

Abstract
Although the crucial role of Ca2+ influx in lymphocyte activation has been well documented, little is known about the properties or expression levels of Ca2+ channels in normal human T lymphocytes. The use of Na+ as the permeant ion in divalent-free solution permitted Ca2+ release-activated Ca2+ (CRAC) channel activation, kinetic properties, and functional expression levels to be investigated with single channel resolution in resting and phytohemagglutinin (PHA)-activated human T cells. Passive Ca2+ store depletion resulted in the opening of 41-pS CRAC channels characterized by high open probabilities, voltage-dependent block by extracellular Ca2+ in the micromolar range, selective Ca2+ permeation in the millimolar range, and inactivation that depended upon intracellular Mg2+ ions. The number of CRAC channels per cell increased greatly from ∼15 in resting T cells to ∼140 in activated T cells. Treatment with the phorbol ester PMA also increased CRAC channel expression to ∼60 channels per cell, whereas the immunosuppressive drug cyclosporin A (1 μM) suppressed the PHA-induced increase in functional channel expression. Capacitative Ca2+ influx induced by thapsigargin was also significantly enhanced in activated T cells. We conclude that a surprisingly low number of CRAC channels are sufficient to mediate Ca2+ influx in human resting T cells, and that the expression of CRAC channels increases ∼10-fold during activation, resulting in enhanced Ca2+ signaling.
Keywords: T lymphocyte, Ca2+ channel, CRAC channel, T cell activation, Ca2+ signaling
Introduction
T lymphocytes play fundamental and diverse roles in the immune response including the recognition of antigens, secretion of cytokines, and killing of foreign or virus-infected cells. T cell receptor (TCR) engagement by specific antigen/major histocompatibility complex proteins on an antigen-presenting cell initiates a program of intracellular signaling within the T cell. By cross-linking surface receptors, the mitogenic lectin phytohemagglutinin (PHA) mimics the early antigen evoked events in a heterogeneous population of resting T cells. A sustained or oscillatory rise in intracellular Ca2+ concentration ([Ca2+]i) following TCR engagement is required to drive many of the subsequent events of lymphocyte activation, including the activation of transcription factors, modulation of cytoskeletal elements and motility, production and release of cytokines, and cell proliferation (Metcalfe et al. 1980; Crabtree 1989; Crabtree and Clipstone 1994; Negulescu et al. 1994, Negulescu et al. 1996; Rao 1994). The rise in [Ca2+]i results initially from IP3-induced Ca2+ release from internal stores and is sustained by Ca2+ influx across the plasma membrane (Donnadieu et al. 1992; Hess et al. 1993; Dolmetsch and Lewis 1994; Lewis and Cahalan 1995; Lewis 1999). In parallel with the Ca2+ signaling pathway, contact with an antigen-presenting cell triggers a PKC-dependent pathway leading to transcriptional activation of additional genes via the Fos/Jun transcription factor AP-1. The Ca2+ and PKC pathways interact at several levels, resulting in amplification of downstream signaling. The crucial role of Ca2+ for lymphocyte activation is underscored by clinical use of the immunosuppressive drug, cyclosporin A (CsA), which inhibits calcineurin, a [Ca2+]i-dependent phosphatase (Schreiber and Crabtree 1992; Rao et al. 1997; Crabtree 1999).
In Jurkat T cells dialyzed with a strong Ca2+ buffer or stimulated acutely with PHA, Ca2+ enters via Ca2+-selective, store-operated channels (Lewis and Cahalan 1989). These channels, also seen in mast cells and named Ca2+ release-activated Ca2+ (CRAC) channels, can be activated by treatment with thapsigargin (Tg) to inhibit Ca2+ pumps located in the ER, thus bypassing proximal receptor signaling and IP3 generation while depleting the Ca2+ store (Hoth and Penner 1992; Zweifach and Lewis 1993). The resulting Ca2+ influx through CRAC channels produces a sustained [Ca2+]i signal that is capable of triggering gene expression (Negulescu et al. 1994; Fanger et al. 1995). CRAC channels are highly selective for Ca2+ under physiological conditions, but exhibit permeability to Na+ and other monovalent cations when extracellular divalent ions are withdrawn, with a size cutoff for permeation of 6Å (Hoth and Penner 1993; Lepple-Wienhues and Cahalan 1996; Kerschbaum and Cahalan 1998). Although the mechanism linking Ca2+store depletion to the opening of CRAC channels remains elusive, progress has been made in characterizing CRAC channels at the single channel level, using Na+ as a charge carrier to amplify the single channel conductance (Kerschbaum and Cahalan 1999). Using this approach, single CRAC channels with 36–40-pS Na+ conductance were observed in Jurkat T cells. The unitary CRAC channel phenotype provides a unique single channel signature for identifying CRAC channels in other cell types. Here, with single channel resolution during whole-cell recording, we describe the properties and regulated expression of CRAC channels in resting and activated T lymphocytes from human donors.
Materials and Methods
Cell Culture
T lymphocytes were purified from freshly drawn peripheral blood of healthy volunteers using a nylon-wool column and placed into culture in modified RPMI 1640 medium in a 5% CO2 incubator at 37°C according to standard procedure (Hess et al. 1993). Purified human T cells (>90% CD3+, containing CD4+ and CD8+ T cells) were pretreated in vitro with 4 μg/ml phytohemagglutinin P (PHA; Difco), 40 nM phorbol 12-myristate 13-acetate (PMA; Calbiochem), or 500 nM ionomycin (Calbiochem) for varying times; cells from the PHA-treated population are referred to as activated T cells. In some experiments, 0.1–1 μM CsA from a 1-mM stock solution in DMSO (Sigma-Aldrich) was added simultaneously with PHA to inhibit activation and proliferation. For electrophysiological and [Ca2+]i-imaging experiments, resting, activated, or PMA-treated T cells were placed on poly-l-lysine–coated glass coverslips and mounted on the stage of an inverted microscope.
Electrophysiology
Whole-cell recording was performed using an EPC-9 amplifier (HEKA). The resistance of sylgard-coated, fire-polished glass microelectrodes varied from 2 to 4 MΩ. Unless otherwise indicated, currents were sampled at 5 kHz and digitally filtered off-line at 1.4 kHz. After establishing the whole-cell recording configuration, cells were held at 0 mV and square pulses to −120 mV for 200 ms or ramp voltages from −120 to +50 mV were applied every 0.7 s. The short-pulse protocol was chosen to avoid the damaging effect of strong hyperpolarization on membrane integrity. In some experiments, long pulses (up to 2,500 ms duration) were applied to voltages ranging from −120 to −10 mV. Liquid junction potentials were measured for all internal and external solutions and corrections were made during data acquisition. All recordings were performed at room temperature (∼21°C). In some experiments, cells were pretreated with 2 μM Tg (Alexis Chemical) in Ringer solution for 15–45 min on the microscope stage. Cell capacitance (C m) was monitored to determine the membrane surface area, based upon a specific C m value of 1 μF/cm2.
