

Abstract
Attachment of many cell types to extracellular matrix proteins triggers cell spreading, a process that strengthens cell adhesion and is a prerequisite for many adhesion-dependent processes including cell migration, survival, and proliferation. Cell spreading requires integrins with intact β cytoplasmic domains, presumably to connect integrins with the actin cytoskeleton and to activate signaling pathways that promote cell spreading. Several signaling proteins are known to regulate cell spreading, including R-Ras, PI 3-kinase, PKCε and Rac1; however, it is not known whether they do so through a mechanism involving integrin β cytoplasmic domains. To study the mechanisms whereby cell spreading is regulated by integrin β cytoplasmic domains, we inhibited cell spreading on collagen I or fibrinogen by expressing tac-β1, a dominant-negative inhibitor of integrin function, and examined whether cell spreading could be restored by the coexpression of either V38R-Ras, p110α-CAAX, myr-PKCε, or L61Rac1. Each of these activated signaling proteins was able to restore cell spreading as assayed by an increase in the area of cells expressing tac-β1. R-Ras and Rac1 rescued cell spreading in a GTP-dependent manner, whereas PKCε required an intact kinase domain. Importantly, each of these signaling proteins required intact β cytoplasmic domains on the integrins mediating adhesion in order to restore cell spreading. In addition, the rescue of cell spreading by V38R-Ras was inhibited by LY294002, suggesting that PI 3-kinase activity is required for V38R-Ras to restore cell spreading. In contrast, L61Rac1 and myr-PKCε each increased cell spreading independent of PI 3-kinase activity. Additionally, the dominant-negative mutant of Rac1, N17Rac1, abrogated cell spreading and inhibited the ability of p110α-CAAX and myr-PKCε to increase cell spreading. These studies suggest that R-Ras, PI 3-kinase, Rac1 and PKCε require the function of integrin β cytoplasmic domains to regulate cell spreading and that Rac1 is downstream of PI 3-kinase and PKCε in a pathway involving integrin β cytoplasmic domain function in cell spreading.
Keywords: cell spreading, integrin, signaling, Rac1, R-Ras
Introduction
Integrins are a family of α/β heterodimeric transmembrane receptors used by cells to interact with their extracellular matrix (Hynes 1992). Integrins form a structural link between the extracellular matrix and the cell's cytoskeletal and signal transduction apparatus. Integrin engagement triggers a myriad of intracellular signals resulting in changes in intracellular pH and [Ca2+], as well as the activation of protein kinases, phosphoinositide kinases, phospholipases, and small GTP binding proteins (Schwartz et al. 1995; Yamada and Miyamoto 1995; Burridge and Chrzanowska-Wodnicka 1996; LaFlamme et al. 1997). Many of the signals activated by integrin engagement regulate the adhesion process itself (Schwartz et al. 1995; Yamada and Miyamoto 1995; Burridge and Chrzanowska-Wodnicka 1996; LaFlamme et al. 1997).
Cell spreading requires actin polymerization and reorganization, the extension of lamellipodia or filopodia, and the formation of new integrin-substrate adhesions (Small et al. 1999). The initial interaction of integrins with their ligand in cell attachment is believed to trigger signaling pathways that regulate the process of cell spreading. PI 3-kinase has been shown to be activated by integrin-mediated cell adhesion, and in some cell types PI 3-kinase activity is required for cell spreading (Khwaja et al. 1997; King et al. 1997). The expression of an activated form of R-Ras (V38R-Ras) has also been shown to positively regulate cell spreading, whereas the expression of dominant negative R-Ras (N43R-Ras) can inhibit cell spreading in CHO cells (Zhang et al. 1996). In addition, both PKCα and PKCε appear to be positive regulators of cell adhesion (Vuori and Ruoslahti 1993; Chun et al. 1996). PKCε has been shown to become activated upon cell attachment before spreading and pharmacological inhibitors of PKC have been shown to inhibit the process of spreading (Vuori and Ruoslahti 1993; Chun et al. 1996). The small GTP binding proteins Rac1 and Cdc42 are also regulators of cell spreading. Integrin-mediated cell adhesion can trigger Rac1 activation (Del Pozo et al. 2000), and expression of dominant negative mutants of Rac1 and Cdc42 have been shown to inhibit cell spreading (Clark et al. 1998; Price et al. 1998; Van Leeuwen et al. 1999). In addition to the requirement for the activities of several signaling proteins, the process of cell spreading is dependent upon intact β subunit cytoplasmic domains on the integrins mediating adhesion (Ylanne et al. 1993); however, the molecular mechanisms by which integrins and various signaling proteins regulate cell spreading are not fully defined.
We and others have previously shown that expression of isolated β cytoplasmic domains connected to the extracellular and transmembrane domains of the small nonsignaling (tac) subunit of the interleukin-2 receptor or other extracellular reporters can inhibit endogenous integrin function in a variety of cellular processes, including cell spreading (Chen et al. 1994; LaFlamme et al. 1994; Lukashev et al. 1994; Smilenov et al. 1994). Recently, we have shown that high levels of expression of the tac-β1 chimera can inhibit both cell attachment to fibronectin and β1 integrin conformations that are favorable to β1 integrin–ligand interactions (Mastrangelo et al. 1999a). Since cells expressing moderate levels of tac-β1 can attach, but are inhibited in cell spreading (LaFlamme et al. 1994; Mastrangelo et al. 1999a), we have used this system to study the molecular mechanisms that regulate the process of cell spreading. In this study, we asked whether activated forms of signaling proteins known to positively regulate cell spreading could restore cell spreading inhibited by the expression of tac-β1. We found that activated forms of PI 3-kinase, R-Ras, Rac1, and PKCε could all restore cell spreading inhibited by tac-β1 in both normal human fibroblasts adherent to collagen I and CHO cells expressing recombinant αIIbβ3 adherent to fibrinogen. Using CHO cells expressing either wild-type recombinant αIIbβ3 or αIIbβ3 with a β3 cytoplasmic domain truncation (αIIbβ3 Δ728), we show that the ability of V38R-Ras, L61Rac1, p110α-CAAX, and myr-PKCε to increase cell spreading in tac-β1–expressing cells is dependent upon the presence of intact β cytoplasmic domains on the integrins mediating adhesion.
Materials and Methods
Antibodies and Matrix Proteins
Tac expression was monitored with FITC-conjugated antibodies to CD25 from Accurate Chemicals and Scientific Corp. and Becton Dickinson. Rabbit and goat polyclonal antibodies to the Myc (9E10) epitope, Flag epitope, and PKCε were purchased from Santa Cruz Biotechnology, Inc. Rhodamine-conjugated goat antibodies to rabbit IgG were purchased from Roche Molecular Biochemicals and Pierce. Rhodamine-conjugated donkey antibodies to goat IgG were provided by Santa Cruz Biotechnology, Inc. The β1 integrin function blocking P4C10 ascites was purchased from GIBCO-BRL and the control ascites from Sigma-Aldrich. The matrix proteins collagen I (Vitrogen 100) and human fibrinogen were purchased from Collagen Biomaterials and Enzyme Research Laboratories, respectively.
cDNAs, Cells, Transfections, and Spreading Assays
The eukaryotic expression vectors have been described previously: V38R-Ras and N43R-Ras by Marte et al. 1996, p110α-CAAX by Wennstrom and Downward 1999, and tac and tac-β1 by LaFlamme et al. 1992. Eukaryotic expression vectors for L61Rac1 and N17Rac1 were generously provided by Dr. Alan Hall (University College, London, UK) and were described previously by Lamarche et al. 1996. Myr-PKCε, PKCε, and W437PKCε were generous gifts from Dr. Alex Toker (Deaconess Medical Center, Boston, MA) and were constructed similarly to the PKCζ constructs as previously described (Chou et al. 1998).
