

Abstract
To investigate the mechanisms by which adhesions form and disperse in migrating cells, we expressed α5 integrin, α-actinin, and paxillin as green fluorescent protein (GFP) fusions. All localized with their endogenous counterparts and did not perturb migration when expressed at moderate levels. α5-GFP also rescued the adhesive defects in CHO B2 cells, which are α5 integrin deficient. In ruffling cells, α5-GFP and α-actinin–GFP localized prominently at the leading edge in membrane protrusions. Of the three GFP fusion proteins that we examined, paxillin was the first component to appear visibly organized in protrusive regions of the cell. When a new protrusion formed, the paxillin appeared to remodel from older to newer adhesions at the leading edge. α-Actinin subsequently entered adhesions, which translocated toward the cell center, and inhibited paxillin turnover. The new adhesions formed from small foci of α-actinin–GFP and paxillin-GFP, which grew in size. Subsequently, α5 integrin entered the adhesions to form visible complexes, which served to stabilize the adhesions. α5-GFP also resided in endocytic vesicles that emanated from the leading edge of protrusions. Integrin vesicles at the cell rear moved toward the cell body. As cells migrated, α5 vesicles also moved from a perinuclear region to the base of the lamellipodium. The α5 vesicles colocalized with transferrin receptor and FM 4-64 dye. After adhesions broke down in the rear, α5-GFP was found in fibrous structures behind the cell, whereas α-actinin–GFP and paxillin-GFP moved up the lateral edge of retracting cells as organized structures and then dissipated.
Keywords: cell adhesion, endocytosis, cytoskeleton, membrane protrusions, vesicle trafficking
Introduction
Cell migration is an integrated process requiring both adhesion to and detachment from the surrounding extracellular matrix (ECM) (Lauffenburger and Horwitz 1996). At the cell front, adhesive complexes form that link the actin cytoskeleton to the ECM through integrins. These sites of adhesion provide the traction necessary to translocate the cell body forward. At the cell rear, the adhesive complexes must be released for the cell to translocate. Although many components of adhesion complexes have been described, the mechanisms by which they assemble and turn over at the cell front and detach at the rear during cell migration are not well understood.
An emerging model for adhesion formation is that adhesive complexes nucleate around a small cluster of ligand-bound integrins with structural and signaling molecules joining the complex in distinct temporal waves. This model is supported by the finding that distinct classes of focal adhesion proteins colocalize with peptides bound to polystyrene beads containing different integrin-binding reagents, that is, those that mediate ligation, clustering, or both (Miyamoto et al. 1995a,Miyamoto et al. 1995b; Yamada and Miyamoto 1995). It is further proposed that small adhesive complexes then merge to form larger complexes, via a Rho-mediated myosin-based contractility (Burridge et al. 1990; Chrzanowska-Wodnicka and Burridge 1996; Schoenwaelder and Burridge 1999). An alternate model envisions integrins associating with and becoming tethered to preassembled cytoskeletal complexes (DePasquale and Izzard 1987; Izzard 1988).
Compared with the process of formation, less is known about adhesion breakdown. One model proposes that molecular interactions fracture in response to contractile forces at relatively specific sites within the ECM integrin–cytoskeletal linkage and thus weaken or release attachments at the rear of the cell. This is supported by observations of the fates of integrins at the cell rear. These studies suggest several possible cleavage sites, whose probability depends on adhesion strength (Regen and Horwitz 1992; Palecek et al. 1996). Other complementary models stress the importance of biochemical alterations involving molecules, such as proteases or signaling molecules that regulate adhesion turnover by phosphorylation/dephosphorylation. When several kinases and phosphatases are inhibited, adhesions become strong and peripheral, and the tails of cells become elongated (Lawson and Maxfield 1995; Richardson et al. 1997; Schoenwaelder and Burridge 1999). With either of these models, the nature of the adhesive breakdown is unclear. For example, it is not known whether subcomponents of the adhesive complex remain intact or disperse within the cell when adhesions turn over.
The formation of adhesions at the front and their dissolution at the cell rear results in the accumulation in the rear of adhesive material derived from the cell front. Thus, it is likely that efficient mechanisms exist to move material from the rear to the front. Previous studies have demonstrated that integrins are rapidly internalized from the cell surface (Bretscher 1989; Dalton et al. 1995). Further, it has been proposed that in migrating cells integrins are endocytosed at the cell rear when adhesions break down and then recycled to the front of the cell and possibly the leading edge (Bretscher 1984; Lawson and Maxfield 1995; Bretscher and Aguado-Velasco 1998; Pierini et al. 2000). In this manner, integrins and perhaps other adhesive components may be supplied to the front of the cell to form new adhesions. In support of this model, integrins were shown to reside in intracellular vesicles that colocalized with a marker of the endocytic-recycling compartment (Pierini et al. 2000). However, it is possible that other pathways exist to supply integrins to the leading edge of the cell, such as directed flow along cytoskeletal elements. In contrast to the proposed model, robust vesicular shuttling of integrins from the cell rear to the leading edge was not detected in migrating fibroblasts using antibody probes (Regen and Horwitz 1992; Palecek et al. 1998).
Since most of the studies examining adhesion formation and breakdown were performed in quiescent cells, it is unclear how these observations apply to migrating cells. When adhesive dynamics were studied in motile fibroblasts, antibody-labeled integrins were rapidly cleared from the cell front, making formation of new adhesions at the leading edge impossible to examine (Regen and Horwitz 1992). A recent study circumvented this problem by generating a reporter containing GFP fused to the membrane-spanning and cytoplasmic domain of the β1 integrin (Smilenov et al. 1999). In stationary fibroblasts, adhesions labeled with the GFP integrin moved toward the cell center or along the cell periphery, whereas in migrating cells little movement of these structures was detected. Interestingly, this study did not report robust vesicular trafficking using this probe. Since only the integrin was labeled, the fates of other cytoskeletal molecules as the adhesions turn over is not known.
To address the mechanisms of adhesion formation and turnover, we generated GFP-labeled fusion proteins of three adhesion components: α5 integrin, paxillin, and α-actinin. This allowed us to visualize the dynamics of three different adhesion-related molecules either singularly or in pairs during migration. These studies produced several interesting observations including (a) the movement of integrin-containing vesicles from the leading lamella to a perinuclear region and trafficking of vesicles from this region to the base of the lamellipodia, (b) a hierarchical mechanism for the formation of adhesions in which paxillin accumulation is followed by organized α-actinin, which in turn is followed by visibly organized α5 integrin, (c) the turnover of paxillin adhesions but not α-actinin at the base of newly forming protrusions, (d) the translocation of α-actinin–containing adhesions, which is inhibited by the presence of visibly organized integrin, and (e) a severing of the integrin–cytoskeletal linkage and the translocation and dispersal of paxillin and α-actinin–containing cytoskeletal complexes at the cell rear. Taken together, these results point to a hierarchical model for the formation of adhesions and multiple integrin-trafficking pathways and suggest that rear release is mediated by contraction and severing of an integrin proximal connection with the cytoskeleton.
Materials and Methods
Cloning
The eukaryotic expression vectors pEGFP-N3, pECFP-N1, and pEYFP-N1 were obtained from CLONTECH Laboratories, Inc. For α5 and paxillin cDNA (provided by L. Reichardt and C.E. Turner, SUNY Upstate Medical University), we inserted a KpnI restriction site before the stop codon using a mutagenic PCR primer with a noncomplementary KpnI site at its 5′ end. A minimal amount of this 3′ cDNA fragment was religated to the original cDNA using either pCR2.1 (Invitrogen) or pCRScript (Stratagene) as a cloning intermediate. We used the KpnI site and one upstream within the original cDNA to ligate each cDNA into the polylinker 5′ to the start codon of the respective GFP variant. The entire PCR fragment and the junction region between the protein and GFP was sequenced. We obtained a similarly prepared α-actinin–GFP construct in the pEGFP-N1 vector from C. Otey and M. Edlund (University of North Carolina, Chapel Hill, NC). This fusion scheme created a 10–amino acid linker between α5 or paxillin (LQAGPGSIAT) and EGFP, a 13–amino acid linker between α5 or paxillin and enhanced yellow fluorescent protein (EYFP) or enhanced cyan fluorescent protein (ECFP) (AAVPRARDPPVAR), and a 20–amino acid linker between α-actinin and EGFP, ECFP, or EYFP (KLRILQSTVPRARDPPVAT). Paxillin and α-actinin ECFP and EYFP were prepared by cloning the cDNAs into the mammalian expression vector pCDNA3.1/Zeo (Invitrogen). The head and rod domains of α-actinin fused to GFP were provided by C. Otey and M. Edlund.