Solutions
CRAC channels were activated by passive Ca2+ store depletion with the Ca2+ chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) included in the pipette solution. The standard internal solution was composed of 128 mM Cs–aspartate, 10 mM Cs–Hepes, 12 mM BAPTA, and 0.9 mM CaCl2. In some experiments, either 3.2 mM Mg2+ or 30 μM IP3 (Sigma-Aldrich) was added to standard solution. To detect single channel CRAC currents, the concentration of external divalent cations was lowered below 1 μM to enable Na+ to permeate and serve as the charge carrier. The standard external solution contained 145 mM Na-methane sulfonate, 5 mM NaCl, 10 mM N-hydroxyethyl-ethylenediamine-triacetic acid (HEDTA), 10 mM Hepes, and 10 mM d-glucose. For some experiments, Na+ was replaced with N-methyl-d-glucamine (NMDG+). Ca2+ currents were detected in Ca2+-containing solution: 155 mM Na-methane sulfonate, 5 mM NaCl, 2 mM Ca-methane sulfonate, 10 mM Hepes, and 10 mM d-glucose. Aliquots from Ca2+-saturated HEDTA solution were added to standard solution to yield the desired concentration of extracellular free Ca2+ (1, 10, and 50 μM). The pH of all solutions was 7.2, and osmolarity was 300–305 mOsm. The HEDTA stock solution saturated with Ca2+ was prepared using a pH-metric method (Neher 1988). Extracellular solutions and drugs were applied using a gravity-driven perfusion system with output tip diameter of ∼50 μm placed ∼50 μm from the cell. Five barrels were inserted close to the output tip, and solution exchange controlled manually by valves. A complete local solution exchange was achieved within 2 s. For [Ca2+]i imaging, cells were bathed in normal Ringer solution consisting of 155 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM d-glucose, 5 mM Hepes, pH 7.4. Ca2+-free Ringer solution contained 1 mM EGTA in place of CaCl2 and a total of 3 mM MgCl2. Ca2+ concentration in normal Ringer solution was lowered to 300 μM to reduce the rate of capacitative Ca2+ influx in some measurements. In K+ Ringer, KCl replaced NaCl.
[Ca2+]i Imaging
Ca2+ imaging experiments were performed as described previously (Fanger et al. 2000). In brief, resting or PHA-activated T cells were incubated for 30 min at 21–24°C with 1 μM fura-2 acetoxymethyl ester (Molecular Probes) in medium. Cells were then washed, resuspended, adhered to poly-l-lysine–coated glass coverslips, and mounted in a chamber permitting rapid (∼1 s) solution exchange by a syringe-driven perfusion system. A Videoprobe Ca2+ imaging system (ETM Systems) was used for [Ca2+]i measurement. Tg was used to deplete Ca2+ stores and elicit Ca2+ influx. Intracellular Ca2+ concentrations were calculated using the standard equation [Ca2+]i = K * (R − R min)/(R max − R), where values of K *, R min, and R max, determined by in vitro calibrations in a glass chamber, were adjusted to the anticipated in vivo values by correction factors derived from T cells dialyzed with known Ca2+ concentration standards (Molecular Probes) as described previously (Fanger et al. 2000).
Cytokine Assay
After 2 d of activation under various conditions, brefeldin A was added for 4–12 h. Cells were fixed, permeabilized, and IL-2 content assessed by staining with FITC–conjugated anti–human IL-2 antibody according to Cytofix/Cytoperm Plus kit instructions (BD PharMingen). Stained cells were analyzed using a FACScan™ (Becton Dickinson) and Cell QUEST™ software.
Data Analysis
Whole-cell currents were initially processed with PULSE™ analysis software (HEKA) and then exported to Microcal Origin 5.0 software (Microcal Software, Inc.) or TAC (Bruxton Corp.) for further analysis. Traces recorded before CRAC channel activation were used as a template for leak subtraction. Initial analyses of imaging data were performed using IGOR Pro software (WaveMetrics, Inc.) with home-written macros and extensions. Two population Student's t test and Mann-Whitney U test were employed for statistical analysis. Data were considered statistically significant at P < 0.01. All statistical data are reported as mean ± SEM, and n = the number of cells tested.
Results
CRAC Channels in Resting and Activated Human T Cells
Fig. 1 shows inward Na+ current through single CRAC channels in a resting T cell. After patch rupture (break in) to initiate whole-cell recording, the first indication of channel activity was the appearance of very brief opening events, <1 ms in duration, within an average of 68 ± 12 s after break in (n = 18 cells; Fig. 1 A, top left trace). Brief openings were typically observed for ∼1 min, and then the first open channel stabilized abruptly to a state characterized by long openings, short closed events, and high open probability (P o = 0.95 at –120 mV). The average latency between break in and stable openings of the first channel was 116.4 ± 11.3 s (n = 33). Additional single channels opened sequentially, with an average time constant of activation (τact) of 85 ± 15 s (n = 12). Amplitude histograms during the activation phase showed equally spaced peaks of 4.8 ± 0.03 pA at −120 mV (n = 23), corresponding to a single channel chord conductance of 41 pS (Fig. 1 B). Fig. 1 C shows the time course of whole-cell current illustrating activation of a total of 16 channels in a resting T cell. The number of open channels gradually declined with a run-down time constant (τr) of 940 ± 430 s (n = 12). Single channel activity at the beginning of activation and at the end of run-down exhibited the same high P o values and open and closed channel lifetimes (Fig. 1A and Fig. D), indicating that changes in the number of open channels underlie both activation and run-down processes. As expected for a store-operated channel, IP3 (30 μM in the pipette) significantly decreased the latency between break in and the onset of channel activation (29.8 ± 15.3 s, n = 13), without affecting the average total number of open channels (Fig. 1 E). In cells pretreated with Tg (2 μM in Ringer for 15–45 min), many channels were already open and current was observed immediately after break in, with an additional increase during prolonged recording (Fig. 1 E). These results indicate that single CRAC channels can be detected in human T cells using Na+ as the permeant ion, permitting channel activation, kinetic properties, and functional expression levels to be investigated with single channel resolution. In resting T cells, the number of CRAC channels simultaneously open during whole cell recording ranged from 2 to 50 per cell.
Figure 1.
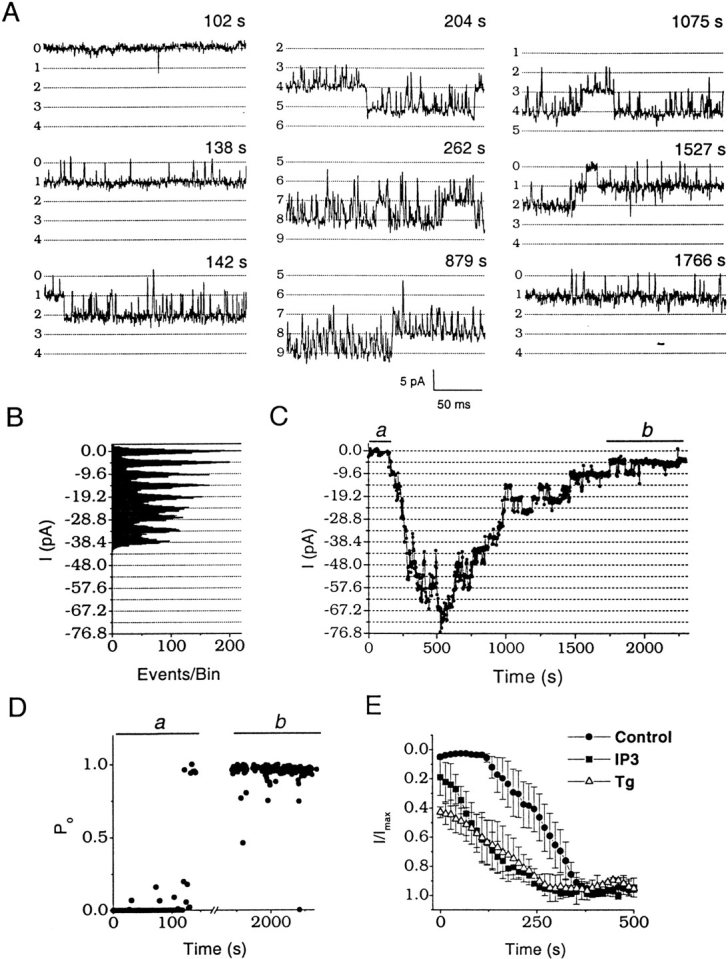
Na+ current through CRAC channels in a resting T cell. All currents recorded in divalent-free solution using a Cs+-aspartate pipette solution containing BAPTA with no Mg2+. (A) Sequence of channel activation displaying currents at −120 mV at the indicated times following break in. Horizontal lines are multiples of 4.8 pA, with the number of open channels shown on the left. Please note baseline shifts in channel numbers for the middle and right columns. (B) All-points amplitude histogram (bin size = 0.2 pA) accumulated during nine consecutive current traces recorded at −120 mV, beginning 135 s after break in. Note that the histogram peaks are equally spaced with intervals of 4.8 pA, corresponding to the sequential and sustained opening of eight channels. (C) Time course of whole-cell current recorded from the same resting T cell. The current amplitude at each time point was measured off-line by averaging 5-ms intervals randomly selected within square-pulse traces. (D) Open probability (P o) obtained from current traces with one channel active at the beginning of current activation (a) and after currents ran down to a single remaining active channel (b). a and b correspond to time intervals marked on C. P o values were measured by integrating current traces containing one active channel. Missing points within the horizontal scale break represent skipped traces with more than one channel active. (E) Activation by passive store dialysis, IP3, and Tg. Average time course of CRAC channel currents from representative resting T cells dialyzed with either normal BAPTA-containing pipette solution (Control, •, n = 4), with 30 μM IP3 added to the normal pipette solution (IP3, ▪, n = 3), or after preincubation with 2 μM Tg for 15–45 min (Tg, ▵, n = 3). All current traces were normalized to maximal current amplitude and then averaged.
