

Abstract
Phagocytosis is a highly localized and rapid event, requiring the generation of spatially and temporally restricted signals. Because phosphatidylinositol 3-kinase (PI3K) plays an important role in the innate immune response, we studied the generation and distribution of 3′ phosphoinositides (3′PIs) in macrophages during the course of phagocytosis. The presence of 3′PI was monitored noninvasively in cells transfected with chimeras of green fluorescent protein and the pleckstrin homology domain of either Akt, Btk, or Gab1. Although virtually undetectable in unstimulated cells, 3′PI rapidly accumulated at sites of phagocytosis. This accumulation was sharply restricted to the phagosomal cup, with little 3′PI detectable in the immediately adjacent areas of the plasmalemma. Measurements of fluorescence recovery after photobleaching were made to estimate the mobility of lipids in the cytosolic monolayer of the phagosomal membrane. Stimulation of phagocytic receptors induced a marked reduction of lipid mobility that likely contributes to the restricted distribution of 3′PI at the cup. 3′PI accumulation during phagocytosis was transient, terminating shortly after sealing of the phagosomal vacuole. Two factors contribute to the rapid disappearance of 3′PI: the dissociation of the type I PI3K from the phagosomal membrane and the persistent accumulation of phosphoinositide phosphatases.
Keywords: Fcγ receptors, PH domain, phosphoinositide, lipid mobility, FRAP
Introduction
Phagocytosis, the engulfment of foreign bodies by specialized myeloid cells, is an essential component of the innate immune response. Invading microorganisms and other particles are ingested by phagocytic cells through a receptor-mediated mechanism that involves extensive cytoskeletal rearrangement and membrane remodelling (Aderem and Underhill 1999; Greenberg 2001). Receptors on the phagocyte surface that recognize surface components of the microorganism, or opsonins attached to it, cluster in the area of contact between the pathogen and phagocyte, triggering signals that initiate localized actin assembly and membrane fusion events (Daeron 1997; Swanson et al. 1999). Prominent among these signals are rapid changes in phosphoinositide metabolism, including the biosynthesis of phosphatidylinositol 3,4,5-trisphosphate (PI[3,4,5]P3) (Araki et al. 1996; Cox et al. 1999) and the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PI[4,5]P2) (Azzoni et al. 1992; Liao et al. 1992). The products of the latter reaction elicit cytosolic calcium transients and the activation of PKC isoforms (Zheleznyak and Brown 1992; Oancea and Meyer 1998).
Activation of type I phosphatidylinositol 3-kinase (PI3K), the enzyme responsible for generation of PI(3,4,5)P3, appears to be essential for the extension and/or fusion of pseudopods around the ingested particle. This view is supported by two lines of evidence: first, inhibition of the kinase with wortmannin or LY294002 virtually eliminates phagocytosis of large particles (Cox et al. 1999), and second forced clustering of a transmembrane chimera that includes the catalytic subunit of PI3K suffices to elicit phagocytosis (Lowry et al. 1998). During phagocytosis mediated by Fcγ receptors, recruitment of PI3K to the membrane is initiated by activation of Syk, a tyrosine kinase. Syk itself is brought to the phagosomal cup by the interaction of its tandem Src homology 2 domains with a pair of appropriately spaced phosphotyrosine residues on the immunoreceptor or on its ancillary γ chain (Crowley et al. 1997; Kiefer et al. 1998). This cascade is thought to be triggered by the clustering of the receptors, which facilitates phosphorylation of their tyrosines by one or more members of the Src family of kinases (Fitzer-Attas et al. 2000; Suzuki et al. 2000; Cox et al. 2001). The cytoskeletal- and membrane-remodelling events that underlie phagocytosis are exquisitely confined to the “phagosomal cup,” the region of the cell in the immediate vicinity of the particle. This local activation implies that the signals generated by the phagocytic receptors convey spatial information to their downstream effectors. The focal recruitment of tyrosine and inositide kinases to the area where receptors cluster provides an anatomical foundation for the generation of localized signals. However, it is less clear whether the signals themselves are restricted to the area of the phagosomal cup. Indeed, phospholipids and products of their metabolism like diacylglycerol would be anticipated to diffuse laterally in the plane of the plasma membrane before phagosomal sealing, which in the case of large particles requires tens of seconds to minutes for completion. However, to date visualization of 3′ phosphoinositides (3′PIs) during the course of phagocytosis has not been reported. This paucity of information stems in all likelihood from our inability to monitor the fate of individual lipids in the inner monolayer of the plasma membrane of live cells.
Very recently, advances in our understanding of protein–lipid interactions have provided a novel approach to analyze the distribution of certain phospholipids in their native environment. Specifically, pleckstrin homology (PH) domains of several proteins were found to interact with varying degrees of affinity and specificity with either PI(3,4,5)P3 or PI(4,5)P2 (Stauffer and Meyer 1997; Hinchliffe et al. 1998; Oancea et al. 1998; Varnai et al. 1999; Botelho et al. 2000). By expressing in mammalian cells chimeras of the appropriate PH domain and green fluorescent protein (GFP), it has become possible to monitor continuously the distribution of specific phosphoinositides in live cells by noninvasive spectroscopic means. In the present report, we used the PH domains of Akt, Gab1, and Btk in conjunction with digital fluorescence imaging to analyze the generation, distribution, and subsequent disappearance of PI(3,4,5)P3 during the course of phagocytosis in a murine monocyte/macrophage cell line. We found that PI(3,4,5)P3 is exquisitely localized to the phagosomal cup with no signs of lateral diffusion to the adjacent areas of the surface membrane. The mechanisms underlying this remarkable spatial confinement were analyzed in detail.
Materials and Methods
Reagents
Sheep red blood cells (SRBCs) and anti-IgG were obtained from ICN Biomedicals. Polystyrene beads (8-μm diameter) were obtained from Bangs Beads. FuGene 6 was obtained from Boehringer. Wortmannin was from Calbiochem. Earl's α-MEM was obtained from Cellgrow. Antibodies to PTEN were raised in rabbits and affinity purified as described (Stambolic et al. 1998). Cy3-labeled secondary antibodies were obtained from Jackson ImmunoResearch Laboratories. Anti–influenza virus hemagglutinin (HA) antibodies were obtained from BabCo. Hepes-buffered RPMI (HPMI), human IgG, and all other reagents were obtained from Sigma-Aldrich.
cDNA Constructs
The Btk-PH–GFP and (R28C)Btk-PH–GFP constructs were prepared as previously described (Varnai et al. 1999). The Akt-PH–GFP construct was prepared as reported (Haugh et al. 2000). peGFP:PM–GFP encodes the 10 amino acid myristoylation/palmitylation sequence from Lyn fused to enhanced GFP (Teruel et al. 1999). The FcγRIIa-GFP, Syk-GFP, and p85-GFP constructs were constructed by subcloning the respective cDNA into pEGFP obtained from CLONTECH Laboratories, Inc. The construct encoding SHIP1 tagged with the HA epitope YPYDVPDYAS was a gift from Dr. J. Penninger (Amgen Institute). The Gab1–GFP construct was provided by Dr. E. Skolnik (Skirball Institute, New York, NY).
Cell Culture and Transfection
The macrophage RAW 264.7 cell line was obtained from the American Type Culture Collection. RAW 264.7 macrophages were cultured in Earl's α-MEM with 10% FCS. Cells were scraped and seeded onto 2.5-cm glass coverslips at ∼30% confluence. After 24 h, the cells were transfected using FuGene 6 according to the manufacturer's directions. For p85-GFP, the cells were transfected 8–10 h before experiments. All other constructs were expressed for 12–24 h.
Phagocytosis Assay
SRBCs and 8-μm polystyrene beads were opsonized for 1 h at 37°C with rabbit anti-SRBC or human IgG, respectively. For phagocytosis, RAW 264.7 cells on 2.5-cm glass coverslips were exposed to opsonized particles in HPMI medium at 37°C. To score phagocytic efficiency, macrophages were incubated with SRBCs for 15 min. External SRBCs were then lysed by brief hypotonic shock, and the cells were fixed with 4% paraformaldehyde overnight at 4°C. The number of phagosomes per cell was counted by differential interference contrast (DIC) microscopy.