Cell Culture
CHO K1 and CHO B2 cells were cultured in DME (GIBCO BRL) supplemented with 10% FBS, 4 mM l-glutamine, 1 mM sodium pyruvate, and nonessential amino acids (Sigma-Aldrich). WI38 cells were maintained in high glucose DME supplemented with 10% FBS and 1 mM sodium pyruvate. Transfections were performed with lipofectamine (GIBCO BRL), and the cells were selected with 1 mg/ml G418 (Sigma-Aldrich) or 0.5 mg/ml zeocin, where appropriate. Stably transfected cells were maintained in 0.5 mg/ml G418 and/or 0.25 mg/ml zeocin and sorted for expression levels by flow cytometry.
Immunocytochemistry
Cells were plated for >4 h in HyQ-CCM1 serum-free medium (Hyclone) on coverslips coated with 20 μg/ml fibronectin (Fn), fixed, and stained as described previously (Sastry et al. 1999). Focal adhesion proteins were detected with the VIN-11-5 antivinculin antibody (Sigma-Aldrich), antipaxillin 165 (Transduction Laboratories), or anti–α-actinin A5044 (Sigma-Aldrich). Human α5 was stained with monoclonal antibody 6F4 and endogenous and expressed α5 were detected with polyclonal anti-α5 Ab1949 (Chemicon). CHO endogenous β1 was detected using monoclonal 7E2 (University of Iowa Developmental Studies Hybridoma Bank, Iowa City, IA) and polyclonal 36E3 (a gift from D. DeSimone, University of Virginia, Charlottesville, VA). The transferrin receptor was stained with monoclonal antibody B65.3 (provided by S. Green, University of Virginia, and I. Trowbridge, Salk Institute, La Jolla, CA). Actin fibers were visualized using rhodamine-phalloidin.
Cell Surface Biotinylation and Immunoprecipitation of α5 Integrin
CHO B2 cells transfected with untagged α5 or α5-GFP were washed twice with EBSS, 25 mM Hepes, pH 7.4 (EBS-H), and biotinylated for 60 min at 4°C with 0.5 mg/ml NHS-biotin (Pierce Chemical Co.). After washing the cultures, the cells were extracted with 100 mM N-octylglucoside in 25 mM Hepes, 150 mM NaCl, pH 7.4 (HBS), with protease inhibitors (aprotinin, leupeptin, and E-64) for 30 min at 4°C and centrifuged for 10 min at 14,000 g. α5 integrin was then immunoprecipitated by incubation with 6F4 monoclonal antibody (5 μg) for 12 h at 4°C and protein G agarose (Santa Cruz Biotechnology, Inc.) for 4 h at 4°C. The samples were washed five times with 25 mM N-octylglucoside in HBS and centrifuged for 10 min at 14,000 g. The immunoprecipitates were subjected to SDS-PAGE on 5% slabs, transferred to nitrocellulose, and detected by Western blot analysis.
Microscopy
Tissue culture dishes were modified to facilitate microscopic observation of living cells as described previously (Palecek et al. 1996). The dishes were coated with Fn for 16 h at 4°C and then blocked with 2% BSA. 105 cells were plated in either CCM1 (Hyclone) or DME/F-12 for 1 h at 37°C. During microscopy, cells were maintained at 37°C, pH 7.4, and illuminated with a halogen lamp. To visualize EGFP, an endow GFP filter cube (ex HQ470/40, em HQ525/50, Q495LP dichroic mirror) was used (Chroma). FM 4-64 was visualized using a rhodamine/TRITC cube (ex BP520-550, barrier filter BA580IF, dichroic mirror DM565). Exposure times ranged from 0.05 to 0.20 s. For two-color fluorescence, ECFP (ex D436/10, em D470/30) and EYFP (ex HQ500/20, em HQ535/30) were positioned in “dual” filter wheels (Ludl Electronic Products), and a JP4 bandpass filter cube was used to visualize CFP/YFP, respectively. Using the 40× (NA 0.75; Nikon) and the 60× (NA 1.40; Nikon) objectives, images were acquired from a cooled CCD camera (Photometrics CH250 or Hammatsu OrcaII) attached to an Olympus IX-70 or Nikon TE-300 inverted microscope. Image acquisition was controlled using the Inovision ISee software program interfaced to a Ludl modular automation controller (Inovision).
Analysis
For migration experiments, cell paths were tracked using the nanotrack tool in ISee, which records the x and y pixel coordinates of the approximate centurion of the somitic cell cortex. The average speed for each cell was determined by computing the average net centroid translation divided by the time interval at each 5- or 10-min time point. This protocol was adapted for calculating rates of movement of paxillin- or α-actinin–containing adhesion complexes.
Online Supplemental Material
Time-lapse images are included as online videos, which further depict Fig. 2 (Videos 1 and 2), 3 (Videos 3 and 4), 5 (Video 5), 6 (Video 6), and 10 (Videos 8–10). Video 7 shows α-actinin adhesions moving inward toward the cell center. Additional time-lapse images for Fig. 3, showing vesicles emanating from membrane protrusions (Video 2) and vesicles moving to and from the base of the lamellipodia (Video 11) are included. The time intervals for each video are as follows: Videos 1, 2, 6, and 10, 5 s; Videos 3, 5, and 8, 10 s; Video 9, 15 s; Video 4, 30 s; Videos 7 and 11, 60s. Supplemental Fig. 1, Fig. 2, and Fig. 3 are also provided. All videos and supplemental figures are available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1.
Figure 2.

α5 integrin and α-actinin localize to membrane protrusions. GFP fluorescence localizes prominently to membrane protrusions in (a) α-actinin–GFP– and (b) α5-GFP–transfected CHO cells. In CHO (c and d) and WI38 cells (e and f), α5-YFP (c and e) and α-actinin–CFP (d and f) colocalized in membrane protrusions along the cell edge (arrows). Videos 1 and 2 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1. Bar, 10 μm.
Figure 3.

α5 integrin resides in vesicle-like structures. (a) In α5-GFP–expressing CHO B2 cells, integrin vesicles depart from membrane protrusions and move toward the cell center. (b) In WI38 cells expressing α5-GFP, vesicles containing integrin moved from a perinuclear region to the base of the lamellipodia. The arrows indicate the vesicles whose paths were tracked in the far right panels. Videos 2, 3, 4, and 11 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1. Bar, 10 μm.
Figure 1.

GFP fused to α5 integrin, paxillin, and α-actinin mimic unlabeled proteins. (a) α5 integrin was immunoprecipitated from biotinylated CHO B2 cells expressing either α5-GFP or untagged α5. The immunoprecipitate was analyzed by SDS-PAGE and stained for biotin (left lanes) or with a polyclonal anti-α5 antibody (right lanes). The α5-GFP band is ∼30-kD larger than that of untagged α5, indicating that GFP was not cleaved. (b) Cell surface levels of α5-GFP integrin were assayed by flow cytometry in transfected CHO B2 cells using monoclonal antibody 6F4. Endogenous (hamster) α5 integrin was assessed in CHO K1 cells with the PB1 monoclonal antibody using saturating concentrations of the antibodies. (c) α5-GFP–, (d) paxillin-GFP–, and (e) α-actinin–GFP–expressing CHO K1 cells were plated on 20 μg/ml Fn for 18 h before fixation and antibody staining. All three fusion protein colocalized with vinculin. Bar, 20 μm.
Results
GFP Fused to α5 Integrin, Paxillin, and α-Actinin Mimic Unlabeled Proteins
To study the formation and breakdown of adhesive structures, we prepared expression constructs that encode α5 integrin, paxillin, and α-actinin as GFP fusion proteins. Previous studies have examined the properties of cells expressing GFP fusions with paxillin and α-actinin; however, α5-GFP has not been characterized (Dabiri et al. 1997; Kiosses et al. 1999; Salgia et al. 1999; Katz et al. 2000; Knight et al. 2000). To demonstrate that GFP was not cleaved in our fusion constructs, cell surface proteins on untagged α5- and α5-GFP–transfected CHO B2 cells were biotinylated, extracted, and immunoprecipitated with the α5 antibody 6F4. Two major bands were biotinylated in each sample, and the larger of these surface-expressed proteins cross-reacted with a polyclonal α5 antibody (Fig. 1 a). The α5-GFP band was ∼30-kD larger than the band precipitated from CHO B2 cells transfected with untagged α5, indicating that GFP was not cleaved (Fig. 1 a). The smaller biotinylated band was ∼98 kD, which is the size expected for coprecipitated β1 integrin (Fig. 1 a). Furthermore, cleaved GFP was not observed in total protein extracts from α5-GFP–expressing CHO B2 and CHO K1 cells (data not shown). Similarly, cleaved GFP was not detected in cells expressing moderate (<2× over expression) levels of α-actinin and paxillin; these cells were subsequently used in our experiments.
We compared cell surface levels of α5 integrin in our transfected CHO B2 and CHO K1 cells by FACS® analysis. As shown in Fig. 1 b, using saturating concentrations of the antibodies, the α5 integrin expression level in the transfected CHO B2 cells was comparable to the level of endogenous α5 integrin in the CHO K1 cells. Thus, these results indicate that α5 integrin is not significantly overexpressed in the CHO B2 cells when compared with the expression of endogenous α5 integrin in CHO K1 cells.
