild-type adults. The particles form a “flame-stitch” pattern across the myofibril, usually centered about the former M line (large arrows). (B) Blank gaps in the filament lattice containing larger globular particles (small arrows) are common at this stage. (C) Z bands have broken apart into Z bodies that are no longer in axial register. Spacing between Z bodies is ∼1.4 μm. Regions with numerous thin filaments but devoid of thick filaments also are seen (arrowhead).
In wild-type Drosophila IFM, myofilaments are in lateral register and Z, A, and M bands are seen in different cross-section levels (Fig. 6, A–C). Cross-sections of IFM of fln0 late pupae show the normal wild-type pattern of ordered Z band, hollow thick filaments in the A band surrounded by six thin filaments and solid thick filaments in the M band (Fig. 6, D–F). That fln0 thick filaments are hollow in the A band (Fig. 6 E) and solid in the M band (F) indicates that flightin is not essential for assembly of an ostensibly normal thick filament shaft. However, fln0 myofibrils have fewer thick filaments across the myofibril diameter (∼17–19), compared to wild-type (∼25–26) at the same stage.
Figure 6.
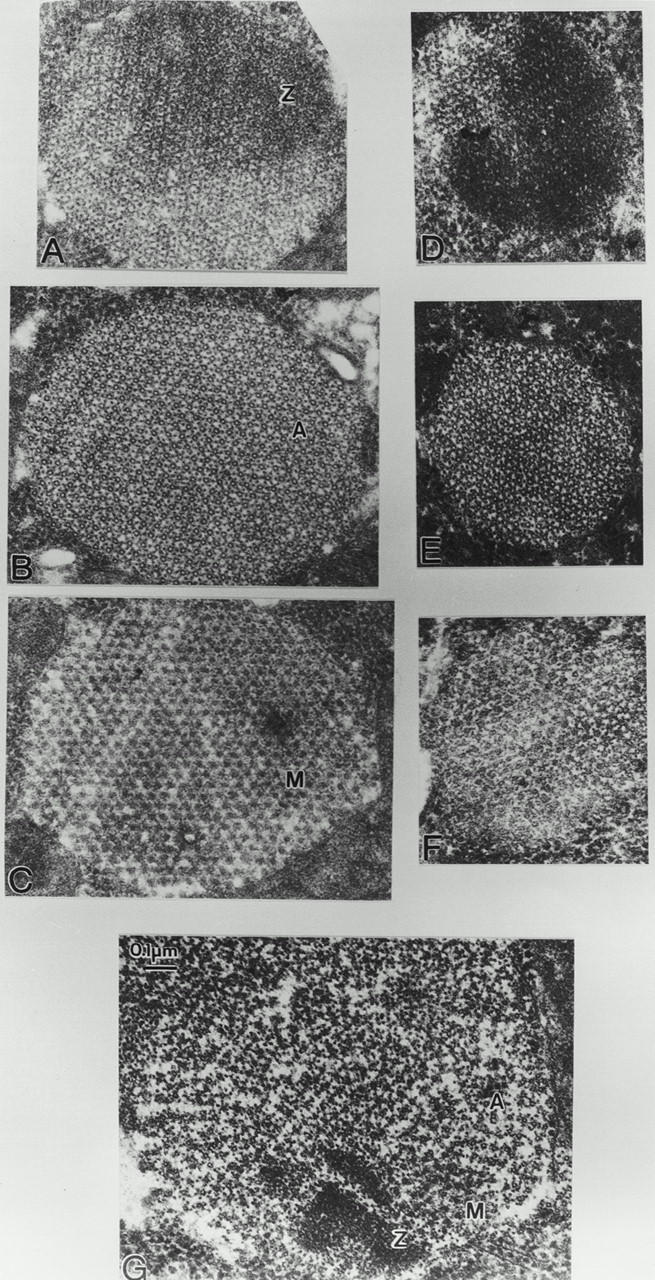
Electron micrographs of cross sections show normal sarcomere assembly of pupal IFM in fln0 and the disruption of adult sarcomeres during the first hours after eclosion. (A–C) Late pupal wild-type (D–F) late pupal fln0, and (G) adult fln0 IFM. Wild-type shows (A) an ordered lattice in Z bands, (B) hollow thick filaments in the A band in a regular hexagonal lattice with thin filaments, and (C) solid thick filament profiles in the M band. fln0 late pupa, although smaller in diameter due to fewer filaments, show normal Z band (D), hollow thick filaments in the A band (E), and solid thick filaments in the M band (F). In adult fln0(G), all sarcomere levels are seen within one section of a single myofibril due to filament misregistration and fragmentation of the sarcomeres. Z bands (Z), A band (A), and M band (M).
Breakdown of Sarcomere Structure in fln0 Adult IFM Seen by Transmission EM
The abnormally long sarcomeres common in fln0 pupa are not seen even in newly eclosed adults; the majority of sarcomeres measure between 2.2 and 2.5 μm. However, adult fln0 IFM show a wide variation in sarcomere length, ranging from 1.4 to 3.3 μm (n = 108) even within a single thorax. This contrast with the uniform length of adult wild-type IFM sarcomeres is consistently between 3.1 and 3.3 μm (Reedy and Beall 1993). IFM sarcomeres have very narrow I bands, both in wild-type and in fln0, so the sarcomere length closely reflects thick filament length. The shorter fln0 sarcomeres are not due to hypercontraction and no double overlap of thin filaments was observed.
Adult fln0sarcomeres become severely disordered, in contrast to the relatively well-ordered sarcomeres in pupa. Initially, a wave of degeneration is seen along the same myofibril, with disorder increasing toward one end (Fig. 7), but as the fly ages the entire IFM degenerates. Fig. 7 shows 25-nm longitudinal sections of adult wild-type (Fig. 7 A) and fln0 (b–d) DLM within 12 h after eclosion. The best ordered sarcomeres in fln0are seen in newly eclosed adults, and even these show complete loss of the M line, signs of Z band fragmentation, focal loss of some thick filaments in the myofibril mid regions, bundles of thin filaments protruding at the periphery, and a few very long (∼10 μm) thick filaments lying alongside the myofibril (Fig. 7 B). Surprisingly, the sarcomere length in these best-ordered regions is close to wild type (∼3.1–3.3 μm). Progressing along the same myofibrils, ∼50-μm shows an increase in disorder of the sarcomeres (Fig. 7 C). The Z bands and filaments separate laterally into smaller bundles and a gradual decrease of Z-band spacing (∼2.0–2.5 μm) is visible. Many of the thick filaments seem to have disappeared, especially from the M band region. Thin filament “cowlicks” project out of the sarcomeres or bow out of the myofibril. These give the impression that thin filaments are not shortening, or not decreasing in length at the same rate as the apparent shortening of the thick filaments. The bundles or cowlicks of actin filaments also suggest that the myosin filaments with which they were interdigitated have been removed. Further along the same myofibril, sarcomeres become severely disordered (Fig. 7 D).
