

Abstract
Vaccinia virus (VV) egress has been studied using confocal, video, and electron microscopy. Previously, intracellular-enveloped virus (IEV) particles were proposed to induce the polymerization of actin tails, which propel IEV particles to the cell surface. However, data presented support an alternative model in which microtubules transport virions to the cell surface and actin tails form beneath cell-associated enveloped virus (CEV) particles at the cell surface. Thus, VV is unique in using both microtubules and actin filaments for egress. The following data support this proposal. (a) Microscopy detected actin tails at the surface but not the center of cells. (b) VV mutants lacking the A33R, A34R, or A36R proteins are unable to induce actin tail formation but produce CEV and extracellular-enveloped virus. (c) CEV formation is inhibited by nocodazole but not cytochalasin D or 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo(3,4-d)pyrimidine (PP1). (d) IEV particles tagged with the enhanced green fluorescent protein fused to the VV B5R protein moved inside cells at 60 μm/min. This movement was stop-start, was along defined pathways, and was inhibited reversibly by nocodazole. This velocity was 20-fold greater than VV movement on actin tails and consonant with the rate of movement of organelles along microtubules.
Keywords: vaccinia virus; green fluorescent protein; actin tails; confocal and electron microscopy; microtubules
Introduction
Vaccinia virus (VV)* replicates in the cytoplasm and forms two distinct infectious virions termed intracellular mature virus (IMV) and extracellular enveloped virus (EEV) (Appleyard et al., 1971; Ichihashi et al., 1971). IMV particles are formed in cytoplasmic factories (Dales and Mosbach, 1968) from where they are transported in a process that requires the VV A27L gene product and microtubules (Sanderson et al., 2000). Subsequently, IMV particles are wrapped by a double membrane (Ichihashi et al., 1971; Morgan, 1976; Payne and Kristensson, 1979) derived from tubular endosomes (Tooze et al., 1993) or the trans-Golgi network (Hiller and Weber, 1985; Schmelz et al., 1994) to form intracellular enveloped virus (IEV). IEV moves to the cell surface where the outer membrane fuses with the plasma membrane to form cell-associated enveloped virus (CEV) that remains on the cell surface (Blasco and Moss, 1992) or EEV that is released from the cell. CEV is important for virus spread between adjacent cells, and EEV mediates the long-range dissemination of virus (Ichihashi et al., 1971; Boulter and Appleyard, 1973; Payne, 1980; Payne and Kristensson, 1985; Blasco and Moss, 1992).
During VV infection, the host cell cytoskeleton undergoes changes, with actin stress fibers disappearing and thickened actin tails becoming visible with virus particles at their tips (Stokes, 1976; Hiller et al., 1979; Krempien et al., 1981; Hiller and Weber, 1982; Blasco et al., 1991; Cudmore et al., 1995). It was proposed that actin tails form on IEV particles and drive these virions to the cell surface (Cudmore et al., 1995, 1996, 1997). This proposal was consistent with the observation that IEV particles are needed for actin tail formation (Cudmore et al., 1995; Wolffe et al., 1997, 1998; Sanderson et al., 1998; Zhang et al., 2000) but inconsistent with the fact that virus mutants lacking the A33R, A34R, or A36R proteins or with mutations in the B5R protein are unable to make actin tails but still produce CEV and EEV and sometimes at levels greater than wild-type virus (McIntosh and Smith, 1996; Wolffe et al., 1997; Mathew et al., 1998; Roper et al., 1998; Sanderson et al., 1998; Wolffe et al., 1998; Röttger et al., 1999). The proposal was also inconsistent with the observation that CEV particles are formed in the presence of cytochalsin D (Payne and Kristensson, 1982). Therefore, an alternative mechanism for IEV transport to the cell surface must exist.
The wrapping of IMV by intracellular membranes requires the interaction of virus protein(s) on the IMV surface and the cytosolic face of the wrapping membranes. Proteins required for wrapping include the A27L protein on IMV (Rodriguez and Smith, 1990; Sanderson et al., 2000) and the EEV proteins F13L (Hirt et al., 1986) and B5R (Engelstad and Smith, 1993; Wolffe et al., 1993). In addition, wrapping is inhibited reversibly by N 1-isonicotinoyl-N 2-3-methyl-4-chlorobenzoylhydrazine (IMCBH) (Payne and Kristensson, 1979). Actin tails mediate efficient cell-to-cell virus spread because mutants deficient in their formation produce a small plaque (Cudmore et al., 1995; Sanderson et al., 1998; Wolffe et al., 1998; Zhang et al., 2000).
Actin tail formation by VV (Cudmore et al., 1995) has similarities with Shigella, Listeria, and Rickettsia (Frischknecht and Way, 2001). However, whereas these bacteria polymerize actin from one end of the bacterium due to the polarized distribution of specific proteins, no VV protein with asymmetrical distribution on the surface of the IEV particles has been reported (Schmelz et al., 1994; Röttger et al., 1999; van Eijl et al., 2000). Indeed, the VV A36R protein that is required for polymerization of actin tails (Sanderson et al., 1998; Wolffe et al., 1998; Röttger et al., 1999) seems evenly distributed on the IEV surface (van Eijl et al., 2000). Therefore, if actin polymerizes on IEV particles it is unknown how this is polarized.
An alternative proposal was that VV-induced actin tails grow beneath the plasma membrane rather than on IEV particles (van Eijl et al., 2000). Immunoelectron microscopy showed that the A36R protein was absent from CEV particles but present on the cytosolic face of the plasma membrane beneath CEV in a position to polymerize actin and drive these virions away from the cell (van Eijl et al., 2000). Moreover, A36R mutagenesis showed that A36R tyrosine phosphorylation by a Src-family kinase is essential for actin tail formation, and this process is inhibited by 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo(3,4-d)pyrimidine (PP1) and mimics receptor tyrosine kinase signaling at the cell surface (Frischknecht et al., 1999).
In this study, we have reexamined the distribution of IEV and CEV particles by confocal, video, and electron microscopy after infection with wild-type and mutant viruses, including a new virus in which the enhanced green fluorescent protein (EGFP) is fused to the outer membrane of IEV particles. These infected cells were also treated with drugs that prevent the wrapping of IMV to form IEV or that disrupt microtubules or actin filaments. We show that IEV particles utilize microtubules to facilitate their intracellular transport to the plasma membrane, and actin tails form from the cell surface beneath CEV particles to aid their dissemination. This represents the first example of a virus that utilizes both microtubules and actin for egress from the cell.
Results
Analysis of actin tail distribution by phase–contrast and confocal microscopy
Phase–contrast microscopy of VV-infected cells reveals phase-dense virus particles and virus-induced actin tails (Fig. 1 A). However, it is not possible to determine if the actin tails are below, within, or above the cell. To investigate their location, infected cells were stained with rhodamine-phalloidin and analysed by confocal microscopy. Fig. 1 B shows a single optical section at the bottom of the cell and three sets of arrows/arrowheads indicate positions of actin tails. Fig. 1, C–E, shows vertical optical sections of this infected cell including the actin tails marked in Fig. 1 B. Panels F and G, show parts of panels B and E at higher magnification, and the two arrows show the position of the same actin tails in the horizontal (Fig. 1 F) and vertical (Fig. 1 G) sections. All actin tails (∼15 in Fig. 1 G) were located at the cell surface or between the cell and the tissue culture support. Notably, all the vertical sections of these and other cells (n = 25) failed to detect actin tails within the cell body away from the cell surface.
Figure 1.