To determine whether the α5-GFP fusion retained integrin function, we performed migration assays and tested its ability to rescue cell spreading of CHO B2 cells, a cell line isolated for its low expression levels of α5 integrin. These cells do not spread on Fn-coated substrates unless α5 is expressed ectopically (Schreiner et al. 1989; Zhang et al. 1993), making this system useful for studies of α5 function (Cao et al. 1998). The GFP fusion had no detectable effect on migration, since cells expressing GFP-tagged α5 and GFP-tagged paxillin and α-actinin migrate indistinguishably from the cells expressing similar levels of the untagged proteins (Table ). Expression of α5-GFP restored the ability of CHO B2 cells to spread similarly to cells expressing untagged α5 integrin (Table ). To confirm that the ectopic expression of α5 integrin rescues CHO B2 spreading on Fn, we used antibodies that perturb the integrin–Fn interaction. This rescue was inhibited by 16G3 Mab (a gift from K. Yamada, National Institutes of Health, Bethesda, MD), which recognizes the RGD sequence in Fn and disrupts the α5–Fn interaction (Table ). The α5 integrin function-blocking antibodies, VD1 and VD10 (provided by R. Isberg, Tufts University, Boston, MA), inhibited spreading of α5-GFP–expressing CHO B2 cells on Fn by 99 ± 2% and 97 ± 3%, respectively. The nonfunction perturbing α5 antibody, 6F4, had no effect on cell spreading (Table ). Thus, the spreading of α5-GFP–transfected CHO B2 cells is mediated by a direct interaction between α5 and Fn.
Table 1.
Migration Rates of CHO Cells on Fn
| 3 μg/ml Fn | 5 μg/ml Fn | |
|---|---|---|
| (μm/h) | ||
| CHO B2 GFP | N/A | N/A |
| CHO B2 untagged α5 | 36 ± 10 | 27 ± 9 |
| CHO B2 α5-GFP | 32 ± 12 | 30 ± 13 |
| CHO K1 GFP | 35 ± 11 | 25 ± 11 |
| CHO K1 α-actinin–GFP | 34 ± 11 | 29 ± 8 |
| CHO K1 paxillin-GFP | 30 ± 11 | 23 ± 7 |
Table 2.
Percentage of CHO Cells Spread on 5 μg/ml Fn
| No antibody | 16G3 | 6F4 | |
|---|---|---|---|
| (%) after 30 min | |||
| CHO B2 GFP | 3 | 0 | 2 |
| CHO B2 untagged α5 | 61 | 2 | 70 |
| CHO B2 α5-GFP | 72 | 2 | 72 |
| CHO K1 | 99 | 1 | 93 |
Each fusion protein reproduced the localization patterns of its untagged counterpart when expressed in CHO B2, CHO K1, and NIH-3T3 cells under conditions that promote formation of prominent focal adhesions, such as plating on Fn concentrations >5 μg/ml or longer than 6 h. When CHO K1 cells expressing α5-GFP, α-actinin–GFP, or paxillin-GFP were allowed to adhere under focal adhesion–promoting conditions, all three fusion proteins colocalized with vinculin in focal adhesions with α-actinin also localizing along fibrous structures (Fig. 1, c–e). This confirms previous studies using paxillin- and α-actinin–GFP (Dabiri et al. 1997; Kiosses et al. 1999; Zamir et al. 1999, Zamir et al. 2000; Katz et al. 2000; Knight et al. 2000; Edlund et al. 2001).
We then compared the kinetics of adhesive assembly of untagged α5- and α5-GFP–expressing cells by allowing cells to adhere for 0.5, 1.5, and 18 h in CCM1 on substrates coated with 5 μg/ml Fn. Cells were stained for endogenous and exogenous α5 and endogenous β1, F-actin, vinculin, paxillin, and α-actinin. Untransfected CHO B2 cells did not adhere to the coverslips even after 18 h of plating, demonstrating that α5 expression is necessary for these cells to adhere to Fn. In CHO B2 cells transfected with either α5-GFP or untagged α5, the integrins, actin, and focal adhesion proteins were visible at the periphery of cells within 30 min of plating (data not shown; Fig. S1, a–c and g–i, available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). When cells were plated for 1.5 h, an increase in organized adhesions was observed (Fig. S1, d–f, available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). However, at both time points the intensity of staining suggested that the integrins were not as highly organized as vinculin. Importantly, the time course of organization was similar regardless of ectopic expression of the different adhesion molecules in all of the cell types studied. Both untagged α5- and α5-GFP–expressing CHO B2 cells organized more slowly than the parental CHO K1 cells, probably reflecting a clonal difference. Integrin α5-GFP–expressing CHO B2 and CHO K1 cells viewed live after plating on Fn concentrations >5 μg/ml or for longer than 6 h exhibited clear focal adhesion–like structures. At lower Fn concentrations or at shorter time points, clearly discernible α5 organization was rarely seen.
Since integrin organization in CHO cells was observed only at longer time points after plating and on higher concentrations of Fn, we complemented our CHO cell studies by examining the adhesive organization in WI38 cells. Organized adhesions as indicated by vinculin and α5 integrin staining were apparent 60 min after plating on 2 μg/ml Fn (Fig. S2, a and b, available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). As with the CHO cells, the intensity of vinculin staining was much greater than that observed for α5 integrin. The GFP fusion had no effect on α5 integrin localization in the WI38 cells since the localization pattern of α5-GFP and endogenous α5 integrin was similar (Fig. S2, a–c, available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1).
α5-GFP and α-Actinin–GFP Localize Prominently at the Leading Edge
To obtain insight into the mechanisms by which adhesions form and stabilize at the cell front, we investigated the dynamics of the GFP probes in protrusive regions under migration-promoting conditions: Fn concentrations <5 μg/ml plated for <2 h. CHO K1 and CHO B2 cells were plated in CCM1 on 1–5 μg/ml Fn for 1–2 h and then observed in fluorescence every 5 or 30 s for 2–10 min. Under these conditions, both cell types are motile and show prominent protrusive activity. α-Actinin–GFP and α5-GFP localized prominently along the cell border in membrane protrusions (Fig. 2, a and b; Videos 1 and 2 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). This localization was observed in ∼70% of the cells viewed and in >90% of the cells that showed protrusive activity. In both CHO and WI38 cells, α-actinin–CFP and α5-YFP colocalized to membrane protrusions along the cell edge (Fig. 2, c–f). α-Actinin–GFP also localized along fibrous-like structures (Fig. 2 a).
α5 Integrin Resides in Vesicles
Previous reports using fixed cells suggest that integrin-containing vesicles are present at the cell rear and gather in a perinuclear region in migrating cells (Regen and Horwitz 1992; Lawson and Maxfield 1995; Palecek et al. 1996; Bretscher and Aguado-Velasco 1998; Pierini et al. 2000). In contrast, our observations in CHO cells revealed prominent large and small α5-GFP vesicle-like structures that emanated from the leading edge in protrusions (Fig. 3 a; Video 3 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). These structures colocalized with the fluorescent lipophilic dye, FM 4-64, indicating that they were membrane-enclosed endocytic vesicles. The α5-containing vesicles moved centripetally toward the cell center and concentrated in a perinuclear region that colocalized with FM 4-64 and the transferrin receptor. The preponderance of GFP-labeled vesicles in CHO cells originated from lamellipodial regions and moved centripetally; this was also observed in WI38 cells in regions where membrane ruffling was apparent. However, in migrating WI38 cells in which minimal membrane ruffling was observed α5-containing vesicles moved from the perinuclear region to the base of the lamellipodium but not into it (Fig. 3 b; Video 4 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). α5 vesicles were also observed in the rear of the cells. These vesicles moved quickly, often along the lateral edge, toward the perinuclear region where we could no longer track them due to the volume of the cell in that region and the large number of vesicular structures that accumulate there. We did not observe the α5-containing vesicles entering the lamellipodium. Interestingly, we observed substantially more vesicles in cells either expressing higher α5 integrin levels or plated on lower substrate concentrations; this coincides with increased membrane protrusive activity. Membrane-bound palmitoylated GFP also emanated from protrusions and moved inward toward the cell body, suggesting membrane internalization.
The observation that integrin-containing vesicles can emanate from the leading lamella prompted us to ask whether the inclusion of α5-GFP in vesicles was caused by fusion of α5 with GFP. Untagged α5-expressing CHO B2 cells were stained for α5 or endogenous β1 and costained for endogenous transferrin receptor. Using deconvolution (not shown) and confocal microscopy, α5 (Fig. 4) and β1 (not shown) were observed to colocalize with the transferrin receptor. After a pulse of FM 4-64, a similar number of FM 4-64–containing vesicles was observed in cells expressing either untagged α5 or α5-GFP. Thus, the vesicular and endosomal localization of α5-GFP is a property of endogenous integrins.