Frustrated Phagocytosis Model
RAW macrophages were transfected 24 h before resuspension in 1 ml of HPMI with 2 mM EDTA. The cells were gently rotated for 1–2 h before the experiments. 100 μl of the suspension was then added to 1 ml of HPMI containing an additional 2 mM of MgCl2 and allowed to sediment on uncoated glass coverslips or coverslips coated with poly-l-lysine or human IgG (10 mg/ml). At the indicated time, the distribution of the fluorescent chimeras was analyzed by confocal microscopy.
FRAP
Cells transfected with Akt-PH–GFP or PM–GFP were allowed to sediment onto poly-lysine– or human IgG–coated or uncoated coverslips for 10–20 min. An initial image of an optical slice in the plane of the adherent (basal) membrane was obtained using the LSM 510 confocal microscope. Next, an ∼3-μm diameter spot was photobleached using the 488-nm laser line at full power. Subsequent images were obtained every 2 s in the case of Akt-PH–GFP or every 10 s in the case of PM–GFP with the laser at ∼1% of full power for excitation. A similar protocol was used to measure FRAP in the phagosomal cup in cells allowed to interact with IgG-opsonized latex beads of 8-μm diameter. Diffusion coefficients were calculated from FRAP as described (Axelrod et al. 1976).
Immunolocalization of SHIP1 and PTEN
RAW cells were fixed with ice-cold 4% paraformaldehyde in PBS between 3 and 8 min after addition of opsonized SRBCs. The fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 20 min before blocking for 1 h with 5% BSA in PBS, followed by staining with the primary antibody for 1 h. Affinity purified rabbit anti-PTEN antibody was used to measure endogenous PTEN (Stambolic et al. 1998). In the case of SHIP1, the localization of ectopically expressed SHIP1–HA (coexpressed with GFP to serve as a transfection marker and internal control) was measured using a monoclonal anti-HA antibody. Appropriate secondary antibodies labeled with Cy3 were used.
Image Analysis
Both live and fixed samples were analyzed by confocal microscopy using a ZEISS LSM 510 laser scanning confocal microscope with a 100× oil immersion objective. GFP-FITC and Cy3 were examined using the conventional standard laser excitation and filter sets. 16-bit digital images were analyzed with Java Image software. The accumulation of soluble proteins such as Btk-PH, Syk, or p85 at the phagosomal cup (p) was normalized by comparing it to the value of the surrounding cytoplasm (c) using the ratio ([p−c]/c). The enrichment of membrane (m)-bound proteins such as PM–GFP or FcγRIIa at the cup was quantified by the ratio (p/m). Digital images were prepared using Adobe Photoshop® 4.0 (Adobe Systems, Inc.) and CorelDraw 8.0 (Corel Corp.).
Results
Accumulation of PI(3,4,5)P3 in the Phagosomal Cup
The distribution of 3-phosphoinositides in resting and stimulated phagocytes was studied by transient transfection of a fusion protein consisting of the PH domain of Akt linked to GFP (Akt-PH–GFP). In otherwise untreated cells, this chimera localized to the cytoplasm and nucleus with little or no association with the plasma membrane (Fig. 1 A, inset). Similar results were obtained using Btk-PH–GFP, Gab1-PH–GFP, or unconjugated GFP, though the cytosol-to-nucleus ratio varied somewhat between constructs (not shown). Initiation of phagocytosis by presentation of IgG-opsonized SRBCs induced a marked and rapid redistribution of Akt-PH–GFP, which accumulated at the phagosomal cup (Fig. 1, A–B). A similar pattern was observed using Gab1-PH–GFP (Fig. 1, C–D) and a weaker but reproducible accumulation of Btk-PH–GFP was also observed (Fig. 1 F, inset), whereas unconjugated GFP remained in the cytosol throughout (not illustrated).
Figure 1.
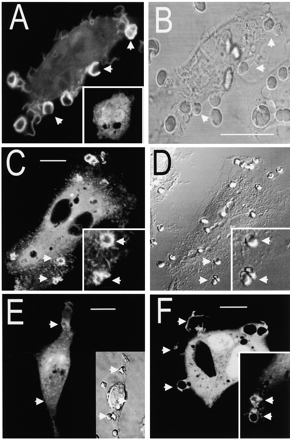
Distribution of PH domain–GFP chimeras in macrophages. RAW 264.7 cells were transfected with chimeras of GFP and the indicated PH domain and then exposed to IgG-opsonized SRBCs to initiate phagocytosis. Confocal fluorescence (A, C, E, and F) and DIC images (B, D, and E, inset) were acquired. (A–B) Cells transfected with Akt-PH–GFP. The inset shows the distribution of Akt-PH–GFP before addition of SRBCs. (C–D) Cells transfected with Gab1-PH–GFP. (E) Cells transfected with Akt-PH–GFP were pretreated with 100 nM wortmannin for 15 min before addition of the SRBCs. (F) The main panel shows cells transfected with (R28C)Btk-PH–GFP, a mutant form of Btk-PH–GFP unable to bind 3′PI in vitro. The inset shows a cell transfected with wild-type Btk-PH–GFP. Arrows indicate sites of attachment of SRBCs. Images are representative of at least three experiments of each type. Bars, 10 μm.
The accumulation of several different PH domains with high affinity for PI(3,4,5)P3 suggests that this lipid concentrates in the phagosomal cup. Two lines of evidence support this notion: first, the redistribution of Akt-PH–GFP was completely eliminated by pretreatment of the macrophages with 100 nM wortmannin, a dose that is predicted to inhibit fully type I PI3K (Ninomiya et al. 1994). Binding of the SRBCs to the macrophage surface persisted following treatment with wortmannin, but Akt-PH–GFP remained in the cytosol and nucleus (Fig. 1 E). Secondly, a mutant form of Btk-PH–GFP lacking the ability to bind PI(3,4,5)P3 (Varnai et al. 1999) similarly failed to redistribute to the phagosome. As shown in the main panel of Fig. 1 F, the inactive (R28C) mutant form of Btk-PH–GFP remained in the cytosol during the course of phagocytosis. Jointly, these observations indicate that PI(3,4,5)P3 and possibly also other 3′PIs accumulate at sites where Fcγ receptors are cross-linked by ligation of opsonized particles. For simplicity, the concentration of Akt-PH–GFP at the membrane will be equated hereafter to accumulation of PI(3,4,5)P3.
Time Course of PI(3,4,5)P3 Redistribution during Phagocytosis
A more detailed analysis of the course of accumulation of PI(3,4,5)P3 at the phagosomal membrane is presented in Fig. 2 and Fig. 3. PI(3,4,5)P3 accumulation was detectable shortly after the plasmalemma established contact with the opsonized particles (Fig. 2, A–B) and became more pronounced as the cup developed (Fig. 2 C). The accumulation was still evident at the time of phagosomal sealing, which we designated arbitrarily as time 0 (Fig. 2 D, 0 s) and declined sharply thereafter (Fig. 2 E).
Figure 2.
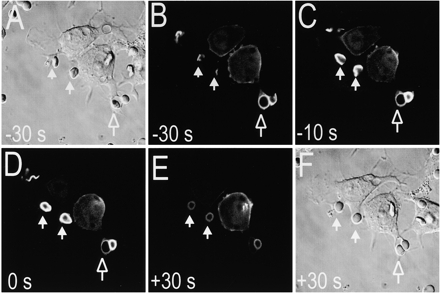
Time course of accumulation of 3′PI in phagosomes. RAW 264.7 cells transfected with Akt-PH–GFP were exposed to IgG-opsonized SRBCs to initiate phagocytosis. Confocal fluorescence (B–E) and DIC images (A and F) were acquired at the indicated times. The time of closure of the two phagosomes noted by solid white arrows was arbitrarily chosen as time 0. A phagosome that had sealed before the acquisition of the first image is shown by an open arrow. Images are representative of six similar time course determinations.
Figure 3.
