

Abstract
We report four children with infantile onset lobular panniculitis, high fever, uveitis, and systemic granulomatous inflammation, recruited through the International Registry of Pediatric Granulomatous Arthritis (PGA)-. Neither CARD15 nor CIAS1 mutations were found. Despite immunosuppressive therapy, disease course was progressive. Response to anti-tumor necrosis factor (TNF) monoclonal antibody in 3 patients is of note.
Keywords: Panniculitis, Systemic Granulomatosis, Uveitis
Non-caseating epitheloid cell granulomas are likely the result of an exaggerated immune-inflammatory response against an unidentified antigen. They can be demonstrated in different chronic inflammatory conditions of which the classic example is sarcoidosis, and are distinguishable from infectious and foreign-body granulomas both histopathologically and biologically (1). In children, granulomatous inflammation is seen in immune deficiencies such as chronic granulomatous disease, systemic vasculitides, and inflammatory disorders such as Crohn disease.
The triad of granulomatous dermatitis, synovitis, and uveitis is characteristic of pediatric granulomatous arthritis (PGA) encompassing Blau syndrome--the familial form and early-onset sarcoidosis; the sporadic form; and that strongly associated with mutations in or near the NACHT domain of CARD15 (2). The International Registry and DNA Repository of PGA have been established recently. By using non-restrictive inclusion criteria, the Registry aims to advance insight into the wide spectrum of pediatric granulomatous diseases both with and without the typical PGA triad (2).
We report four children recruited through the registry who presented with infantile onset of widespread lobular panniculitis, panuveitis, arthropathy, and severe systemic illness. In all patients, granulomatous inflammation was demonstrated in involved organs and tissues.
Methods and Results
Four infants came to medical attention with prolonged high fever, systemic illness, and histologically documented lobular panniculitis. All patients had anemia and hepatosplenomegaly. With time, uveitis and arthritis became manifest, and granulomatous inflammation was demonstrated in a variety of organs and tissues (Table). Patient 4 showed hypercalcemia and hypercalciuria. For patient 1, disease course was progressive despite cortico-steroid and cyclosporine treatment, with pulmonary involvement and death from respiratory insufficiency at age 14. In patients 2, 3, and 4, treatment with anti-TNF monoclonal antibody allowed better disease control.
Table.
Clinical, laboratory and pathologic manifestations, and CARD15 analysis in 4 patients
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | |
|---|---|---|---|---|
| Age at onset | 6 weeks | 12 months | 1 week | 4 months |
| Painful panniculitis | + | + | + | + |
| Fever | + | + | + | + |
| Hepatosplenomegaly | + | + | + | + |
| Anemia,
Thrombocytopenia |
+
+ |
+
− |
+
+ |
+
− |
| Serum Level of angiotension converting enzyme (ACE) | Normal | Increased | Normal | Increased |
| Uveitis | Panuveitis, retinal detachment | Panuveitis | Anterior uveitis, optic nerve edema | Anterior uveitis |
| Arthritis | Oligoarticular | Polyarticular + tenosynovitis | Oligoarticular | Polyarticular + tenosynovitis |
| Lipoatrophy | + | − | − | − |
| Epitheloid cell granulomata | Liver | Dermis, synovium | Lymph node, salivary gland, colonic submucosa, subcutaneous fat | Dermis, lung |
| CARD15 mutation | No | No | No | No |
| Treatment | Cortico-steroids, cyclosporine, IGIV | Cortico-steroids, MTX, TNF-antagonists | Cortico-steroids, IGIV, TNF-antagonists | Cortico-steroids, colchicine, cyclosporine, soluble TNF receptor, thalidomide |
| Course | Respiratory failure and death | Response to anti-TNF MoAb | Moderate response to anti-TNF MoAb | Response to anti-TNF MoAb |
Histopathologically, all had numerous infiltrating histiocytes with lymphocytes and neutrophils in the subdermal fat, constituting a non-vasculitic, non-lipophagic, non-cytophagic lobular panniculitis. Patient 3 had diffuse histiocytic lobular panniculitis (Figure 1 A, B) and typical granuloma on followup biopsy documenting the evolving character of the process (Figure 1 C, D). Multiple granulomas composed of epitheloid cells, lymphocytes, and multinucleated giant cells were present in the liver (patient 1), synovium (patient 2), salivary gland, lymph node, colon (patient 3), dermis, and lung (patient 4). Immunohistochemical analysis showed histiocytes within a nodule and lymph node (patient 3) to be strongly positive for CD68, and negative for S100. Acid-fast and fungal stains were negative, as were cultures for bacteria, mycobacteria, and fungi.
Figure.