Figure 7.

Rapid, progressive sarcomere degeneration in fln0 adult IFM. Electron micrographs of (A) wild-type sarcomeres (B–D) fln0 sarcomeres along the same myofibril in longitudinal 25-nm sections of adult IFM within 12 h after eclosion. In contrast to the relatively well-ordered sarcomeres in pupa (see Fig. 5), adult fln0 sarcomeres are severely disordered. There appears to be a gradual increase in disorder toward one end of a fiber. (B) Even in the best-ordered fln0adult sarcomeres, the M line has disappeared and the Z band tends to fragment. Peripheral bundles of thin filaments (arrowhead) and very long thick filaments (∼10 μm, arrows) are evident. These well-ordered sarcomeres have already shortened to ∼3.1-μm long, about the same length as wild type. (C) Same myofibril as B, but ∼50 μm closer to one end. Note increase in disorder. Z bands are more fragmented and sarcomeres are shorter (∼2.0 μm). In some regions, thick filaments are missing or appear fragmented. Thin filament “cowlicks” project out of the sarcomere (arrowhead). (D) Even more disordered sarcomeres are found further along the same myofibril.
Slightly thicker 60-nm longitudinal sections of adult fln0 IFM reveal an abundance of dense particles, similar to those observed in pupa, associated with the myofibril (Fig. 8 A). The particles form a “flame-stitch” pattern across the myofibril, usually centered near the former M line. This distribution of particles is never seen in wild-type myofibrils. Occasionally, the particles coincide with blank gaps in the filament lattice (Fig. 8 B), which contain several larger globular particles. To determine whether the particles seen associated with gaps in the filament lattice are proteosomes, we performed Western blots on whole fln0 and wild-type fibers from different age adults using antibodies that recognize the α and C8 proteosome subunits (Covi et al. 1999). We were not able to detect expression of either subunit in wild-type or fln0 IFM (data not shown).
Further along the same myofibrils, the sarcomeric pattern becomes disordered. Z bands break apart into Z bodies that are not in axial register, and Z-body spacing becomes very short, down to 1.4 μm. Even on sarcomeres this shortened, significant numbers of thick filaments are present. Remarkably, these disordered sarcomeres maintain cohesion as myofibrils with well-defined periphery, separated by mitochondria (Fig. 8 C). We note also that thick filaments are consistently seen emerging from Z bands or Z bodies, even in the most disordered areas. Thick filaments appear to be missing primarily from the middle of the sarcomere, creating boundaries where sarcomeres seem to split. The variable and preferential shortening of thick filaments seems to be responsible for the presence of mini-sarcomeres and sarcomeres of variable length.
Cross-sections of adult fln0 IFM show all sarcomere levels in one section of a single myofibril, due to filament misregistration, misorientation, and lattice disarray (Fig. 6 G). Hollow thick filament profiles remain prevalent, and solid profiles of thick filaments characteristic of M bands can still be detected in patches (Fig. 6 G), as expected if groups of only a few thick filaments remain laterally aligned. The solid profiles are noteworthy because the M band is consistently absent in adult fln0 IFM, but even the shortened thick filaments still have a solid mid region. This suggests that the antiparallel arrangement of myosin molecules is preserved in fln0 even though thick filament length and stability are altered. It also suggests that myosin is not being removed primarily from the bare zone of the thick filaments.
To determine whether the absence of flightin prevents actomyosin interaction even under high affinity conditions in the absence of ATP, fln0 IFM was glycerinated in situ and placed under rigor conditions to promote maximal affinity of myosin for actin (Reedy et al. 1989). As seen in Fig. 9, thin filaments appeared decorated by myosin everywhere, but only in well-ordered areas was it possible to see that rigor chevrons or arrowheads pointed toward the middle of the sarcomere (toward the missing M line) and that crossbridge polarity was consistent for long stretches along a single thin filament. These results indicate that myosin polarity was appropriately reversed somewhere along the thick filament. However, thick and thin filament arrangement and/or structure was disturbed sufficiently that chevrons sometimes showed opposite polarity on adjacent thin filaments and polarity did not reverse uniformly at mid sarcomere. Frequently, crossbridges pointed in one direction well past the midpoint of the sarcomere. However, chevron polarity was always reversed close to opposite Z bands delineating an individual sarcomere. This suggests that, by an otherwise random process, thick filaments are being shortened in the mid region of each half sarcomere, leaving normal length actin filaments extending into what previously was the opposite half sarcomere.
Figure 9.
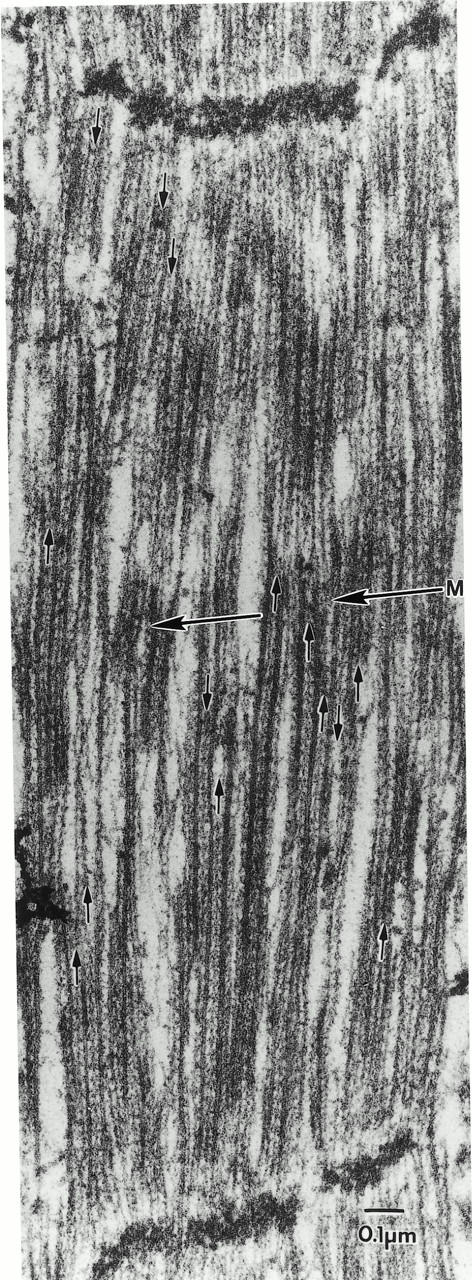
Absence of flightin does not impair rigor crossbridge formation. Rigor was induced in glycerinated adult fln 0 IFM by ATP washout. Thin filaments appear decorated throughout by myosin but rigor chevrons (arrowheads) can be seen pointing towards the missing M line only in well-ordered areas. The level of the missing M line is indicated by a transverse arrow. Arrows on thin filaments are ahead of a series of crossbridges on a thin filament and indicate the direction in which the crossbridge arrowheads are pointing. In normal IFM, all rigor crossbridges within a half sarcomere point uniformly towards the M line (Dickinson et al. 1997).