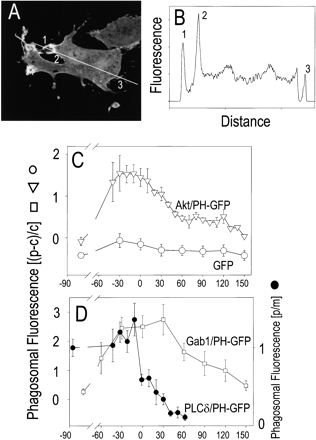
Quantitation of the time course of 3′PI accumulation. RAW 264.7 cells transfected with PH domain–GFP chimeras or with soluble GFP were exposed to IgG-opsonized SRBCs to initiate phagocytosis, and confocal images were acquired. A representative image of a cell transfected with Akt-PH–GFP is shown in A. To quantify the accumulation of Akt-PH–GFP at the phagosomal membrane, lines were drawn through the digitized images, and the fluorescence intensity of individual pixels was determined. The line scan corresponding to A is shown in B; for reference, numbers have been assigned to the intersections with the phagosomal (1, 2) and contralateral plasma membrane (3). In C, the time course of accumulation of Akt-PH–GFP (▿) at the phagosomal membrane is compared with that of free GFP (○). In D, the course of association of Gab1-PH–GFP (□) and PLCδ/PH-GFP (•) are shown. The time of closure of the phagosomes was arbitrarily chosen as time 0 as in the legend to Fig. 2. The left ordinate, which applies to the open symbols, shows the fluorescence of the phagosomal membrane (p) minus the cytosolic fluorescence (c; subtracted to discount the free fluorophore present in the immediate vicinity of the phagosome) divided by c to normalize for expression level. The right ordinate, which applies to the solid circles (•), shows the fluorescence of the phagosomal membrane (p) divided by the fluorescence of a nonphagosomal region of the plasma membrane (m). Data are mean ± standard error of three to six experiments.
Densitometry of line scans was used to quantify more precisely the course and extent of accumulation of Akt-PH–GFP at the phagosomal membrane. A representative experiment is illustrated in Fig. 3A and Fig. B. In Fig. 3 C, the density of the chimera at the phagosome was corrected by subtracting the cytosolic fluorescence and was normalized with respect to the expression level to allow comparison among cells and between experiments. As shown in Fig. 3 C (▿), which represents the average of six experiments, maximal accumulation of Akt-PH–GFP was attained nearly 30 s before phagosomal sealing and decayed rapidly after the phagosome pinched off from the plasmalemma, reaching near baseline levels 1–2 min later. Similar results were obtained using Gab1-PH–GFP (Fig. 3 D), whereas no significant concentration of unconjugated GFP was observed at the cup at any time (Fig. 3 C, ○).
Fig. 3 D compares the course of accumulation of PI(3,4,5)P3 with the changes in PI(4,5)P2 detected using the PH domain of PLCδ. As reported previously (Botelho et al. 2000), PI(4,5)P2 undergoes a biphasic change: a small transient increase followed immediately by its virtual disappearance from the phagosomal membrane. The formation of PI(3,4,5)P3 precedes the changes in PI(4,5)P2. Accumulation of PI(3,4,5)P3 proceeds during the phase of PI(4,5)P2 disappearance, implying that formation of the former likely contributes to the disappearance of PI(4,5)P2, although activation of PLC is also known to occur in this setting (Greenberg 2001). The rapid association and dissociation of PLCδ-PH–GFP indicates that the kinetics of Akt-PH–GFP or Gab1-PH–GFP binding to the phagosomal membrane is not limited by diffusion through the actin meshwork that lines the phagosomal cup.
In Fig. 4, the temporal course of PI(3,4,5)P3 accumulation is compared with that of the Fcγ receptor clustering (Fig. 4 A) and with the recruitment of Syk (Fig. 4 B) and PI3K (Fig. 4 C). In all cases, GFP–chimeric constructs were transfected into RAW 264.7 cells, which were challenged with SRBCs, as described for the PH domain chimeras (as described in Materials and Methods). The receptors clustered rapidly under the adherent particle, and although their density decreased somewhat by the time of phagosomal closure, receptor concentration in the sealed phagosomal membrane clearly exceeded that of the surface membrane. Accumulation of receptors under the phagocytic particle resulted from their lateral displacement along the plane of the bilayer and not from localized membrane ruffling, since an acylated form of GFP (PM–GFP) used as a nonspecific membrane marker (Teruel et al. 1999) accumulated only marginally at the cup (Fig. 4 A). Maximal receptor clustering in nascent phagosomes preceded the peak accumulation of PI(3,4,5)P3, consistent with a cause–effect relationship.
Figure 4.
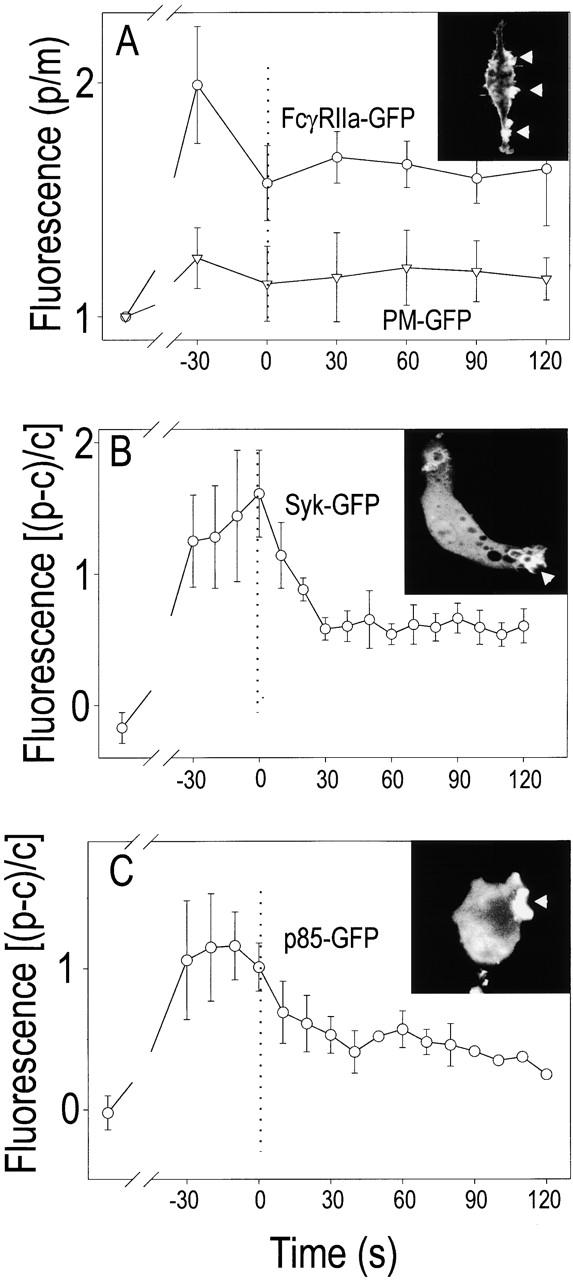
Quantitation of the time course of accumulation of Fc receptors, Syk, and PI3K on phagosomes. RAW 264.7 cells transfected with either FcγRIIa-GFP (A, ○), PM–GFP (A, ▿), Syk-GFP (B), or p85-GFP (C) were exposed to IgG-opsonized SRBCs to initiate phagocytosis, and confocal images were acquired. Representative images are shown in the insets. The time of closure of the phagosomes was arbitrarily defined as time 0. Quantitation in B and C was as in the legend to Fig. 2. In A, the ordinate refers to the fluorescence intensity of the phagosomal membrane (p) relative to the extraphagosomal (usually contralateral) plasmalemma (m). Data are mean ± standard error of at least three experiments.
The tyrosine kinase Syk and the p85 regulatory subunit of the type I PI3K also accumulated at the phagosomal cup before membrane sealing (Fig. 4B and Fig. C). By contrast, soluble GFP remained in the cytosol throughout the phagocytic sequence (Fig. 3 C, ○), confirming the specificity of the measurements. Binding of Syk and recruitment of PI3K are thought to be downstream of receptor clustering and to precede the de novo biosynthesis of PI(3,4,5)P3. The site and time of recruitment of these proteins described in Fig. 4 are compatible with the notion that the accumulation of the PH domain–GFP chimeras is indeed an accurate reflection of the localized biosynthesis of PI(3,4,5)P3.