Primary human foreskin fibroblasts (Vec Technologies) were maintained in DMEM supplemented with 10% fetal bovine serum and 2 mM l-glutamine. The stable CHO cell lines A5 (αIIbβ3) and ETC12 (αIIbβ3Δ728) (Ylanne et al. 1995) were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and MEM nonessential amino acids. Cells were transfected by electroporation (Mastrangelo et al. 1999b) with 20 μg of tac or tac-β1 expression vectors together with 40 μg of expression vector for the various signaling proteins. For the kinase-dead PKCε experiment, comparable expression of the PKCε proteins was observed by immunofluorescence when 30 μg of PKCε and 60 μg of kinase-dead W437PKCε expression vectors were transfected. For spreading assays, human fibroblasts were harvested by trypsinization with cold PBS ∼16 h after electroporation and were allowed to recover for 40 min at 37°C in suspension. Approximately 4 × 105 cells were plated onto glass coverslips coated with 20 μg/ml collagen I and were allowed to spread for 1 h at 37°C in serum-free medium. Spreading assays using A5 and ETC12 CHO cell lines were performed similarly; however, cells were allowed to recover for 1 h before plating onto glass coverslips coated with 15 μg/ml fibrinogen, and spreading was assayed after 30 min (Ylanne et al. 1995). To determine whether cell spreading on collagen I was dependent on endogenous β1 integrins, the β1 function blocking P4C10 ascites (1:1,000 dilution) or control ascites (1:1,000 dilution) was added 10 min after plating, at which time >90% of the cells had attached, but not spread. To determine the requirement for PI 3-kinase activity, transiently transfected cells were incubated for 40 min in suspension with LY294002 (Calbiochem) at a final concentration of 50 μM, and subsequently analyzed for spreading as described above.
Immunofluorescence and Quantification of Cell Area and Fluorescence Intensity
Adherent cells were fixed and processed for immunofluorescence as previously described (LaFlamme et al. 1992). Cells were stained for cell surface expression of the tac epitope with FITC-conjugated antibodies to CD25 before permeabilization. Cells cotransfected with R-Ras, Rac1, and myr-PKCε signaling proteins were subsequently permeabilized with PBS containing 0.4% Triton X-100 for 5 min at room temperature, incubated with rabbit polyclonal antibodies to the epitope tag of the signaling proteins, and then with rhodamine-conjugated secondary antibodies. Cells cotransfected with wild-type PKCε and kinase-dead W437PKCε were permeabilized with PBS containing 0.1% Triton X-100 and 3.7% formaldehyde for 15 min at room temperature and stained as described previously. Microscopy was performed using an Olympus BX60 microscope equipped with phase contrast and epifluorescence and attached to a Spot low light camera interfaced with a PC computer (Diagnostic Instruments). Spot software was used to acquire the fluorescence and interference reflection microscopy (Izzard and Lochner 1976) images of transfected cells. Cell area and fluorescence intensity were measured using Image Pro-Plus software (Media Cybernetics). Image Pro-Plus software was calibrated with an Applied Micro Stage micrometer (Applied Image, Inc.). The accuracy of the area measurements was confirmed with a TSM measurement slide that contains etched squares of known dimensions. Preliminary studies indicated that cells that are round and have not begun to spread have cell areas <560 μm2. Hence, we define those cells with areas >560 μm2 as spread and cells with areas <560 μm2 as not spread. The conditions used to acquire the fluorescence images minimized the influence of cell shape; however, for very small cells (areas <320 μm2), our analysis may underestimate the expression level of recombinant proteins.
Results
Activated V38R-Ras, myr-PKCε, L61Rac1, or p110α-CAAX Increase the Spreading of Cells Expressing Tac-β1
Previous studies have shown that the expression of tac-β1 can inhibit cell spreading on a variety of extracellular matrix proteins (LaFlamme et al. 1994). In the present study, we tested whether the expression of activated forms of signaling proteins known to positively regulate cell spreading could restore tac-β1–inhibited spreading. Normal fibroblasts were transiently transfected with tac-β1 or the control tac receptor alone, or cotransfected with tac-β1 and either activated R-Ras, PKCε, Rac1, or PI 3-kinase. Transfected cells were plated onto collagen I and allowed to spread for 1 h. The cells adherent to collagen I were immunostained for tac expression and the expression of the cotransfected signaling protein where appropriate. Cell area and fluorescence intensity were quantified for positive and double positive cells using Image Pro-Plus software as described in Materials and Methods.
In agreement with previous studies (LaFlamme et al. 1994), we found that the majority of the tac-β1 positive cells had cell areas <560 μm2, indicating that they were inhibited in cell spreading (Fig. 1 B). In contrast, the majority of cells expressing the control tac receptor had cell areas >560 μm2, indicating that they were not inhibited in spreading (Fig. 1 B). When we tested the ability of activated R-Ras (V38R-Ras) and membrane-anchored PKCε (myr-PKCε) to restore tac-β1–inhibited cell spreading, we found that the coexpression of tac-β1 with either V38R-Ras or myr-PKCε resulted in an increase in cell area as compared with cells expressing tac-β1 alone (Fig. 1 B). Quantitation of the spreading assay revealed that 35% of cells coexpressing myr-PKCε and 52% of cells coexpressing V38R-Ras were spread, compared with 12% of cells expressing tac-β1 alone. Coexpression of constitutively active Rac1 (L61Rac1) also increased spreading of tac-β1–expressing cells (Fig. 1 C). Of the cells coexpressing tac-β1 and L61Rac1, 86% were spread, compared with 13% expressing tac-β1 alone. Cotransfection of the membrane-anchored catalytic subunit of PI 3-kinase also increased the area of tac-β1–expressing cells (Fig. 1 D). When we quantitated the spreading of cells cotransfected with tac-β1 and p110α-CAAX as a function of tac epitope expression only (Fig. 1 D), we found that 59% of the tac-β1 and p110α-CAAX cotransfected cells were spread compared with only 9% of the cells transfected with tac-β1 alone. In this instance, we were unable to quantitate p110α-CAAX expression by immunofluorescence. However, we have found in our assays that ∼80% of transfected cells coexpress cotransfected plasmids (data not shown). Thus, as an additional control, we analyzed the percentage of transfected cells that spread as a function of tac epitope expression in four separate experiments and found that 57 ± 9% of the cells cotransfected with tac-β1 and p110α-CAAX were spread compared with 12 ± 5% of the cells transfected with tac-β1 alone (quantitation is the mean ± SD).
Figure 1.