CRAC channels in activated T cells opened in a similar sequential manner, with a shorter delay before observing the first channel activity in comparison to resting T cells. Channel opening and stabilization typically began within seconds after break in, followed by rapid activation of several channels. Discrete inward current steps of 4.8 pA at −120 mV were resolved at the beginning of the experiment, similar to those observed in resting T cells (Fig. 2A and Fig. B). CRAC channels in activated T cells also exhibited very high open probabilities (P o = 0.95 at −120 mV), and similar rates of macroscopic current activation (τact = 114 ± 9 s, n = 24; Fig. 2C and Fig. D), followed by slow run-down (τr = 1940 ± 490 s, n = 11). Peak currents typically represented the summed activity of 50–500 CRAC channels, averaging tenfold higher than in resting T cells.
Figure 2.

Na+ currents through CRAC channels in an activated T cell. Same recording conditions as described in the legend to Fig. 1 A. (A) Sequence of channel activation at the indicated times. Horizontal lines are multiples of 4.8 pA, with the number of open channels shown on the left. Again, please note baseline shifts that indicate the number of open channels. (B) Amplitude histogram showing equally spaced peaks corresponding to the first nine channels that opened. (C and D) Time course of CRAC channel activation. C represents the initial time course marked with bar on D on an expanded time scale. Horizontal lines represent equally spaced intervals of 4.8 pA to illustrate the stepwise activation of channels.
Ion Permeation and Divalent Block
Several features of ion permeation and divalent block demonstrate that the single channel activity reported here in human T cells is closely similar to CRAC channels described previously in Jurkat T cells (Lepple-Wienhues and Cahalan 1996; Kerschbaum and Cahalan 1998, Kerschbaum and Cahalan 1999). Upon development of macroscopic Na+ currents representing the activation of many CRAC channels, I–V curves rectified weakly and had a reversal potential of 0 mV (Fig. 3 A). Introduction of physiological levels of external Ca2+ resulted in much smaller macroscopic Ca2+ currents with more pronounced inward rectification and a positive reversal potential typical of I crac. As expected for current through a common channel, Na+ and Ca2+ current amplitudes in different cells were strongly correlated (Fig. 3 B), with a slope determined by linear regression of ∼80, implying a much larger single channel conductance for Na+ traversing the CRAC channel compared with Ca2+ (measured at 2 mM external [Ca2+]). External Ca2+ ions in the micromolar range blocked Na+ current through CRAC channels. In Fig. 3 C, with a single stably open CRAC channel, buffering extracellular Ca2+ to 50 μM produced burst-like (“flickering”) block and unblock events at −120 mV. Ca2+ block reduced the mean open time to <1 ms (n = 3), while closed times did not change significantly. The rapid blocking events reflect Ca2+ entry into the open channel. Fig. 3 D shows that CRAC channels in resting and activated T cells exhibit identical sensitivity to block by Ca2+. The voltage dependence of block was quantitatively assessed through normalization of I–V curves in the presence and absence of external Ca2+ (Fig. 3 E). This procedure revealed that Ca2+ block is relieved at either strongly hyperpolarized or depolarized potentials. Na+ currents inactivated if Mg2+ (3.6 mM) was present internally (Fig. 3 F). Although Na+ permeates readily in the absence of external divalent ions, NMDG+ did not carry measurable inward current (n = 8, not shown). We conclude that the permeation properties and divalent block of CRAC channels in human T cells are indistinguishable from those described previously in Jurkat T cells (Table ).
Figure 3.

Ionic selectivity, rectification, and divalent block of CRAC channels in human T cells. (A) Na+ and Ca2+ currents recorded during a voltage ramp from −120 mV to 50 mV in the absence and presence of 2 mM extracellular Ca2+ in an activated T cell. Note the 10-fold difference in scales for I Na and I Ca. External solutions were changed rapidly between Ca2+-free and Ca2+-containing external solutions. (B) Ratio of Na+ to Ca2+ current. Na+ current amplitudes measured immediately after application of Ca2+ -free solution plotted against Ca2+ current amplitude (2 mM external Ca2+). Each point was obtained from an individual PHA-activated cell (n = 14) from different donors. The straight line is a linear regression fit with a slope factor of 81.4 ± 2.4 and a correlation coefficient of 0.99. (C) Example of single channel currents recorded at −120 mV from a resting T cell. Top trace recorded in Ca2+-free external solution (10 mM HEDTA, no Ca2+ added). Bottom trace recorded after application of 50 μM Ca2+ external solution. (D) External Ca2+ blocks Na+ current through CRAC channels in resting and activated T cells. Currents at different external Ca2+ concentrations were normalized to the current in Ca2+-free external solution. Averaged data from several resting (open symbols) and activated (closed symbols) T cells (n = 4–11 cells), fitted with the equation: Fractional Current = 1 / (1 + [Ca2+]o / K d). Apparent K d values were 4 μM and 20 μM at −90 mV and −120 mV, respectively. (E) Voltage dependence of Ca2+ block. I–V curves recorded in the presence and absence of 10 μM external Ca2+ were divided point by point and fitted with the Boltzmann equation: Fractional Current = 1 / {1 + exp [(V h − V) / k]}, with V h = −7.3 mV and k = 13.3 mV. The steepness factor k is equivalent to movement of a single Ca2+ ion 93% of the distance from the outside across the electric field of the membrane to the blocking site within the channel. (F) Ca2+ and Na+ currents recorded at −120 mV from a cell dialyzed with 3.6 mM Mg2+. External solution containing 2 mM Ca2+ was exchanged with Ca2+-free HEDTA-containing solution as indicated. Note the reversible inactivation of Na+ current.
Table 1.