Figure 4.

α5 integrin colocalizes with the transferrin receptor. CHO B2 cells transfected with untagged α5 were fixed and coimmunostained for endogenous transferrin receptor (false-colored red) and α5 integrin (false-colored green). As seen in the overlay, the integrin colocalized with transferrin receptor. Bar, 5 μm.
Dynamics of Adhesive Components during Formation and Stabilization of Adhesions
The leading edge of a protrusion is a site where adhesions form and stabilize. Using either GFP- or CFP- and YFP-tagged α5, α-actinin, and paxillin-transfected CHO B2 or CHO K1 cells, we observed the differing dynamics of these molecules. Of the three fusion proteins examined, paxillin appeared first. Paxillin was observed initially in a wave of diffuse fluorescence and then localized in small clusters (∼1 μm) near the leading edge of the lamellipodium (Fig. 5 a). These small clusters rapidly reached maximum intensity in ∼300 s. Once formed, individual foci did not join together to make larger adhesive complexes.
Figure 5.

Paxillin localizes in clusters near the leading edge of the lamellipodium and turns over as new adhesions form. (a) The intensity of paxillin adhesions at the base of new protrusions diminished (thin arrows) and eventually disappeared as adhesions formed in newly protruded region of the lamellipodium (compare t = 600 and t = 0, thick arrows). (b) Plots of the relative intensity of paxillin (from panel a) in the original adhesions (1–3, indicated with thin arrows) and the newly forming adhesions (1′ and 2′) are shown. CHO K1 cells expressing paxillin-GFP were plated on 2 μg/ml Fn. The cells were then fixed and viewed in epifluorescence (c) or by total internal reflection microscopy (d). Note that the paxillin clusters were visible by total internal reflection microscopy, demonstrating their proximity to the substrate. Video 5 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1. Bars, 10 μm.
Paxillin-containing adhesions at the base of new protrusions were highly dynamic and tended to turn over. We viewed numerous rounds of adhesion, protrusion, and reformation in paxillin-GFP–transfected cells. In nearly every observation, the intensity of the paxillin clusters at the base of the new protrusion diminished and often disappeared as adhesions formed in a newly protruded region of the lamellipodium (Fig. 5 a; Video 5 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). The rates at which the intensity decreased paralleled an increase of intensity in new adhesions (Fig. 5 b). Interestingly, these paxillin clusters were visible by total internal reflection microscopy, demonstrating their proximity to the substrate (Fig. 5 d).
Organized α-actinin was either not apparent or very weak in the highly dynamic paxillin adhesions. This may be due to differential turnover of α-actinin and paxillin in the adhesions or the lack of visibly organized α-actinin in the newly forming paxillin adhesions. To distinguish between these, CHO K1 cells were transfected with α-actinin–CFP and paxillin-YFP. Clearly organized α-actinin was not detected or was very weak in the paxillin adhesions that turned over (Fig. 6, a–c; Fig. S3 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). Furthermore, the paxillin adhesions that formed at the lamellipodial tip as old adhesions broke down also lacked organized α-actinin (Fig. 6, a–c).
Figure 6.

α-Actinin localizes in small foci at the edge of the lamellipodium after protrusive activity subsides. CHO K1 cells were transfected with α-actinin–CFP (a; false color red in panel c) and paxillin-YFP (b; false color green in panel c). (c) As shown in the overlay, α-actinin was observed in the ruffles but not in the paxillin adhesions near the leading edge of the protrusion. (d) After the bulk of the α-actinin departs from a protrusion, small foci are left behind (t = 60 s at arrows), which grow larger and from which α-actinin–containing filaments extend toward the cell body. Video 6 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1. Bars, 10 μm.
However, when protrusive activity ceased, α-actinin–GFP began to localize in small ∼0.5-μm foci at the edge of the former lamellipodium. These structures developed and persisted as the protrusive activity, as seen in edge-enriched localization, dissipated (Fig. 6 d; Video 6 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). The individual foci did not join together to make larger adhesive complexes but often grew gradually in size and intensity and extended filamentous structures toward the cell body. When visibly organized α-actinin entered the adhesions, the individual clusters tended to move centripetally from their original location, and further intensification was observed. Unlike the highly dynamic paxillin adhesions, α-actinin–containing adhesions did not turn over but instead tended to move inward toward the cell body. These results suggest paxillin dynamics differed from that of α-actinin and that some adhesive components at the lamellipodial base are turned over as adhesions form at the leading edge.
In CHO cells, we were unable to detect visibly organized α5 integrin colocalizing with α-actinin in newly forming adhesions (Fig. 7 a). It is unlikely that another integrin is involved, since clearly organized αv and β1 integrins were also not observed in the new adhesions. In addition, αvβ3 was not detected in the CHO cells, which is consistent with previous reports that these cells express little β3 or other integrins (Ylanne et al. 1993; Takagi et al. 1997). A more likely possibility is that the α5 integrin, although present in new adhesions, is not highly concentrated and thus is not readily observed. Comparing the intensity of integrin staining with that of other adhesion molecules supports this hypothesis, since even in cells with highly organized adhesions the integrin staining is considerably less intense than that of paxillin or α-actinin.
Figure 7.

Organized α5 integrin was not observed in the forming α-actinin adhesions, but when present the adhesions did not translocate. (a) CHO K1 cells were transfected with α5 integrin-YFP (false color red) and α-actinin–CFP (false color green) and allowed to adhere for 1 h on 5 μg/ml Fn. α5 integrin was not visible in the α-actinin adhesions near the cell edge. WI38 cells expressing α5 integrin-YFP (b, d, f, and h) and α-actinin–CFP (c, e, g, and i) were plated on 1 μg/ml Fn for 1 h. A line is drawn to indicate the relative positions of the α-actinin adhesions. The α-actinin adhesions lacking visibly organized α5 integrin moved inward toward the cell center (c and e), whereas the adhesions containing organized α5 integrin did not move (g and i). Video 7 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1. Bars, 15 μm.
Since endogenous α5 was more visibly organized in the WI38 cells than in the CHO cells, we examined the dynamics of the fusion proteins as these cell migrated. Paxillin-GFP was observed near the leading edge and turned over in regions where new protrusions formed, which is similar to our observations in the CHO cells. As membrane ruffling dissipated, organized α-actinin and subsequently α5 integrin were observed in the adhesions. Unlike the highly dynamic paxillin-containing adhesions, the α5-containing adhesions were stable and did not turn over. In WI38 cells expressing α5-YFP and α-actinin–CFP, some α-actinin adhesions slide inward from the cell perimeter (Fig. 7, b–e; Video 7 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1); however, those that also contained α5 integrin remained stationary (Fig. 7, f–i). Thus, in these cells highly organized α5 was not visible in all adhesions, and when it was the adhesions did not translocate.
Since clearly organized integrin was not observed in the forming adhesions, we examined the role of α5 ligation in the formation of paxillin clusters. When paxillin-GFP–expressing CHO K1 cells were plated on poly-l-lysine, paxillin-GFP–containing adhesions were not detected; however, paxillin clusters were observed when the cells were plated on 2 μg/ml Fn (Fig. 8, a and b). Paxillin clusters were no longer apparent after addition of the function-blocking antibody, VD1, which disrupts integrin binding to Fn (Fig. 8e and Fig. f). The nonfunction-blocking antibody, 6F4, had no effect on paxillin adhesions (Fig. 8c and Fig. d). These results suggest that the integrin–Fn interaction is necessary for paxillin clustering.
Figure 8.
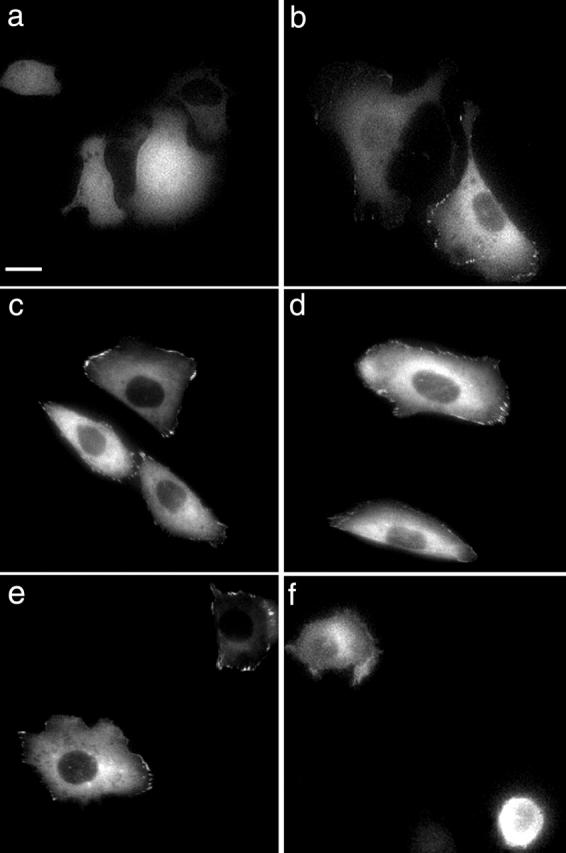
α5 integrin ligation is necessary for the formation of paxillin adhesions. Paxillin-GFP–containing adhesions were not detected when paxillin-GFP–expressing CHO K1 cells were plated on poly-l-lysine (a); however, they were observed when the cells were plated on 2 μg/ml Fn (b). CHO K1 cells expressing paxillin-GFP were plated on 2 μg/ml Fn for 1 h, and then images were obtained (c and e). The cells were then treated with the nonfunction–blocking antibody, 6F4 (d), or the function-blocking antibody, VD1 (f). Images were taken 30 min after treatment. Bar, 10 μm.