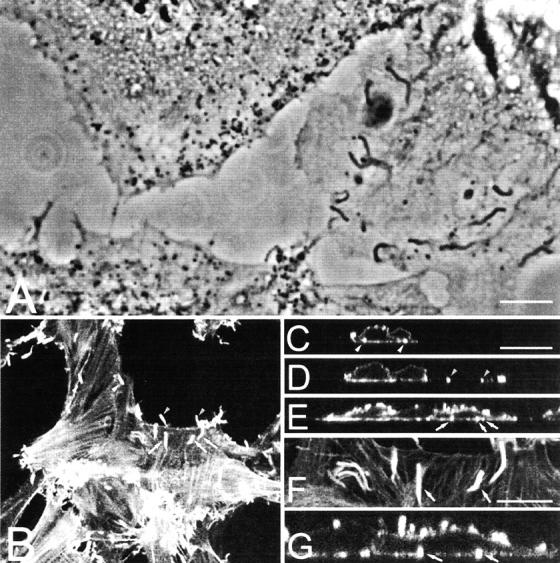
Distribution of actin tails in infected cells. (A) Phase–contrast microscopy. BS-C-1 cells were infected with VV at 1 PFU/cell and at 12 hpi fixed with methanol and photographed under phase–contrast. The actin tails with dense particles at their tips are visible. (B) Confocal microscopy. RK13 cells were infected with VV as in A and fixed and permeabilized at 9 hpi. Cells were stained with rhodamine-conjugated phalloidin, and a single optical section at the base of the cell is shown. Arrows denote pairs of actin tails that are also illustrated in the vertical optical section shown in C–E. F is a magnification of B, showing two actin tails that are shown in the vertical section in G. Bars, 5 μm.
The three-dimensional distribution of virus particles and actin tails was studied in more detail by collecting horizontal optical sections starting at the basal lateral cell surface and moving in 0.5-μm steps up through the cell. When the images were condensed as a single plane (Fig. 2 A), phalloidin-stained actin tails appeared constrained within the cell boundary and projecting away from the cell. Representative z-sections from Fig. 2 A are shown in Fig. 2, B–E, with one cell boundary outlined. Actin tails are visible close to the bottom (Fig. 2 B) and top of the cell (Fig. 2 E) or near the cell periphery (Fig. 2 D) but not within the cell lumen midway through the z-series (Fig. 2 C). A vertical section (Fig. 2 F) shows two actin tails (arrows) that are also marked in panel B. One tail is at the bottom of the cell, whereas the other protrudes away from the cell. As in Fig. 1, actin tails were not observed within the cytoplasm separated from the plasma membrane. Fig. 2 G is a stereo image of the z-sections of Fig. 2 A in which the three-dimensional organization of actin fibers and virus-induced actin tails can be observed more clearly.
Figure 2.
VV-induced actin tails are formed from the cell surface. RK13 cells were infected with VV, stained with phalloidin, and analyzed by confocal microscopy as described in the legend to Fig. 1 B. A shows a z-series reconstruction (17 sections), and G shows this projection as a stereo anaglif. B–E show projections of individual optical sections moving from the basolateral (B) to the apical surface (E). The outline of the cell is drawn with a dotted line. F shows a vertical section of the cell illustrating the positions of the actin tails shown in B. Arrows in B and F mark the same actin tails. Bar, 5 μm.
Analysis of actin tail distribution by EM
The optical sections taken close to the basolateral cell membrane (Fig. 1, B and F, and Fig. 2 B) showed actin tails, but it was not evident if these and others were within or beneath the cell. This was studied by EM of vertical sections of infected cells. Fig. 3 A shows some electron-dense IEV and CEV particles and an actin tail protruding into the space between the culture support and the cell. In this example, the distinctive actin tail does not possess a virus particle at its tip, perhaps because it is located in another plane or has been released already. Fig. 3 B shows additional examples of actin tails beneath the cell, in some cases with a CEV at their tip. Such actin tails have little space into which to extend and so might push back into the cell. A virus particle at the tip of such an actin tail would appear to be within the cell, and an image of an intracellular virus-tipped actin tail has been published (Cudmore et al., 1995). However, a virion at the tip of an actin tail that has reentered the cell can be distinguished from an actin tail that polymerized from an IEV particle within a cell by whether or not it was associated with additional cellular membranes. An IEV particle that polymerized actin would have no membrane bordering the actin tail. In contrast, an actin tail that had grown from the cell surface and then reentered the cell would be surrounded by two membranes, representing the plasma membranes acquired during actin tail formation and during protrusion back into the cell. Fig. 3 C shows a virus-tipped actin tail protruding from one cell (bottom) into another cell (top) and membranes down either side of the tail. In addition, the outer membrane encloses the CEV particle at the tip of the actin tail. Fig. 3, D–F, shows an additional example taken from deeper within the cell. In Fig. 3 D, the number of membranes surrounding the virus particle and tail are not well discerned. However, when the sample is tilted ± 35° in the electron beam (Fig. 3, E and F) the membranes down either side of the actin tail become clear. The insets show a higher magnification of the multiple membranes on either side of this CEV particle. The example shown in Fig. 3, D–F, illustrates the rare situation where a CEV particle has reentered the cell and appears as an intracellular particle associated with an actin tail. In contrast, it was easy to find numerous IEV particles, none of which were associated with actin tails, and numerous CEV particles that were associated with actin tails (for examples see Figs. 3 and 4) .
Figure 3.
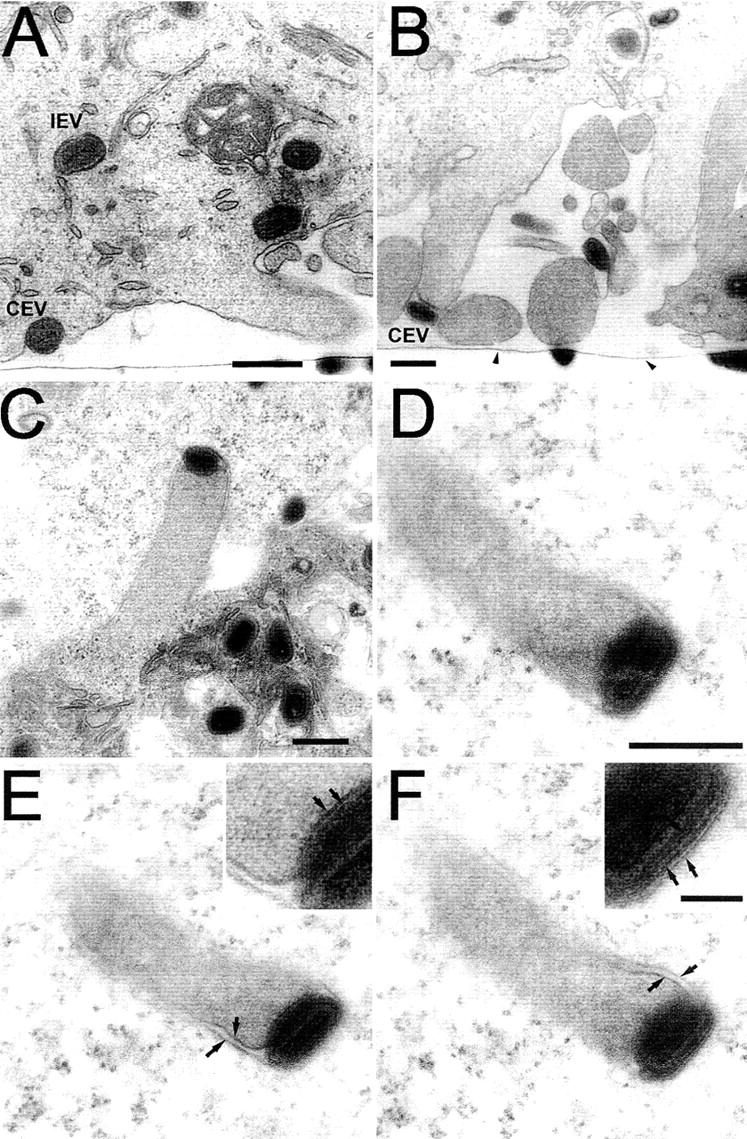
EM of VV-tipped actin tails. HeLa cells were infected with VV at 1 PFU/cell and processed for thin section transmission electron microscopy at 9 hpi. (A and B) Vertical section through the infected cell; note the tissue culture plastic (arrowheads). (A) Basolateral region showing IEV and CEV particles and an actin tail protruding into the space between the cell surface and the tissue culture plastic. (B) Further examples of actin tails and CEV particles beneath the cell. (C–F) CEV particles pushing into neighboring cells on actin tails. In E and F, the sample shown in D has been tilted ± 35°, respectively, to bring the membranes into focus. Arrows in the magnified inserts highlight the membranes surrounding the actin tail and CEV particle. Bars: (A) 1 μm; (B–D) 500 nm; (F, inset) 100 nm.