CRAC Channel Properties in Resting and Activated Human T Lymphocytes and in Jurkat T Cells
| Property | Resting T cells | Activated T cells | Jurkat T cells |
|---|---|---|---|
| Activation by passive store depletion | Yes | Yes | Yes |
| IP3 accelerates activation | Yes | ND | Yes |
| Single channel Na+conductance | 41 pS | 41 pS | 38–40 pS |
| Open probability (−120 mV) | >0.95 | >0.95 | >0.94 |
| Ratio of Na+to Ca2+ current | ND | 81.4 ± 2.4 (2 mM Ca2+) | 27 (20 mM Ca2+) |
| External Ca2+ block (−90 mV) | K d = 4 μM | K d = 4 μM | K d = 5 μM |
| Mg2+-dependent inactivation | Yes | Yes | Yes |
| Permeability to NMDG+ | <0.01 | <0.01 | <0.01 |
Single Channel Kinetics
Resting T cells were favorable for detailed kinetic analysis of single CRAC channels. Single channel recordings during voltage ramps (Fig. 4 A) and continuous recordings at various potentials (Fig. 4 B) suggested that the lifetimes of both open and closed channel states increase upon membrane depolarization, resulting in fewer event transitions. Single channel closed times (τc) were consistently <1 ms at −120 mV in all cells tested, and most cells had open times (τo) averaging 11.2 ± 0.9 ms, (n = 11). Some channels exhibited longer channel open lifetimes (τo ∼40 ms, n = 9), and, less frequently, channels with a flickery gating mode were also observed; here we restrict our analysis to the most commonly observed channel behavior. Fig. 4 C shows histograms of closed and open times for a single channel at two potentials. A single exponential function provides an adequate fit to the lifetime distributions, consistent with a simple two-state channel gating scheme with voltage-dependent rate constants connecting a closed state C and an open state O, C↔O. Membrane depolarization from −120 to −20 mV increased both mean closed and open lifetimes (Fig. 4 D). Exponential functions describe the voltage dependence of both open and closed channel lifetimes. The measured increases in τo and τc predict a weak voltage dependence of P o from the equation P o = τo/(τo+τc). Computed values of P o determined from lifetime measurements are in excellent agreement with direct measurements of P o obtained by integrating single channel traces (Fig. 4 E). Although not strongly voltage-dependent, the gating process of CRAC channels would contribute to the channel's inward rectification. I–V relationships of single channel Na+ current obtained during voltage ramps (Fig. 4 A) or from unitary currents at different potentials (Fig. 4 E) exhibited weak inward rectification, a reversal potential near 0 mV, a chord conductance of 41 pS at −120 mV, and a calculated slope conductance of 55 pS between −120 mV and −80 mV.
Figure 4.

Voltage-dependent properties of single channel currents in resting T cells. (A) Single channel activity during a voltage ramp from −120 to 50 mV. Note the very brief transitions that occur between –120 and –80 mV and longer transitions at more depolarized potentials. (B) Single channel currents recorded at −120, −80, and −40 mV. Again, note that depolarization promotes longer open and closed events. (C) Distributions of open (τo) and closed (τc) times at −120 and −40 mV. For single channel analysis, traces with stable opening of only one channel were selected. The kinetics of transitions between the open and closed levels were idealized by fitting manually controlled cursors to the two levels and setting a discriminator at 50% of the current between levels. Time histograms computed from a minimum of 60 intervals. Histograms of open and closed event durations were fitted by single Gaussian distributions; two or more components did not provide a significantly better fit. Same cell as in B. (D) Voltage dependence of mean open (τo, ○) and mean closed (τc, ▪) times (n = 4). Values of τo and τc for individual cells were calculated by fitting duration histograms with a single Gaussian function, as described in C. Data are fitted with a single exponential function with a steepness factor k = 35 mV for open times and 50 mV for closed times. (E) Single channel current amplitudes (▪) and open probabilities (P o, ○) at varying potentials. Averages from four cells are shown; if not shown, standard error bars are smaller than symbol size. The smooth curve through P o data was obtained from exponential fits of τc and τo (smooth lines on D) according to equation P o = τo/(τo+ τc).
Upregulated CRAC Channel Expression and Capacitative Ca2+ Influx in Activated T Cells
Properties of single CRAC channels were identical in resting and activated T cells, but the number of CRAC channels increased substantially following treatment with PHA to induce T cell activation. We confirmed that this treatment also activated IL-2 production by flow cytometric analysis of fixed and permeabilized T cells (data not shown). To evaluate the number and surface density of CRAC channels, peak currents were normalized to membrane capacitance (C m) in cells selected from resting and PHA-treated T cell populations. Measurements from 15 different donors were combined. Capacitance values were 1.7 ± 0.1 pF in 43 resting T cells and 3.6 ± 0.2 pF in 44 PHA-activated T cells. CRAC channel current density in PHA-treated T cells increased by a factor of four compared with resting T cells (Fig. 5 A). The number of channels per cell increased from 14.9 ± 2.4 (n = 40), in resting T cells, to 136.0 ± 19.2 (n = 37), in activated T cells (Fig. 5 B). CsA (1 μM) substantially inhibited this increase in functional CRAC channel expression, while also inhibiting PHA-stimulated cell enlargement (C m = 2.3 ± 0.3 pF, n = 9), cell proliferation assessed by uptake of tritiated thymidine (90% block), and IL-2 production (90% block). At lower concentration (100 nM), CsA was less effective in blocking IL-2 production and CRAC channel upregulation. Acute application of PHA or CsA during whole-cell recording had no effect on the amplitude of monovalent CRAC current (n = 4, data not shown). These data demonstrate that PHA stimulation to induce T cell activation increases functional expression of CRAC channels. Pretreatment of resting T cells with the phorbol ester PMA (40 nM for 2–4 d) to stimulate PKC-dependent pathways also increased the CRAC channel current density (Fig. 5 A) and the average number of channels per cell (63.6 ± 13.0, n = 12), while causing moderate cell enlargement with C m values averaging 2.8 ± 0.2 pF in the cells selected for recording.
Figure 5.

PHA induces functional upregulation of CRAC channels in human T cells. (A) Average CRAC channel Na+ current densities in resting, PHA-activated, PHA + CsA-treated, and PMA-treated T cells. Resting T cells were maintained in culture for 24–72 h (n = 40). Activated T cells were incubated with PHA for 48–96 h (n = 37). Combined data are presented from cells incubated with PHA + CsA for 75 and 96 h (n = 9). PMA-treated cells were incubated with 40 nM PMA for 48–96 h (n = 12). Between 48 and 96 h, there was no statistically significant difference in CRAC channel expression. Average current densities in resting and PHA + CsA-treated cells are significantly different than those in PHA-activated or PMA-treated T cells (Student's t test, P < 0.001). (B) Distributions of the number of CRAC channels per cell in resting (top) and PHA-activated (bottom) cell populations. Note the difference in horizontal scales. Maximal currents were divided by the amplitude of single channel current at −120 mV (4.8 pA).