α5-GFP But Not Paxillin-GFP or α-Actinin–GFP Remains on the Substrate at the Cell Rear
As adhesions break down, a fraction of the integrins can be left behind the cell in tracks (Regen and Horwitz 1992; Palecek et al. 1996, Palecek et al. 1998); however, the fate of the cytoskeletal proteins is not known. Thus, we examined the fate of adhesive components at the cell rear as adhesions broke down. When α5-GFP integrin-expressing CHO B2 and CHO K1 cells were plated on 2.5–5 μg/ml Fn, we observed fluorescent fibers extending behind retracting cell regions (Fig. 9 a). A network of “retraction” fibers greater than a cell length was also observed when α5-GFP–expressing WI38 cells were plated on 0.5–2 μg/ml Fn. In some cases, the fibers were left behind as the cells migrated. The length of these fibers was variable. These fibrous structures were membranous, since similar structures were seen behind cells transfected with a palmitoylated membrane-bound GFP (Moriyoshi et al. 1996), but FM 4-64 was not observed in fibers. When CHO K1 cells expressing similar levels of soluble GFP were plated under the same conditions, “retraction” fibers were not observed, indicating that the cytoplasmic volume of these structures is small. Paxillin-CFP and α-actinin–CFP do not localize with α5-YFP in these fibrous networks (Fig. 9, b–e). This suggests that the adhesion is breaking down closer to the integrin than to the actin.
Figure 9.
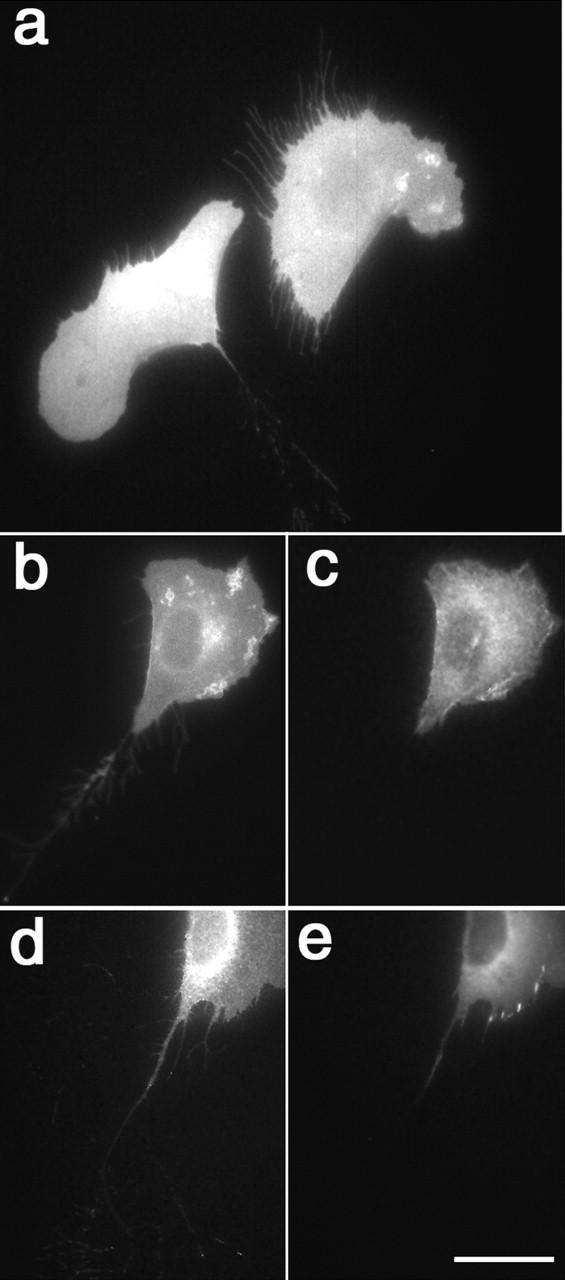
α5 integrin remains in fibrous structures left behind migrating cells. (a) When α5-GFP–expressing CHO cells were plated on Fn, fluorescent fibers containing the integrin remained behind the migrating cells on the substratum. In cells cotransfected with α5-YFP (b and d) and α-actinin-CFP (c) or paxillin-CFP (e), α5 integrin was found in fibers, whereas neither α-actinin nor paxillin was observed in these structures. Bar, 25 μm.
Intracellular Adhesive Structures Move along the Edges of Retracting Tails
Previous studies have also reported the movement of aggregates of integrins along the cell edge (Regen and Horwitz 1992; Palecek et al. 1996). One interpretation is that the weakening or release of integrin–ligand interactions can be an early event in the process of adhesion breakdown. Thus, one expects to see organized cytoplasmic adhesion complexes remaining. We observed clusters of paxillin-GFP and α-actinin–GFP moving along the lateral edge of the cell (Fig. 10, a and b; Videos 8 and 9 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). These clusters averaged 1–2 μm in length and moved at ∼30 μm/h from the cell rear toward the cell front, although there was variation in speed between individual clusters. The clusters remained intact for over 30 min, indicating their stability (Fig. 10 c; Video 10 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1). Small clusters would often appear to coalesce and move up the tail as it retracted behind them. The clusters would gradually decrease in intensity and eventually disperse. Paxillin-YFP and α-actinin–CFP colocalized in sliding adhesive structures on the ends of α-actinin–containing fibers. The connection between stress fibers and paxillin and α-actinin complexes suggests that they remained connected to actin, which likely mediated their movement. Since visibly organized α5 adhesions were seldom seen at these early times in CHO cells, we also examined the breakdown of adhesive structures in WI38 cells. When WI38 cells were transfected with α-actinin–CFP and α5-YFP, α-actinin–containing adhesive structures slide inward toward the cell center as the rear of the cell retracted; however, the α5 integrin remained associated with the substrate.
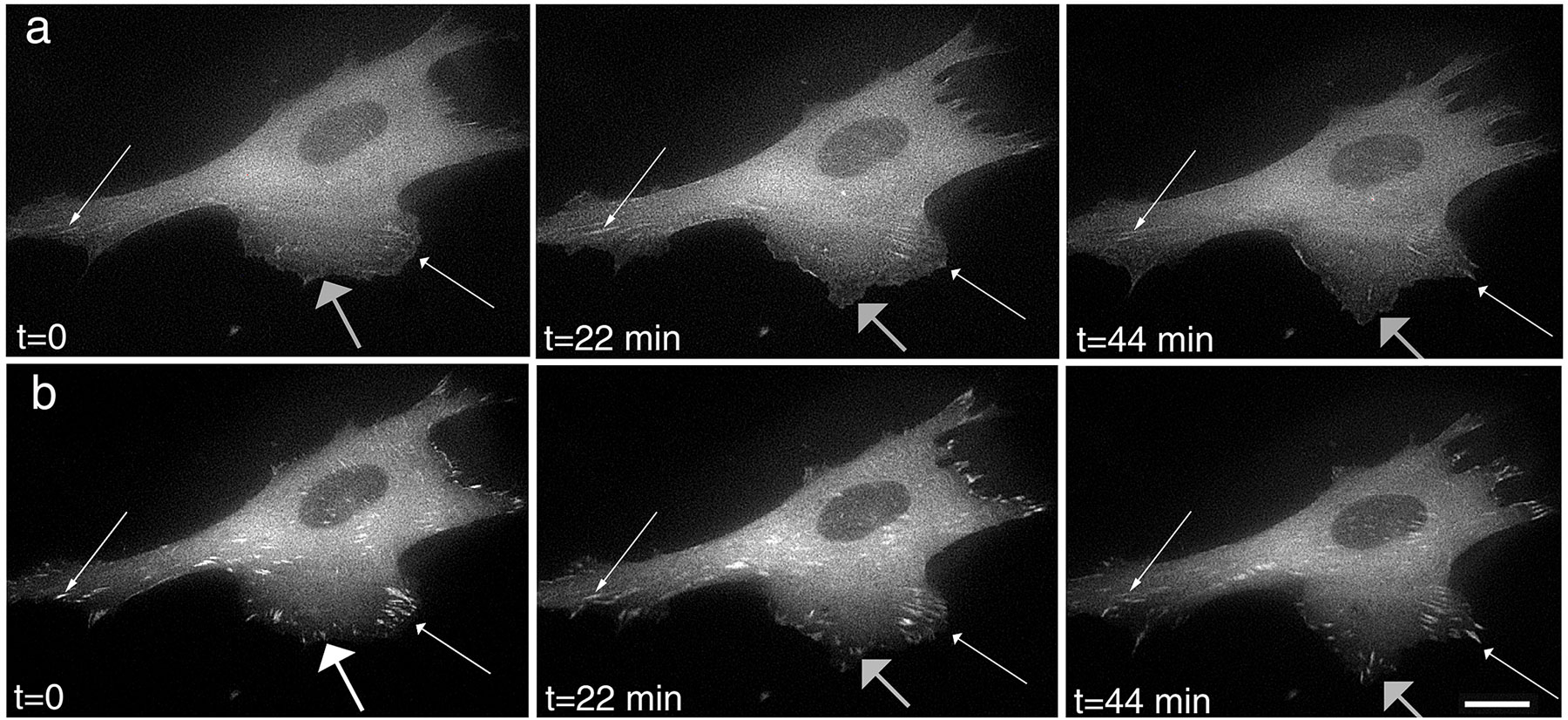
Figure 10.