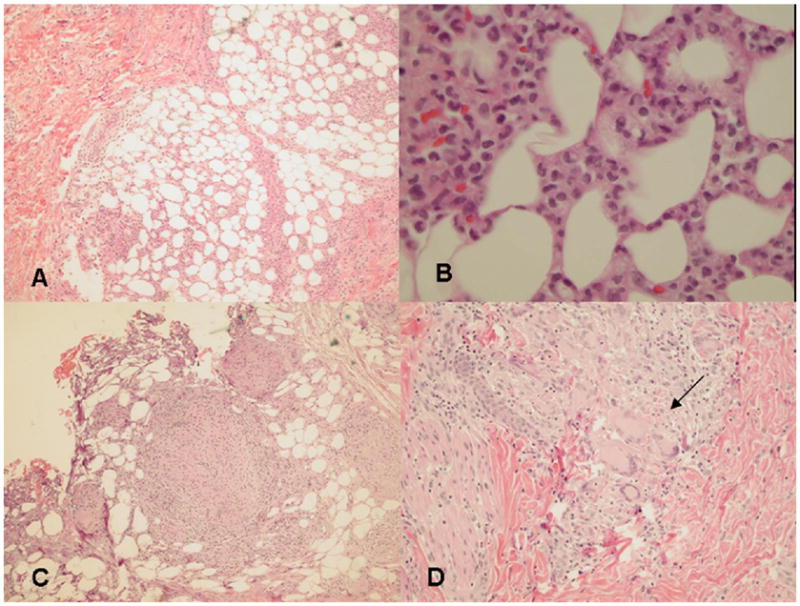
A and B: Histopathologic features of histiocytic lobular panniculitis. A. Low-power view: a mostly lobular panniculitis. B. Higher magnification: dense infiltration with lymphocytes and histiocytes.
C and D: Later stage panniculitis lesion (patient 3). C. Low-power view: granulomas within the fat lobule. D. Higher magnification: numerous lymphocytes, epitheloid cells, and multinucleated giant cells (arrow) in a granuloma extending into the dermis. (A, B, C and D, Hematoxylin-eosin stain; magnifications A, C × 20, B × 60, D × 40).
Extensive investigations ruled out infections, pancreatic disease, α1-antitrypsin deficiency, autoimmune disease, complement deficiency, hemophagocytosis or neutrophil oxidase deficiency. At presentation, immunoglobulin levels and lymphocyte counts were normal in all patients. Patient 1 developed hypogammaglobulinaemia and lymphopenia at the age of 3 years possibly related to cortico-steroid treatment from a very young age (3). CARD15 and CIAS-1 gene analyses revealed no disease-associated mutations.
Discussion
These four infants have what appears to be a previously unrecognized syndrome consisting of febrile lobular panniculitis associated with arthritis, uveitis, and widespread granulomatous inflammation. Our patients showed a severe phenotype including significant visual impairment in 2 cases and widespread granulomatous inflammation with fatal outcome in one patient. The disease is not associated with a mutation in CARD15.
Panniculitis was chronic, recurrent, lobular, lymphohistiocytic, non-lipophagic, non-cytophagic, non-vasculitic, and in one instance with documented evolution to granuloma formation. Epitheloid giant cell granulomas were documented in subcutaneous fat tissue, dermis, synovium, salivary gland, lymph node, liver, colon, and lung parenchyma confirming the systemic nature of this disease. Hypercalcemia and hypercalciuria (as seen in patient 4) is a well-known feature of granulomatous disorders reportedly due to endogenous overproduction of 1,25-dihydroxy vitamin D3 by activated macrophages (4).
Panniculitis in children is uncommon and is a cardinal feature of a wide range of disorders from the relatively common erythema nodosum, to rare entities such as idiopathic relapsing febrile lobular panniculitis (historically known as Weber Christian disease) and fatal cytophagic histiocytic panniculitis (5). Histologically, panniculitis lesions can be predominantly septal (e.g. erythema nodosum) or lobular depending on the location of the infiltrate (6). All our patients showed lobular histiocytic panniculitis and none showed vasculitis as with the lobular panniculitis of polyarteritis nodosa (5). Granulomas can be seen in septal panniculitides associated with tuberculosis, fungal infections, and Crohn disease but are rare in lobular panniculitides (5). In patient 4, histiocytic panniculitis exhibited granuloma formation at a later stage, underscoring the dynamic nature of the process as reported (6,7).
The very young age at onset of panniculitis in our patients, and the association with systemic features might raise suspicion of a cytophagic histiocytic syndrome within the spectrum of hemophagocytic disorders (5). However, a hemophagocytic syndrome was excluded by the absence of cytophagia and S100 staining in the subcutaneous tissue and absence of hemophagocytosis in bone marrow, liver, and lymph nodes. Although granulomas rarely can be observed in association with a hemophagocytic syndrome (8), widespread granuloma formation is not a characteristic feature of these disorders (9).