A Model of Frustrated Phagocytosis to Analyze Lipid Mobility
The experiments in Fig. 1 and Fig. 2 illustrate the fact that the PI(3,4,5)P3 generated at the nascent phagosome is precisely confined to the small region of the membrane apposed to the particle and fails to diffuse noticeably to the contiguous regions of the plasma membrane, despite their physical continuity before phagosomal closure. This observation suggests that the PI(3,4,5)P3 generated at the phagosomal cup has a limited ability to diffuse laterally. This idea could in principle be tested by estimating the lateral mobility of phagosomal lipids by FRAP.
The size (≤4 μm), shape, and dynamic nature of phagosomes formed around SRBCs constrained our ability to accurately measure FRAP. To circumvent these limitations, we implemented a system where the clustering of Fcγ receptors by IgG occurred on a planar surface, extending the area and the time available for measurement of fluorescence, since under these conditions internalization of the opsonized surface is precluded. Human IgG was adhered to a glass coverslip and suspended RAW 264.7 cells transfected with Akt-PH–GFP were allowed to land and spread on this surface while the process was monitored continuously by DIC microscopy. As controls for nonspecific interactions with the surface, uncoated, polylysine- or BSA-coated coverslips were also used. The typical morphology of a cell allowed to spread on IgG for 20 min is illustrated in Fig. 5 A. Such cells adhered firmly and spread wide and thin lamellipodia on the coated surface. Importantly, Akt-PH–GFP concentrated greatly on the membrane apposed to the IgG with no obvious accumulation elsewhere. As in the case of bona fide phagosomes, the accumulation of Akt-PH–GFP (that is, PI[3,4,5]P3) was confined to the area of the membrane in direct contact with IgG with little evidence for lateral spreading for at least 10 min. By contrast, cells that adhered to either glass or poly-lysine for comparable periods remained more rounded and extended shorter lamellipodia (Fig. 5 C). Only a slight accumulation of Akt-PH–GFP at sites of contact was detected (Fig. 5 D). In five determinations, the concentration at the membrane was 70% higher than the cytosol compared with 230% higher in the case of IgG. These findings suggest that the recruitment of Akt-PH–GFP to the IgG-coated surface reflects the specific receptor-induced accumulation of PI(3,4,5)P3 and that this system is an adequate surrogate for the phagosomal cup.
Figure 5.

Distribution of Akt-PH–GFP in cells adhering to IgG-coated versus uncoated glass. RAW 264.7 cells transfected with Akt-PH–GFP were detached by resuspension in HPMI with 2 mM EDTA and then allowed to sediment on either IgG-coated (A and B) or uncoated glass (C and D) for ∼10–20 min. DIC (A and C) and confocal fluorescence images were acquired, and vertical (x versus z) sections of Akt-PH–GFP are illustrated (B and D). Images are representative of five similar determinations.
Lipid Mobility in Control and IgG-stimulated Cells
The complex formed between PI(3,4,5)P3 and Akt-PH–GFP is reversible, which confounds measurements of lateral mobility by FRAP. Following bleaching, the recovery of fluorescence will be effected at least in part by dissociation and replacement of a bleached Akt-PH–GFP by fluorescent cytosolic Akt-PH–GFP rather than by lateral displacement of the lipid. Because reversibly bound PH domains are inappropriate for the measurement of PI(3,4,5)P3 mobility and covalently labeled fluorescent derivatives of this lipid are not suitable, we chose instead to measure the mobility of a model lipid. Since the lipid distribution and properties of the two monolayers of the plasma membrane differ greatly, we felt it was important to measure lipid mobility in the inner monolayer. This was accomplished by transfecting the cells with a vector encoding the 10 amino acid myristoylation/palmitylation sequence from Lyn fused to GFP (PM–GFP). This acylated form of GFP partitions preferentially to the inner monolayer of the plasma membrane (Teruel et al. 1999), although small and varying amounts are often seen in endomembranes (Botelho et al. 2000). A typical vertical (x versus z) section of a macrophage transfected with PM–GFP and adhered to IgG-coated glass is shown in Fig. 6 A, illustrating the predominantly plasmalemmal distribution of this probe. FRAP experiments were performed in such cells, and typical results are presented in Fig. 6 B.
Figure 6.

Measurement of PM–GFP FRAP. RAW 264.7 cells transfected with PM–GFP were detached by gentle scraping and then allowed to sediment on either IgG-coated or uncoated glass coverslips. (A) Confocal fluorescence images of a cell plated on IgG were acquired, and reconstruction of a vertical (x versus z) section is illustrated. (B) Confocal (x versus y) images of the adherent membrane were acquired before (Pre), immediately after (0 s), and at the indicated intervals following photobleaching of a spot near the edge of the membrane. In C, the rate of recovery after bleaching was quantified and compared for cells plated on glass (○) or IgG (□). Data are mean ± standard error of six individual experiments.
In adherent yet unstimulated cells, PM–GFP diffused rapidly on the plane of the membrane: the mobile fraction of the probe approached 65%, and when the bleached area averaged 4 μm in diameter the t 1/2 for recovery was ∼10 s (Fig. 6 C), corresponding to a diffusion coefficient of ∼3.5 × 10−9 cm2/s, comparable to that reported for lipids in the inner monolayer of the plasma membrane (Morrot et al. 1986; el Hage Chahine et al. 1993). Remarkably, the mobile fraction of the acylated GFP decreased markedly when the Fcγ receptors were engaged: only ∼20% of the fluorescence was restored after 30 s (Fig. 6 C). This finding indicates that lipid mobility is restricted in the stimulated cells, possibly accounting for the confinement of PI(3,4,5)P3 within the phagosomal cup.
To ensure that the observed changes in lipid mobility were not unique to the frustrated phagocytosis model in which IgG-coated coverslips are used, experiments were performed using large (8-μm) IgG-coated poly styrene beads. Such beads are effectively internalized albeit more slowly than SRBCs, facilitating determination of FRAP. As in the case of frustrated phagocytosis, the mobility of PM–GFP at the phagosomal cup was greatly reduced compared with that estimated before presentation of the beads (not illustrated).
Rapid Dissociation/Reassociation of Akt-PH–GFP
The decreased lipid mobility observed in Fig. 6 is consistent with the restricted diffusion of PI(3,4,5)P3 observed using the PH domains (for example, Fig. 2). However, it was conceivable that the latter was not an intrinsic property of the lipid but was instead a consequence of its interaction with Akt-PH–GFP. This concern was also addressed using FRAP. A typical determination is illustrated in Fig. 7A–D. The rapid (t 1/2 ∼ 5 s) and virtually complete (80%) recovery of the fluorescence in the bleached region is noteworthy. This rapid recovery contrasts markedly with the persistence of differential fluorescence intensity between the adherent (basal) membrane and the nonadherent regions of the plasma membrane, even after much longer periods (Fig. 5 B). These observations may be interpreted to mean that PI(3,4,5)P3 is corralled within the area of contact with the IgG-coated surface, being capable of movement within but not across this boundary. However, it is more likely that the data reflect the rapid dissociation and reassociation of Akt-PH–GFP with the headgroup of PI(3,4,5)P3. This was concluded after analyzing the shape of the photobleaching pattern as a function of time (Fig. 7 E). The optical features of the system used for illumination rendered a Gaussian photobleaching pattern. As shown in Fig. 7 E, the peak of the Gaussian curve diminishes as the bleaching recovers; yet, the width of the curve remains unaltered. A widening of the Gaussian curve would be anticipated if lateral motion of the lipid–probe complex contributes significantly to the recovery process (for details see Oancea et al. 1998). Failure of the pattern to widen measurably implies that the lipid moved slowly in these IgG-activated membranes and that recovery was due instead to exchange of bleached (bound) Akt-PH–GFP with soluble fluorescent Akt-PH–GFP. These findings indicate that the restricted localization of the complex formed between PI(3,4,5)P3 and Akt-PH–GFP cannot be attributed to tight poorly reversible binding of the latter and must reflect instead a property of the phospholipid.
Figure 7.