Clusters of paxillin and α-actinin translocate along the lateral edge of the cell in areas of cell retraction. (a) In retracting regions of the cell, paxillin-GFP clusters move centripetally along the edge of the cell (thin arrows). Strong lateral clusters (thick arrow) strengthen as smaller adhesive structures incorporate into them but move slower than the smaller clusters (thin arrows). (b) α-Actinin–GFP also resides in clusters along the cell edge that move centripetally (thin arrows). The smaller clusters move faster than the larger clusters (thick arrow). (c) Paxillin-GFP clusters remain intact for over 30 min as they translocate along the cell edge. Compare the original location of the cluster marked with a thin arrow to the new location marked with a thick arrow. Videos 8–10 available at http://www.jcb.org/cgi/content/full/153/7/1427/DC1. Bar: (a) 5 μm; (b) 4.5 μm; (c) 8.7 μm.
Discussion
Rapid cell migration requires the efficient regulated formation and breakdown of adhesions and cycling of components from the rear to the front. Several models have been proposed for adhesion formation, but less is known about the breakdown of adhesions. One set of studies suggest a hierarchical model for adhesive assembly (Miyamoto et al. 1995a,Miyamoto et al. 1995b; Yamada and Miyamoto 1995), whereas other studies suggest that large preformed cytoskeletal complexes are stabilized by their association with integrins bound to the substratum (DePasquale and Izzard 1987; Izzard 1988). Adhesion breakdown may occur by a reversal of the mechanisms for assembly, specific enzymatic modifications, or mechanical stresses that lead to the fracture of specific interactions in the cytoskeletal–integrin linkage. Mechanisms have been proposed for cycling components that accumulate at the rear to the cell front, including directed vesicle trafficking, movement of adhesive complexes, and directed molecular movements.
In this study, we evaluated the relative contributions of these mechanisms to adhesion dynamics by directly visualizing α5 integrin-, paxillin-, and α-actinin–GFP as adhesions formed and dispersed in migrating cells. Our data support hierarchical models for the formation of initial adhesive complexes. We provide evidence that classes of adhesive components enter adhesions serially. Our observations further suggest that signaling components such as paxillin enter adhesions early and turn over readily with prominent accumulations of structural molecules such as α-actinin subsequently joining the adhesion. We also support nucleation rather than the clustering of smaller minicomplexes, as reported previously for the formation of E-cadherin junctions (Adams et al. 1998), as the mechanism by which small adhesions (putative focal complexes) form. By contrast, we do not have evidence supporting the stabilization of large preformed cytoplasmic complexes. After adhesions broke down in the rear, α5-GFP was found in fibrous structures behind the cell, whereas α-actinin–GFP and paxillin-GFP moved up the lateral edge of retracting cells as organized structures and then dispersed.
The leading edge of membrane protrusions is a site where new adhesions form. Of the three fusion proteins that we examined, paxillin was the first component to appear visibly organized in protrusive regions of the cell near the leading edge. It appeared in a wave of fluorescent intensity and then concentrated in visible focal complex–like structures. Interestingly, α5 integrin and prominent α-actinin though present at the leading edge are not detectable in these paxillin-rich complexes. Thus, paxillin recruitment to these contact sites is an early event in the formation of adhesions. Since paxillin serves an adaptor function in recruiting several signaling components to the membrane, it follows that these newly forming adhesions likely serve signaling roles. Consistent with this hypothesis, other studies have suggested that tyrosine phosphorylation of paxillin occurs early in focal adhesion assembly (Richardson et al. 1997). Tyrosine phosphorylation of paxillin can create at least two SH2 domains, which can function as binding sites for other signaling molecules such as members of the Crk family (Bellis et al. 1995; Schaller and Parsons 1995; Richardson et al. 1997).
The absence of clearly visible α5 integrin in these complexes suggests that either the α5 integrin is not involved in the formation of new adhesions or newly forming adhesions are initiated and/or nucleated by α5 concentrations that are too low to be detected as discrete visible complexes in the light microscope. Although it is possible that other molecules, including other integrins, layilin, or syndecan play this role, it is also clear that these cells require α5 to adhere and migrate (Borowsky and Hynes 1998; Longley et al. 1999). The CHO B2 cells, which have almost no endogenous α5 integrin, do not adhere and migrate unless they ectopically express α5 integrin. In addition, adhesion-perturbing antibodies directed against the α5 integrin or Fn inhibited the organization of paxillin, which was also not seen in cells plated on poly-l-lysine. Despite reports that there are few other integrins in these cells, we stained for αv, β1, and β3 subunits and were unable to detect organized adhesions. We repeated all of these studies in WI38 cells, which tend to have more highly organized adhesions. The result was similar in that the forming adhesions show clearly organized paxillin but not α5 integrin. It is also evident that α5 binding to Fn is necessary for the formation of these adhesions since they were not observed when the integrin–Fn interaction was disrupted with a function-blocking antibody. Finally, as visualized with antibody staining or GFP probes in cells with well-developed focal adhesions the amount of α5 in the adhesions is considerably less than that seen for cytoskeletal markers such as paxillin or vinculin. Furthermore, not all adhesions that stain with cytoskeletal markers colocalize with visibly organized integrin. From this, we conclude that the newly forming adhesions are initiated by α5 engagement, since it is required for organization of paxillin. These putative α5 integrin-containing nucleation sites are not organized as large visible complexes, though present in a concentration sufficient to stimulate robust paxillin recruitment. As with other amplification cascades that typify many other signaling pathways, ligand activation of only a small number of integrins may be sufficient to stimulate the assembly of a large macromolecular adhesive complex.
Once protrusions stabilized, α-actinin began to colocalize with paxillin in small foci at the edge of the former lamellipodium. These small α-actinin–containing foci grew in size and extended small fiber-like structures toward the cell body, which is consistent with a recent study (Edlund et al. 2001). Once formed, some of the α-actinin adhesions slid inward from the cell perimeter and stabilized the paxillin, which no longer turned over. Thus, both paxillin and α-actinin appeared to enter newly forming adhesions before visible α5 integrin. However, once visibly organized α5 integrin entered the adhesions they remained relatively stationary and did not move toward the cell center; these adhesions also contained paxillin and α-actinin. Taken together, this suggests that not all adhesions contain clearly visible α5 integrin, but when the integrin was present the adhesions remained stationary. Previous studies have also reported a sliding of adhesions in cells (Smilenov et al. 1999; Pankov et al. 2000; Zamir et al. 2000). In one study, the membrane-spanning and cytoplasmic domain of β1 integrin was fused to GFP such that the GFP region resided on the outside of the cell (Smilenov et al. 1999). Interestingly, they reported that in stationary cells adhesions moved toward the cell center, whereas in migrating cells the adhesions were stationary (Smilenov et al. 1999). Although we observed adhesions moving toward the cell center in migrating cells, those with visible α5 integrin complexes were stationary.