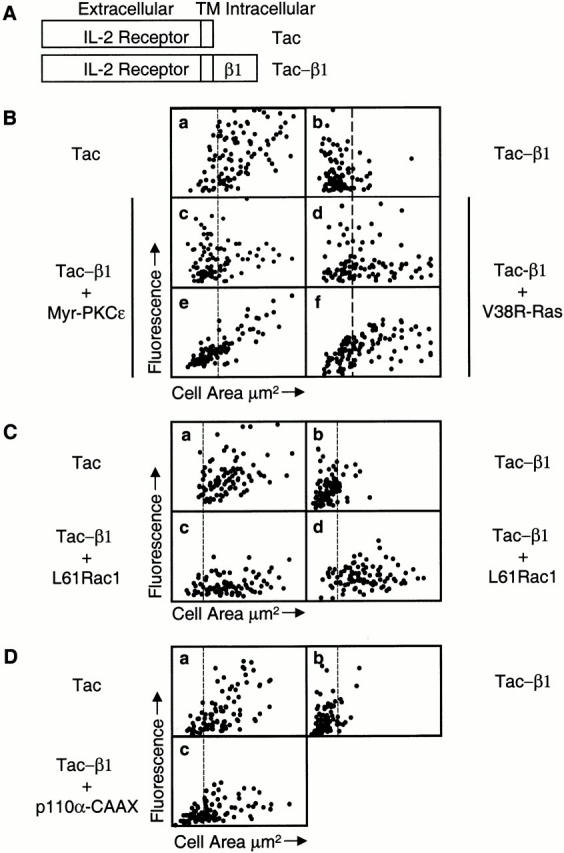
Coexpression of either myr-PKCε, V38R-Ras, L61Rac1, or p110α-CAAX with tac-β1 can restore cell spreading on collagen I. (A) Diagram of the control tac receptor containing the extracellular and transmembrane domains of the small (tac) subunit of the human interleukin-2 receptor and the tac-β1 chimera containing the same domains of the interleukin-2 receptor fused to the human integrin β1A cytoplasmic domain. (B) Fibroblasts were transfected with tac (a) or tac-β1 (b), or cotransfected with tac-β1 and myr-PKCε-Flag (c and e) or tac-β1 and myc-V38R-Ras (d and f). Cell area for 100 randomly sampled positively transfected cells is plotted as a function of either tac epitope expression (a–d), myr-PKCε-Flag expression (e) for the same cells shown in c, or myc-V38R-Ras expression (f) for the same cells shown in d. The x axis is a linear scale of cell area from 0 to 1,600 μm2, the y axis is a linear scale of either FITC fluorescence (tac epitope expression) units defined by Image Pro-Plus from 0 to 2.4 × 104 (a–d) or rhodamine fluorescence (Flag or myc epitope expression) from 0 to 1.2 × 105 (e and f). (C) Fibroblasts were transfected with tac (a) or tac-β1 (b), or cotransfected with tac-β1 and myc-L61Rac1 (c and d). Cell area for 90 randomly sampled cells expressing tac, tac-β1, or coexpressing tac-β1 and myc-L61Rac1 is plotted as a function of tac epitope expression (a–c) or myc epitope expression (d) for the cells shown in c. The x axis is a linear scale of cell area from 0 to 2,400 μm2, the y axis is a linear scale of FITC fluorescence (tac epitope expression) from 0 to 2.4 × 104 (a–c) or rhodamine fluorescence (myc epitope expression) from 0 to 1.2 × 105 (d). (D) Fibroblasts transfected with the control tac receptor (a) or tac-β1 (b) or tac-β1 and p110α-CAAX (c) were analyzed for cell-surface expression of the tac epitope and cell area, as described in Materials and Methods. Cell area for 98 randomly sampled positively transfected cells is plotted as a function of tac epitope expression. The x axis is a linear scale of cell area from 0 to 2,400 μm2 and the y axis is a linear scale of FITC fluorescence (tac epitope expression) from 0 to 3.2 × 104 (a) or 0 to 1.6 × 104 (b and c). (B–D) The vertical line positioned at a cell area of 560 μm2 indicates the separation of spread (right) and not spread (left) cells. It is important to note that our spreading assays primarily analyze cells expressing moderate to low levels of tac-β1, since cell attachment to collagen I is inhibited by the expression of high levels of tac-β1 (data not shown). This observation is consistent with our recent studies showing that high levels of tac-β1 inhibit cell attachment to fibronectin (Mastrangelo et al. 1999a). The range of FITC fluorescence represents the range of tac-β1 expression detected in the adherent transfected cells. These experiments were performed three times and similar results were obtained.
We also demonstrated that the rescue of cell spreading by R-Ras, PI 3-kinase, PKCε, and Rac1 was dependent upon their activation state or kinase activity. The rescue of cell spreading by R-Ras and Rac1 was a GTP-dependent process, since the coexpression of dominant-negative R-Ras (N43R-Ras) or dominant-negative Rac1 (N17Rac1) failed to increase the area of tac-β1–expressing cells (compare Fig. 2g, Fig. i, and Fig. k). In addition, the kinase activities of PKCε and PI 3-kinase were required to increase the cell area of tac-β1–expressing cells, since the coexpression of kinase-dead PKCε (W437PKCε) failed to rescue cell spreading and LY294002 inhibited the ability of p110α-CAAX to restore cell spreading (Fig. 3g and Fig. i, and see Fig. 8 A).
Figure 2.

Coexpression of either N43R-Ras or N17Rac1 with tac-β1 does not restore cell spreading on collagen I. Transiently transfected cells were plated on collagen I and analyzed for cell area (x axis) and expression of recombinant proteins (y axis): cells transfected with tac (a) or tac-β1 alone (b) were analyzed for cell area and tac epitope expression; cells cotransfected with tac and myc-N43R-Ras were analyzed for cell area and tac (c) or myc (d) epitope expression; cells cotransfected with tac and myc-N17Rac1 were analyzed for cell area and tac (e) or myc (f) epitope expression; cells cotransfected with tac-β1 and myc-V38R-Ras were analyzed for cell area and tac (g) or myc (h) epitope expression; cells cotransfected with tac-β1 and myc-N43R-Ras were analyzed for cell area and tac epitope (i) or myc (j) epitope expression; and cells cotransfected with tac-β1 and myc-N17Rac1 were analyzed for cell area and tac (k) or myc (l) epitope expression. In each instance, cell area and epitope expression was analyzed for 100 randomly sampled cells. The x axis is a linear scale of cell area from 0 to 2.5 × 103 μm2. The y axis is a linear scale of FITC fluorescence (tac epitope expression) from 0 to 105 (a, c, and e) and from 0 to 2.5 × 104 (b, g, i, and k), or rhodamine fluorescence (myc epitope expression) from 0 to 8.0 × 104 (d, f, h, j, and l). The vertical line positioned at a cell area of 560 μm2 indicates the separation of spread (right) and not spread (left) cells. This experiment was performed twice and similar results were observed.
Figure 3.

The kinase activity of PKCε is required to increase the spreading of cells coexpressing tac-β1. Transiently transfected cells were plated on collagen I and analyzed for cell area (x axis) and expression of recombinant proteins (y axis): cells expressing tac (a) or tac-β1 (b) were analyzed for cell area and tac epitope expression; cells coexpressing tac and PKCε were analyzed for cell area and tac (c) or PKCε (d) expression; cells coexpressing tac and W437PKCε were analyzed for cell area and tac (e) or PKCε (f) expression; cells coexpressing tac-β1 and PKCε were analyzed for cell area and tac-β1 (g) or PKCε (h) expression; and cells coexpressing tac-β1 and W437PKCε were analyzed for cell area and tac-β1 (i) or PKCε (j) expression. In each instance, 100 randomly sampled positively transfected cells were analyzed. The x axis is a linear scale of cell area from 0 to 4.0 × 103 μm2. The y axis is a linear scale of FITC fluorescence (tac epitope expression) from 0 to 5.0 × 104 (a–c, e, g, and i), or rhodamine fluorescence (PKCε expression) from 0 to 3.0 × 105 (d, f, h, and j). The vertical line positioned at a cell area of 560 μm2 indicates the separation of spread (right) and not spread (left) cells. This experiment was performed twice and similar results were observed.
Figure 8.

PI 3-kinase activity is required for V38R-Ras, but not for L61Rac1 or myr-PKCε, to rescue cell spreading. Normal fibroblasts were transfected with either tac alone (A, a and e, and B, a and c) or cotransfected with tac-β1 and either V38R-Ras (B, b and d), p110α-CAAX (A, b and f), L61Rac1 (A, c and g), or myr-PKCε (A, d and h), and the morphology of cells adherent to collagen was analyzed in the presence of the PI 3-kinase inhibitor, LY294002 (A, e–h, and B, c and d), or DMSO (A, a–d, and B, c and d) as described in Materials and Methods. In each case, 100 (A) or 95 (B) randomly sampled transfected cells were analyzed for tac epitope expression and cell area as described previously. The x axis is a linear scale of cell area from 0 to 2.4 × 103 μm2 (A) or 0 to 3.2 × 103 μm2 (B), and the y axis is a linear scale of FITC fluorescence (tac epitope expression) from 0 to 4.8 × 104 (A, a and e) or 0 to 2.4 × 104 (A, b–d and f–h, and B, b, d) or 0 to 8.0 × 104 (B, a and c). The vertical line positioned at 560 μm2 indicates the separation of spread (right) and not spread (left) cells. These experiments were performed three times and similar results were obtained.