Myosin Undergoes Site-specific Proteolysis in fln0 Adult IFM
The shortened thick filaments of fln0 adult IFM could result from myosin proteolysis that promotes disassembly and sarcomeric dysgenesis. To test this possibility, we examined MHC in pupal and adult IFM for evidence of proteolytic cleavage. Fig. 10 A shows a Western blot of skinned fiber proteins from adults before (0–2 h after eclosion) and after (2–4 d) fiber degeneration. The blot was probed with an anti–MHC mAb that recognizes an epitope in the heavy meromyosin region of myosin. In all samples tested, the antibody recognizes a prominent protein band that corresponds to full-length MHC. However, in 2–4-d fln0and Mhc13 fibers, proteolytic cleavage was evidenced by an additional, faster migrating band of ∼150 kD. The 150-kD band in Mhc13 has been shown to correspond to the NH2-terminal region of MHC that results from proteolytic cleavage at a site near the hinge region at the S2–light meromyosin (LMM) junction (Kronert et al. 1995). Note that the 150-kD MHC peptide is not present in 2–4-d heldup2 (hdp2) fibers. hdp2 carries a point mutation in the troponin I (TnI) gene that has little or no effect on myofibril assembly but leads to IFM degeneration during pupal-to-adult transition (Beall and Fyrberg 1991). These results suggest that fln0IFM myosin is subject to proteolysis at a site near or identical to the proteolytic site in Mhc13 IFM myosin. Cleavage of myosin near the hinge region is not a widespread trait of muscle degeneration, as evidenced by its absence in the TnI mutant, whose sarcomeres are severely disrupted.
Figure 10.

Myosin undergoes proteolysis in IFM of fln0adults. (A) Immunoblot analysis of skinned IFM fiber proteins separated on a 10% SDS-PAGE. The blot was incubated with an anti–MHC mAb. Lanes b are IFM proteins from adult flies aged 2–4 d; lanes a are IFM proteins from newly eclosed adults, <5 h old. Full-length MHC is detected in all samples. However, note the presence of the ∼150-kD peptide (arrow) in the aged Mhc13 and fln0, but not in the OR (wild-type) and hdp2 samples. The absence of site-specific myosin proteolysis in hdp2 is significant since this TnI mutation leads to severe fiber degeneration and sarcomere breakdown. (B) In vitro proteolysis of IFM skinned fibers with endoproteinase Arg-C. Skinned fibers from <15-min-old adults were incubated without (−) or with (+) endoproteinase Arg-C and partial digestion products separated on a 10% SDS-PAGE. An immunoblot incubated with anti–MHC mAb 3E8 is shown. A 150-kD peptide appears in Mhc13- and fln0-treated fibers, but not in OR- or hdp2-treated fibers. Since flightin is absent in Mhc13and fln0, it may protect an MHC protease-sensitive region in vivo.
Myosin from adult Mhc13 IFM and adult fln0IFM appears to be cleaved at the same site and both mutants lack flightin (Kronert et al. 1995). This suggests that in normal IFM, flightin protects a protease-sensitive site in the MHC rod that is exposed in the absence of flightin. To test this possibility, we conducted in vitro proteolysis of skinned fibers from <15-min-old adults (before proteolysis is detected in fln0 in vivo) to establish whether MHC from Mhc13 and fln0is more susceptible to proteolysis than MHC from wild-type or hdp2 fibers. We used endoproteinase Arg-C since the in vivo cleavage site in Mhc13 lies at the carboxylic side of an arginine residue (Kronert et al. 1995). Limited proteolysis of fln0and Mhc13 fibers produces an MHC NH2-terminal peptide that comigrates with the in vivo 150-kD MHC peptide seen in 2–4-d adults. The same proteolytic treatment of wild-type and hdp2 fibers did not produce the 150-kD peptide (Fig. 10 B).
Absence of Flightin Results in Partial MHC Solubility
To determine whether the absence of flightin affects the incorporation of myosin into thick filaments and/or the stability of assembled molecules, we tested for the presence of soluble myosin in commonly used solutions. In normal muscle (<30-min-old adult), myosin is not solubilized when the fiber is treated with a skinning or relaxing solution of physiologic ionic strength (Fig. 11). In contrast, a small pool of soluble MHC is recovered from fibers of <30-min-old fln0adults under the same physiological conditions. Treatment of skinned fibers with a relaxing solution of higher ionic strength (0.1 M KCl) solubilizes ∼50% of the myosin from fln0fibers, but considerably less from wild-type fibers (Fig. 11). None of these treatments results in solubilization of actin from either wild-type or fln0fibers, suggesting that the absence of flightin does not affect the integrity of thin filaments.
Figure 11.

The absence of flightin results in partial solubility of myosin. Skinned IFM fibers from wild-type (sample 1) or fln0 (sample 2) were first incubated in ATP relaxing solution followed by a 0.1 M KCl extraction solution (see Materials and Methods). (Left) 10% SDS-PAGE stained with Coomassie blue, and (right) corresponding Western blot incubated with anti–MHC mAb 3E8. Fractions S correspond to proteins extracted with skinning solution (YMG), fractions R contain proteins extracted with relaxing solution, fractions E contain proteins extracted with 0.1 M KCl myosin extraction buffer, and P is the remaining pellet after all extractions. MHC is detected in both skinning and relaxing soluble fractions from fln0but not from wild type. Also note that a larger proportion of MHC is extracted by 0.1 M KCl from fln0 than from wild type. Myosin solubility appears to be specific as judged by the absence of actin from all soluble fractions.
Discussion
One of the most remarkable features of striated muscle is the assembly to a particular and uniform thick filament length within a muscle. Several models for thick filament length determination have been proposed (reviewed in Barral and Epstein 1999). Virtually all models include the assistance of accessory and/or scaffolding proteins to enable the correct assembly of myosin into thick filaments in striated muscle. Our results establish that flightin is a thick filament–associated protein that is likely to interact with the myosin rod. In the absence of flightin, thick filaments assemble to abnormally long lengths during pupation and become unstable immediately upon initiation of contractile activity in adults. This is followed by a rapidly progressing degeneration and retraction of the IFM and concomitant site-specific cleavage of myosin.