To confirm that upregulated expression of CRAC channels produces larger Ca2+ influx upon re-stimulation of intact T cells, we measured the rate of capacitative Ca2+ influx in resting and activated T cells using Tg to deplete intracellular Ca2+ stores. Fig. 6 A shows averaged [Ca2+]i traces upon removal of external Ca2+, application of Tg, and reintroduction of external Ca2+; for these experiments, extracellular [Ca2+] was reduced to 300 μM so that the upstroke of the [Ca2+]i signal could be resolved clearly. [Ca2+]i signaling in PHA-activated T cells was enhanced, relative to resting T cells, in three respects. First, Tg-induced release from intracellular Ca2+ stores in the absence of external Ca2+ was increased, from a release transient peaking at 190 ± 2.7 nM (n = 320) in resting T cells to 375 ± 16.5 nM (n = 163) in activated T cells, suggesting an increased capacity of intracellular Ca2+ stores in activated T cells. Second, upon reintroduction of extracellular Ca2+, the rate of rise in [Ca2+]i was 1.60 ± 0.05 times larger in activated than in resting T cells (Fig. 6A and Fig. C). Third, plateau levels of [Ca2+]i were also significantly higher in activated T cells (775 ± 22 nM for resting T cells, and 1150 ± 30 nM for activated T cells; Fig. 6 A). Expression levels of both voltage-gated and Ca2+-activated K+ channels are known to increase in activated versus resting T cells (Grissmer et al. 1993). To confirm that the observed changes in Ca2+ signaling were caused by upregulation of CRAC channels rather than changes in expression of K+ channels or membrane potential, we repeated these experiments in the presence of K+ Ringer solution (159 mM K+) to null out a possible contribution of the membrane potential to Ca2+ influx. As expected for a depolarized condition, mean influx rates were much lower in K+ Ringer than in normal Ringer (9.24 ± 0.25 nM/s in K+ Ringer and 24.4 ± 0.9 nM/s in normal Ringer for resting cells). Interestingly, the rate of Ca2+ influx in activated cells was still 1.65 ± 0.05 times that observed in resting cells (Fig. 6B and Fig. C). In addition, Ca2+-imaging experiments confirmed that stimulation with PMA alone (40 nM), but not ionomycin alone (200 nM), caused upregulation of CRAC channels. These results are summarized in Fig. 6 C. We conclude that the increased functional expression of CRAC channels results in a significant increase in capacitative Ca2+ influx.
Figure 6.
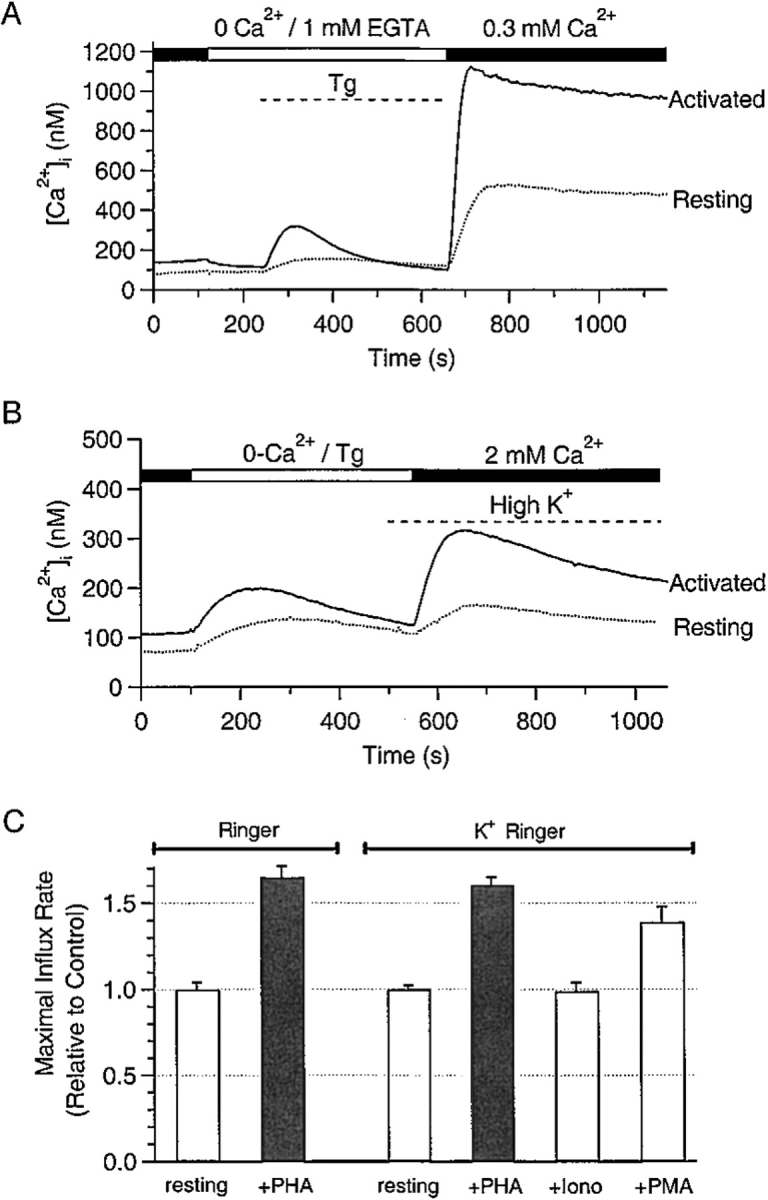
Enhanced capacitative Ca2+ entry in activated human T cells. (A) Average time course of changes in [Ca2+]i upon removal of external Ca2+, and consequent reintroduction of 300 μM external Ca2+ in resting (n = 56, dashed line) and PHA-activated (72 h in PHA; n = 32, solid line) T cells. Tg (1 μM) was applied in Ca2+-free solution as indicated. Results shown are typical of three to four experiments for each condition with two donors. (B) In this experiment, extracellular solutions were changed to K+-Ringer at the end of the store depletion transient, as indicated. Results shown are from 65 activated and 127 resting cells in a typical experiment. (C) The maximal rate of initial Ca2+ influx was calculated for each cell by finding the maximal slope between each pair of data points. To prevent bias from donor to donor variability, influx rates for activated cells were normalized to the mean influx rate for resting cells from the same donor on the same day. Influx rates in activated cells and in cells stimulated with PMA alone (but not ionomycin alone) are significantly different from those of resting cells (P < 0.01, Mann-Whitney U test). Average maximal influx rates in normal Ringer solution from 320 resting and 163 activated T cells (two donors), and in K+ Ringer solution from 452 resting, 382 activated (five donors), 210 ionomycin-treated (two donors), and 72 PMA-treated T cells (one donor) were compiled for this graph.
Discussion
CRAC Channels in Human T Lymphocytes
Store-operated Ca2+ channels play important roles in the regulation of diverse cellular functions in a wide variety of cells. A highly Ca2+-selective type of store-operated channel, the CRAC channel, was originally described in Jurkat T cells, mast cells, and the related RBL cell line (for reviews see Lewis and Cahalan 1995; Parekh and Penner 1997; Lewis 1999). CRAC channels mediate Ca2+ influx following receptor stimulation, resulting in [Ca2+]i signaling that in turn regulates gene transcription and secretion of biologically active molecules in hematopoietic cells (Lewis and Cahalan 1989; Negulescu et al. 1994; Fanger et al. 1995; Zhang and McCloskey 1995). Ca2+ currents through CRAC channels were reported previously in human T cells and shown to be lacking in a patient suffering from a primary immunodeficiency (Partiseti et al. 1994). However, no data have been available on the single channel properties or functional expression levels of CRAC channels, or any other Ca2+ channels, in normal human T lymphocytes. The use of Na+ as the permeant ion permitted CRAC channels to be investigated in Jurkat T cells with single channel resolution (Kerschbaum and Cahalan 1999). In this study, we demonstrate that Ca2+ store depletion in human T cells activates unitary Na+ currents through CRAC channels with biophysical properties similar to those described previously in Jurkat T cells (Table ). Single channels with a conductance of 41 pS were visualized during whole-cell recording, using Na+ as the permeant ion in divalent-free external solution. External Ca2+ in the micromolar range blocked the monovalent current, and at millimolar concentrations exhibited selective permeation with small current magnitudes. The ratio of Na+ to Ca2+ current was constant from cell to cell, indicating a common channel mechanism. In addition, Na+ current exhibited inactivation in the presence of intracellular Mg2+. A bulky cation, NMDG+, was unable to carry current, as shown previously for CRAC channel currents in Jurkat T cells. These results demonstrate that human T cells express CRAC channels with identical biophysical characteristics to those of Jurkat T cells.