Figure 4.
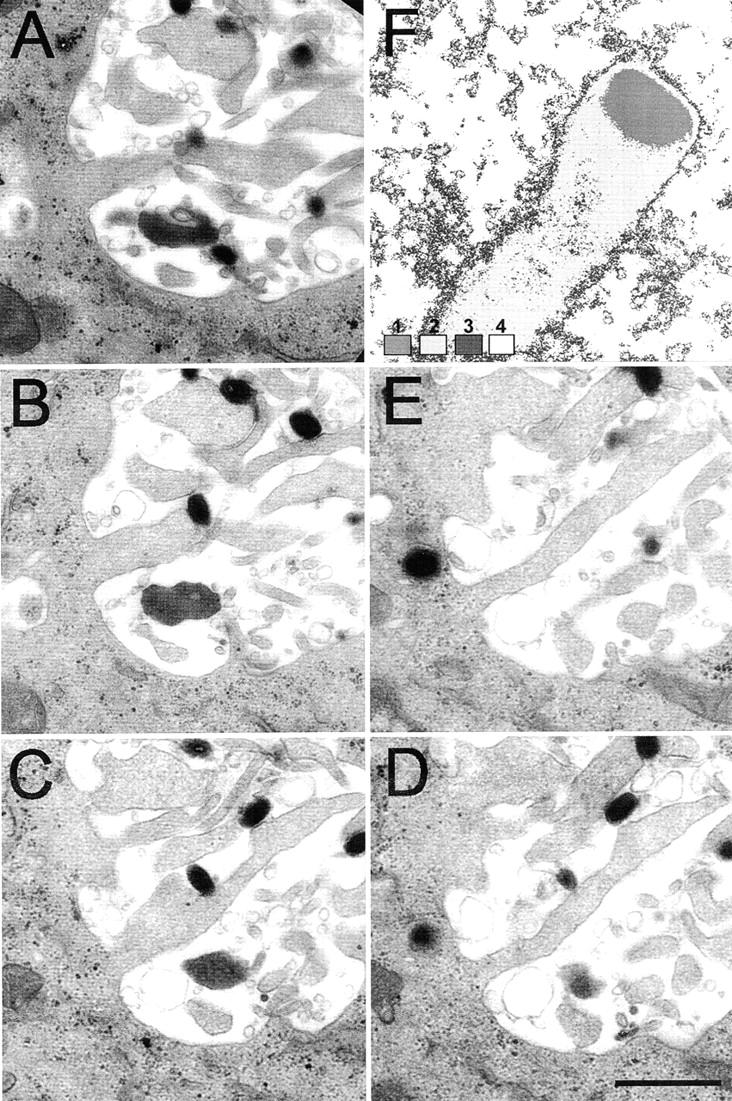
EM of serial sections. HeLa cells were infected with VV and processed for EM as described in the legend to Fig. 3. Consecutive sections (80-nm apart) from the basolateral surface (A) going up into the cell (ending in E) are shown. F shows an electron density scan of the image shown in Fig. 3 D to illustrate the uniform electron density of the actin tail that is distinct from that of the surrounding cytosol. 8-Bit gray scale image coded for gray values 0–86 = 1, 87–170 = 2, 171–190 = 3, and 191–255 = 4. Bar, 1 μm.
The characteristic and uniform electron density of the actin tails compared with the cell cytoplasm (Fig. 4 F) provides additional evidence that actin tails are growing from the cell surface (Figs. 3 and 4). If an actin tail was polymerizing from an IEV particle within a cell, the characteristic electron density of the actin tail would extend from deeper within that cell and not just from the cell surface as occurs in Fig. 3 C. This was examined further in Fig. 4, A–E, in which serial sections were cut to search for actin tails that might extend from deeper within the cell in a different section plane. The images show several actin tails emerging from a similar location on the cell surface. These are of different lengths outside the cell, and in several of the sections a CEV particle is visible at the tip of the actin tail. However, in no case (n = 100) was there evidence of actin tails extending into the granular cytoplasm of the cell.
Actin tail formation by CEV particles requires microtubule-dependent IEV particle egress
Since actin tails are formed from the cell surface, we investigated if microtubules facilitate IEV egress by using the microtubule-depolymerizing drug nocodazole (Storrie et al., 1998). However, because microtubules are needed for the movement of IMV particles from the virus factory to the site of wrapping to form IEV particles (Sanderson et al., 2000), nocodazole would prevent the formation of IEV and CEV particles and thereby actin tails indirectly. Therefore, we used the drug IMCBH that arrests morphogenesis, reversibly, after IMV but before IEV formation (Payne and Kristensson, 1979; Schmutz et al., 1991) and studied actin tail formation after IMCBH washout in the presence or absence of nocodazole (Fig. 5) . Without IMCBH treatment, actin tails are evident, protruding from VV-infected cells (Fig. 5 A), whereas in the presence of IMCBH actin tails are not formed, and there are a greater number of actin stress fibers (Fig. 5 B), a feature characteristic of virus mutants unable to make actin tails (Sanderson et al., 1998). These data are consistent with previous observations (Cudmore et al., 1995). Within 60 min of IMCBH washout, numerous actin tails are evident (Fig. 5 C). However, if IMCBH is washed out in the presence of nocodazole, actin tails are not formed (Fig. 5 D).
Figure 5.
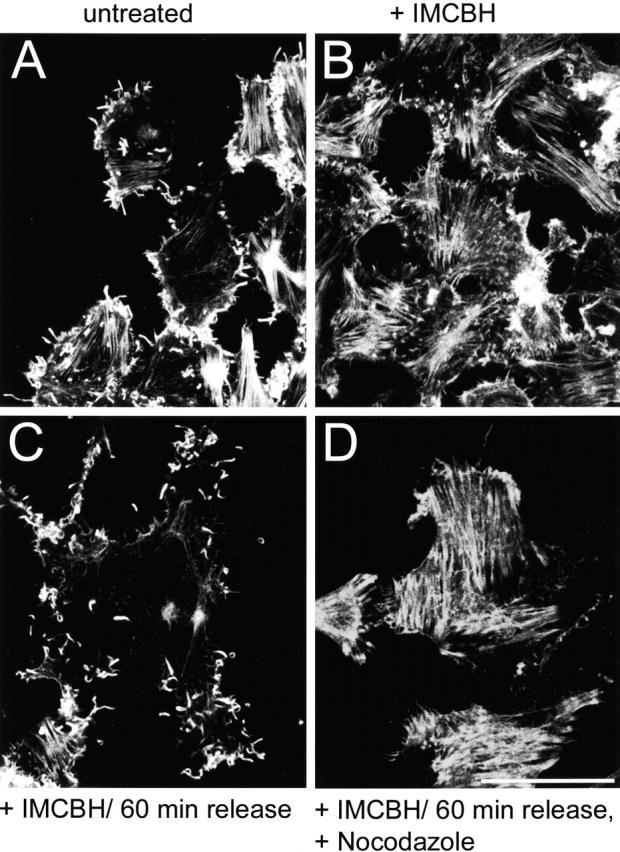
Microtubules and IEV particles are required for the formation of actin tails. RK13 cells were infected with VV at 10 PFU/cell in either the absence (A) or presence (B–D) of IMCBH. At 8 hpi, the cells in C and D were washed three times in DME (C) or in DME containing 33 μM nocodazole (D). 1 h later, all cells were fixed and stained with rhodamine-conjugated phalloidin as described in Materials and methods. Samples were analyzed by confocal microscopy, and the reconstructed z-series are shown. Bar, 10 μm.