Flightin Is Associated with IFM Thick Filaments
In wild-type IFM, light immunofluorescence microscopy (Vigoreaux et al. 1993) and immuno EM (this study) showed flightin distributed throughout the thick filament–thin filament overlap region, except for ∼0.3 μm flanking the Z bands. The recovery of flightin in the myosin-enriched fraction from a high salt extraction reported here, the absence of flightin from Mhc7 (Vigoreaux 1994) and Mhc13 (Kronert et al. 1995) IFM, and the recovery of flightin in the cytoskeletal fraction of the actin null KM88 IFM (Vigoreaux 1994) support the association of flightin with myosin. Our immuno EM of KM88 IFM shows clearly that flightin labeling occurs along the thick filaments, with gaps around the Z lines and at the M band. These gaps in labeling (of the Z lines and M bands) is consistent with our observation that Z lines, M bands, and the hexagonal packing of myofilaments appear normal well into late stages of pupal development in the absence of flightin in fln0. Note that flightin expression does not begin until ∼60 h of pupal development (Vigoreaux et al. 1993). Our observation that fln0 thick filaments are hollow in the A band and solid through the M band region establishes that the absence of flightin does not induce large scale malformation of thick filament structure. On the other hand, the enhanced solubility of fln0myosin indicates that flightin is important in establishing and/or maintaining the cohesiveness of the thick filament. Because solubilization of myosin precedes detectable proteolysis (Fig. 11), it is possible that sarcomeres in fln0adults are initially shortening by disassembly of thick filaments by release of intact myosin molecules. It is significant that the heterozygote flightin mutant Df(3L)fln showed disturbed lattice arrangement at the myofibril periphery, but also displayed ostensibly normal thick filament structure. The loosely organized peripheral myofilaments in Df(3L)fln heterozygotes were also solubilized by normal skinning procedures, leaving a halo of low density around the myofibril core. The solubility was likely due to the reduced amount of flightin in mutant Df(3L)fln fibers (Vigoreaux et al. 1998).
Flightin Is Essential for Thick Filament Length Determination In Vivo
Myosin II can self-assemble into “synthetic filaments” in vitro through interactions of its coiled-coil (rod) region. The formation of ordered paracrystals in vitro requires the assembly competence domain, a 29-residue sequence in the COOH-terminal region of the LMM that, in addition, confers assembly properties in vivo (Sohn et al. 1997). This region is necessary but not sufficient for proper thick filament formation since the in vitro paracrystals are not of uniform length. The formation of synthetic filaments that resemble in vivo filaments requires the presence of myosin-associated proteins (Barral and Epstein 1999; Winegrad 1999).
In the absence of flightin's association with the thick filaments, the only structural feature that appears abnormal in pupal fln0 IFM is filament length. Sarcomere and thick and thin filament lengths are uniformly longer than normal by the end of pupation in fln0. Because flightin expression begins at ∼60 h after pupation, well after thick filament assembly has begun, and peaks at the end of pupation, the role of flightin may be associated with termination of thick filament assembly at the specific wild-type length. Consistent with this, in ∼2-h-old adult fln0 IFM, very long (∼10 μm) single isolated thick filaments are often seen alongside myofibrils.
A termination of assembly role for flightin contrasts with the roles modeled for other accessory proteins proposed to be involved in length determination (Whiting et al. 1989; Barral and Epstein 1999). Titin is suggested to act as a molecular template for myofibril assembly (Gregorio et al. 1999) and its A-band region as a “protein ruler” that determines the length of vertebrate thick filaments (Whiting et al. 1989; Trinick and Tskhovrebova 1999). A potential Drosophila titin (D-titin) has been identified recently and shown to be expressed in IFM (Machado et al. 1998; for alternate explanations, see Hakeda et al. 2000; Kolmerer et al. 2000). The increase in length (from 1.6 to 3.0 μm) of Drosophila IFM thick filaments during pupation, in contrast to vertebrate striated muscle thick filaments that remain at their initial assembled length of 1.6 μm, argues against a mechanism whereby an ∼2.0-μm fixed-length titin ruler is a primary determinant of Drosophila IFM thick filament length. Because Drosophila thick filament length changes significantly during pupation, an additional mechanism may be required to determine uniform filament length. Flightin may be a key accessory protein that serves this purpose.
The high degree of regulation implied by the coordinated, uniform elongation of both thick and thin filaments in Drosophila IFM suggests multiple proteins and interactions are probably involved in filament length regulation. Although thick and thin filaments can assemble independently of one another (e.g., Beall et al. 1989; Holtzer et al. 1997), uniform filament length and sarcomere regularity appear to require actin–myosin interactions. The absence of thick filaments in Drosophila IFM interferes with the length definition of thin filaments (Chun and Falkenthal 1988). The thick filaments and sarcomeres in the IFM of transgenic Drosophila that express a “headless” myosin are normal in many respects except for variable filament and sarcomere lengths (Cripps et al. 1999). Classic studies of vertebrate muscle development showed that actin-myosin interactions between adjacent thick and thin filaments brought filaments into lateral alignment in uniform length sarcomeres (Shimada and Obinata 1977). All of these studies provide evidence that interactions between thick filaments and thin filaments play a significant role in length determination and the formation of ordered sarcomeres. A requirement for thick and thin filament interactions in determining filament lengths offers a possible explanation as to why a primary effect of the absence of flightin on thick filament length secondarily affects thin filament length.
Flightin Is Necessary for Sarcomere Stability in Active Muscle
In the absence of flightin, adult IFM undergoes rapid and severe degeneration. In ∼2-h-old adult fln0 IFM, thick filaments incorporated into sarcomeres have already shortened to ∼3 μm, which approximates the wild-type length. By 24 h of adult life, complete breakdown of sarcomere structure is evident. This breakdown is characterized by disintegration of M lines and Z bands, shortening of thick filaments, and myosin proteolysis. One possibility is that the absence of flightin uncovers a protease-sensitive site in myosin that leads to myosin proteolysis and sarcomere breakdown. Hence, myosin's heightened sensitivity to proteolysis in fln0 is likely to result from an indigenous structural defect of the thick filament.
The MHC 150-kD peptide that appears in myosin rod mutant Mhc13 from adult IFM results from proteolysis at a site near the LMM hinge, a region far removed from where the mutant amino acid is found (Kronert et al. 1995). Because the comigrating MHC peptide detected in fln0 also originates from the NH2-terminal region of myosin, we believe this peptide and the one in Mhc13 IFM are the same. Mhc13shares additional characteristics with fln0: (a) both mutations engender defects primarily (Mhc13) or exclusively (fln0) in the IFM, (b) both mutations lead to severe sarcomere degeneration in the adult despite the presence of well-defined sarcomeres throughout pupal myofibrillogenesis, (c) adult fibers of both mutants exhibit a similar “bunched” phenotype, and (d) both mutations prevent flightin accumulation. Kronert et al. 1995 discussed the possibility that the abnormalities manifested in Mhc13arise from the absence of flightin, rather than from some primary effect of the rod mutation upon thick filament structure or stability. The convergent phenotypes, despite distinct molecular etiology, of Mhc13and fln0 lends strong support to this idea. Proteolytic cleavage at the myosin hinge is not a normal occurrence in mutant or disordered IFM. It is absent in degenerated hdp2 IFM, a point mutation in TnI that leads to severe sarcomere breakdown in young adults (Beall and Fyrberg 1991). Thick filaments and M lines appear stable in the actin null KM88 IFM (Okamoto et al. 1986) and dissolution of IFM thin filaments with gelsolin does not affect the thick filament/connecting filament lattice (Granzier and Wang 1993). These observations suggest that myosin hinge cleavage is not an automatic casualty of breakdown but is a pathogenic condition dictated by the absence of flightin.