Single Channel Kinetics: Activation and Steady-State Gating
The small number of CRAC channels present in resting T cells provided increased observation time for detailed kinetic investigation of single channel properties, compared with Jurkat or activated human T cells. During whole-cell recording, CRAC channels first exhibited very short-duration openings that later stabilized to an open state with intrinsic short-closing events. In resting T cells dialyzed with BAPTA to deplete Ca2+ stores passively, the lag time before the first channel opening was relatively long (∼1 min), suggesting that the latency to the first channel opening event is rate-limited by the number of functional channels, rather than by the rate of BAPTA dialysis or Ca2+ store depletion, which should occur more rapidly in smaller resting T cells. Strong intracellular Ca2+ buffering, addition of IP3 to the pipette, or pretreatment of intact cells with Tg activated the same population of channels by depleting Ca2+ stores, similar to results for Ca2+ current through CRAC channels (Hoth and Penner 1992; Zweifach and Lewis 1993). IP3 significantly reduced the latency to current activation (Fig. 1 E), similar to that described for I crac (Parekh et al. 1997). In addition, CRAC channels opened by Tg pretreatment were detected immediately following break in to initiate whole-cell recording. Thus, Na+ current through CRAC channels exhibits normal activation kinetics upon depletion of Tg-sensitive intracellular Ca2+ stores.
The mechanism of CRAC channel activation upon Ca2+ store depletion remains uncertain. Recent evidence on other store-operated Ca2+ channels favors a mechanism that involves close physical contact between the store membrane and the surface membrane, with activation resulting either from channel delivery by vesicle fusion (Patterson et al. 1999; Yao et al. 1999), or by conformational coupling between IP3 receptors in the store and channels in the surface membrane (Kiselyov et al. 1998; Ma et al. 2000). In principle, a channel delivery mechanism might deliver several channels simultaneously upon vesicle fusion. However, as observed previously in Jurkat T cells (Kerschbaum and Cahalan 1999), CRAC channel current increased in unitary stepwise increments as individual CRAC channels opened. In these experiments, we never observed the simultaneous initial activation of more than one channel. Furthermore, during the period of CRAC channel activation, no consistent changes in C m were observed in resting or activated T cells; on average, C m changed by <5%, or <0.1 pF, a value equivalent to <10 μm2. Clearly, major changes in membrane area are not associated with the activation of CRAC channels. Thus, our data do not provide support for the vesicle fusion mechanism, although such a mechanism involving vesicles containing no more than a single CRAC channel, in combination with small vesicle size or a mechanism for membrane retrieval, cannot be excluded.
After channel stabilization, the mean open time was 10 ms, and the mean closed time was <1 ms at −120 mV, resulting in a very high P o value of >0.95. At more depolarized potentials, both mean open and mean closed times increased, and the open probability declined to ∼0.85 at −40 mV. Are these channel gating events intrinsic to the channel protein, or might they be the result of ionic block? With external Ca2+ in the micromolar range, individual block and unblock events were visualized as a rapid process that significantly reduced mean open times (<1 ms at 50 μM Ca2+ at −120 mV). Maximal block was observed near −40 mV, with relief of block by depolarization indicating a binding site deep within the channel accessed by external Ca2+, and relief of block at hyperpolarized potentials suggesting “punch-through” of bound Ca2+ to the inside induced by the strong electric field within the channel. These results suggest the possibility that trace concentrations of external Ca2+, or another polyvalent cation, could produce channel block at moderate membrane potentials, perhaps even in the presence of a strong Ca2+ chelator like HEDTA, and account for channel gating. However, in this case, we would expect a decrease in mean open time at the potentials where divalent block is maximal. Instead, in the absence of external Ca2+ and with 10 mM HEDTA outside, both open and closed channel lifetimes increased monotonically with depolarization, with P o remaining high over a broad potential range, suggesting that inward rectification is not the result of ionic block. Open and closed channel lifetimes are weakly voltage dependent, but current amplitudes are strongly dependent on voltage because changes in the electrical driving force affect the unitary current amplitude. Thus, voltage is a dominant fast regulatory mechanism controlling the amount of Ca2+ entry through open CRAC channels that exhibit weakly voltage-dependent gating.
CRAC Channel Expression and Ca2+ Influx Rates in Resting and Activated T Cells
Ca2+ influx through CRAC channels underlies [Ca2+]i signaling in Jurkat T cells, and, by extension, is thought to trigger antigen-induced activation of resting human T cells. Whole-cell recordings with single channel resolution enable the number of CRAC channels per cell to be determined in resting and activated cells. Despite their importance for lymphocyte activation, CRAC channels in resting T cells are expressed at surprisingly low copy number, averaging 15 functional CRAC channels per cell. Is this small number of channels sufficient to account for tracer Ca2+ influx measurements and for the rate of rise of [Ca2+]i during activation? To address this question, we calculated the expected influx of Ca2+ under physiological conditions during a stimulus that opens all of the available CRAC channels (Fig. 7). Macroscopic Na+ currents were consistently ∼80-fold larger than Ca2+ currents recorded with 2 mM external [Ca2+], leading to an estimated single channel conductance of 0.5 pS at −120 mV for Ca2+ current through the CRAC channel with physiological levels of external Ca2+. This estimate is based upon the assumption that the number of open channels remains constant during rapid solution exchange. To calculate Ca2+ influx in the range of membrane potentials found normally in intact T cells, we scaled the measured single channel Na+ current at –60 mV by the factor 80 and converted to flux units. In a cell with 15 activated CRAC channels, the calculated Ca2+ influx at a resting membrane potential of −60 mV would be 1.9 amol/s, sufficient to account for previous measurements of 45Ca uptake in resting T cells upon mitogen activation, which ranged from 0.4 to 2.2 amol/s (Metcalfe et al. 1980; Hesketh et al. 1983). Can this amount of Ca2+ influx produce the upstroke of the [Ca2+]i signal? The expected rise in [Ca2+]i due to Ca2+ influx depends upon cell volume and buffer capacity. Cell volume was estimated to be 200 fl (200 × 10−15 liter) from measured values of membrane capacitance. An endogenous Ca2+ buffer was assumed to capture 99% of the Ca2+ entering the cell (Neher and Augustine 1992; Neher 1995). Given these estimates and scaling external [Ca2+] to 0.3 mM for comparison with our measurements, the Ca2+ influx through 15 CRAC channels would produce a rise in [Ca2+]i of 28 nM/s in resting T cells, in close agreement with direct measurements of capacitative Ca2+ influx in resting T cells averaging 24.4 ± 0.9 nM/s. Thus, despite their small number, CRAC channels would carry sufficient Ca2+ into resting T cells to account for measured tracer Ca2+ influx and the rates of rise of [Ca2+]i.
Figure 7.

Channel expression and calculated Ca2+ influx in resting and activated T lymphocytes. The two cells depicted in cartoon form represent typical resting (top) and activated (bottom) T cells. Diameters (d), surface areas (s), and volumes (v) are representative of cells selected for recording. Within each cell, a signal transduction pathway leads from antigen binding to the TCR, activating tyrosine kinases (TK), phosphorylating phospholipase C (PLC), generating IP3, and releasing Ca2+ from the store. Ca2+ influx through CRAC channels is activated by the depletion of Ca2+ from the IP3-sensitive intracellular Ca2+ store via an unknown mechanism. Functional expression levels are represented by the average number of channels per cell (in parentheses), for three channel types: voltage-gated K+ channels encoded by Kv1.3 (DeCoursey et al. 1984; Deutsch et al. 1986), Ca2+-activated K+ channels encoded by IKCa1 together with preassociated calmodulin (CaM) (Grissmer et al. 1993; Fanger et al. 1999; Ghanshani et al. 2000), and CRAC channels (this paper, Fig. 5 B). The calculated Ca2+ influx (ions/sec and amol/s) is based upon the number of CRAC channels per cell, the measured single CRAC channel conductance for Na+, the measured ratio of I Ca/I Na, and an assumed membrane potential of −60 mV. The maximal rate of [Ca2+]i increase (d[Ca2+]i/dt in nM/s), resulting from the Ca2+ influx with external Ca2+ of 2 mM is based upon the estimated cell volume and the buffer capacity.