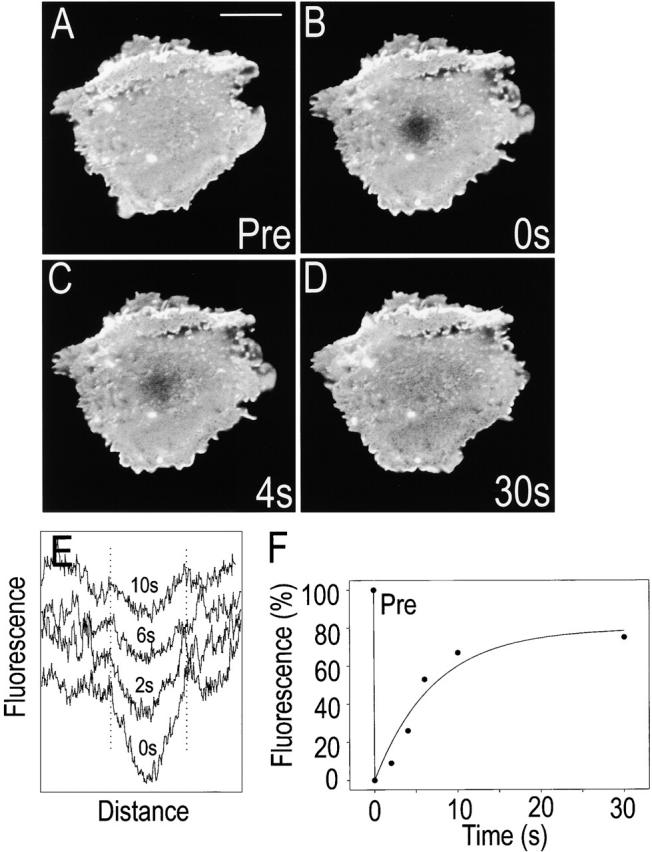
Measurements of Akt-PH–GFP FRAP. RAW 264.7 cells transfected with Akt-PH–GFP were resuspended and allowed to sediment on IgG-coated coverslips as in the legend to Fig. 5. Confocal images of the adherent membrane were acquired before (A, Pre), immediately after (B, 0 s), and at the indicated intervals following photobleaching of a spot near the middle of the membrane (C–D). In E, the fluorescence intensity along line scans traversing the photobleached area was determined at the indicated times after photobleaching. The width of the initial bleached area is demarcated by the dotted lines. In F, the relative fluorescence intensity at the center of the area selected for analysis is plotted versus time after bleaching. Data are representative of five experiments.
Contribution of Phosphatases to the Restricted Accumulation of PI(3,4,5)P3
Although reduced lipid mobility appears to influence the distribution of PI(3,4,5)P3, other mechanisms may also contribute to its restricted localization. One conceivable mechanism involves the combination of focal synthesis with rapid degradation of the lipid. If the catabolic enzymes are present outside the area of synthesis, the rate of degradation would outstrip the (modest) diffusion of the lipid out of the restricted area of biosynthesis, limiting its distribution. Such a “focal source-peripheral degradation” mechanism could contribute to PI(3,4,5)P3 localization at the phagosomal cup only if the turnover of the lipid is high, that is, if its rate of hydrolysis during the course of phagocytosis is significant. This was evaluated by measuring the rate of disappearance of PI(3,4,5)P3 from the phagosomal cup when biosynthesis was suddenly interrupted by addition of wortmannin. Large polystyrene beads were used in these experiments to slow the internalization process thereby extending the period of accumulation of PI(3,4,5)P3 at the cup. Fig. 8 shows that PI(3,4,5)P3 accumulated at the base of the cup decreases modestly over 80 s when the vehicle, dimethylsulfoxide, is added. In contrast, the inositide disappeared rapidly (t 1/2 ∼ 50 s) following the addition of the PI3K inhibitor. This is a low estimate of the rate of ongoing PI(3,4,5)P3 degradation because inhibition of PI3K is unlikely to occur instantaneously upon addition of wortmannin, which must enter the cell and react covalently with the enzyme. Moreover, hydrolysis of PI(3,4,5)P3 by a 5-phosphatase would yield phosphatidylinositol 3,4-bisphosphate (PI[3,4]P2), which would still retain Akt-PH–GFP. Fig. 8 also illustrates the distribution of actin in cells treated with or without wortmannin during the course of phagocytosis. Note that the actin cup formed under nascent phagosomes persists for ≥1 min, despite inhibition of PI3K. The rapid dissociation of the chimera under these conditions indicates that the accumulation and restricted mobility of Akt-PH–GFP are not attributable to its nonspecific binding or trapping by the actin cytoskeleton.
Figure 8.
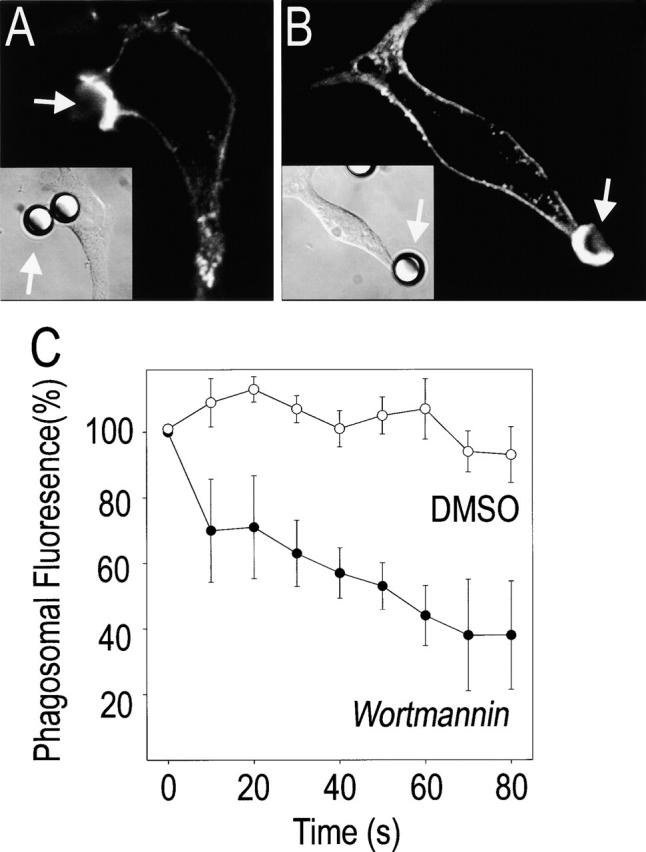
Reversal of 3′PI accumulation by wortmannin. (A and B) RAW 264.7 cells were allowed to interact with IgG-opsonized polystyrene beads (8-μm diameter), and when phagocytosis commenced the cells were treated with either 100 nM wortmannin or with an equivalent volume of the vehicle (DMSO). After 60 s, the cells were fixed, and F-actin was stained with rhodamine-phalloidin. A and B show F-actin distribution, and insets show the corresponding DIC image. (A) DMSO-treated; (B) wortmannin-treated. In C, the cells were transfected with Akt-PH–GFP ∼24 h before exposure to the beads. The concentration of the chimera at the phagosomal cup was monitored by fluorescence microscopy, and when a sizable accumulation occurred the samples were treated with wortmannin or DMSO. The persistence of Akt-PH–GFP was monitored thereafter at 10 s intervals. Data were normalized to the fluorescence at the time of addition of the inhibitor to facilitate comparison. Data are mean ± standard error of three experiments.
In the immunological synapse of lymphocytes, receptors and the associated molecules are arranged concentrically, generating microdomains where signaling is restricted (Monks et al. 1998). It is conceivable that a similar situation exists in the phagosomal cup, perhaps contributing to the observed restricted lipid distribution. Specifically, phosphoinositide phosphatases may accumulate in the periphery of the phagosome, preventing diffusion of PI(3,4,5)P3 out of the phagosomal boundary. This notion was tested experimentally. The distribution of phosphatases capable of removing the phosphate group at positions 3 and 5 of the inositol ring of PI(3,4,5)P3 was analyzed during the course of phagocytosis (Fig. 9). We initially studied the distribution of the 3-phosphatase PTEN (Stambolic et al. 1998) in cells at rest and during phagocytosis. Immunostaining of the endogenous PTEN using an affinity purified antibody revealed a coarse punctate distribution throughout the cytoplasm and substantial accumulation in the nucleus with no evidence of plasmalemmal association. This distribution was not altered during the course of phagocytosis and association of PTEN with the phagosomal cup or with the formed phagosome was never observed (Fig. 9A and Fig. B).