There are several examples where instability of one myofilament type does not affect the accumulation of the other filament types during disruption or degeneration of muscle structure. In cardiac myocytes, overexpression of a recombinant construct encoding the N2-B region of titin led to thin filament dissolution, but left thick filaments intact (Linke et al. 1999). Increasing or decreasing the expression of tropomodulin in cultured cardiomyocytes leads to shortening or lengthening, respectively, of thin filament length (Sussman et al. 1998), but does not affect thick filaments. In C. elegans, the product of UNC-45, a thick filament protein that colocalizes with MHC B, is required for the stability of MHC-B in the thick filament (Ao and Pilgrim 2000), but does not affect thin filament stability. Activation of the tyrosine kinase v-src in quail myotubes leads to rapid breakdown and reorganization of actin filaments, while thick filaments remain for 24 h in intact sarcomeric arrays (Castellani et al. 1996).
Myosin proteolysis and sarcomere breakdown in the absence of flightin may reflect a critical role for this protein in structural reinforcement of the sarcomere. Force transmission along the muscle fiber axis depends on the parallel arrangement of thick and thin filaments and assembly of myosin into filaments of specific and uniform length. The wavy fibers typical of fln0 late pupa/young adults, resulting from over-long and then variable length thick filaments, may prevent the propagation of tension evenly along the axis of the fiber and unbalance the forces experienced by the myofibrils. The absence of flightin may weaken the thick filament shaft, especially if flightin compensates for the low paramyosin content in the core of Drosophila IFM thick filaments. Mechanical damage to weakened fln0 thick filaments during chaotic contractions may trigger waves of myosin proteolysis, particularly if the absence of flightin leaves a protease-sensitive site near the myosin hinge unprotected. Rupture of IFM fibers at the myotendon junction may foster more protease activity. This latter possibility is supported by studies in Calliphora in which surgical cutting of the tendon/guide cells that anchor fibers to the cuticle lead to muscle retraction very similar to that observed in fln0 (Houlihan and Newton 1979).
The high degree of myofibrillar deterioration that occurs in the absence of flightin is consistent with the idea that flightin plays a structural role in the myofilament lattice. During flight, IFM fibers continuously oscillate between lengthening and shortening cycles at ∼200/s. The cell cytoskeleton must accommodate incoming stress and outgoing contractile force and instantaneously switch from a force-bearing to a force-producing mode. Given this continuous assault of mechanical forces, it should not be surprising if IFM has evolved specialized cytoskeletal reinforcements to protect the cell from mechanical damage.
Flightin may serve to reinforce the IFM filament lattice by forming part of a protein link connecting the thick and thin filaments. A possible association between flightin and the thin filaments is suggested by the observation that in the actin null (KM88) phosphorylation of flightin is affected (Vigoreaux 1994) and by studies showing genetic interactions between flightin mutants and actin mutants (Vigoreaux, J., U. Nongthomba, M. Cummins, and J.C. Sparrow. 1999. American Society for Cell Biology Annual Meeting. 1424[Abstr.]; our unpublished data). The coincidence of phosphorylation with final assembly suggests that phosphorylation of flightin may promote formation of an interfilament link and hence provide stability. Interestingly, a recent study showed that the actin cross-linker ABP 280 is phosphorylated in response to external mechanical force (Glogauer et al. 1998). This modification apparently serves to reinforce the actin lattice that is assembled at the site of force application and hence protects the cell from mechanical damage. It would be interesting to establish whether an analogous mechanism exists in flies whereby external mechanical tension (e.g., from wing expansion shortly after eclosion) triggers phosphorylation of flightin and subsequent reinforcement of the myofibrillar lattice by cross-linking. Unlike the phosphorylation of ABP280, which is dynamic in response to applied tension, flightin may achieve a steady state level of phosphorylation early in adult life. Having a permanently reinforced lattice is essential for escape response in which the fly must take off without warning. In this light, flightin may play a function analogous to the mechanoprotective role proposed for phosphorylated MyBP-C in cardiac muscle (Winegrad 1999) that, like IFM, is myogenic and oscillatory.
A Model for Flightin Function in Thick Filament Length Determination
The formation of an adult (full-length) fiber occurs through gradual and uniform growth in sarcomere length and width via the incorporation of actin, myosin, and associated proteins to pre-existing myofilaments (Reedy and Beall 1993). Sarcomeres ∼1.7-μm long have assembled throughout the developing IFM ∼42 h after pupation. By the end of pupal development (100 h after the beginning of pupation), the sarcomeres have grown to ∼2.8–3.0 μm.
The process by which myosin molecules are added to a growing filament is likely to depend largely on self-assembly properties, guided by specific sequences within the rod (e.g., the assembly-competent domain; Sohn et al. 1997) or by specific features of the rod (e.g., charge distribution; McLachlan and Karn 1982).
The final length of 3.2 μm is attained during the first few hours after eclosion (Reedy and Beall 1993). Our results suggest that in addition to self assembly properties, such as specific myosin rod sequences (Sohn et al. 1997) or charge distribution along the rod (McLachlan and Karn 1982), that flightin plays an essential role in the elongation of the thick filaments in IFM. Flightin accumulation begins at ∼60 h after pupation and continues throughout pupal development (Vigoreaux et al. 1993). Phosphorylation of flightin begins during the final stages of pupal development; however, hyperphosphorylation of flightin is not achieved until 2–5 h into adulthood (Vigoreaux and Perry 1994). Given this scenario, we propose a model of how flightin may participate in thick filament length determination (Fig. 12). The initial assembly of thick filaments occurs independently of flightin. As nonphosphorylated flightin begins to accumulate in the IFM, it associates with pre-existing filaments. The nearly homogenous distribution of flightin throughout the A band in the adult muscle, together with our preliminary quantitative measurements, suggest that flightin associates ∼1:1 with myosin molecules along most of the filament. The immunolabeling of wild-type IFM shows that flightin is not present in thick filaments at the end of the A band. Flightin may be prevented from binding to myosin molecules by projectin, which is associated with thick filaments close to the Z bands.
Figure 12.