CRAC channel properties were identical in resting and activated T cells. However, activated T cells expressed substantially more functional channels, averaging 136 CRAC channels per cell. This functional upregulation of CRAC channels in human T lymphocytes paralleled the growth of small lymphocytes to T cell blasts. In our experiments, average membrane capacitance values increased from 1.7 pF in resting T cells to 3.6 pF in activated T cells, indicating an increase in volume to 700 fl. As calculated for resting cells above, the expected maximal rise in [Ca2+]i due to Ca2+ entry via CRAC channels in a typical activated T cell was 73 nM/s, roughly twofold larger than, but still in reasonable agreement with, the average rate of 43.7 ± 1.6 nM/s obtained by direct measurements of [Ca2+]i rise induced by capacitative Ca2+ influx. A higher Ca2+ buffering capacity or increased pump activity in activated T cells may account for the difference between those numbers. Thus, the increase in number of functional channels in activated T cells not only compensates for cell enlargement but also enhances Ca2+ signaling in activated T cells.
Significance of Channel Upregulation for T Cell Function
During T cell activation, numerous transcription factors are activated, resulting in changes in the expression level of many different genes (Lanzavecchia and Sallusto 2000). Some gene products, such as IL-2 and other secreted lymphokines, help to carry out the effector functions of T cells, and others, such as the Ca2+-activated K+ (KCa) channel encoded by IKCa1, help to augment [Ca2+]i signaling and activation responses upon future restimulation. Increased expression of IL-2 is regulated by several transcription factors, but can be inhibited by the immunosuppressive drug CsA at doses that prevent dephosphorylation and nuclear translocation of the transcription factor NF-AT by the phosphatase calcineurin (Rao et al. 1997; Crabtree 1999). In contrast, enhanced expression of KCa channels is transcriptionally regulated by AP-1 and Ikaros-2 sites in the promoter region and is independent of NF-AT, since enhanced expression can be stimulated by PMA alone and is unaffected either by CsA treatment or by deletion of an NF-AT-binding element in the promoter region of the IKCa1 gene (Ghanshani et al. 2000). The increased number of CRAC channels in activated T cells may also result from transcriptional upregulation of the unknown gene or genes that encode CRAC channels. Both CRAC and KCa channel expression increase greatly in T cells stimulated for at least 24 h with PHA or PMA alone, but not with ionomycin alone (Fig. 5 Fig. 6 Fig. 7), suggesting a similar mechanism. In addition, CsA, at a concentration reported to block calcineurin-mediated nuclear translocation of NF-AT (Rao et al. 1997), did not inhibit CRAC channel upregulation. However, PHA-induced IL-2 secretion, T cell proliferation, and upregulation of CRAC channels were inhibited by CsA at a concentration of 1 μM, a level that may have nonspecific effects. Thus, a possible direct role of calcineurin and NF-AT in CRAC channel upregulation requires further investigation. A detailed understanding of the mechanism awaits molecular identification of the CRAC channel.
Two levels of positive feedback involving CRAC and KCa channels augment [Ca2+]i signaling, as suggested by Fig. 7. Before TCR engagement, [Ca2+]i is low (∼100 nM), CRAC and KCa channels are closed, and the resting membrane potential is maintained by voltage-gated Kv1.3 channels. After TCR engagement, [Ca2+]i begins to rise, initially due to IP3-induced release of Ca2+. The depletion of IP3-sensitive Ca2+ stores opens CRAC channels, further elevating [Ca2+]i. As soon as [Ca2+]i reaches 200–300 nM, KCa channels begin to open, hyperpolarizing the membrane and thereby increasing Ca2+ influx through CRAC channels, in turn augmenting the rise in [Ca2+]i and opening the remaining KCa channels. Thus, rapid positive feedback among [Ca2+]i, KCa channels, and CRAC channels contributes to the upstroke of the Ca2+ signal. In addition, slow positive feedback involving changes in gene expression is initiated by the rise in [Ca2+]i, along with other signaling pathways inside the cell. Activated T cells exhibit enhanced Ca2+ signaling in response to either TCR engagement or Tg stimulation (Hess et al. 1993; Verheugen et al. 1997) (Fig. 6). Increased expression of CRAC channels in activated T cells occurs in parallel with a comparable increase in expression of Ca2+-activated K+ channels encoded by IKCa1 (Grissmer et al. 1993; Ghanshani et al. 2000). Relative to resting T cells, Tg-induced Ca2+ influx is augmented in activated T cells by the same amount in normal Ringer and in K+ Ringer (1.65 ± 0.06-fold upregulation in normal Ringer vs. 1.60 ± 0.05-fold upregulation in K+ Ringer). This implies that upregulation of CRAC channels augments Ca2+ signaling, and that parallel KCa channel upregulation is adequate to provide the necessary driving force and to compensate for the increase in cell size. Without accompanying KCa channel upregulation, Ca2+ influx through the increased number of CRAC channels would depolarize the membrane, causing Ca2+ influx to be self-limiting.
Amplification of the Ca2+ signal may be responsible for increased sensitivity to TCR stimulation observed previously in secondary T cell activation (Manger et al. 1985; Byrne et al. 1988). In addition, upregulation of Ca2+ influx through CRAC channels may sustain mitogenesis. The number of CRAC channels in activated T cells (∼140 per cell) is close to that in proliferating Jurkat T cells (100–400 per cell), suggesting a requirement for larger numbers of CRAC channels for clonal expansion. Sustained [Ca2+]i elevation is required for calcineurin-dependent translocation of NF-AT into the nucleus, resulting in activation of NF-AT–dependent transcription of lymphokines, and presumably other immune response genes (Rao et al. 1997; Crabtree 1999). Therefore, functional upregulation of CRAC channels may play a crucial role for continued cell proliferation and more vigorous secondary T cell responses.
Acknowledgments
We thank Dr. Lu Forrest for excellent technical support and Dr. George Chandy for helpful suggestions.
This work was supported by National Institutes of Health grants NS14609 and GM41514 to M.D. Cahalan and the American Heart Association grant 0030275N to A.F. Fomina.
Footnotes
Abbreviations used in this paper: BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid; [Ca2+]i, intracellular calcium concentration; CRAC, Ca2+ release-activated Ca2+; CsA, cyclosporin A; HEDTA, N-hydroxyethyl-ethylenediamine-triacetic acid; KCa, Ca2+-activated K+; NF-AT, nuclear factor of activated T cells; NMDG+, N-methyl d-glucamine; PHA, phytohemagglutinin; TCR, T cell receptor; Tg, thapsigargin.