Figure 9.
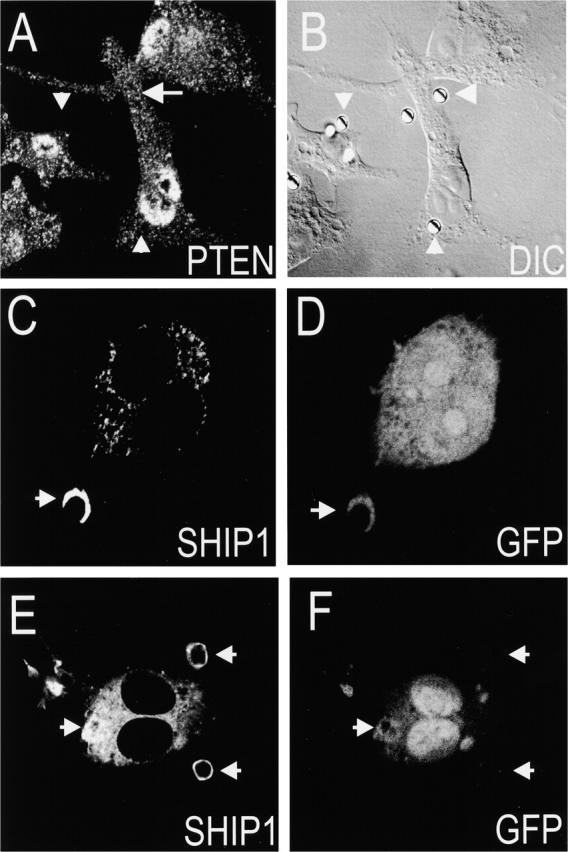
Distribution of phosphoinositide phosphatases during phagocytosis. (A and B) Cells were exposed to human IgG-opsonized latex beads to initiate phagocytosis. After fixation and permeabilization, the distribution of endogenous PTEN was revealed by immunostaining using an affinity purified polyclonal antibody. Fluorescence (A) and DIC (B) images are shown. (C–F) RAW 264.7 cells were transfected with epitope-tagged SHIP1 and GFP and then exposed to opsonized SRBCs for either ∼6 (C–D) or ∼8 min (E–F). After fixation and permeabilization, the localization of SHIP1 was revealed by immunostaining using anti-HA monoclonal antibodies (C and E). The fluorescence of GFP is also shown (D and F). Arrows indicate the location of nascent (A–D) or sealed phagosomes (E–F). Results are representative of at least five experiments of each type.
The distribution of the 5′-phosphatase SHIP1 was studied by transfection of an epitope-tagged form of the enzyme. As described recently by Cox et al. 2001, we find that heterologous expression of SHIP1 produces partial inhibition of phagocytosis (not illustrated). SHIP1 was also distributed throughout the cytoplasm with little plasmalemmal association, but unlike PTEN it was excluded from the nucleus. Importantly, SHIP1 accumulated greatly on the phagosomal cup (Fig. 9 C) and remained associated with the phagosome after closure, likely contributing to the disappearance of PI(3,4,5)P3 that follows completion of phagocytosis (Fig. 9 E). These results are consistent with those of Cox et al. 2001 who localized the endogenous SHIP1 of primary macrophages. Of note, lateral sections of forming phagosomes, such as that in Fig. 9 C, failed to reveal any preferential accumulation of SHIP1 at the periphery of cup, showing instead comparable or even higher levels of the phosphatase at the center of the cup. Therefore, although the accumulation of SHIP1 may account for the fast turnover of PI(3,4,5)P3, we found no evidence that a circumferential distribution of this phosphatase contributes to the restricted distribution of the phosphoinositide.
Discussion
Probe Specificity
Two lines of evidence indicate that the PH domain–GFP chimeras used in our studies are suitable indicators of the distribution of 3-phosphoinositides in intact macrophages. First, their accumulation at the phagosome was eliminated by treatment with wortmannin (Fig. 1) or LY294002 (not shown), which are potent inhibitors of PI3Ks (Cox et al. 1999; Walker et al. 1999). Second, unlike the wild-type Btk PH domain, a mutated form that lacks the ability to bind 3-phosphoinositides in vitro failed to associate with the phagosomal membrane (Fig. 1).
The main probe used in our studies, namely the PH domain of Akt, binds with comparable affinity to PI(3,4,5)P3 and PI(3,4)P2 (Rameh et al. 1997; Kavran et al. 1998). However, we believe that PI(3,4,5)P3 was a major contributor to our measurements, since the PH domains of Gab1 and Btk also concentrated at the phagosome. The affinity of these PH domains for the inositol headgroup phosphorylated only on positions 3 and 4 is much lower than that for the 3, 4, and 5 phosphorylated species (Rodrigues et al. 2000). Nevertheless, accumulation of PI(3,4)P2 at sites of phagocytosis cannot be ruled out and may have contributed to our determinations when using Akt-PH–GFP.
It is also noteworthy that the probes did not alter the function of the transfected cells. Phagocytic efficiency in cells expressing Akt-PH–GFP was indistinguishable from untransfected controls. This observation is compatible with the notion that the probe is in a fast dynamic equilibrium with the phosphoinositides, as suggested by the pattern of recovery after photobleaching (Fig. 7) and by the ability of phosphatases to access and rapidly degrade PI(3,4,5)P3 following addition of wortmannin (Fig. 8).
Time Course of PI(3,4,5)P3 Accumulation
Accumulation of PI(3,4,5)P3 at sites of phagocytosis was an early event detectable during the course of pseudopod extension before closure of the phagosomal vacuole (Fig. 2). The appearance of PI(3,4,5)P3 coincides with the equally localized disappearance of PI(4,5)P2 from the base of the cup (Fig. 3; Botelho et al. 2000). Although degradation of PI(4,5)P2 is due in part to hydrolysis by PLC, conversion to PI(3,4,5)P3 is also likely to contribute to its disappearance.
The early stages of the phagocytic process involve active actin remodelling and focal exocytosis of endomembranes. It is likely that the generation of PI(3,4,5)P3 plays a role in these processes, although the requirement for PI3K in actin polymerization remains controversial. On one hand, clustering of membrane-bound type I PI3K suffices to induce actin assembly and phagocytosis (Lowry et al. 1998). Yet, although phagocytosis of large particles is eliminated when PI3K activity is inhibited by wortmannin at least some actin polymerization persists at the sites of frustrated phagocytosis (Cox et al. 1999). A precise quantitation of F-actin under the latter conditions will be required to resolve the apparent inconsistencies.
The accumulation of PI(3,4,5)P3 at the phagosome is transient, becoming undetectable within 2–3 min of phagosomal sealing. The disappearance of the phosphoinositide follows closely the detachment of type I PI3K, suggesting the presence of active catabolic enzymes at the phagosome. Accordingly, SHIP1 was found to accumulate on the phagosomal membrane, where it persisted throughout the period of PI(3,4,5)P3 degradation. Interestingly, the time of disappearance of PI(3,4,5)P3 coincides approximately with the disassembly of phagosomal F-actin and with the onset of maturation. The earliest identified stages of phagosomal maturation are thought to be controlled by Rab5 (Duclos et al. 2000; Roberts et al. 2000), which purportedly catalyzes the fusion of endosomes via EEA1 (McBride et al. 1999). The latter protein attaches to the membrane in part by interaction with phosphatidylinositol 3-phosphate (PI[3]P) (Gaullier et al. 2000). Preliminary experiments using the FYVE domain of EEA1 indicate that PI(3)P accumulates greatly in phagosomes, but only after sealing, at a time when PI(3,4,5)P3 is disappearing (our unpublished observations). PI(3)P can be synthesized directly by type III PI3K but can also be generated in principle by degradation of the products of the type I PI3K. In this regard, the hydrolysis of PI(3,4,5)P3 by SHIP1 would contribute to the generation of PI(3,4)P2, which could in turn serve as a substrate of a 4-phosphatase to produce PI(3)P. Whether degradation of type I PI3K products contributes to phagosomal maturation remains to be established.