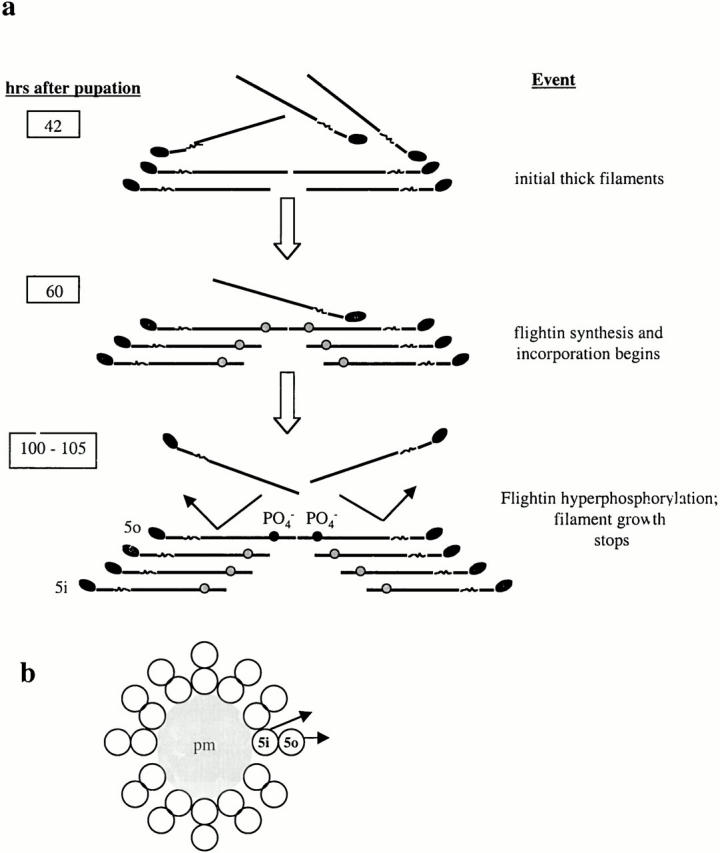
A model of how flightin may participate in determination of thick filament length in Drosophila IFM. (a) Nascent thick filaments are organized in sarcomeres by 42 h post pupation (pp). At ∼60 h pp, flightin (gray circles) begins to accumulate and we show it associating with preassembled filaments. The rod region defined by Mhc13 represents the primary flightin binding site in the thick filament. Phosphorylation of flightin (100–105 h) may add enough negative charges to the surface of the filament so that incoming myosin molecules are repelled and filament growth stops. Alternatively, phosphorylation of flightin on the surface of the thick filament may serve to align thick and thin filaments as part of the mechanism for establishing final length (not shown). (b) Schematic cross section of an insect flight muscle thick filament according to Beinbrech et al. 1988, Beinbrech et al. 1990. Each circle represents the rod region of a myosin molecule. According to this model, a thick filament consists of 12 subfilaments surrounding a paramyosin core (pm). Each subfilament is composed of two myosin molecules, an outer myosin (e.g., 5o) facing the thin filament and an inner myosin (e.g., 5i) sandwiched between the paramyosin core and the outer myosin. The crossbridges that emanate from each subfilament (arrows) are staggered axially. For simplicity, only single heads from one subfilament are shown. We propose that flightin bound to inner myosin remains unphosphorylated and contacts the hinge-LMM junction (shown as a kink in a) of its outer myosin partner in the subfilament. Flightin bound to outer myosin is facing the thin filament. Phosphorylation of these outer flightin molecules is involved in length regulation.
We show flightin binding directly to the LMM heptad repeat that harbors the Mhc13 mutation. Flightin bound to the LMM of one myosin could interact with the hinge region of a second molecule staggered axially, especially if these two molecules form one subfilament (Beinbrech et al. 1988, Beinbrech et al. 1990). The outer myosin of the subfilament (Fig. 12, Fig. 5o) would face the thin filament and the inner myosin (5i) would face the neighboring outer myosin. The association of flightin with myosin molecules on the surface of the filament may prevent the incorporation of incoming myosins by blocking rod sequences that are important for myosin–myosin interactions. Alternatively, phosphorylation of flightin may place enough of a negative charge on the surface of the filament such that repulsive forces dominate over the electrostatic attractions of the 28-residue charge repeat along the rod (McLachlan and Karn 1982). The charge possibility is attractive for several reasons: (a) final sarcomere length is achieved early in adult life, when flightin hyperphosphorylation is taking place, (b) thick filament length is variable in KM88 actin null IFM in which the timing of phosphorylation of flightin is disrupted, (c) repulsive forces have been proposed as a mechanism for regulating filament length (Davis 1986), and (d) myosin II filament assembly in Dictyostelium is regulated by phosphorylation of three residues in the LMM region, where the addition of the negative charges caused a change in the conformation of myosin, rendering it incompetent for assembly (Liang et al. 1999).
The model also explains the susceptibility of myosin to proteolysis in the absence of flightin. In a subfilament, flightin bound to the inner myosin would be juxtaposed to the hinge region of the outer myosin partner and protect the proteolytic cleavage site exposed in Mhc13and fln0. In adult IFM, ∼50% of flightin is phosphorylated (Vigoreaux and Perry 1994). One possibility is that flightin bound to the outer myosin is an accessible substrate to protein kinases, while flightin bound to the inner myosin is not. The difference in phosphorylation states may define two functional classes: (a) inner, nonphosphorylated flightin may be involved in cross-linking and/or stabilizing myosin pairs into subfilaments, and (b) outer, hyperphosphorylated flightin may be involved in establishing final thick filament length either by a repulsion mechanism or by interacting with the thin filament as part of an interfilament cross-link.
Our observations of the flightin null IFM do not provide direct evidence about the possible role of flightin phosphorylation on modulating actin–myosin interactions. The availability of fln0 will allow further testing of the function of flightin by permitting specific mutations of flightin to be expressed in transgenic flies that do not express a wild-type flightin. Substitution of phosphorylated residues for nonphosphorylatable residues is the next step in elucidating flightin function.
Acknowledgments
We thank Carmen Hernandez, Eugene Valsky, Allison Cox, and Victoria Dugay for technical assistance, Donald Myckles for providing antibodies to proteosome proteins, and John Sparrow, Sanford Bernstein, and Ted Homyk for providing flies. We are grateful to Charles Ferguson for the immuno-electron microscopy, to Kevin Leonard for the histogram in Figure 2, and to Gretchen Ayer for the expression and purification of recombinant flightin. We acknowledge David Maughan, Gretchen Ayer, and members of the Vigoreaux lab who provided valuable comments to the manuscript. J.O. Vigoreaux is indebted to John Sparrow, Margarita Cervera, and Roberto Marco, in whose labs part of this work was conducted.
This study was supported by the National Science Foundation.
Footnotes
Abbreviations used in this paper: DLM, dorsolongitudinal muscle; ELC, myosin essential light chain; IFM, indirect flight muscles; LMM, light meromyosin; MHC, myosin heavy chain; RLC, myosin regulatory light chain.