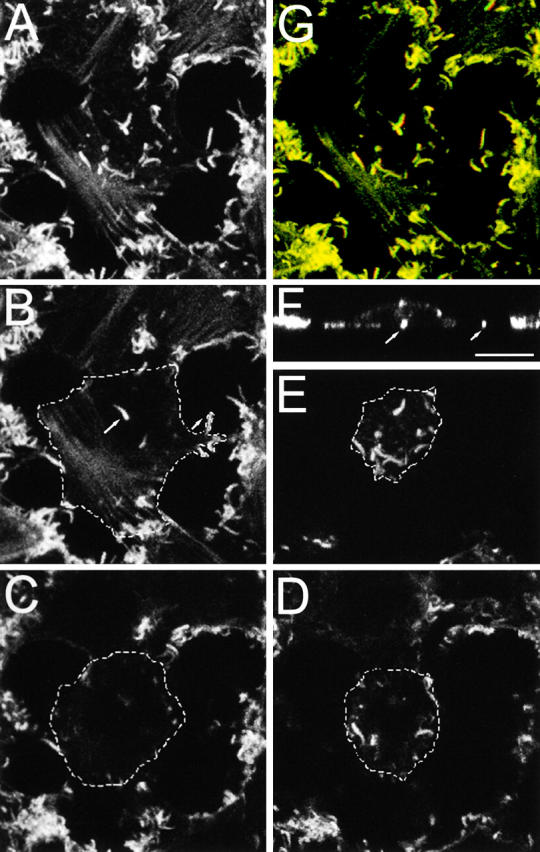
The effect of nocodazole on the distribution of VV particles was investigated by immunofluorescence. Cells were infected for 11.5 h and then incubated with or without drug for 1.5 h and stained for VV protein B5R (Fig. 6) . Without nocodazole clusters of VV, particles are seen in peripheral regions of the cell (Fig. 6 A), and this clustering is inconsistent with the movement of IEV particles driven by actin tails, which would be random. In contrast, in the presence of nocadazole VV particles were distributed randomly in the cytoplasm consistent with the fragmentation of the microtubular network (Fig. 6 B).
Figure 6.
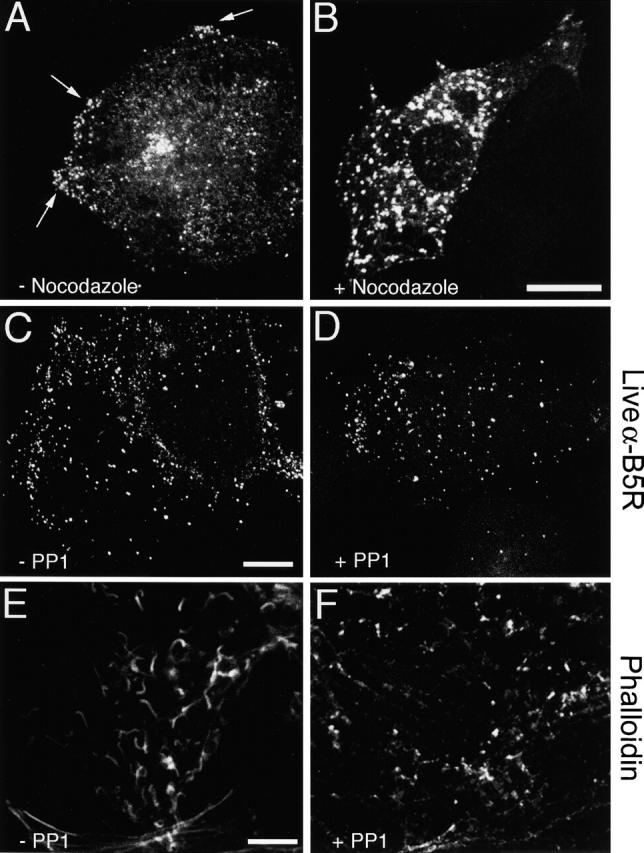
Confocal microscopy of VV-infected cells. (A–B) Microtubules are needed for IEV transport to the cell periphery. RK13 cells were infected with VV at 1 PFU/cell for 11.5 h. The medium was replaced by either fresh DME (A) or DME containing 33 μm nocodazole (B). At 13 hpi, the cells were fixed, permeabilized, and stained with Mab 19C2 (anti-B5R). Arrows indicate clusters of virus particles near the cell surface. (C–F) PP1 blocks the formation of actin tails but not CEV particles. RK13 cells were infected with VV at 1 PFU/cell, and at 5 hpi PP1 was added to a final concentration of 20 μM. (C and D) At 11 hpi, the medium was replaced with DME containing mAb 19C2 (anti-B5R) (live cells), and the cells were incubated for 1 h before fixation and processing for fluorescent microscopy. (E and F) Cells were fixed at 12 hpi and stained with and rhodamine-conjugated phalloidin. Bars, 10 μm.
Inhibition of tyrosine phosphorylation by PP1 blocks actin tail but not CEV particle formation
Drug PP1 inhibits tyrosine phosphorylation of the A36R protein and thereby prevents actin tail formation (Frischknecht et al., 1999). Here we used PP1 to investigate whether CEV particles were formed in the absence of A36R phosphorylation and actin tail formation. At 5 hours postinfection (hpi), cells were incubated in the presence or absence of PP1 for 6 h before staining live cells with monoclonal antibody (mAb) 19C2 to detect surface B5R protein. Similar densities of cell surface CEV particles were seen in the presence and absence of PP1, indicating that the inhibition of actin tail formation did not prevent movement of enveloped virions to the cell surface (Fig. 6, C and D). Staining of infected cells with phalloidin (Fig. 6, E–F) confirmed that actin tail formation was inhibited by PP1.
CEV particle formation requires microtubules but neither the A36R protein nor actin tails
To confirm that the ability of nocodazole and PP1 to inhibit actin tail formation was not an indirect effect of these drugs on virus morphogenesis, cells were analyzed by EM. In wild-type VV-infected cells that had been treated with IMCBH followed by drug washout in the presence of nocodazole, numerous IEV particles were present (Fig. 7 A), but CEV particles were not found at the plasma membrane. However, if IMCBH was washed out in the presence of cytochalasin D numerous CEV particles were visible (Fig. 7 B). Similarly, in PP1-treated infected cells IEV were found inside the cell and CEV particles were present on the plasma membrane (Fig. 7 C). Thus, although nocodazole and PP1 both prevent actin tail formation, they inhibit virus egress at different stages: nocodazole does not prevent IEV particle formation but inhibits movement of IEV particles to the cell surface, and PP1 prevents actin tail formation from the cell surface beneath CEV particles. Cytochalasin D did not inhibit IEV transport.
Figure 7.