The α5-GFP and α-actinin–GFP localized prominently at the leading edge in membrane ruffles and protrusions. Since membrane-bound but not soluble GFP was also seen in ruffles, this reflects their membrane localization. However, α-actinin is an intracellular molecule that has no membrane-targeting signal and thus must rely on intermolecular interactions to localize it to the membrane. Since neither soluble GFP nor paxillin was observed at the leading edge, the interactions mediating α-actinin targeting are specific. A possible mechanism for α-actinin targeting to membrane protrusions is through its interaction with integrins, since α-actinin binds directly to the cytoplasmic domain of the β1 subunit in vitro (Otey et al. 1990). Consistent with this mechanism, α-actinin colocalized with α5 integrin in the membrane ruffles. When the head and rod domains of α-actinin fused to GFP were expressed in the CHO K1 cells, the rod domain, which contains the β1 integrin–binding site (Otey et al. 1993), localized to membrane protrusions; however, the head domain, which contains the actin-binding site, appeared in highly organized fiber-like structures. This observation is consistent with a mechanism by which an interaction with integrins may function to localize α-actinin to membrane protrusions, but cytotoxicity precludes a firm conclusion. Alternatively, other α-actinin–binding partners such as phosphatidylinositol bisphosphate and phosphatidylinositol (3,4,5)-trisphosphate may play a role in its recruitment to membrane protrusions (Fukami et al. 1994; Greenwood et al. 2000).
The movement of the cell over stable adhesions suggests that adhesive components will tend to concentrate away from the leading edge toward the cell rear. One hypothesis proposes that integrins are recycled from the rear of the cell to the leading edge, thus providing a supply of integrins to newly forming adhesions (Bretscher 1989; Dalton et al. 1995; Lawson and Maxfield 1995; Pierini et al. 2000). The most direct observation supporting this mechanism comes from staining integrins in fixed neutrophils (Lawson and Maxfield 1995; Pierini et al. 2000). In these studies, vesicles are seen at the rear in cells whose migration was inhibited by calcium buffering and in a polarized perinuclear area in migrating cells. However, these cells are too small to discern spatially the biosynthetic and recycling compartments from the base of the protrusion. Furthermore, some mechanisms used by neutrophils are not readily apparent in other cells such as fibroblasts and are specific for certain integrins (Lawson and Maxfield 1995; Pierini et al. 2000). Our studies with larger cells and direct visualization at high temporal resolution intervals provide a far more complete picture.
In CHO cells with robust protrusive activity, we observed α5 integrin in vesicle-like structures that emanated from membrane protrusions and congregated in a perinuclear region where it colocalized with endogenous transferrin receptor. Although the fate of these internalized integrins is not known, this observation suggests that at least a fraction of these molecules are delivered to a large recycling compartment. Alternatively, some of the internalized integrin vesicles may be degraded in lysosomal compartments. A small fraction of the vesicles moved from the perinuclear area toward the cell front, but they disappeared at or before reaching the lamellipodial base; none were observed in the lamellipodium or at the leading edge. Cells migrating under conditions in which they exhibited minimal membrane ruffling presented a complementary picture. In these cells, α5 vesicles moved from the perinuclear region to the base of the lamellipodia, whereas vesicles moving from the front were seen only infrequently. In all cells, we observed vesicles moving from the cell rear to the perinuclear area in agreement with previous observations in fibroblasts and neutrophils (Regen and Horwitz 1992; Lawson and Maxfield 1995; Palecek et al. 1996; Pierini et al. 2000). However, vesicles moving from the rear to the front were not observed possibly due to the density of fluorescent material in the perinuclear area.
Thus, two endocytic pathways may be used by integrins. One may function to remove unligated integrin from the membrane in highly protrusive regions of the cell. In support of this, fewer vesicles were observed emanating from membrane protrusions as the substrate concentration increased. A second pathway removes integrins at the cell rear and delivers them either to the lysosomal compartment or to the cell front for formation of new adhesions. The movement of integrin-containing vesicles from the perinuclear area to the base of protrusions is consistent with previous studies (Lawson and Maxfield 1995; Pierini et al. 2000). It also complements particle-tracking studies that reported the directed movement of integrins to the leading edge in lamellipodia (Schmidt et al. 1993). Finally, we do not observe α-actinin on any of the α5-containing vesicles, suggesting that the integrins are not trafficked in α-actinin–containing complexes.
Cleavage of the linkage between integrin and other cytoskeletal components may initiate the release of adhesions. The integrins are seen in fibers behind migrating cells without visible α-actinin or paxillin, whereas adhesive complexes containing α-actinin and paxillin without highly organized integrin translocate from the rear by sliding along the cell edge. Unlike adhesion formation, α-actinin and paxillin were not observed to depart the adhesive clusters serially, but instead the complexes were seen to disperse. This suggests that adhesion breakdown is not simply a reversal of the mechanisms of formation.
We propose the following working model based on our observations of adhesion formation and turnover in migrating cells. Integrin, membrane-bound α-actinin (possibly complexed to the integrin), and cytoplasmic paxillin are all present in new protrusions. The binding of integrins to the ECM initiates, perhaps in conjunction with other receptors, the recruitment of signaling molecules such as paxillin to newly forming contact sites. Although substrate-bound integrins may also serve as the nucleation sites for these new adhesions, it is also possible that other molecules serve this role. The unligated integrins are rapidly endocytosed and traffic to a perinuclear region. These paxillin-rich sites are highly dynamic and tend to turn over at the base of the protrusion and cycle to the leading edge as new adhesions form. Structural molecules like α-actinin are subsequently recruited to this site, although small quantities may reside with the initial putative integrin foci. These developing adhesion complexes grow in size and molecular complexity as α-actinin first enters them and then forms stress fiber–like extensions that grow toward the cell body. The presence of α-actinin serves to stabilize the paxillin, which does not turn over in adhesions containing prominent α-actinin; its presence also coincides with the centripetal movement of adhesions. Subsequently, visible concentrations of integrin enter the adhesive complex and function to stabilize the centripetal movement. At the cell rear, cleavage of the integrin–cytoskeletal linkage at a site proximal to the integrin is a prominent mechanism to initiate breakdown of adhesions. The remaining paxillin- and α-actinin–containing complexes move toward the cell body and then disperse rapidly. Although some integrin is left behind on the substrate, some integrin also appears in vesicles that move toward the cell body where they are either degraded or cycled to the cell front for incorporation into new adhesions.
Supplemental Material
Acknowledgments
We thank James Casanova for advice and helpful suggestions, and Tom Parsons, Doug DeSimone, Lukas Tamm, Ken Yamada, Ralph Isberg, Magnus Edlund, Carol Otey, and Chris Turner for reagents and suggestions.
This work was supported by National Institutes of Health grant GM23244 and the University of Virginia Cancer Center. D.J. Webb was supported by National Institutes of Health postdoctoral training grant HD07528-01.
Footnotes
The online version of this paper contains supplemental material.
C.M. Laukaitis and D.J. Webb contributed equally to this work.
Abbreviations used in this paper: CFP, cyan fluorescent protein; ECFP, enhanced CFP; ECM, extracellular matrix; EGFP, enhanced GFP; EYFP, enhanced YFP; Fn, fibronectin; GFP, green fluorescent protein; YFP, yellow fluorescent protein.