References
- Ao W., Pilgrim D. Caenorhabditis elegans UNC-45 is a component of muscle thick filaments and colocalizes with myosin heavy chain B, but not myosin heavy chain A. J. Cell Biol. 2000;148:375–384. doi: 10.1083/jcb.148.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral J.M., Epstein H.F. Protein machines and self assembly in muscle organization. BioEssays. 1999;21:813–823. doi: 10.1002/(SICI)1521-1878(199910)21:10<813::AID-BIES3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Beall C.J., Fyrberg E. Muscle abnormalities in Drosophila melanogaster heldup mutants are caused by missing or aberrant troponin-I isoforms. J. Cell Biol. 1991;114:941–951. doi: 10.1083/jcb.114.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall C.J., Sepanski M.A., Fyrberg E.A. Genetic dissection of Drosophila myofibril formationeffects of actin and myosin heavy chain null alleles. Genes Dev. 1989;3:131–140. doi: 10.1101/gad.3.2.131. [DOI] [PubMed] [Google Scholar]
- Beinbrech G., Ashton F.T., Pepe F. The invertebrate myosin filamentsubfilament arrangement in the wall of tubular filaments of insect flight muscles. J. Mol. Biol. 1988;201:557–565. doi: 10.1016/0022-2836(88)90637-7. [DOI] [PubMed] [Google Scholar]
- Beinbrech G., Ashton F.T., Pepe F.A. Orientation of the backbone structure of myosin filaments in relaxed and rigor muscles of the houseflyevidence for non-equivalent crossbridge positions at the surface of thick filaments. Tissue Cell. 1990;22:803–810. doi: 10.1016/0040-8166(90)90045-b. [DOI] [PubMed] [Google Scholar]
- Bennett P.M., Furst D.O., Gautel M. The C-protein (Myosin binding protein C) familyregulators of contraction and sarcomere formation? Rev. Physiol. Biochem. Pharmacol. 1999;138:203–234. doi: 10.1007/BFb0119628. [DOI] [PubMed] [Google Scholar]
- Bernstein S.I., O'Donnell P.T., Cripps R.M. Molecular genetic analysis of muscle development, structure and function in Drosophila . Int. Rev. Cytol. 1993;143:63–152. doi: 10.1016/s0074-7696(08)61874-4. [DOI] [PubMed] [Google Scholar]
- Castellani L., Reedy M., Airey J.A., Gallo R., Ciotti M.T., Falcone G., Alema S. Remodeling of cytoskeleton and triads following activation of v-Src tyrosine kinase in quail myotubes. J. Cell Sci. 1996;109:1335–1346. doi: 10.1242/jcs.109.6.1335. [DOI] [PubMed] [Google Scholar]
- Chun M., Falkenthal S. Ifm(2)2 is a myosin heavy chain allele that disrupts myofibrillar assembly only in the indirect flight muscle of Drosophila melanogaster . J. Cell Biol. 1988;107:2613–2621. doi: 10.1083/jcb.107.6.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covi J.A., Belote J.M., Mykles D.L. Subunit composition and catalytic properties of proteosomes from developmental temperature-sensitive mutants of Drosophila melanogaster . Arch. Biochem. Biophys. 1999;368:85–97. doi: 10.1006/abbi.1999.1294. [DOI] [PubMed] [Google Scholar]
- Cripps R.M., Suggs J.A., Bernstein S.I. Assembly of thick filaments and myofibrils occurs in the absence of the myosin head. EMBO (Eur. Mol. Biol. Organ.) J. 1999;18:1793–1804. doi: 10.1093/emboj/18.7.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.S. A model for length-regulation in thick filaments of vertebrate skeletal myosin. Biophys. J. 1986;50:417–422. doi: 10.1016/S0006-3495(86)83477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J.S. Interaction of C-protein with pH 8.0 synthetic thick filaments prepared from the myosin of vertebrate skeletal muscle. J. Muscle Res. Cell Motil. 1988;9:174–183. doi: 10.1007/BF01773739. [DOI] [PubMed] [Google Scholar]
- Dickinson M.H., Hyatt C.J., Lehmann F.-O., Moore J.R., Reedy M.C., Simcox A., Tohtong R., Vigoreaux J.O., Yamashita H., Maughan D.W. Phosphorylation-dependent power output of transgenic fliesan integrated study. Biophys. J. 1997;73:3122–3134. doi: 10.1016/S0006-3495(97)78338-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert R., Kelly M.G., Mikawa T., Fischman D.A. The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A band of striated muscle. J. Cell Sci. 1996;109:101–111. doi: 10.1242/jcs.109.1.101. [DOI] [PubMed] [Google Scholar]
- Glogauer M., Arora P., Chou D., Janmey P.A., Downey G.P., McCulloch C.A. The role of actin-binding protein 280 in integrin-dependent mechanoprotection. J. Biol. Chem. 1998;273:1689–1698. doi: 10.1074/jbc.273.3.1689. [DOI] [PubMed] [Google Scholar]
- Granzier H.L.M., Wang K. Interplay between passive tension and strong and weak binding cross-bridges in insect indirect flight muscle. J. Gen. Physiol. 1993;101:235–270. doi: 10.1085/jgp.101.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio C.C., Granzier H., Sorimachi H., Labeit S. Muscle assemblya titanic achievement? Curr. Opin. Cell Biol. 1999;11:18–25. doi: 10.1016/s0955-0674(99)80003-9. [DOI] [PubMed] [Google Scholar]
- Hakeda S., Endo S., Saigo K. Requirements of Kettin, a giant muscle protein highly conserved in overall structure in evolution, for normal muscle function, viability, and flight activity of Drosophila . J. Cell Biol. 2000;148:101–114. doi: 10.1083/jcb.148.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, C.J. 1998. A deficiency spanning the flightin gene, a putative flightin null mutation and a genetic study of the 76D-E region of Drosophila melanogaster. Ph.D. thesis, University of Vermont.