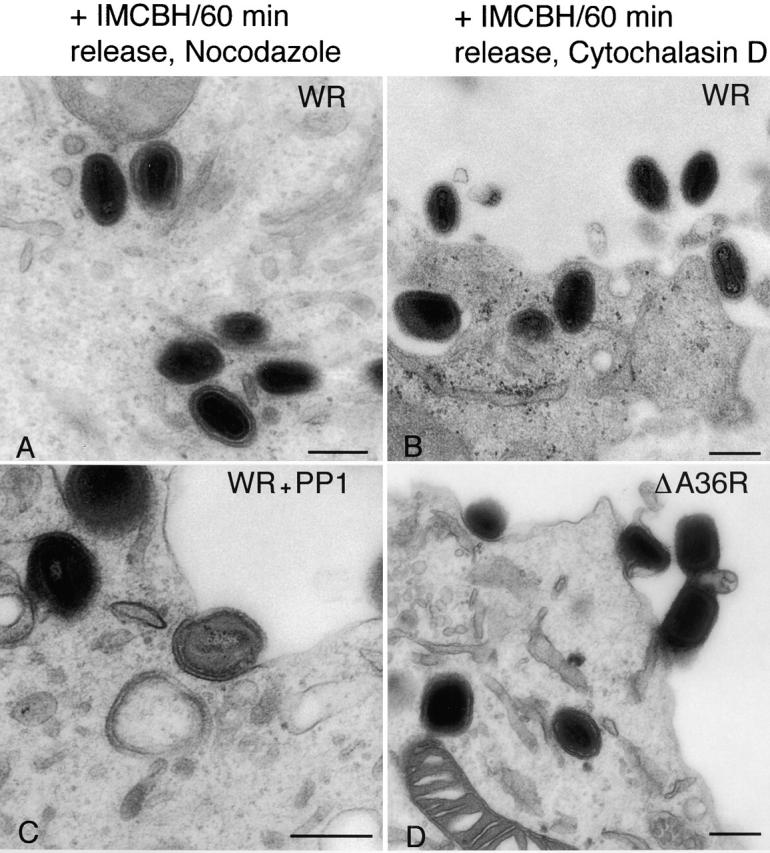
EM of infected cells. RK13 cells were infected with VV (A–C) or vΔA36R (D) at 1 PFU/cell. Cells in A and B were treated with IMCBH and either nocodazole (A) or cytochalasin D (B) as described in the legend to Fig. 5. In C, PP1 was present from 5 to 11 hpi. In D, vΔA36R-infected cells were harvested at 12 hpi. All cells were processed for thin section transmission electron microscopy. Bars, 250 nm.
The vΔA36R virus mutant (Parkinson and Smith, 1994) that lacks the A36R protein and does not make actin tails was also studied. EM of vΔA36R-infected cells showed that CEV particles are present on the cell surface (Fig. 7 D) consistent with a previous report (Wolffe et al., 1998) and our own confocal data from live cells (unpublished data). This demonstrated independently that actin tail formation is not necessary for the movement of enveloped particles to the cell sur-face.
Time-lapse photography of VV transport
To study the movement of IEV particles in live cells, we constructed a VV mutant in which the IEV particles were tagged with EGFP fused to the B5R protein. Mutagenesis of the B5R protein had shown that the transmembrane and cytoplasmic domains of this type I membrane protein are sufficient to direct antigens onto EEV (Katz et al., 1997), and deleting the extracellular domain of B5R did not prevent IEV formation (Herrera et al., 1998; Mathew et al., 1998). Therefore, the extracellular domain of B5R was replaced by EGFP, and the chimeric gene was inserted into VV strain Western reserve (WR), replacing the natural B5R gene. Cells infected with this virus synthesized a 40-kD protein that was recognized by an anti-EGFP antibody (unpublished data). EM showed that virus morphogenesis was unaltered by this change (Fig. 8, A, C, and E) , and immunoelectron microscopy showed that the B5R-EGFP protein was incorporated into IEV and CEV but not IMV particles (Fig. 8, B, D, and F). Staining of infected cells with rhodamine-phalloidin revealed that virus-induced actin tails were not made (unpublished data), but nonetheless CEV particles were detected by EM (Fig. 8 E).
Figure 8.
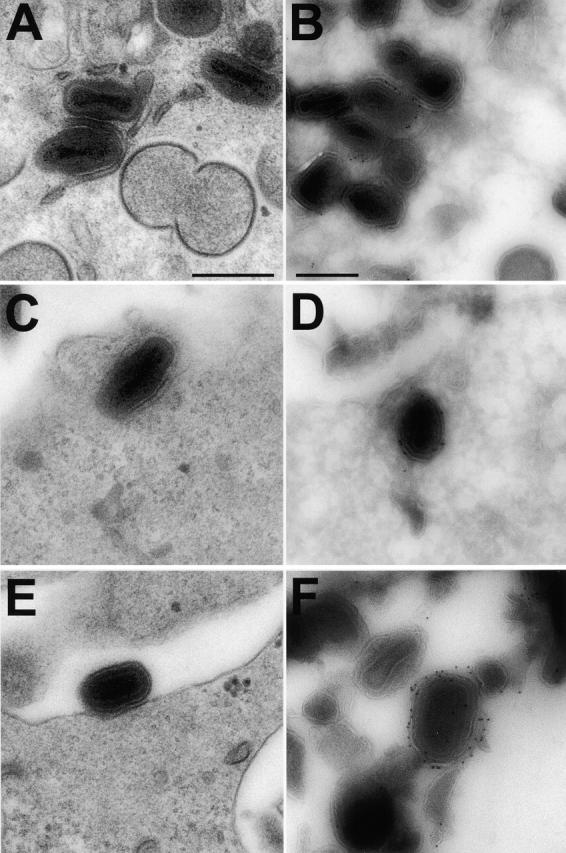
VV morphogenesis in cells infected with vB5R-GFP. RK13 cells were infected with vB5R-EGFP and processed for conventional EM after fixation at 12 hpi (A, C, and E). Alternatively, samples were processed for cryo-immunoelectron microscopy and labeled with anti-GFP and protein A gold (B, D, and F). Bars, 300 nm.
Live cells infected with vB5R-EGFP were analysed by time-lapse confocal microscopy to study transport of IEV particles to the cell surface. In Fig. 9 , the position of a single IEV particle is shown at 3-s intervals, and measurement of the distance travelled indicated that this particle moved at 40 μm/min. Measurements of 10 other particles gave similar velocities (range, 40–98 μm/min; mean, 60). This velocity is similar to that mediated by microtubules but not actin (Trinczek et al., 1999; Sodeik, 2000). In addition, treatment of cells with nocodazole blocked IEV particle movement, and this was reversed by washout of the drug (unpublished data).
Figure 9.
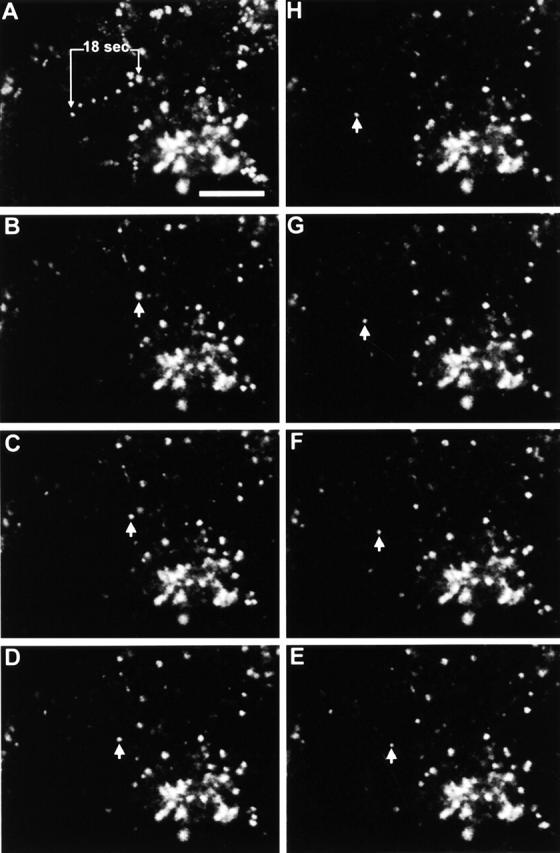
Real time movement of CEV particles in infected cells. Images from a single optical plane of a cell 8 hpi with vB5R-EGFP were taken at 3-s intervals. (A) Projection of movement of vB5R-EGFP over 18 s. B–H show the position of the single particle (arrowhead) at 3-s intervals. Calculated speed of movement 40 μm/min. Bar, 10 μm.
Lastly, the relative locations of microtubules and IEV particles were examined (Fig. 10) . Virions were identified by DIC microscopy (Fig. 10 A) and stained with DAPI (Fig. 10 C), IEV particles were visualized by virtue of their EGFP (Fig. 10 B), and microtubules were stained with an antitubulin mAb (Fig. 10, D–F). Although the microtubule network was reported to be disrupted by VV infection (Ploubidou et al., 2000), we found a normal distribution of microtubules (Fig. 10 E) late during infection after new virus particles were disseminated. When the α-tubulin and EGFP images were merged, many of the IEV particles were coincident with microtubules (Fig. 10 E), and this is more apparent at higher magnification (Fig. 10, F and G). This provides direct evidence for colocalization of IEV particles and microtubules.