We next examined the morphology of cells coexpressing tac-β1 and each of the signaling proteins. As expected, cells expressing tac had a normal fibroblast morphology, whereas cells expressing tac-β1 were not spread (Fig. 4 A). Cells expressing tac-β1 and either p110α-CAAX or myr-PKCε were spread and had many membrane projections (Fig. 4 A). In some instances, tac-β1 was observed in focal adhesions of myr-PKCε–expressing cells (Fig. 4 B). The morphology of cells coexpressing tac-β1 and either p110α-caax or myr-pkcε was similar to many of the untransfected cells or the cells expressing tac alone, indicating that this is a common morphology for fibroblasts in our assay. Many of the cells coexpressing tac-β1 and V38R-Ras exhibited large polarized lamellipodia (Fig. 4 A) with prominent focal adhesion staining of tac-β1 (Fig. 4 B). Cells coexpressing tac-β1 and L61Rac1 were symmetrically spread and exhibited membrane ruffling (Fig. 4 A) and tac-β1 localization in focal adhesions at the cell periphery (Fig. 4 B). Thus, expression of activated forms of PI 3-kinase, PKCε, R-Ras, and Rac1 increased cell spreading through distinct cell morphologies. Interestingly, cells expressing L61Rac1 alone or V38R-Ras alone had cell morphologies very similar to cells coexpressing tac-β1, which tended to have smaller cell areas (Fig. 4 A and 5). Cells expressing either myr-PKCε or p110α-CAAX alone had cell morphologies similar to untransfected cells or cells transfected with tac alone (Fig. 5).
Figure 4.


The morphology of cotransfected cells adherent to collagen I. (A) The morphology of cells expressing tac or tac-β1 alone or cells coexpressing tac-β1 and either V38R-Ras, L61Rac1, or myr-PKCε. Shown is tac epitope expression (FITC fluorescence) for representative cells from the quantitative experiment shown in Fig. 1. Also included is tac-β1 expression of a representative cell from the cotransfection of tac-β1 and p110α-CAAX. Fluorescence images were obtained with Spot software and composites were generated in adobe photoshop. Scale bar: 10 μm. (B) Coexpression of either V38R-Ras, L61Rac1, or myr-PKCε in addition to rescuing cell spreading also restores the localization of tac-β1 to the focal contact. Fibroblasts were cotransfected with tac-β1 and either V38R-Ras, L61Rac1, or myr-PKCε, and cells adherent to collagen were costained for tac and signaling protein expression as described previously. The tac-FITC staining (right) and corresponding interference reflection pattern (left) of coexpressing cells is shown. Scale bar: 10 μm.
Figure 5.

The morphology of cells expressing either p110α-CAAX, L61Rac1, N17Rac1, V38R-Ras, N43R-Ras, or myr-PKCε. Normal fibroblasts were transfected with tac alone or each of the signaling proteins, and subsequently replated onto collagen I as described in Materials and Methods. Adherent cells were immunostained for expression of the tac epitope in cells transfected with the control tac receptor alone or cotransfected with the control tac receptor and p110α-CAAX. Cells were immunostained for expression of the flag epitope in cells transfected with myr-PKCε-Flag, or the myc epitope in the case of myc-L61Rac1, myc-N17Rac1, myc-V38R-Ras, and myc-N43R-Ras. Staining was visualized using rhodamine-conjugated secondary antibodies. Fluorescence images were obtained as described in Fig. 4. Scale bar: 10 μm.
We also examined the morphology of cells expressing dominant-negative forms of Rac1 (N17Rac1) and R-Ras (N43R-Ras). Expression of N17Rac1 resulted in cells that were round and inhibited in cell spreading (Fig. 5). This is in agreement with previously published observations by others in different cell types (Clark et al. 1998; Price et al. 1998). In contrast to N17Rac1, the expression of N43R-Ras did not significantly affect cell morphology in our assay (Fig. 5). This observation differs from a previously published report that N43R-Ras inhibits CHO cell spreading (Zhang et al. 1996). Quantitation of the effect of expressing the kinase-dead or dominant-negative signaling proteins on the extent of cell spreading confirmed our morphological observations and indicated that the expression of kinase-dead PKCε did not significantly alter the extent of cell spreading (Fig. 2 and Fig. 3).
Although Cdc42 has also been suggested as a potential regulator of cell spreading (Clark et al. 1998; Price et al. 1998), we did not test the ability of activated Cdc42 (L61Cdc42) to rescue tac-β1–inhibited cell spreading. Expression of L61Cdc42 in normal human fibroblasts resulted in cells with small areas, but which had prominent focal adhesions and stress fibers (data not shown). Thus, scoring a rescue of cell spreading by L61Cdc42 based on cell area would have been difficult. However, expression of dominant-negative Cdc42 (N17Cdc42) inhibited cell spreading similar to the expression of N17Rac1 (data not shown).
Endogenous Integrin Extracellular and Cytoplasmic Domain Function Is Required for p110α-CAAX, myr-PKCε, V38R-Ras, and L61Rac1 to Restore Cell Spreading
To determine whether the rescue of cell spreading on collagen I induced by the coexpression of activated signaling proteins was a β1 integrin-dependent process, β1 function blocking antibodies were added to the spreading assay after cell attachment had occurred, but before cell spreading. Comparison of cell spreading of control and anti–β1-treated cells indicated that fibroblast spreading on collagen I and the rescue of spreading by these signaling proteins were inhibited by the β1 blocking antibodies (Fig. 6). Thus, the rescue of cell spreading by V38R-Ras, myr-PKCε, p110α-CAAX, and L61Rac1 is dependent upon the extracellular function of endogenous β1 integrins. In the case of cells coexpressing either R-Ras or Rac1, we also examined the cell-surface expression of tac-β1 and the endogenous β1 subunit. We found that the rescue of cell spreading by constitutively active R-Ras and Rac1 did not involve changes in the cell-surface expression of either tac-β1 or the endogenous β1 subunit (data not shown).
Figure 6.

The rescue of tac-β1–inhibited cell spreading on collagen I is a β1 integrin–dependent process. Human fibroblasts transfected with the control tac receptor alone (A, a and e, and B, a and c) or cotransfected with tac-β1 and either p110α-CAAX (A, b and f), myr-PKCε (A, c and g), V38R-Ras (A, d and h), or L61Rac1 (B, b and d) were analyzed for spreading on collagen I in the presence of control ascites (A, a–d, and B, a and b) or β1 function blocking P4C10 ascites (A, e–h, and B, c and d) as described in Materials and Methods. The extent of cell spreading and the expression levels of the tac epitope were quantified for 100 (A) or 80 (B) randomly sampled positively transfected cells, and the results are shown by dot plot. The x axis is a linear scale of cell area from 0 to 3.2 × 103 μm2 (A and B). The y axis is a linear scale of FITC fluorescence (tac epitope expression) from 0 to 2.4 × 104 (A and B). The vertical line positioned at a cell area of 560 μm2 indicates the separation of spread (right) and not spread (left) cells. These experiments were performed twice and similar results were obtained.
We subsequently tested whether the ability of these signaling proteins to restore cell spreading required intact integrin β cytoplasmic domains on the integrins mediating cell adhesion. For these studies, we used CHO cells stably expressing either wild-type recombinant αIIbβ3 (A5 cells) or αIIbβ3Δ728 containing a β3 cytoplasmic domain truncation (ETC12 cells) (Ylanne et al. 1993). Previous studies had demonstrated that A5 cells expressing wild-type αIIbβ3 spread on fibrinogen, whereas ETC12 cells expressing the β cytoplasmic domain truncation are inhibited in cell spreading (Ylanne et al. 1993). Thus, if the expression of an activated signaling protein can induce cell spreading independent of integrin β cytoplasmic domain function, then this signaling protein should do so when expressed in ETC12 cells plated on fibrinogen.