- Holtzer H., Hijikata T., Lin Z.X., Zhang Z.Q., Holtzer S., Protasi F., Franzini-Armstrong C., Sweeney H.L. Independent assembly of 1.6 microns long bipolar MHC filaments and I-Z-I bodies. Cell Struct. Funct. 1997;22:83–93. doi: 10.1247/csf.22.83. [DOI] [PubMed] [Google Scholar]
- Houlihan D.F., Newton J.R.L. Sarcomere formation and longitudinal growth in the developing flight muscles of Calliphora. J. Insect Physiol. 1979;25:879–893. [Google Scholar]
- Huxley H.E. Electron microscope studies on the structure of natural and synthetic protein filaments from striated muscle. J. Mol. Biol. 1963;7:281–308. doi: 10.1016/s0022-2836(63)80008-x. [DOI] [PubMed] [Google Scholar]
- Kolmerer B., Clayton J., Benes V., Allen T., Ferguson C., Leonard K., Weber U., Knekt M., Ansorge W., Labeit S., Bullard B. Sequence and expression of the kettin gene in Drosophila melanogaster and Caenorhabditis elegans . J. Mol. Biol. 2000;296:435–448. doi: 10.1006/jmbi.1999.3461. [DOI] [PubMed] [Google Scholar]
- Kronert W.A., O'Donnell P.T., Fieck A., Lawn A., Vigoreaux J.O., Sparrow J.C., Bernstein S.I. Defects in the Drosophila myosin rod permit sarcomere assembly but cause flight muscle degeneration. J. Mol. Biol. 1995;249:111–125. doi: 10.1006/jmbi.1995.0283. [DOI] [PubMed] [Google Scholar]
- Lakey A., Ferguson C., Labeit S., Reedy M., Larkins A., Butcher G., Leonard K., Bullard B. Identification and localization of high molecular weight proteins in insect flight and leg muscle. EMBO (Eur. Mol. Biol. Organ.) J. 1990;9:3459–3467. doi: 10.1002/j.1460-2075.1990.tb07554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W., Warrick H.M., Spudich J.A. A structural model for phosphorylation control of Dictyostelium myosin II thick filament assembly. J. Cell Biol. 1999;147:1039–1047. doi: 10.1083/jcb.147.5.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke W.A., Rudy D.E., Centner T., Gautel M., Witt C., Labeit S., Gregorio C.C. I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J. Cell Biol. 1999;146:631–644. doi: 10.1083/jcb.146.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Barral J.M., Bauer C.C., Ortiz I., Cook R.G., Schmid M.F., Epstein H.F. Assemblases and coupling proteins in thick filament assembly. Cell Struct. Funct. 1997;22:155–162. doi: 10.1247/csf.22.155. [DOI] [PubMed] [Google Scholar]
- Machado C., Sunkel C.E., Andrew D.J. Human autoantibodies reveal titin as a chromosomal protein. J. Cell Biol. 1998;141:321–334. doi: 10.1083/jcb.141.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maughan D.W., Vigoreaux J.O. An integrated view of insect flight musclegenes, motor molecules, and motion. News Physiol. Sci. 1999;14:87–92. doi: 10.1152/physiologyonline.1999.14.3.87. [DOI] [PubMed] [Google Scholar]
- McLachlan A.D., Karn J. Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature. 1982;299:226–231. doi: 10.1038/299226a0. [DOI] [PubMed] [Google Scholar]
- Okamoto H., Hiromi Y., Ishikawa E., Yamada T., Isoda K., Maekawa H., Hotta Y. Molecular characterization of mutant actin genes which induce heat-shock proteins in Drosophila flight muscles. EMBO (Eur. Mol. Biol. Organ.) J. 1986;5:589–596. doi: 10.1002/j.1460-2075.1986.tb04251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reedy M.C., Beall C. Ultrastructure of developing flight muscle in Drosophila. I. Assembly of myofibrils. Dev. Biol. 1993;160:443–465. doi: 10.1006/dbio.1993.1320. [DOI] [PubMed] [Google Scholar]
- Reedy M.C., Beall C., Fyrberg E. Formation of reverse rigor chevrons by myosin heads. Nature. 1989;339:481–483. doi: 10.1038/339481a0. [DOI] [PubMed] [Google Scholar]
- Saide J.D., Chin-Bow S., Hogan-Sheldon J., Busquets-Turner L., Vigoreaux J.O., Valgeirsdottir K., Pardue M.L. Characterization of components of Z-bands in the fibrillar flight muscle of Drosophila melanogaster . J. Cell Biol. 1989;109:2157–2167. doi: 10.1083/jcb.109.5.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada Y., Obinata T. Polarity of actin filaments at the initial stage of myofibril assembly in myogenic cells in vitro. J. Cell Biol. 1977;72:777–785. doi: 10.1083/jcb.72.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn R.L., Vikstrom K.L., Strauss M., Cohen C., Szent-Gyorgyi A.G., Leinwand L.A. A 29 residue region of the sarcomeric myosin rod is necessary for filament formation. J. Mol. Biol. 1997;266:317–330. doi: 10.1006/jmbi.1996.0790. [DOI] [PubMed] [Google Scholar]
- Squire J.M. Muscledesign, diversity, and disease 1986. Benjamin/Cummings Publishing Co; Menlo Park, CA: pp. 381 pp [Google Scholar]
- Starr R., Offer G. Preparation of C-protein, H-protein, X-protein, and phosphofructokinase. Methods Enzymol. 1982;85:130–138. doi: 10.1016/0076-6879(82)85016-7. [DOI] [PubMed] [Google Scholar]
- Sussman M.A., Baque S., Uhm C.S., Daniels M.P., Price R.L., Simpson D., Terracio L., Kedes L. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circ. Res. 1998;82:94–105. doi: 10.1161/01.res.82.1.94. [DOI] [PubMed] [Google Scholar]
- Trinick J. Titin as a scaffold and spring. Cytoskeleton. Curr. Biol. 1996;6:258–260. doi: 10.1016/s0960-9822(02)00472-4. [DOI] [PubMed] [Google Scholar]
- Trinick J., Tskhovrebova L. Titina molecular control freak. Trends Cell Biol. 1999;9:377–380. doi: 10.1016/s0962-8924(99)01641-4. [DOI] [PubMed] [Google Scholar]
- van Genderen I., van Meer G., Slot J., Geuze H., Voorhout W. Subcellular localization of Forssman glycolipid in epithelial MDCK cells by immuno-electronmicroscopy after freeze-substitution. J. Cell Biol. 1991;115:1009–1019. doi: 10.1083/jcb.115.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigoreaux J.O. Alterations in flightin phosphorylation in Drosophila flight muscles are associated with myofibrillar defects engendered by actin and myosin heavy chain mutant alleles. Biochem. Genet. 1994;32:301–314. doi: 10.1007/BF00555832. [DOI] [PubMed] [Google Scholar]
- Vigoreaux J.O., Hernandez C., Moore J., Ayer G., Maughan D. A genetic deficiency that spans the flightin gene of Drosophila melanogaster affects the ultrastructure and function of the flight muscles. J. Exp. Biol. 1998;201:2033–2044. doi: 10.1242/jeb.201.13.2033. [DOI] [PubMed] [Google Scholar]
- Vigoreaux J.O., Perry L.M. Multiple isoelectric variants of flightin in Drosophila stretch-activated muscles are generated by temporally regulated phosphorylations. J. Muscle Res. Cell Motil. 1994;15:607–616. doi: 10.1007/BF00121068. [DOI] [PubMed] [Google Scholar]
- Vigoreaux J.O., Saide J.D., Valgeirsdottir K., Pardue M.L. Flightin, a novel myofibrillar protein of Drosophila stretch-activated muscles. J. Cell Biol. 1993;121:587–598. doi: 10.1083/jcb.121.3.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting A., Wardale J., Trinick J. Does titin regulate the length of muscle thick filaments? J. Mol. Biol. 1989;205:263–268. doi: 10.1016/0022-2836(89)90381-1. [DOI] [PubMed] [Google Scholar]
- Winegrad S. Cardiac myosin binding protein C. Circ. Res. 1999;84:1117–1126. doi: 10.1161/01.res.84.10.1117. [DOI] [PubMed] [Google Scholar]