Figure 10.
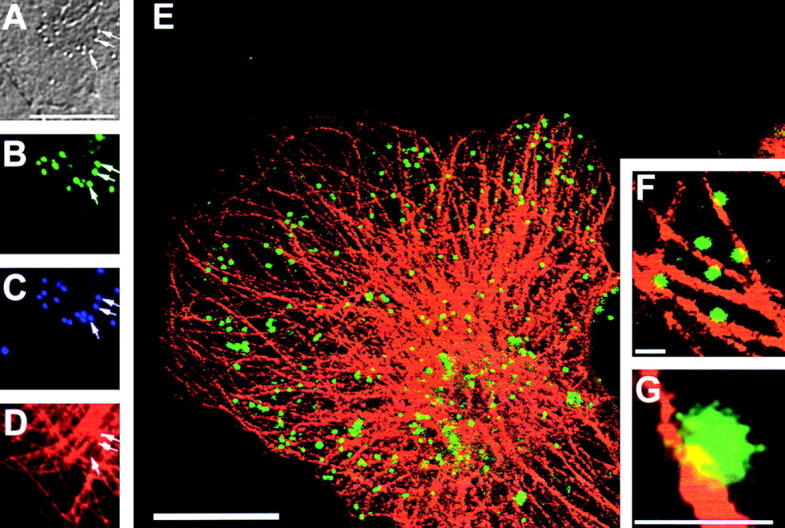
Colocalization of microtubules and IEV particles. Cells were infected with vB5R-EGFP and at 8 hpi fixed and stained with a mAb against α-tubulin (D) or with DAPI (C). In B, the positions of VV particles containing EGFP are shown, and the same virions were imaged by DIC microscopy (A). A merged image of α-tubulin and EGFP images is shown in E. (F and G) Higher magnification images of part of E. Bars: (A–E) 10 μm; (F–G) 1 μm.
Discussion
VV infection induces virus-tipped actin tails that were proposed to form on IEV particles and propel these virions to the cell surface at a rate of 2.8 μm/min (Cudmore et al., 1995, 1996, 1997). At the cell surface, actin tails drive CEV particles away from the cell or into neighboring cells at the same rate. There is substantive evidence that actin tails are needed for efficient cell-to-cell spread of virus, since mutant viruses that lack proteins A33R, A34R, A36R, B5R, F12L, or F13L produce few if any tails and form a small plaque. However, the site of actin tail formation was less clear, and in this paper we demonstrate that actin tails form beneath CEV particles at the cell surface rather than on IEV particles intracellularly. Instead of virions moving to the cell surface in an actin-dependent manner, we demonstrate that this process requires microtubules.
Several observations are inconsistent with the view that actin tails form on IEV particles. First, it was reported that cytochalasin D did not prevent the formation of CEV particles (Payne and Kristensson, 1982). Second, some virus mutants that are unable to form actin tails can release enhanced levels of EEV (McIntosh and Smith, 1996; Wolffe et al., 1997; Mathew et al., 1998; Sanderson et al., 1998). Third, no virus protein needed for actin tail formation has an asymmetric distribution on IEV particles such that actin would polymerize on only one side of the virion. Lastly, the A36R protein, which is essential for actin tail formation, is located beneath CEV particles on the cytosolic face of the plasma membrane in a position to induce actin tails from the cell surface (van Eijl et al., 2000). These data prompted us to reinvestigate the site of actin tail formation and how IEV particles move to the cell surface.
The original reports proposing actin tails formed on IEV particles (Cudmore et al., 1995, 1996) did not distinguish between IEV and CEV particles moving on actin tails. Under the light microscope, although virus particles and actin tails are visible (Fig. 1) it is impossible to determine their exact location because of the lack of resolution. Therefore, we used confocal microscopy that can section cells vertically or horizontally and thereby analyze where actin tails are located. These analyses demonstrated that actin tails were located close to the cell periphery at the top, bottom, or edge of the cell but not within the cytoplasm distinct from the cell plasma membrane (Figs. 1 and 2).
EM also provided data consistent with this view. Although a virus-tipped actin tail was sometimes found within an infected cell, these are CEV particles that have reentered the cell driven by an actin tail rather than an IEV particle that induced actin polymerization. This distinction was straightforward: the actin tail was bordered by two membranes derived from the plasma membrane as the CEV left and then reentered the cell (Fig. 3), whereas an actin tail on an IEV particle would lack these membranes. Such membranes were not observed previously, probably because the HeLa cells were permeabilized with streptolysin O and then treated with S1 myosin before analysis by EM (Cudmore et al., 1995, 1996). The cellular ultrastructure, including membranes, is poorly conserved by this technique.
EM showed also that the characteristic electron density of the actin tail started from the cell surface rather than from deeper within the cell. To be sure that actin tails that might have extended from deeper within the cell cytoplasm to the cell surface had not been missed in another plane, the samples were subjected to serial section analysis (Fig. 4). Although many actin tails of differing lengths extended from the cell surface, in no case were actin tails observed extending from deep within the cell. The presence of actin tails of differing lengths suggested that the failure to observe actin tails within the cytoplasm was not due to depolymerization of these structures.
The site of actin tail formation was also investigated in cells infected with wild-type VV and then treated with specific drugs or after infection with mutant virus. IMCBH blocks the wrapping of IMV particles by cellular membranes. However, this process is restored after removal of the drug. When the drug was washed out in the presence of nocodazole, actin tails were not formed (Fig. 5). Under these conditions, EM showed that IEV particles were still formed, but these were not transported to the cell surface (Fig. 7). Moreover, immunofluorescent microscopy showed that the clustering of virus particles near or on the cell surface was prevented (Fig. 6). In contrast, washout of IMCBH in the presence of cytochalasin D did not prevent CEV formation (Fig. 7). Collectively, these data show a requirement for microtubules and not actin for the movement of IEV particles to the cell surface.
In another approach, we used the drug PP1 that inhibits tyrosine phosphorylation, since tyrosine phosphorylation of the A36R protein is essential for actin tail formation (Frischknecht et al., 1999). After treatment of cells with PP1, CEV particles were seen on the cell surface by fluorescent and electron microscopy, yet under the same conditions actin tails were not formed (Figs. 6 and 7). Similarly, the VV mutant lacking the A36R protein (Parkinson and Smith, 1994) does not make actin tails (Sanderson et al., 1998; Wolffe et al., 1998; Röttger et al., 1999), but nevertheless numerous CEV particles were seen at the cell surface (Fig. 7 D).
Lastly, we constructed and analysed a VV mutant that expresses EGFP fused to B5R on the surface of IEV particles. Time-lapse photography showed that individual virus particles moved in infected cells at rates of 40–98 μm/min, (mean, 60; SEM = 5; n = 10). This speed is similar to that for microtubular transport and is 20-fold greater than VV movement on actin tails (2.8 μm/min) (Cudmore et al., 1995). Furthermore, the movement of IEV particles was stop-start in nature, was along defined pathways rather than being random in the cytosol, and was inhibited reversibly by nocodazole. Finally, we showed that EGFP-positive IEV particles colocalized with microtubules in infected cells and that after IEV particles had been produced the microtubule network remained intact, in contrast to a previous report (Ploubidou et al., 2000).
The transport of IMV particles away from virus factories requires microtubules and the A27L gene product (Sanderson et al., 2000). Thus, VV requires microtubules at two stages during virus egress, and the revised model for VV egress is summarized in Fig. 11 . Although, a VV protein on the surface of the IMV particle has been identified that is necessary for the microtubule-mediated movement of IMV particles, no such protein on the surface of IEV particles has been reported. In all cases where IEV- or EEV-specific genes have been deleted, the wrapping of IMV particles is either inhibited (B5R or F13L) or IEV particles are formed and transported to the cell surface. A mutant that forms IEV particles that are not transported would be useful for investigation of IEV–microtubule interactions.