A5 and ETC12 cells expressing the control tac receptor exhibited differences in spreading on fibrinogen consistent with the requirement for the β cytoplasmic domain in cell spreading. The majority of A5 cells expressing the control tac receptor had spread after 30 min, whereas the majority of the tac expressing ETC12 cells had cell areas <560 μm2 and were not spread (Fig. 7, a and b). Both A5 and ETC12 cells expressing tac-β1 were inhibited in spreading with >97% of the cells having cell areas <560 μm2 (Fig. 7c and Fig. d). Thus, expression of tac-β1 also inhibited CHO cell spreading on fibrinogen mediated by αIIbβ3. V38R-Ras, p110α-CAAX, myr-PKCε, and L61Rac1 were each able to restore CHO cell spreading inhibited by tac-β1; however, this increase in cell area was restricted to the A5 cells and was not observed in ETC12 cells (Fig. 7). More than 30% of the A5 cells transfected with tac-β1 and p110α-CAAX spread on fibrinogen, whereas >98% of the ETC12 cells transfected with tac-β1 and p110α-CAAX were not spread (Fig. 7e and Fig. f). Similarly, comparison of the spreading of A5 and ETC12 cells expressing both tac-β1 and L61Rac1 revealed that none of the ETC12 cells spread, whereas >15% of the A5 cells spread on fibrinogen (Fig. 7i and Fig. j). Finally, >40% of the A5 cells transfected with tac-β1 and myr-PKCε spread on fibrinogen. In contrast, >98% of the ETC12 cells transfected with tac-β1 and myr-PKCε were not spread and exhibited a round cell morphology (Fig. 7k and Fig. l). Thus, the enhancement of cell spreading by V38R-Ras, p110α-CAAX, myr-PKCε, and L61Rac1 requires intact β cytoplasmic domains on the integrins mediating cell adhesion.
Figure 7.
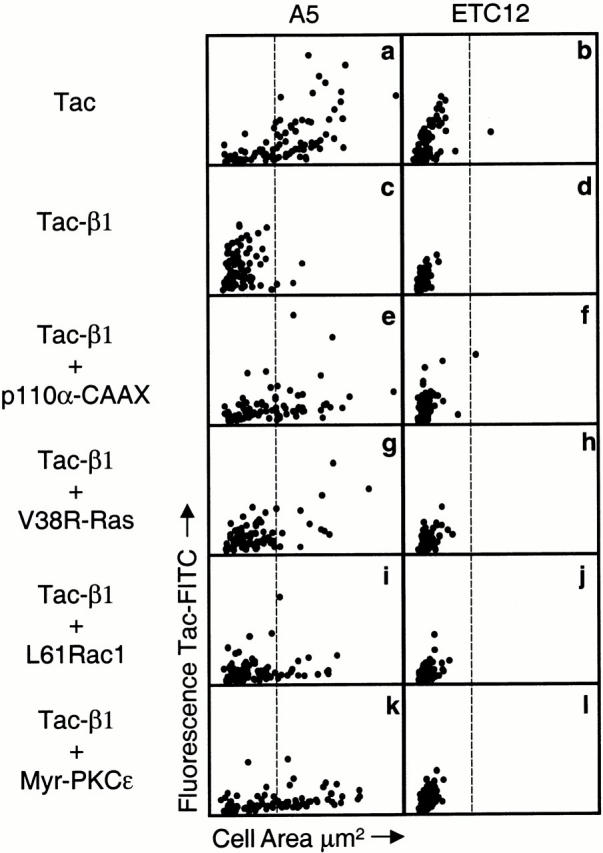
The rescue of cell spreading by p110α-CAAX, V38R-Ras, L61Rac1, and myr-PKCε is dependent upon integrin β cytoplasmic domains. Stable CHO cell lines A5 (left) and ETC12 (right) were each transfected with either tac (a and b) or tac-β1 alone (c and d) and the extent of cell spreading on fibrinogen was analyzed. In addition, A5 (left) and ETC12 (right) cells were cotransfected with tac-β1 and either p110α-CAAX (e and f), V38R-Ras (g and h), L61Rac1 (i and j), or myr-PKCε (k and l) and the extent of cell spreading on fibrinogen was analyzed. In each case, 90 randomly sampled positively transfected cells expressing both tac-β1 and the signaling protein where appropriate were analyzed for tac epitope expression and cell area. The data are shown on the dot plots of tac epitope expression versus cell area. The x axis is a linear scale of cell area from 0 to 1.6 × 103 μm2, and the y axis is a linear scale of FITC fluorescence (tac epitope expression) from 0 to 4.8 × 104 (a and b) or 0 to 3.2 × 104 (c–l). These experiments were performed three times and similar results were obtained.
To ensure that the inability of signaling proteins to rescue ETC12 cell spreading on fibrinogen was due to the lack of intact β3 cytoplasmic domains on the fibrinogen binding integrins, and not to some peculiarity of the ETC12 clone, we tested the ability of L61Rac1 and myr-PKCε to restore tac-β1–inhibited ETC12 cell spreading on fibronectin. Although ETC12 cells are not capable of spreading on fibrinogen, they are able to spread on fibronectin (Ylanne et al. 1995). We found that ETC12 cells spread on fibronectin, and this spreading was inhibited by tac-β1 and restored by the coexpression of either L61Rac1 or myr-PKCε (data not shown).
V38R-Ras Requires PI 3-Kinase Activity to Rescue Cell Spreading, whereas L61Rac1 and myr-PKCε Do Not
R-Ras has been shown to activate PI 3-kinase (Marte et al. 1996), suggesting that PI 3-kinase may be downstream of R-Ras in regulating cell spreading. In addition, PIP3, a product of PI 3-kinase, has been shown to be involved in the activation of PKCε (Toker et al. 1994; Moriya et al. 1996), suggesting that PI 3-kinase may function upstream of PKCε activation during cell spreading. PI 3-kinase activity has also been reported in signaling pathways containing Rac1. Since PI 3-kinase has been positioned both upstream and downstream of Rac1 activation (Hawkins et al. 1995; Nobes et al. 1995; Parker 1995; Keely et al. 1997; Missy et al. 1998), the role of PI 3-kinase in Rac1 signaling during cell spreading is not certain.
To determine the requirement for PI 3-kinase activity in the rescue of cell spreading by V38R-Ras, myr-PKCε, and L61Rac1, the ability of cotransfected cells to spread on collagen I was assayed in the presence and absence of the PI 3-kinase inhibitor LY294002 (Fig. 8). Initial experiments demonstrated a role for PI 3-kinase in normal fibroblast cell spreading. When fibroblasts transiently expressing the control tac receptor were assayed for their ability to spread on collagen I in the presence of the PI 3-kinase inhibitor (LY294002), >50% of the tac-expressing cells were inhibited in spreading, compared with <20% in the presence of DMSO alone (Fig. 8A, a and e, and B, a and c). Thus, normal human fibroblasts require PI 3-kinase activity for cell spreading on collagen I.
We then tested the ability of LY294002 to impair cell spreading in cells cotransfected with tac-β1 and the various constitutively activated signaling proteins. Greater than 60% of cells cotransfected with tac-β1 and p110α-CAAX spread in the presence of DMSO, whereas, in the presence of LY294002, <15% of these transfected cells had areas >560 μm2, suggesting that p110α-CAAX requires PI 3-kinase activity to enhance cell spreading (Fig. 8 A, b and f). The V38R-Ras–triggered rescue was also found to require PI 3-kinase activity: >80% of the cotransfected cells treated with LY294002 were not spread (Fig. 8 B, b and d), compared with 44% of cotransfected cells treated with DMSO. In contrast, cells coexpressing tac-β1 and L61Rac1 were spread when treated with either LY294002 or DMSO alone. Of the cells coexpressing tac-β1 and L61Rac1, 78% were spread when treated with DMSO and 63% were spread when treated with LY294002 (Fig. 8 A, c and g). This was also true for cells coexpressing tac-β1 and myr-PKCε. In cells coexpressing tac-β1 and myr-PKCε, 60% of the DMSO-treated cells and 45% of the LY294002-treated cells were spread (Fig. 8 A, d and h). These results suggest that endogenous PI 3-kinase activity is required for V38R-Ras, but not for L61Rac1 or myr-PKCε, to restore cell spreading in tac-β1–expressing cells.