The similarity of this phenotype with pediatric granulomatous arthritis (PGA), a disease strongly associated with CARD15 mutations and characterized by granulomatous uveitis and arthritis (2) is intriguing. Although the function of CARD15 is not yet completely understood, an uncontrolled pro-inflammatory state and/or an apoptosis defect may be involved in granuloma formation (10). One could postulate a defect in related proteins involved in pathways of inflammation and/or apoptosis to be involved in the present disorder. Although one of our patients had documented granulomas in the synovium (patient 2), the clinical presentation in all 4 cases differed from classic PGA by prominent panniculitis, early diffuse systemic involvement, and absence of mutation in CARD15.
Anti-TNF monoclonal antibody therapy has been used successfully in granulomatous inflammatory diseases including Crohn’s and adult sarcoidosis (11). In our experience, the administration of anti-TNF agents allowed better disease control with tapering of systemic cortico-steroids. Failure to control uveitis, as reported elsewhere is of note (12).
Our report on four infants with lobular panniculitis, fever, and widespread granulomatous inflammation, underscores the complexity of the entities associated with panniculitis. We propose “Infantile Onset Panniculitis with Systemic Granulomatosis” as a new clinicopathologic entity and part of the spectrum of pediatric granulomatous inflammatory diseases.
Acknowledgments
Genetic analyses were performed by Tammy Martin PhD at Casey Eye Institute Oregon Health & Science University, Portland, OR (patients 1–3) and by Severine Drunat PhD at Hôpital Robert Debré (CARD15) and Laurence Cuisset PhD at Hôpital Cochin (CIAS1), Paris for patient 4. We thank Katrina Conard MD, pediatric pathologist from duPont Children’s Hospital, Wilmington, DE, for providing micrographs of fat tissue biopsy specimen.
Dr. Martin’s work was supported by a Career Development Award from The Research to Prevent Blindness, by NIH grant EY013139, and by the Gerlinger Award, Oregon Health Foundation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Majno G, Joris I. Chronic Inflammation. Cells, Tissues, and Diseases. Principles of General Pathology. Oxford University Press; 2004. pp. 442–476. [Google Scholar]
- 2.Rose CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, Martin TM. Pediatric Granulomatous Arthritis. An International Registry. Arthritis Rheum. 2006;54:3337–3344. doi: 10.1002/art.22122. [DOI] [PubMed] [Google Scholar]
- 3.Cupps TR, Elgar Lc, Thomas CA, Fauci AS. Multiple mechanisms of B cell immunoregulation in man after administration of in vivo corticosteroids. J Immunol. 1984;132:170–175. [PubMed] [Google Scholar]
- 4.Sharma OP. Hypercalcemia in granulomatous disorders: a clinical review. Curr Opin Pulm Med. 2000;6:442–7. doi: 10.1097/00063198-200009000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Goldsmith PC, Black MM. Erythema nodosum and other forms of panniculitis. Pediatric Dermatology. 2000;9.4:616–627. [Google Scholar]
- 6.Cascajo CD, Borghi S, Weyers W. Panniculitis. Definition of Terms and Diagnostic Strategy. The American Journal of Dermopathology. 2000;22:430–549. doi: 10.1097/00000372-200012000-00009. [DOI] [PubMed] [Google Scholar]
- 7.Requena L, Sanchez E. Panniculitis. J Am Acad Dermatol. 2001;45:163–83. doi: 10.1067/mjd.2001.114736. [DOI] [PubMed] [Google Scholar]
- 8.Brinkmeyer T, Reuter T, Metze D, Frosch PJ, Herbst RA. Disseminated hyperkeratotic and granulomatous nodules in a child with fatal Epstein-Barr-virus-associated hemophagocytic lymphohistiocytosis. Pediatr Dermatol. 2006;23:35–8. doi: 10.1111/j.1525-1470.2006.00167.x. [DOI] [PubMed] [Google Scholar]
- 9.Henter JL, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004. Diagnostic and Therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2005 doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 10.Inohara, Chamaillard, McDonald C, Nunez G. NOD proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–83. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 11.Baughman RP, Drent M, Kavuru M, Judson MA, Costabel U, du Bois R, Albera C, Brutsche M, Davis G, Donohue JF, Muller-Quernheim J, Schlenker-Herceg R, Flavin S, Lo KH, Oemar B, Barnathan ES. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006:13. doi: 10.1164/rccm.200603-402OC. [DOI] [PubMed] [Google Scholar]
- 12.Saurenmann RK, Levin AV, Rose JB, Parker S, Rabinovitch T, Tyrrell PN, Feldman BM, Laxer RM, Schneider R, Silverman ED. Tumor necrosis factor-α inhibitors in the treatment of childhood uveitis. Rheumatology (Oxford) 2006;45:982–9. doi: 10.1093/rheumatology/kel030. [DOI] [PubMed] [Google Scholar]