Figure 11.

Model for VV egress. IMV produced in cytoplasmic factories are transported on microtubules to sites of wrapping by a double layer of membrane from the trans-Golgi or early endosomal membranes to form IEV. IEV particles are then transported by microtubules to the cell surface where the outer membrane fuses with the plasma membrane forming CEV. Actin tails are formed beneath CEV particles. EEV may be released either before or after actin tail formation.
The revised model of VV egress presented here (Fig. 11) shows that VV and intracellular bacteria differ in the utilization of actin polymerization for their dissemination. Whereas these bacteria polymerize actin from one end of the bacterium within the cytosol and use the growing actin tails for movement within and out of the cells, VV polymerizes actin tails from the plasma membrane beneath virus particles on the cell surface. These virions are then driven away from the cell to infect new cells.
The polymerization of actin beneath CEV particles provides a plausible explanation for the existence of CEV. Initially, it might seem surprising that the majority of virus that reaches the cell surface is retained rather than released. Indeed several viruses, for example influenza virus, express receptor-destroying enzymes to prevent aggregation of new virions on the cell surface or with each other. However, VV needs to retain enveloped virus on the cell surface long enough for the polymerization of actin to take place and drive the virions into surrounding cells.
In conclusion, we show that microtubules but not actin tails are used to facilitate transport of IEV particles to the cell surface, and the actin tails form beneath CEV particles at the cell surface to drive virus particles away from the cell. Although other viruses such as adenovirus (Suomalainen et al., 1999), herpes simplex virus (Sodeik et al., 1997), and African swine fever virus (Carvalho et al., 1988) use microtubules for intracellular transport, VV egress is unique in exploiting both microtubules and actin for dissemination.
Materials and methods
Cells, viruses, and drugs
BS-C-1, HeLa, and RK13 cells were grown in MEM supplemented with 10% FBS (GIBCO BRL). VV strain WR and a mutant lacking the A36R gene (Parkinson and Smith, 1994) were used. IMCBH was provided by R. Wittek (University of Lausanne, Lausanne, Switzerland). Nocodazole (Sigma-Aldrich), cytochalasin D (Sigma-Aldrich), and PP1 (Alexis Biochemicals) were added to a final concentration of 33, 1, and 20 μM, respectively.
Phase–contrast and DIC microscopy
A Zeiss Axioplan II microscope with a 100× objective was used, and images were acquired with a Eastman Kodak Co. Spot 2 CCD camera.
Fluorescent microscopy
Cells growing on glass coverslips (Chance Proper, Ltd.) were infected with VV for 1 h at 1 PFU/cell and at the indicated time postinfection were fixed in paraformaldehyde as described (van Eijl et al., 2000). Cells were blocked and permeabilized for 5 min at room temperature in PBS containing 0.1% saponin and 10% FBS. Alternatively, infected cells were fixed for 5 min at −20°C in methanol. To identify VV particles, cells were incubated with mAb 19C2 (Schmelz et al., 1994) that recognizes the VV B5R protein. Bound mAb was detected by FITC-conjugated goat anti–rat IgG (mouse-adsorbed) antibody (Stratech Scientific) (diluted 1:200). F-actin was stained with TRITC-phalloidin (Sigma-Aldrich) (diluted 1/100). Cells were analyzed using a Bio-Rad Laboratories MicroRadiance confocal laser scanning microscope as described (Sanderson et al., 1998). Images were collected and processed using Lasersharp and Adobe Photoshop® software. Live cells were stained with mAb 19C2 as described (van Eijl et al., 2000). Microtubules were identified by staining fixed cells with an antitubulin rat mAbYOL-1/34 (Serotech) (diluted 1:50) and rhodamine-conjugated donkey anti–rat IgG (mouse-adsorbed) antibody (diluted 1:100) (Stratech Scientific).
Live cell imaging
BS-C-1 cells were infected with vB5R-EGFP for 2 h at 10 PFU/cell. At 8 hpi, time-lapse images of the infected cells maintained at 37°C on a heated microscope stage were recorded using a Bio-Rad Laboratories 1024 confocal laser scanning microscope. Images were collected every 3 s using Lasersharp software and processed using Adobe Photoshop® software.
Preparation of samples for EM
BS-C-1, HeLa, and RK13 cells were infected with VV at 1 PFU/cell and at the indicated times after specific treatments and processed for thin section transmission electron microscopy as described (Hollinshead et al., 1999). For immunoelectron microscopy, ultrathin cryosections were labeled with anti-GFP (CLONTECH) (diluted 1:10). All digital images were captured with the integrated SIS image analysis package (Soft Imaging Software) and processed using Adobe Photoshop® software.
Construction of recombinant virus expressing EGFP
To follow the movement of IEV particles, a recombinant VV was constructed in which EGFP was fused to the transmembrane and cytoplasmic domains of protein B5R. The chimeric gene was assembled in a plasmid vector by PCR and splicing by overlap extension (Horton et al., 1989). Plasmid pSTH2 (Engelstad et al., 1992) containing the VV WR B5R gene and flanking sequences cloned into pUC13 was used as template for PCRs. Oligonucleotides (1) 5′-TCATTTAAGCTTCCTTCTTTCGTGAAATGC-3′ and (2) 5′-CTCGCCCTTGCTCACTGTTGAATAAACAAC-3′ generated a fragment containing 322 bp upstream of the B5R ORF and B5R amino acids 1–20, including the signal peptide. Oligonucleotides (3) 5′-GACGAGCTGTACAAGGAAGAATTTGATCCA-3′ and (4) 5′-GTACTCAAGCTTGCTTACAGAAACATCGCGTT-3′ generated a fragment encoding B5R amino acids 242–317, including the transmembrane and cytoplasmic domains and 337 nucleotides downstream. The EGFP ORF was amplified by PCR using pEGFPC1 (CLONTECH) as template and oligonucleotides (5) 5′-GTGAGCAAGGGCGAG-3′ and (6) 5′-CTTGTACAGCTC-3′. Oligonucleotides (1) and (4) introduced HindIII restriction sites (underlined), whereas primers (2) and (3) contained EGFP sequences, enabling the individual fragments to be assembled into a single 1680-bp gene. This was digested with HindIII and cloned into HindIII-digested pSJH7 (Hughes et al., 1991) to form pB5R-EGFP. The fidelity of the cloned PCR product was confirmed by sequencing.
The recombinant VV vB5R-EGFP was constructed by transient dominant selection (Falkner and Moss, 1990). Cells infected with VV strain WR at 0.1 PFU/cell were transfected with pB5R-EGFP and a virus in which the wild-type B5R gene was replaced with EGFP-B5R, selected using published methods (Parkinson and Smith, 1994), and called vB5R-EGFP.
Acknowledgments
This work was supported by Programme Grant P8901790 from the United Kingdom Medical Research Council and an equipment grant from The Wellcome Trust. M. Law is funded by the Croucher Foundation Scholarship, Hong Kong Special Administrative Region, China. G.L. Smith is a Wellcome Trust principal research fellow.
G. Rodger's and G.L. Smith's current address is The Wright-Fleming Institute, Imperial College School of Medicine, St. Mary's Campus, London W2 1PG, UK.
Footnotes
Abbreviations used in this paper: CEV, cell-associated enveloped virus; EEV, extracellular enveloped virus; EGFP, enhanced green fluorescent protein; hpi, hours postinfection; IEV, intracellular enveloped virus; IMCBH, N 1-isonicotinoyl-N 2-3-methyl-4-chlorobenzoylhydrazine; IMV, intracellular mature virus; mAb, monoclonal antibody; PP1, 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo(3,4-d)pyrimidine; VV, vaccinia virus; WR, Western reserve.