Dominant-Negative N17Rac1 Blocks the p110α-CAAX and myr-PKCε Rescue of Cell Spreading
Both L61Rac1 and myr-PKCε rescued tac-β1–inhibited cell spreading independent of PI 3-kinase activity, suggesting that PKCε and Rac1 may function downstream of PI 3-kinase in the rescue of cell spreading. To determine whether PI 3-kinase and PKCε require Rac1 activity, we tested the ability of p110α-CAAX and myr-PKCε to restore spreading in cells expressing dominant-negative Rac1 (N17Rac1). Fibroblasts were cotransfected with tac-β1 and either p110α-CAAX or myr-PKCε together with N17Rac1, and the extent of cell spreading was examined. Expression of N17Rac1 inhibited cell spreading in all cases tested (Table ). Greater than 90% of cells that coexpressed N17Rac1, tac-β1, and either myr-PKCε or p110α-CAAX were round and inhibited in cell spreading (Table ), indicating that dominant-negative N17Rac1 blocks the ability of both myr-PKCε and p110α-CAAX to rescue cell spreading. These results suggest that Rac1 may function downstream of PKCε and PI 3-kinase in a pathway requiring integrin β cytoplasmic domain function in cell spreading.
Table 1.
N17Rac1 Blocks the p110α-CAAX and myr-PKCε Rescue of Cell Spreading
| Transfected recombinant protein(s) | Percentage of cells not spread |
|---|---|
| Tac + Empty Vector | 23 ± 2‡ |
| Tac + N17Rac1 | 94 ± 4 |
| Tac + N17Rac1 + Empty Vector | 91 ± 5 |
| Tac-β1 + Empty Vector | 57 ± 14 |
| Tac-β1 + myr-PKCε | 32 ± 4 |
| Tac-β1 + myr-PKCε + Empty Vector | 31 ± 3 |
| Tac-β1 + myr-PKCε + N17Rac1§‖ | 99 ± 1 |
| Tac-β1 + myr-PKCε + N17Rac1§¶ | 92 ± 4 |
| Tac-β1 + p110α-CAAX | 26 ± 6 |
| Tac-β1 + p110α-CAAX + Empty Vector | 26 ± 2 |
| Tac-β1 + p110α-CAAX + N17Rac1‖ | 91 ± 4 |
Discussion
Cell spreading is a fundamental cellular process in many cell types, which is initiated by integrin-mediated cell attachment and requires integrin β subunit cytoplasmic domain function. Although the activation of R-Ras, PI 3-kinase, Rac1, and PKCε has been shown to positively regulate cell spreading (Chun et al. 1996; Zhang et al. 1996; King et al. 1997; Khwaja et al. 1997; Clark et al. 1998; Price et al. 1998), the molecular mechanisms involved for the most part remain undefined. In this study, we asked whether the integrin β cytoplasmic domain is involved in the regulation of cell spreading by these signaling proteins. The experimental approach we used was to inhibit cell spreading by the expression of tac-β1, and then to test whether the overexpression of activated forms of these signaling proteins could overcome the inhibitory effect of tac-β1. We found that inhibition of cell spreading by tac-β1 can be rescued by the coexpression of activated forms of either R-Ras, PI 3-kinase, Rac1, or PKCε. The rescue of cell spreading by each of these signaling proteins required endogenous integrin extracellular domain function and intact β subunit cytoplasmic domains on the adhering integrins.
The coexpression of each of these activated signaling proteins with tac-β1 lead to significant increases in cell area. Constitutively active R-Ras was by far the most potent at reversing tac-β1–inhibited cell spreading. Coexpression of V38R-Ras resulted in cells with similar sizes and morphologies to untransfected or control tac-transfected cells. The ability of V38R-Ras to rescue cell spreading was dependent upon PI 3-kinase activity, which is consistent with previous reports indicating that activated R-Ras can associate with and activate PI 3-kinase (Marte et al. 1996). Thus, V38R-Ras may rescue cell spreading by triggering pathways normally activated by integrins such as PI 3-kinase.
Interestingly, activated V38R-Ras has also been shown to rescue high affinity ligand binding of αIIbβ3 inhibited by the constitutive activation of the ERK-MAPK cascade; however, in this instance, R-Ras regulated integrin activity by a pathway independent of PI 3-kinase (Sethi et al. 1999). Thus, R-Ras may modulate integrin function via multiple downstream effectors. Additionally, stable expression of activated R-Ras has been shown to activate α2β1-dependent cell migration and invasion of carcinoma cells by a mechanism requiring the α2 cytoplasmic domain, suggesting that R-Ras might also specifically target individual integrin heterodimers by regulating protein interactions with their α subunit cytoplasmic domains (Keely et al. 1999). However, the role of individual α subunit cytoplasmic domains in the rescue of cell spreading by V38R-Ras is not known at present.
Little data has been published regarding how R-Ras is activated under physiological conditions and there is currently no evidence linking integrin engagement to R-Ras activation. Our result that expression of dominant-negative N43R-Ras does not inhibit cell spreading suggests that integrin-triggered activation of R-Ras is not required for spreading, at least in normal human fibroblasts. This result contradicts earlier studies demonstrating that N43R-Ras can inhibit CHO cell spreading (Zhang et al. 1996); however, the reason for these differences is not clear at this time. Nonetheless, the ability of V38R-Ras to regulate cell spreading in our system suggests that the activation of endogenous R-Ras by signals initiated by other cell surface receptors may regulate the spreading of some cell types.
Interestingly, although PI 3-kinase activity is required for V38R-Ras to rescue tac-β1–inhibited cell spreading, the membrane targeted form of PI 3-kinase, p110α-CAAX, only partially restored cell spreading in tac-β1–expressing cells compared with V38R-Ras. One possible explanation is that V38R-Ras may lead to more robust PI 3-kinase signaling compared with the expression of p110α-CAAX. Alternatively, V38R-Ras may activate additional pathways that enhance cell spreading in conjunction with PI 3-kinase.
PI 3-kinase is known to be activated by integrin engagement and was found to be important in normal fibroblast cell spreading in the present study. The pathways linking integrins to activation of PI 3-kinase are not yet fully defined. The ability of integrins to activate FAK and H-Ras provides two potential pathways, since both have been shown to interact with and activate PI 3-kinase (Chen and Guan 1994; Kodaki et al. 1994; Rodriguez-Viciana et al. 1994, Rodriguez-Viciana et al. 1996; Guan 1997; Giancotti and Ruoslahti 1999). However, the relative contribution of FAK and H-Ras to integrin activation of PI 3-kinase is currently unknown. Potential downstream effectors of PI 3-kinase in the regulation of cell spreading are PKCε and Rac1 (Toker et al. 1994; Moriya et al. 1996; Rodriguez-Viciana et al. 1997; Missy et al. 1998). It is known that growth factor activation of PI 3-kinase can trigger membrane ruffling through a Rac1-dependent pathway and roles for PI 3-kinase in the activation of Rac1 have been described (Hawkins et al. 1995; Nobes et al. 1995; Parker et al., 1995). Our observations that N17Rac1 prevents the ability of p110α-CAAX to increase cell spreading and that L61Rac1 can restore cell spreading independent of PI 3-kinase are consistent with PI 3-kinase activation occurring either upstream or independent of Rac1 in the regulation of cell spreading. The rescue of cell spreading by myr-PKCε is also independent of PI 3-kinase, but inhibited by N17Rac1, similarly suggesting that PKCε may reside downstream of PI 3-kinase and upstream of Rac1 in a pathway regulating cell spreading.
The mechanism by which Rac1 regulates integrin-mediated cell spreading is not completely understood. It is assumed to do so by triggering actin polymerization and membrane extension (lamellipodia) required for the increase in cell area that accompanies cell spreading. We have not yet identified signaling proteins that function downstream of Rac1 to regulate cell spreading. PAK1, which can be activated by Rac1, is a potential candidate (Sells and Chernoff 1997). Activated PAK1 mutants have been shown to induce lamellipodia (Sells et al. 1997), and dominant-negative mutants of PAK1 have been found to inhibit cell spreading (Price et al. 1998). However, whether PAK1 functions downstream of Rac1 in the induction of lamellipodia is unclear since Rac1-induced lamellipodia can occur independent of Rac1-PAK1 interactions (Joneson et al. 1996; Lamarche et al. 1996; Westwick et al. 1997).