Mechanism of Focal Accumulation of PI(3,4,5)P3
The diffusion coefficients estimated for a variety of lipids in biological membranes, including 3′PI, range from 2–30 × 10−9 cm2/s (Morrot et al. 1986; el Hage Chahine et al. 1993; Gilmanshin et al. 1994; Haugh et al. 2000). It is possible on this basis to predict the mean distance traveled by a phospholipid along the membrane over the period required for completion of phagocytosis (30–60 s for SRBCs). We calculate that PI(3,4,5)P3 generated at the early stages of phagocytosis could travel up to ∼25 μm from the site of phagocytosis before sealing of the vacuole. This distance is much greater than the circumference of the phagosomal cup, and noticeable diffusion of PI(3,4,5)P3 out of the phagosomal boundary would be predicted on this basis. This prediction was clearly not fulfilled, and two mechanisms that could account for the unexpected behavior were contemplated. On one hand, we considered a model of “focal source–peripheral degradation,” whereby diffusion of PI(3,4,5)P3 synthesized focally at the cup would be limited by degradative enzymes (likely phosphatases) localized peripherally. In an extreme scenario, the phosphatases would have a circumferential distribution delimiting the phagosomal cup. This model requires that PI3K be accumulated at the site of particle attachment and that PI(3,4,5)P3 turn over rapidly. As shown in Fig. 4 and Fig. 8, both of these conditions were met. However, we failed to detect a phosphatase located in the periphery of the phagosome: PTEN was not found at the plasma membrane, and SHIP1 appeared to concentrate at the phagosome but with comparable density throughout. SHIP1 is known to associate with the immunoreceptor tyrosine inhibition motif of FcγIIB receptors (Lesourne et al. 2001), dampening the formation of polyphosphoinositides and modulating the immune response (Cox et al. 2001). Clustering of FcγRIIB by the opsonized particles, which is expected to occur homogeneously throughout the area of contact with the particle, is likely to account for the accumulation of SHIP1 at the phagocytic cup. Despite our failure to detect peripheral phosphatases, the “focal source–peripheral degradation” model cannot be disregarded and remains an attractive possibility. Microdomains of SHIP1 or PTEN may have escaped our detection and other phosphatases not tested (for example, SHIP2 or other heretofore unidentified enzymes) may distribute in the periphery of the phagosome.
A second mechanism that could contribute to the restricted distribution of PI(3,4,5)P3 is the reduction in lipid mobility observed when Fc receptors are engaged. Though 3′PI mobility could not be measured directly, we found that a di-acylated GFP construct that, like PI(3,4,5)P3, inserts into the inner leaflet of the plasmalemma could be used reliably for FRAP measurements. Although the majority of this probe (95%) diffused freely before stimulation of the cells with a coefficient in the range reported for other lipids in biological membranes (∼3.5 × 10−9 cm2/s), we found that its mobility was severely curtailed upon cross-linking of Fc receptors. Aggregation of lipid rafts could account for this phenomenon, since proteins acylated with saturated chains (such as our PM–GFP probe) and polyphosphoinositides (Laux et al. 2000) are known to partition preferentially into cholesterol- and glycolipid-rich domains. However, the evidence that lipid rafts are involved in opsonic phagocytosis is controversial (Gatfield and Pieters 2000; Peyron et al. 2000), and their involvement in lipid immobilization at the phagosomal cup will require detailed analysis. Alternatively, the alteration in lipid mobility may result from redistribution of lipids in a raft-independent manner or from the establishment of associations with proteins that are redistributed by interaction of Fc receptors with the opsonized particle. The receptors and/or associated proteins may bind to or “fence in” lipids within regions of restricted mobility. Localized ionic changes or cytoskeletal rearrangements may also contribute to reduce lipid mobility.
Regardless of the underlying mechanism, the occurrence of focal accumulation of lipids that serve messenger roles has important implications. PI(3,4,5)P3 and/or other 3′PIs are expected to recruit to the phagosomal cup a variety of proteins containing PH domains, the same way they direct the accumulation of the GFP probes. Proteins such as Vav and Arf nucleotide exchange factors, which possess such PH domains, are likely to play a critical role in actin and membrane remodelling. In addition, kinases like Akt and Btk will influence the activity of phospholipases and other downstream effectors. Many if not all of these processes likely require precise spatial coordinates that are provided by the restricted distribution of 3′PI. The subsequent degradation of the inositides may signal the termination of some of these early events and in addition may provide secondary messengers, such as PI(3)P or other derivatives, to initiate phagosomal maturation.
In summary, formation of phagosomes appears to depend on the accurate spatial and temporal control of the distribution of inositides. The mechanisms responsible for such fine control cannot be easily studied by conventional biochemical means and will require the application and further development of noninvasive approaches to study lipid metabolism.
Acknowledgments
This work was supported by the Medical Research Council of Canada, the Arthritis Society of Canada, the National Sanatorium Association, and by National Institutes of Health grant HL28207. J.G. Marshall is supported by a fellowship from the Research Institute of the Hospital for Sick Children. J.W. Booth is the recipient of a fellowship from Canadian Cystic Fibrosis Foundation. S. Grinstein is an International Scholar of the Howard Hughes Medical Institute and the current holder of the Pitblado Chair in Cell Biology.
Footnotes
Abbreviations used in this paper: DIC, differential interference contrast; GFP, green fluorescent protein; HA, influenza virus hemagglutinin; PH, pleckstrin homology; PI(3)P, phosphatidylinositol 3-phosphate; PI(3,4)P2, phosphatidylinositol 3,4-bisphosphate; PI(3,4,5)P3, phosphatidylinositol 3,4,5-trisphosphate; PI(4,5)P2, phosphatidylinositol 4,5-bisphosphate; PI3K, phosphatidylinositol 3-kinase; SRBC, sheep red blood cell; 3′PI, 3′ phosphoinositide.