References
- Byrne J.A., Butler J.L., Cooper M.D. Differential activation requirements for virgin and memory T cells. J. Immunol. 1988;141:3249–3257. [PubMed] [Google Scholar]
- Crabtree G.R. Contingent genetic regulatory events in T lymphocyte activation. Science. 1989;243:355–361. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- Crabtree G.R. Generic signals and specific outcomessignaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611–614. doi: 10.1016/s0092-8674(00)80571-1. [DOI] [PubMed] [Google Scholar]
- Crabtree G.R., Clipstone N.A. Signal transmission between the plasma membrane and nucleus of T lymphocytes. Ann. Rev. Biochem. 1994;63:1045–1083. doi: 10.1146/annurev.bi.63.070194.005145. [DOI] [PubMed] [Google Scholar]
- DeCoursey T.E., Chandy K.G., Gupta S., Cahalan M.D. Voltage-gated K+ channels in human T lymphocytesa role in mitogenesis? Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- Deutsch C., Krause D., Lee S.C. Voltage-gated potassium conductance in human T lymphocytes stimulated with phorbol ester. J. Physiol. (Lond.) 1986;372:405–423. doi: 10.1113/jphysiol.1986.sp016016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch R.E., Lewis R.S. Signaling between intracellular Ca21 stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. J. Gen. Physiol. 1994;103:365–388. doi: 10.1085/jgp.103.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnadieu E., Cefai D., Tan Y.P., Paresys G., Bismuth G. Imaging early steps of human T cell activation by antigen-presenting cells. J. Immunol. 1992;148:2643–2653. [PubMed] [Google Scholar]
- Fanger C.M., Hoth M., Crabtree G.R., Lewis R.S. Characterization of T cell mutants with defects in capacitative calcium entrygenetic evidence for the physiological roles of CRAC channels. J. Cell Biol. 1995;131:655–667. doi: 10.1083/jcb.131.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanger C.M., Ghanshani S., Logsdon N.J., Rauer H., Kalman K., Zhou J., Beckingham K., Chandy K.G., Cahalan M.D., Aiyar J. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1 . J. Biol. Chem. 1999;274:5746–5754. doi: 10.1074/jbc.274.9.5746. [DOI] [PubMed] [Google Scholar]
- Fanger C.M., Neben A.L., Cahalan M.D. Differential Ca2+ influx, KCa channel activity and Ca2+ clearance distinguish Th1 and Th2 lymphocytes. J. Immunol. 2000;164:1153–1160. doi: 10.4049/jimmunol.164.3.1153. [DOI] [PubMed] [Google Scholar]
- Ghanshani S., Wulff H., Miller M.J., Rohm H., Neben A., Gutman G.A., Cahalan M.D., Chandy K.G. Up-regulation of the IKCa1 potassium channel during T-cell activationmolecular mechanism and functional consequences J. Biol. Chem. 10.1074jbc.m0039412002000. [DOI] [PubMed] [Google Scholar]
- Grissmer S., Nguyen A.N., Cahalan M.D. Calcium activated potassium channels in resting and activated human T lymphocytes. J. Gen. Physiol. 1993;102:601–630. doi: 10.1085/jgp.102.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesketh T.R., Smith G.A., Moore J.P., Taylor M.V., Metcalfe J.C. Free cytoplasmic calcium concentration and the mitogenic stimulation of lymphocytes. . J. Biol. Chem. 1983;258:4876–4882. [PubMed] [Google Scholar]
- Hess S.D., Oortgiesen M., Cahalan M.D. Calcium oscillations in human T and natural killer cells depend upon membrane potential and calcium influx. J. Immunol. 1993;150:2620–2633. [PubMed] [Google Scholar]
- Hoth M., Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M., Penner R. Calcium release-activated calcium current in rat mast cells. J. Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschbaum H.H., Cahalan M.D. Monovalent permeability, rectification, and ionic block of store-operated calcium channels in Jurkat T lymphocytes. J. Gen. Physiol. 1998;111:521–537. doi: 10.1085/jgp.111.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschbaum H.H., Cahalan M.D. Single-channel recording of a store-operated Ca2+ channel in Jurkat T lymphocytes. Science. 1999;283:836–839. doi: 10.1126/science.283.5403.836. [DOI] [PubMed] [Google Scholar]
- Kiselyov K., Xu X., Mozhayeva G., Kuo T., Pessah I., Mignery G., Zhu X., Birnbaumer L., Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A., Sallusto F. From synapses to immunological memorythe role of sustained T cell stimulation. Curr. Opin. Immunol. 2000;12:92–98. doi: 10.1016/s0952-7915(99)00056-4. [DOI] [PubMed] [Google Scholar]
- Lepple-Wienhues A., Cahalan M.D. Conductance and permeation of monovalent ions through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophys. J. 1996;71:787–794. doi: 10.1016/S0006-3495(96)79278-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis R.S. Store-operated calcium channels. Adv. Second Messenger Phosphoprotein Res. 1999;33:279–307. doi: 10.1016/s1040-7952(99)80014-7. [DOI] [PubMed] [Google Scholar]
- Lewis R.S., Cahalan M.D. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis R.S., Cahalan M.D. Potassium and calcium channels in lymphocytes. Annu. Rev. Immunol. 1995;13:623–653. doi: 10.1146/annurev.iy.13.040195.003203. [DOI] [PubMed] [Google Scholar]
- Ma H.-T., Patterson R.L., van Rossum D.B., Birnbaumer L., Mikoshiba K., Gill D.L. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Nature. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- Manger B., Weiss A., Weyand C., Goronzy J., Stobo J.D. T cell activationDifferences in the signals required for IL 2 production by nonactivated and activated T cells. J. Immunol. 1985;135:3669–3673. [PubMed] [Google Scholar]
- Metcalfe J.C., Pozzan T., Smith G.A., Hesketh T.R. A calcium hypothesis for the control of cell growth. Biochem. Soc. Symposia. 1980;45:1–26. [PubMed] [Google Scholar]
- Neher E. The influence of intracellular calcium concentration on degranulation of dialysed mast cells from rat peritoneum. J. Physiol. 1988;395:193–214. doi: 10.1113/jphysiol.1988.sp016914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. The use of Fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- Neher E., Augustine G.J. Calcium gradients and buffers in bovine chromaffin cells. J. Physiol. 1992;450:273–304. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negulescu P.A., Krasieva T.B., Khan A., Kerschbaum H.H., Cahalan M.D. Polarity of T cell shape, motility and sensitivity to antigen. Immunity. 1996;4:421–430. doi: 10.1016/s1074-7613(00)80409-4. [DOI] [PubMed] [Google Scholar]
- Negulescu P.A., Shastri N., Cahalan M.D. Intracellular calcium dependence of gene expression in single T lymphocytes. Proc. Natl. Acad. Sci. USA. 1994;91:2873–2877. doi: 10.1073/pnas.91.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A.B., Penner R. Store depletion and calcium influx. Physiol. Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Parekh A.B., Fleig A., Penner R. The store-operated calcium current I(CRAC)nonlinear activation by InsP3 and dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Partiseti M., Le Deist F., Hivroz C., Fischer A., Korn H., Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J. Biol. Chem. 1994;269:32327–32335. [PubMed] [Google Scholar]
- Patterson R.L., van Rossum D.B., Gill D.L. Store-operated Ca2+ entryevidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- Rao A. NF-ATpa transcription factor required for the co-ordinate induction of several cytokine genes. Immunol. Today. 1994;15:274–281. doi: 10.1016/0167-5699(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Rao A., Luo C., Hogan P.G. Transcription factors of the NFAT familyregulation and function. Ann. Rev. of Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Schreiber S.L., Crabtree G.R. The mechanism of action of cyclosporin A and FK506. Immunol. Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- Verheugen J.A.H., Le Deist F., Devignot V., Korn H. Enhancement of calcium signaling and proliferation responses in activated human T lymphocytes. Cell Calcium. 1997;21:1–17. doi: 10.1016/s0143-4160(97)90092-0. [DOI] [PubMed] [Google Scholar]
- Yao Y., Ferrer-Montiel A.V., Montal M., Tsien R.Y. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- Zhang L., McCloskey M.A. Immunoglobulin E receptor-activated calcium conductance in rat mast cells. J. Physiol. 1995;483:59–66. doi: 10.1113/jphysiol.1995.sp020567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A., Lewis R.S. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. USA. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]