POR1 is another potential downstream effector of Rac1 that may regulate cell spreading (Van Aelst et al. 1996). POR1 has been shown to localize to lamellipodia and dominant-negative forms of POR1 have been shown to inhibit Rac1-induced lamellipodia (Van Aelst et al. 1996). In addition, mutations in Rac1 that inhibit Rac1-POR1 interactions also inhibit lamellipodia formation (Joneson et al. 1996), suggesting that POR1 is a good candidate. P160Rock may also be a good candidate. P160Rock interacts with Rac1 in a GTP-dependent manner (Lamarche et al. 1996) and Rac1 mutations that inhibit p160Rock-Rac1 interactions also inhibit lamellipodia formation (Lamarche et al. 1996). Our future studies will determine whether these effectors regulate integrin function in cell spreading.
We propose three models for the inhibition and rescue of cell spreading. Our first model is that expression of tac-β1 may inhibit cell spreading by titrating a β cytoplasmic domain binding protein from endogenous integrin β cytoplasmic domains that may be required for the adhesion-triggered activation of Rac1 (Fig. 9 a). Expression of activated Rac1 may bypass the block in integrin-triggered Rac1 activation. However, even if this is the case, β cytoplasmic domain function is also required downstream of Rac1 activation, since L61Rac1 could not rescue the spreading of CHO cells attached to fibrinogen via αIIbβ3Δ728. So, in our first model, expression of tac-β1 inhibits the function of the β cytoplasmic domain upstream but not downstream of Rac1 activation.
Figure 9.

Models for the rescue of tac-β1–inhibited cell spreading. (a, Model I) Expression of tac-β1 results in the titration of cellular factors from the endogenous β cytoplasmic domains that trigger the activation of signaling proteins required for cell spreading. The coexpression of activated recombinant signaling proteins with tac-β1 may bypass this block in integrin signaling and restore cell spreading. Tac-β1 does not inhibit integrin β cytoplasmic domain function that is required downstream of these signaling proteins. (b, Model II) Integrin-mediated cell attachment activates signaling proteins that regulate the binding of proteins to endogenous β cytoplasmic domains that are required for cell spreading. Expression of tac-β1 titrates these adhesion-induced protein interactions with the integrin β cytoplasmic domains. Coexpression of activated signaling proteins with tac-β1 increases the pool of proteins that bind the integrin β cytoplasmic domain and restores endogenous integrin function in cell spreading. (Model III). Tac-β1 inhibits the adhesion-induced activation of these signaling proteins and additionally inhibits protein interactions with the β cytoplasmic domain that are triggered by the signaling proteins (not shown).
In our second model, we propose that Rac1 signaling may regulate protein interactions with integrin β cytoplasmic domains that are required for cell spreading (Fig. 9 b). In this model, integrin-induced Rac1 activation is not inhibited by the expression of tac-β1, instead tac-β1 inhibits cell spreading by titrating these Rac1-induced integrin-binding proteins from the endogenous integrin β cytoplasmic domains. Expression of L61Rac1 may rescue cell spreading by increasing the cytoplasmic pool of proteins that can bind to integrin β cytoplasmic domains. These interactions may be required for Rac1 to initiate actin polymerization and lamellipodia formation. A third model is also possible in which tac-β1 inhibits integrin activation of Rac1 as well as the ability of Rac1 signaling to regulate integrin β cytoplasmic domain function. Future studies will explore these possibilities.
Acknowledgments
We thank Drs. J. Sottile and M. Dipersio for critically reading this manuscript, Dr. J. Depasquale for valuable advice in using the Image Pro-Plus software to accurately measure cell area and flourescence intensity, and R. Martinez for skillful technical assistance. We also thank Drs. Hall and Toker for providing us with important reagents used in these studies.
This work was supported by National Institutes of Health grants GM51540, T32HL07529, and T32GM07033, and the American Heart Association grant 0020180T.
References
- Burridge K., Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu. Rev. Cell Dev. Biol. 1996;12:463–519. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Chen H.-C., Guan J.-L. Association of focal adhesion kinase with its potential substrate phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA. 1994;91:10148–10152. doi: 10.1073/pnas.91.21.10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-P., O'Toole T.E., Shipley T., Forsyth J., LaFlamme S.E., Yamada K.M., Shattil S.J., Ginsberg M.H. “Inside-out” signal transduction inhibited by isolated integrin cytoplasmic domains. J. Biol. Chem. 1994;269:18307–18310. [PubMed] [Google Scholar]
- Chou M.M., Hou W., Johnson J., Graham L.K., Lee M.H., Chen C.-S., Newton A.C., Schaffhausen B.S., Toker A. Regulation of protein kinase C ζ by PI 3-kinase and PDK-1. Curr. Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- Chun J.-S., Ha M.-J., Jacobson B.S. Differential translocation of protein kinase C ε during HeLa cell adhesion to a gelatin substratum. J. Biol. Chem. 1996;271:13008–13012. doi: 10.1074/jbc.271.22.13008. [DOI] [PubMed] [Google Scholar]
- Clark E.A., King W.G., Brugge J.S., Symons M., Hynes R.O. Integrin-mediated signals regulated by members of the Rho Family of GTPases. J. Cell Biol. 1998;142:573–586. doi: 10.1083/jcb.142.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Pozo M.A., Price L.S., Alderson N.B., Ren X.-D., Schwartz M.A. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO (Eur. Mol. Biol. Organ.) J. 2000;19:2008–2014. doi: 10.1093/emboj/19.9.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti F.G., Ruoslahti E. Integrin Signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Guan J.-L. Focal adhesion kinase in integrin signaling. Matrix Biol. 1997;16:195–200. doi: 10.1016/s0945-053x(97)90008-1. [DOI] [PubMed] [Google Scholar]
- Hawkins P.T., Eguinoa A., Qiu R.-G., Stokoe D., Cooke F.T., Walters R., Wennstrom S., Claesson-Welsh L., Evans T., Symons M., Stephens L. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr. Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- Homan S.M., Mercurio A.M., LaFlamme S.E. Endothelial cells assemble two distinct α6β4-containing vimentin-associated structuresroles for ligand binding and the β4 cytoplasmic domain. J. Cell Sci. 1998;111:2717–2728. doi: 10.1242/jcs.111.18.2717. [DOI] [PubMed] [Google Scholar]
- Hynes R.O. Integrinsversatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Izzard C.S., Lochner L.R. Cell-to-substrate contacts in living fibroblastsand interference-reflexion study with an evaluation of the technique. J. Cell Sci. 1976;21:129–159. doi: 10.1242/jcs.21.1.129. [DOI] [PubMed] [Google Scholar]
- Joneson T., McDonough M., Bar-Sagi D., Van Aelst L. Rac regulation of actin polymerization and proliferation by a pathway distinct from Jun kinase. Science. 1996;274:1374–1376. doi: 10.1126/science.274.5291.1374. [DOI] [PubMed] [Google Scholar]
- Keely P.J., Rusyn E.V., Cox A.D., Parise L.V. R-Ras signals through specific integrin α cytoplasmic domains to promote migration and invasion of breast epithelial cells. J. Cell Biol. 1999;145:1077–1088. doi: 10.1083/jcb.145.5.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keely P.J., Westwick J.K., Whitehead I.P., Der C.J., Parise L.V. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Science. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- Khwaja A., Rodriguez-Viciana P., Wennstrom S., Warne P.H., Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/AKT cellular survival pathway. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2783–2793. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King W.G., Mattaliano M.D., Chan T.O., Tsichlis P.N., Brugge J.S. Phosphatidylinositol 3-kinase is required for integrin-stimulated AKT and raf-1/mitogen-activated protein kinase pathway activation. Mol. Cell. Biol. 1997;17:4406–4418. doi: 10.1128/mcb.17.8.4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaki T., Woscholski R., Hallberg B., Rodriguez-Viciana P., Downward J., Parker P.J. The activation of phosphatidylinositol 3-kinase by Ras. Curr. Biol. 1994;4:798–806. doi: 10.1016/s0960-9822(00)00177-9. [DOI] [PubMed] [Google Scholar]