References
- Adams C., Chen Y.-T., Smith S., Nelson W. Mechanisms of epithelial cell–cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin–green fluorescent protein. J. Cell Biol. 1998;142:1105–1119. doi: 10.1083/jcb.142.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellis S., Miller J., Turner C. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J. Biol. Chem. 1995;270:17437–17441. doi: 10.1074/jbc.270.29.17437. [DOI] [PubMed] [Google Scholar]
- Borowsky M., Hynes R. Layilin, a novel talin-binding transmembrane protein homologous with C-type lectins, is localized in membrane ruffles. J. Cell Biol. 1998;143:429–442. doi: 10.1083/jcb.143.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher M. Endocytosisrelation to capping and cell locomotion. Science. 1984;224:681–686. doi: 10.1126/science.6719108. [DOI] [PubMed] [Google Scholar]
- Bretscher M. Endocytosis and recycling of the fibronectin receptor in CHO cells. EMBO J. 1989;8:1341–1348. doi: 10.1002/j.1460-2075.1989.tb03514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher M., Aguado-Velasco C. Membrane traffic during cell locomotion. Curr. Opin. Cell Biol. 1998;10:537–541. doi: 10.1016/s0955-0674(98)80070-7. [DOI] [PubMed] [Google Scholar]
- Burridge K., Nuckolls G., Otey C., Pavalko F., Simon K., Turner C. Actin-membrane interaction in focal adhesions. Cell Differ. Dev. 1990;32:337–342. doi: 10.1016/0922-3371(90)90048-2. [DOI] [PubMed] [Google Scholar]
- Cao Z., Huang K., Horwitz A. Identification of a domain on the integrin alpha5 subunit implicated in cell spreading and signaling. J. Biol. Chem. 1998;273:31670–31679. doi: 10.1074/jbc.273.48.31670. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M., Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabiri G.A., Turnacioglu K.K., Sanger J.M., Sanger J.W. Myofibrillogenesis visualized in living embryonic cardiomyocytes. Proc. Natl. Acad. Sci. USA. 1997;94:9493–9498. doi: 10.1073/pnas.94.17.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton S., Scharf E., Briesewitz R., Marcantonio E., Assoian R. Cell adhesion to extracellular matrix regulates the life cycle of integrins. Mol. Biol. Cell. 1995;6:1781–1791. doi: 10.1091/mbc.6.12.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePasquale J.A., Izzard C.S. Evidence for an actin-containing cytoplasmic precursor of the focal contact and the timing of incorporation of vinculin at the focal contact. J. Cell Biol. 1987;105:2803–2809. doi: 10.1083/jcb.105.6.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund M., Lotano M.A., Otey C.A. Dynamics of α-actinin in focal adhesions and stress fibers visualized with α-actinin-green fluorescent protein. Cell Motil. Cytoskeleton. 2001;48:190–200. doi: 10.1002/1097-0169(200103)48:3<190::AID-CM1008>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Fukami K., Endo T., Imamura M., Takenawa T. α-Actinin and vinculin are PIP2-binding proteins involved in signaling by tyrosine kinase. J. Biol. Chem. 1994;269:1518–1522. [PubMed] [Google Scholar]
- Greenwood J.A., Theibert A.B., Prestwich G.D., Murphy-Ullrich J.E. Restructuring of focal adhesion plaques by PI 3-kinase. Regulation by PtdIns (3,4,5)-p(3) binding to alpha-actinin. J. Cell Biol. 2000;150:627–641. doi: 10.1083/jcb.150.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzard C.S. A precursor of the focal contact in cultured fibroblasts. Cell. Motil. Cytoskeleton. 1988;10:137–142. doi: 10.1002/cm.970100118. [DOI] [PubMed] [Google Scholar]
- Katz B.-Z., Zamir E., Bershadsky A., Kam Z., Yamada K., Geiger B. Physical state of the extracellular matrix regulates the structure and molecular composition of cell-matrix adhesions. Mol. Biol. Cell. 2000;11:1047–1060. doi: 10.1091/mbc.11.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiosses W., Daniels R., Otey C., Bokoch G., Schwartz M. A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 1999;147:831–844. doi: 10.1083/jcb.147.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight B., Laukaitis C., Akhtar N., Hotchin N., Edlund M., Horwitz A. Visualizing muscle cell migration in situ. Curr. Biol. 2000;10:576–585. doi: 10.1016/s0960-9822(00)00486-3. [DOI] [PubMed] [Google Scholar]
- Lauffenburger D., Horwitz A. Cell migrationa physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- Lawson M., Maxfield F. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377:75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- Longley R., Woods A., Fleetwood A., Cowling G., Gallagher J., Couchman J. Control of morphology, cytoskeleton and migration by syndecan-4. J. Cell Sci. 1999;112:3421–3431. doi: 10.1242/jcs.112.20.3421. [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Akiyama S., Yamada K. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function Science 267 1995. 883 885a [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Teramoto H., Coso O., Gutkind J., Burvbelo P., Akiyama S., Yamada K. Integrin functionmolecular hierarchies of cytoskeletal and signaling molecules J. Cell Biol. 131 1995. 791 805b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyoshi K., Richards L., Akazawa C., O'Leary D., Nakanishi S. Labeling neural cells using adenoviral gene transfer of membrane-targeted GFP. Neuron. 1996;16:255–260. doi: 10.1016/s0896-6273(00)80044-6. [DOI] [PubMed] [Google Scholar]
- Otey C., Pavalko F., Burridge K. An interaction between alpha-actinin and the beta 1 integrin subunit in vitro. J. Cell Biol. 1990;111:721–729. doi: 10.1083/jcb.111.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otey C., Vasquez G., Burridge K., Erickson B. Mapping of the α-actinin binding site within the β1 integrin cytoplasmic domain. J. Biol. Chem. 1993;268:21193–21197. [PubMed] [Google Scholar]
- Palecek S., Schmidt C., Lauffenburger D., Horwitz A. Integrin dynamics on the tail region of migrating fibroblasts. J. Cell Sci. 1996;109:941–952. doi: 10.1242/jcs.109.5.941. [DOI] [PubMed] [Google Scholar]
- Palecek S., Huttenlocher A., Horwitz A., Lauffenburger D. Physical and biochemical regulation of integrin release during rear detachment of migrating cells. J. Cell Sci. 1998;111:929–940. doi: 10.1242/jcs.111.7.929. [DOI] [PubMed] [Google Scholar]
- Pankov R., Cuckierman E., Katz B.-Z., Matsumoto K., Lin D., Lin S., Hahn C., Yamada K. Integrin dynamics and matrix assemblytensin-dependent translocation of alpha(5)beta(1) integrins promotes early fibronectin fibrillogenesis. J. Cell Biol. 2000;148:1075–1090. doi: 10.1083/jcb.148.5.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierini L., Lawson M., Eddy R., Hendey B., Maxfield F. Oriented endocytic recycling of α5β1 in motile neutrophils. Blood. 2000;95:2471–2480. [PubMed] [Google Scholar]
- Regen C., Horwitz A. Dynamics of beta 1 integrin-mediated adhesive contacts in motile fibroblasts. J. Cell Biol. 1992;119:1347–1359. doi: 10.1083/jcb.119.5.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A., Malik R., Heldebrand J., Parsons J. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src of catalytically inactive FAKa role for paxillin tyrosine phosphorylation. Mol. Cell. Biol. 1997;17:6906–6914. doi: 10.1128/mcb.17.12.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgia R., Li J.-L., Ewaniuk D., Wang Y.-B., Sattler M., Chen W.-C., Richards W., Pisick E., Shapiro G., Rollins B. Expression of the focal adhesion protein paxillin in lung cancer and its relation to cell motility. Oncogene. 1999;18:67–77. doi: 10.1038/sj.onc.1202273. [DOI] [PubMed] [Google Scholar]
- Sastry S., Lakonishok M., Wu S., Truong T., Huttenlocher A., Turner C., Horwitz A. Quantitative changes in integrin and focal adhesion signaling regulate myoblast cell cycle withdrawal. J. Cell Biol. 1999;144:1295–1309. doi: 10.1083/jcb.144.6.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller M., Parsons J. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol. Cell. Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C., Horwitz A., Lauffenburger D., Sheetz M. Integrin–cytoskeletal interactions in migrating fibroblasts are dynamic, asymmetric, and regulated. J. Cell Biol. 1993;123:977–991. doi: 10.1083/jcb.123.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenwaelder S., Burridge K. Evidence for a calpeptin-sensitive protein-tyrosine phosphatase upstream of the small GTPase Rho. A novel role for the calpain inhibitor calpeptin in the inhibition of the protein-tyrosine phosphatases. J. Biol. Chem. 1999;274:14359–14367. doi: 10.1074/jbc.274.20.14359. [DOI] [PubMed] [Google Scholar]
- Schreiner C., Bauer J., Danilov Y., Hussein S., Sczekan M., Juliano R. Isolation and characterization of Chinese hamster ovary cell variants deficient in the expression of fibronectin receptor. J. Cell Biol. 1989;109:3157–3167. doi: 10.1083/jcb.109.6.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilenov L., Mikhailov A., Pelham R., Marcantonio E., Gundersen G. Focal adhesion motility revealed in stationary fibroblasts. Science. 1999;286:1172–1174. doi: 10.1126/science.286.5442.1172. [DOI] [PubMed] [Google Scholar]
- Takagi J., Kamata T., Meredith J., Puzon-McLaughlin W., Takada Y. Changing ligand specificities of αvβ1 and αvβ3 integrins by swapping a short diverse sequence of the β subunit. J. Biol. Chem. 1997;272:19794–19800. doi: 10.1074/jbc.272.32.19794. [DOI] [PubMed] [Google Scholar]
- Yamada K., Miyamoto S. Integrin transmembrane signaling and cytoskeletal control. Curr. Opin. Cell Biol. 1995;7:681–689. doi: 10.1016/0955-0674(95)80110-3. [DOI] [PubMed] [Google Scholar]
- Ylanne J., Chen Y., O'Toole T., Loftus J., Takada Y., Ginsberg M. Distinct functions of integrin alpha and beta subunit cytoplasmic domains in cell spreading and formation of focal adhesions. J. Cell Biol. 1993;122:223–233. doi: 10.1083/jcb.122.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamir E., Katz B.-Z., Aota S.-i., Yamada K., Geiger B., Kam Z. Molecular diversity of cell-matrix adhesions. J. Cell Sci. 1999;112:1655–1669. doi: 10.1242/jcs.112.11.1655. [DOI] [PubMed] [Google Scholar]
- Zamir E., Katz M., Posen Y., Erez N., Yamada K., Katz B.-Z., Lin S., Lin D., Bershadsky A., Kam Z. Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts. Nat. Cell Biol. 2000;2:191–197. doi: 10.1038/35008607. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Morla A., Vuori K., Bauer J., Juliano R., Ruoslahti E. The alpha v beta 1 integrin functions as a fibronectin receptor but does not support fibronectin matrix assembly and cell migration on fibronectin. J. Cell Biol. 1993;122:235–242. doi: 10.1083/jcb.122.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}