References
- Aderem A., Underhill D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- Araki N., Johnson M.T., Swanson J.A. A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 1996;135:1249–1260. doi: 10.1083/jcb.135.5.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D., Koppel D.E., Schlessinger J., Elson E., Webb W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzoni L., Kamoun M., Salcedo T.W., Kanakaraj P., Perussia B. Stimulation of Fc gamma RIIIA results in phospholipase C-gamma 1 tyrosine phosphorylation and p56lck activation. J. Exp. Med. 1992;176:1745–1750. doi: 10.1084/jem.176.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho R.J., Teruel M., Dierckman R., Anderson R., Wells A., York J.D., Meyer T., Grinstein S. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D., Tseng C.C., Bjekic G., Greenberg S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension. J. Biol. Chem. 1999;274:1240–1247. doi: 10.1074/jbc.274.3.1240. [DOI] [PubMed] [Google Scholar]
- Cox D., Dale B.M., Kashiwada M., Helgason C.D., Greenberg S. A regulatory role for Src Homology 2 domain-containing inositol 5′-phosphatase (SHIP) in phagocytosis mediated by Fcγ receptors and complement receptor 3 (α(M)β(2); CD11b/CD18) J. Exp. Med. 2001;193:61–72. doi: 10.1084/jem.193.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley M.T., Costello P.S., Fitzer-Attas C.J., Turner M., Meng F., Lowell C., Tybulewicz V.L., DeFranco A.L. A critical role for Syk in signal transduction and phagocytosis mediated by Fc gamma receptors on macrophages. J. Exp. Med. 1997;186:1027–1039. doi: 10.1084/jem.186.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daeron M. Fc receptor biology. Annu. Rev. Immunol. 1997;15:203–234. doi: 10.1146/annurev.immunol.15.1.203. [DOI] [PubMed] [Google Scholar]
- Duclos S., Diez R., Garin J., Papadopoulou B., Descoteaux A., Stenmark H., Desjardins M. Rab5 regulates the kiss and run fusion between phagosomes and endosomes and the acquisition of phagosome leishmaniacidal properties in RAW 264.7 macrophages. J. Cell Sci. 2000;113:3531–3541. doi: 10.1242/jcs.113.19.3531. [DOI] [PubMed] [Google Scholar]
- el Hage Chahine J.M., Cribier S., Devaux P.F. Phospholipid transmembrane domains and lateral diffusion in fibroblasts. Proc. Natl. Acad. Sci. USA. 1993;90:447–451. doi: 10.1073/pnas.90.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzer-Attas C.J., Lowry M., Crowley M.T., Finn A.J., Meng F., DeFranco A.L., Lowell C.A. Fc gamma receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J. Exp. Med. 2000;191:669–682. doi: 10.1084/jem.191.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfield J., Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 2000;288:1647–1650. doi: 10.1126/science.288.5471.1647. [DOI] [PubMed] [Google Scholar]
- Gaullier J.M., Ronning E., Gillooly D.J., Stenmark H. Interaction of the EEA1 FYVE finger with phosphatidylinositol 3-phosphate and early endosomes. Role of conserved residues. J. Biol. Chem. 2000;275:24595–24600. doi: 10.1074/jbc.M906554199. [DOI] [PubMed] [Google Scholar]
- Gilmanshin R., Creutz C.E., Tamm L.K. Annexin IV reduces the rate of lateral diffusion and changes the fluid phase structure of the lipid bilayer when it binds to negatively charged membranes. Biochemistry. 1994;33:8225–8232. doi: 10.1021/bi00193a008. [DOI] [PubMed] [Google Scholar]
- Greenberg S. Diversity in phagocytic signalling. J. Cell Sci. 2001;114:1039–1040. doi: 10.1242/jcs.114.6.1039. [DOI] [PubMed] [Google Scholar]
- Haugh J.M., Codazzi F., Teruel M., Meyer T. Spatial sensing in fibroblasts mediated by 3′ phosphoinositides. J. Cell Biol. 2000;151:1269–1279. doi: 10.1083/jcb.151.6.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchliffe K.A., Ciruela A., Irvine R.F. PIPkins1, their substrates and their productsnew functions for old enzymes. Biochim. Biophys. Acta. 1998;1436:87–104. doi: 10.1016/s0005-2760(98)00140-4. [DOI] [PubMed] [Google Scholar]
- Kavran J.M., Klein D.E., Lee A., Falasca M., Isakoff S.J., Skolnik E.Y., Lemmon M.A. Specificity and promiscuity in phosphoinositide binding by pleckstrin homology domains. J. Biol. Chem. 1998;273:30497–30508. doi: 10.1074/jbc.273.46.30497. [DOI] [PubMed] [Google Scholar]
- Kiefer F., Brumell J., Al-Alawi N., Latour S., Cheng A., Veillette A., Grinstein S., Pawson T. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol. Cell. Biol. 1998;18:4209–4220. doi: 10.1128/mcb.18.7.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laux T., Fukami K., Thelen M., Golub T., Frey D., Caroni P. GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J. Cell Biol. 2000;149:1455–1472. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesourne R., Bruhns P., Fridman W.H., Daeron M. Insufficient phosphorylation prevents Fc{gamma}RIIB from recruiting the SH2 domain-containing protein tyrosine phosphatase SHP-1. J. Biol. Chem. 2001;276:6327–6336. doi: 10.1074/jbc.M006537200. [DOI] [PubMed] [Google Scholar]
- Liao F., Shin H.S., Rhee S.G. Tyrosine phosphorylation of phospholipase C-gamma 1 induced by cross-linking of the high-affinity or low-affinity Fc receptor for IgG in U937 cells. Proc. Natl. Acad. Sci. USA. 1992;89:3659–3663. doi: 10.1073/pnas.89.8.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry M.B., Duchemin A.M., Coggeshall K.M., Robinson J.M., Anderson C.L. Chimeric receptors composed of phosphoinositide 3-kinase domains and FCgamma receptor ligand-binding domains mediate phagocytosis in COS fibroblasts. J. Biol. Chem. 1998;273:24513–24520. doi: 10.1074/jbc.273.38.24513. [DOI] [PubMed] [Google Scholar]
- McBride H.M., Rybin V., Murphy C., Giner A., Teasdale R., Zerial M. Oligomeric complexes link Rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell. 1999;98:377–386. doi: 10.1016/s0092-8674(00)81966-2. [DOI] [PubMed] [Google Scholar]
- Monks C.R., Freiberg B.A., Kupfer H., Sciaky N., Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- Morrot G., Cribier S., Devaux P.F., Geldwerth D., Davoust J., Bureau J.F., Fellmann P., Herve P., Frilley B. Asymmetric lateral mobility of phospholipids in the human erythrocyte membrane. Proc. Natl. Acad. Sci. USA. 1986;83:6863–6867. doi: 10.1073/pnas.83.18.6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya N., Hazeki K., Fukui Y., Seya T., Okada T., Hazeki O., Ui M. Involvement of phosphatidylinositol 3-kinase in Fc gamma receptor signaling. J. Biol. Chem. 1994;269:22732–22737. [PubMed] [Google Scholar]
- Oancea E., Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- Oancea E., Teruel M.N., Quest A.F., Meyer T. Green fluorescent protein (GFP)-tagged cysteine-rich domains from protein kinase C as fluorescent indicators for diacylglycerol signaling in living cells. J. Cell Biol. 1998;140:485–498. doi: 10.1083/jcb.140.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron P., Bordier C., N'Diaye E. N., Maridonneau-Parini I. Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol-anchored proteins. J. Immunol. 2000;165:5186–5191. doi: 10.4049/jimmunol.165.9.5186. [DOI] [PubMed] [Google Scholar]
- Rameh L.E., Arvidsson A., Carraway K.L., III, Couvillon A.D., Rathbun G., Crompton A., VanRenterghem B., Czech M.P., Ravichandran K.S., Burakoff S.J. A comparative analysis of the phosphoinositide binding specificity of pleckstrin homology domains. J. Biol. Chem. 1997;272:22059–22066. doi: 10.1074/jbc.272.35.22059. [DOI] [PubMed] [Google Scholar]
- Roberts R.L., Barbieri M.A., Ullrich J., Stahl P.D. Dynamics of rab5 activation in endocytosis and phagocytosis. J. Leukoc. Biol. 2000;68:627–632. [PubMed] [Google Scholar]
- Rodrigues G.A., Falasca M., Zhang Z., Ong S.H., Schlessinger J. A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol. Cell. Biol. 2000;20:1448–1459. doi: 10.1128/mcb.20.4.1448-1459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V., Suzuki A., de la Pompa J.L., Brothers G.M., Mirtsos C., Sasaki T., Ruland J., Penninger J.M., Siderovski D.P., Mak T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Stauffer T.P., Meyer T. Compartmentalized IgE receptor-mediated signal transduction in living cells. J. Cell Biol. 1997;139:1447–1454. doi: 10.1083/jcb.139.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T., Kono H., Hirose N., Okada M., Yamamoto T., Yamamoto K., Honda Z. Differential involvement of Src family kinases in Fc gamma receptor-mediated phagocytosis. J. Immunol. 2000;165:473–482. doi: 10.4049/jimmunol.165.1.473. [DOI] [PubMed] [Google Scholar]
- Swanson J.A., Johnson M.T., Beningo K., Post P., Mooseker M., Araki N. A contractile activity that closes phagosomes in macrophages. J. Cell Sci. 1999;112:307–316. doi: 10.1242/jcs.112.3.307. [DOI] [PubMed] [Google Scholar]
- Teruel M.N., Blanpied T.A., Shen K., Augustine G.J., Meyer T. A versatile microporation technique for the transfection of cultured CNS neurons. J. Neurosci. Methods. 1999;93:37–48. doi: 10.1016/s0165-0270(99)00112-0. [DOI] [PubMed] [Google Scholar]
- Varnai P., Rother K.I., Balla T. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in single living cells. J. Biol. Chem. 1999;274:10983–10989. doi: 10.1074/jbc.274.16.10983. [DOI] [PubMed] [Google Scholar]
- Walker E.H., Perisic O., Ried C., Stephens L., Williams R.L. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- Zheleznyak A., Brown E.J. Immunoglobulin-mediated phagocytosis by human monocytes requires protein kinase C activation. Evidence for protein kinase C translocation to phagosomes. J. Biol. Chem. 1992;267:12042–12048. [PubMed] [Google Scholar]