- LaFlamme S.E., Akiyama S.K., Yamada K.M. Regulation of fibronectin receptor distribution. J. Cell Biol. 1992;117:437–447. doi: 10.1083/jcb.117.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFlamme S.E., Homan S.M., Bodeau A.L., Mastrangelo A.M. Integrin cytoplasmic domains as connectors to the cell's signal transduction apparatus. Matrix Biol. 1997;16:153–163. doi: 10.1016/s0945-053x(97)90003-2. [DOI] [PubMed] [Google Scholar]
- LaFlamme S.E., Thomas L.A., Yamada S.S., Yamada K.M. Single subunit chimeric integrins as mimics and inhibitors of endogenous integrin functions in receptor localization, cell spreading and migration, and matrix assembly. J. Cell Biol. 1994;126:1287–1298. doi: 10.1083/jcb.126.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarche N., Tapon N., Stowers L., Burbelo P.D., Aspenstrom P., Bridges T., Chant J., Hall A. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell. 1996;87:519–529. doi: 10.1016/s0092-8674(00)81371-9. [DOI] [PubMed] [Google Scholar]
- Lukashev M.E., Sheppard D., Pytela R. Disruption of integrin function and induction of tyrosine phosphorylation by the autonomously expressed β1 integrin cytoplasmic domain. J. Biol. Chem. 1994;269:18311–18314. [PubMed] [Google Scholar]
- Marte B.M., Rodriguez-Viciana P., Wennstrom S., Warne P.H., Downward J. R-Ras can activate the phosphoinositide 3-kinase but not the MAP kinase arm of the Ras effector pathways. Curr. Biol. 1996;7:63–70. doi: 10.1016/s0960-9822(06)00028-5. [DOI] [PubMed] [Google Scholar]
- Mastrangelo A.M., Homan S.M., Humphries M.J., LaFlamme S.E. Amino acid motifs required for isolated β cytoplasmic domains to regulate ‘in trans’ β1 integrin conformation and function in cell attachment J. Cell Sci 112 1999. 217 229a [DOI] [PubMed] [Google Scholar]
- Mastrangelo A.M., Bodeau A.L., Homan S.M., Berrier A.L., LaFlamme S.E. The use of chimeric receptors in the study of integrin signaling Guan J.-L. Signaling Through Cell Adhesion Molecules 1999. 3 17 CRC Press; New York, NY: b [Google Scholar]
- Missy K., Van Poucke V., Raynal P., Viala C., Mauco G., Plantavid M., Chap H., Payrastre B. Lipid products of phosphoinositide 3-kinase interact with Rac1 GTPase and stimulate GDP dissociation. J. Biol. Chem. 1998;273:30279–30286. doi: 10.1074/jbc.273.46.30279. [DOI] [PubMed] [Google Scholar]
- Moriya S., Kazlauskas A., Akimoto K., Hirai S.-I., Mizuno K., Takenawa T., Fukui Y., Watanabe Y., Ozaki S., Ohno S. Platelet-derived growth factor activates protein kinase Cε through redundant and independent signaling pathways involving phospholipase Cγ or phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA. 1996;93:151–155. doi: 10.1073/pnas.93.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes C.D., Hawkins P., Stephens L., Hall A. Activation of the small GTP-binding proteins Rho and Rac by growth factor receptors. J. Cell Sci. 1995;108:225–233. doi: 10.1242/jcs.108.1.225. [DOI] [PubMed] [Google Scholar]
- Parker P.J. PI 3-kinase puts GTP on the Rac. Curr. Biol. 1995;5:577–579. doi: 10.1016/s0960-9822(95)00113-8. [DOI] [PubMed] [Google Scholar]
- Price L.S., Leng J., Schwartz M.A., Bokoch G.M. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol. Biol. Cell. 1998;9:1863–1871. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Warne P.H., Dhand R., Vanhaesebroeck B., Gout I., Fry M.J., Waterfield M.D., Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Warne P.H., Khwaja A., Marte B.M., Pappin D., Das P., Waterfield M.D., Ridley A., Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P., Warne P.H., Vanhaesebroeck B., Waterfield M.D., Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- Schwartz M.A., Schaller M.D., Ginsberg M.H. Integrinsemerging paradigms of signal transduction. Annu. Rev. Cell Dev. Biol. 1995;11:549–599. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- Sells M.A., Chernoff J. Emerging from the Pakthe p21-activated protein kinase family. Trends Cell Biol. 1997;7:162–167. doi: 10.1016/S0962-8924(97)01003-9. [DOI] [PubMed] [Google Scholar]
- Sells M.A., Knaus U.G., Bagrodia S., Ambrose D.M., Bokoch G.M., Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Sethi T., Ginsberg M.H., Downward J., Hughes P.E. The small GTP-binding protein R-Ras can influence integrin activation by antagonizing a Ras/Raf-initiated integrin suppression pathway. Mol. Biol. Cell. 1999;10:1799–1809. doi: 10.1091/mbc.10.6.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small J.V., Rottner K., Kaverina I. Functional design in the actin cytoskeleton. Curr. Opin. Cell Biol. 1999;11:54–60. doi: 10.1016/s0955-0674(99)80007-6. [DOI] [PubMed] [Google Scholar]
- Smilenov L., Briesewitz R., Marcantonio E.E. Integrin β1 cytoplasmic domain dominant negative effects revealed by lysophosphatidic acid treatment. Mol. Biol. Cell. 1994;5:1215–1223. doi: 10.1091/mbc.5.11.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker A., Meyer M., Reddy K.K., Falck J.R., Aneja R., Aneja S., Parra A., Burns D.J., Ballas L.M., Cantley L.C. Activation of protein kinase C family members by the novel polyphosphoinositides PtdIns-3,4-P2 and PtdIns-3,4,5-P3 . J. Biol. Chem. 1994;269:32358–32367. [PubMed] [Google Scholar]
- Van Aelst L., Joneson T., Bar-Sagi D. Identification of a novel Rac1-interacting protein involved in membrane ruffling. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3778–3786. [PMC free article] [PubMed] [Google Scholar]
- Van Leeuwen F.N., Van Delft S., Kain H.E., Van der Kammen R.A., Collard J.G. Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nat. Cell Biol. 1999;1:242–248. doi: 10.1038/12068. [DOI] [PubMed] [Google Scholar]
- Vuori K., Ruoslahti E. Activation of protein kinase C precedes α5β1 integrin-mediated cell spreading on fibronectin. J. Biol. Chem. 1993;268:21459–21462. [PubMed] [Google Scholar]
- Wennstrom S., Downward J. Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol. Cell. Biol. 1999;19:4279–4288. doi: 10.1128/mcb.19.6.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwick J.K., Lambert Q.T., Clark G.J., Symons M., Van Aelst L., Pestell R.G., Der C.J. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol. Cell. Biol. 1997;17:1324–1335. doi: 10.1128/mcb.17.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K.M., Miyamoto S. Integrin transmembrane signaling and cytoskeletal control. Curr. Opin. Cell Biol. 1995;7:681–689. doi: 10.1016/0955-0674(95)80110-3. [DOI] [PubMed] [Google Scholar]
- Ylanne J., Chen Y., O'Toole T.E., Loftus J.C., Takada Y., Ginsberg M.H. Distinct functions of integrin α and β subunit cytoplasmic domains in cell spreading and formation of focal adhesions. J. Cell Biol. 1993;122:223–233. doi: 10.1083/jcb.122.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylanne J., Huuskonen J., O'Toole T.E., Ginsberg M.H., Virtanen I., Gahmberg C.G. Mutation of the cytoplasmic domain of the integrin β3 subunit. Differential effects on cell spreading, recruitment to adhesion plaques, endocytosis, and phagocytosis. J. Biol. Chem. 1995;270:9550–9557. doi: 10.1074/jbc.270.16.9550. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Vuori K., Wang H.-G., Reed J.C., Ruoslahti E. Integrin activation by R-Ras. Cell. 1996;85:61–69. doi: 10.1016/s0092-8674(00)81082-x. [DOI] [PubMed] [Google Scholar]