References
- Appleyard, G., A.J. Hapel, and E.A. Boulter. 1971. An antigenic difference between intracellular and extracellular rabbitpox virus. J. Gen. Virol. 13:9–17. [DOI] [PubMed] [Google Scholar]
- Blasco, R., N.B. Cole, and B. Moss. 1991. Sequence analysis, expression, and deletion of a vaccinia virus gene encoding a homolog of profilin, a eukaryotic actin-binding protein. J. Virol. 65:4598–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco, R., and B. Moss. 1992. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J. Virol. 66:4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter, E.A., and G. Appleyard. 1973. Differences between extracellular and intracellular forms of poxviruses and their implications. Prog. Med. Virol. 16:86–108. [PubMed] [Google Scholar]
- Carvalho, Z.G., A.A.P. De Matos, and C. Rodrigues-Pousada. 1988. Association of African swine fever virus with the cytoskeleton. Virus Res. 11:175–192. [DOI] [PubMed] [Google Scholar]
- Cudmore, S., P. Cossart, G. Griffiths, and M. Way. 1995. Actin-based motility of vaccinia virus. Nature. 378:636–638. [DOI] [PubMed] [Google Scholar]
- Cudmore, S., I. Reckmann, G. Griffiths, and M. Way. 1996. Vaccinia virus: a model system for actin-membrane interactions. J. Cell Sci. 109:1739–1747. [DOI] [PubMed] [Google Scholar]
- Cudmore, S., I. Reckmann, and M. Way. 1997. Viral manipulations of the actin cytoskeleton. TIBS. 5:142–148. [DOI] [PubMed] [Google Scholar]
- Dales, S., and E.H. Mosbach. 1968. Vaccinia as a model for membrane biogenesis. Virology. 35:564–583. [DOI] [PubMed] [Google Scholar]
- Engelstad, M., S.T. Howard, and G.L. Smith. 1992. A constitutively expressed vaccinia gene encodes a 42-kDa glycoprotein related to complement control factors that forms part of the extracellular virus envelope. Virology. 188:801–810. [DOI] [PubMed] [Google Scholar]
- Engelstad, M., and G.L. Smith. 1993. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology. 194:627–637. [DOI] [PubMed] [Google Scholar]
- Falkner, F.G., and B. Moss. 1990. Transient dominant selection of recombinant vaccinia viruses. J. Virol. 64:3108–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht, F., V. Moreau, S. Röttger, S. Gonfloni, I. Rechmann, G. Superti-Furga, and M. Way. 1999. Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature. 401:926–929. [DOI] [PubMed] [Google Scholar]
- Frischknecht, F., and M. Way. 2001. Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol. 11:30–38. [DOI] [PubMed] [Google Scholar]
- Herrera, E., M. del Mar Lorenzo, R. Blasco, and S.N. Isaacs. 1998. Functional analysis of vaccinia virus B5R protein: essential role in virus envelopment is independent of a large portion of the extracellular domain. J. Virol. 72:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller, G., and K. Weber. 1982. A phosphorylated basic vaccinia virus virion polypeptide of molecular weight 11,000 is exposed on the surface of mature particles and interacts with actin-containing cytoskeletal elements. J. Virol. 44:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller, G., and K. Weber. 1985. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 55:651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller, G., K. Weber, L. Schneider, C. Parajsz, and C. Jungwirth. 1979. Interaction of assembled progeny pox viruses with the cellular cytoskeleton. Virology. 98:142–153. [DOI] [PubMed] [Google Scholar]
- Hirt, P., G. Hiller, and R. Wittek. 1986. Localization and fine structure of a vaccinia virus gene encoding an envelope antigen. J. Virol. 58:757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinshead, M., A. Vanderplasschen, G.L. Smith, and D.J. Vaux. 1999. Vaccinia virus intracellular mature virions contain only one lipid membrane. J. Virol. 73:1503–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton, R.M., Z.L. Cai, S.N. Ho, and L.R. Pease. 1989. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques. 8:528–535. [PubMed] [Google Scholar]
- Hughes, S.J., L.H. Johnston, A. de Carlos, and G.L. Smith. 1991. Vaccinia virus encodes an active thymidylate kinase that complements a cdc8 mutant of Saccharomyces cerevisiae. J. Biol. Chem. 266:20103–20109. [PubMed] [Google Scholar]
- Ichihashi, Y., S. Matsumoto, and S. Dales. 1971. Biogenesis of poxviruses: role of A-type inclusions and host cell membranes in virus dissemination. Virology. 46:507–532. [DOI] [PubMed] [Google Scholar]
- Katz, E., E.J. Wolffe, and B. Moss. 1997. The cytoplasmic and transmembrane domains of the vaccinia virus B5R protein target a chimeric human immunodeficiency virus type 1 glycoprotein to the outer envelope of nascent vaccinia virions. J. Virol. 71:3178–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krempien, U., L. Schneider, G. Hiller, K. Weber, E. Katz, and C. Jungwirth. 1981. Conditions for pox virus-specific microvilli formation studied during synchronized virus assembly. Virology. 113:556–564. [DOI] [PubMed] [Google Scholar]
- Mathew, E., C.M. Sanderson, M. Hollinshead, and G.L. Smith. 1998. The extracellular domain of vaccinia virus protein B5R affects plaque phenotype, extracellular enveloped virus release, and intracellular actin tail formation. J. Virol. 72:2429–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh, A.A., and G.L. Smith. 1996. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J. Virol. 70:272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, C. 1976. Vaccinia virus reexamined: development and release. Virology. 73:43–58. [DOI] [PubMed] [Google Scholar]
- Parkinson, J.E., and G.L. Smith. 1994. Vaccinia virus gene A36R encodes a M(r) 43-50 K protein on the surface of extracellular enveloped virus. Virology. 204:376–390. [DOI] [PubMed] [Google Scholar]
- Payne, L.G. 1980. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia virus. J. Gen. Virol. 50:89–100. [DOI] [PubMed] [Google Scholar]
- Payne, L.G., and K. Kristensson. 1979. Mechanism of vaccinia virus release and its specific inhibition by N 1-isonicotinoyl-N 2-3-methyl-4-chlorobenzoylhydrazine. J. Virol. 32:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne, L.G., and K. Kristensson. 1982. The effect of cytochalasin D and monensin on enveloped vaccinia virus release. Arch. Virol. 74:11–20. [DOI] [PubMed] [Google Scholar]
- Payne, L.G., and K. Kristensson. 1985. Extracellular release of enveloped vaccinia virus from mouse nasal epithelial cells in vivo. J. Gen. Virol. 66:643–646. [DOI] [PubMed] [Google Scholar]
- Ploubidou, A., V. Moreau, K. Ashman, I. Reckmann, C. Gonzalez, and M. Way. 2000. Vaccinia virus infection disrupts microtubule organization and centrosome function. EMBO J. 19:3932–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, J.F., and G.L. Smith. 1990. IPTG-dependent vaccinia virus: identification of a virus protein enabling virion envelopment by Golgi membrane and egress. Nucl. Acids Res. 18:5347–5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper, R.L., E.J. Wolffe, A. Weisberg, and B. Moss. 1998. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J. Virol. 72:4192–4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röttger, S., F. Frischknecht, I. Reckmann, G.L. Smith, and M. Way. 1999. Interactions between vaccinia virus IEV membrane proteins and their roles in IEV assembly and actin tail formation. J. Virol. 73:2863–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson, C.M., F. Frischknecht, M. Way, M. Hollinshead, and G.L. Smith. 1998. Roles of vaccinia virus EEV-specific proteins in intracellular actin tail formation and low pH-induced cell-cell fusion. J. Gen. Virol. 79:1415–1425. [DOI] [PubMed] [Google Scholar]
- Sanderson, C.M., M. Hollinshead, and G.L. Smith. 2000. The vaccinia virus A27L gene is needed for the microtubule-dependent transport of intracellular mature virus particles. J. Gen. Virol. 81:47–58. [DOI] [PubMed] [Google Scholar]
- Schmelz, M., B. Sodeik, M. Ericsson, E. Wolffe, H. Shida, G. Hiller, and G. Griffiths. 1994. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J. Virol. 68:130–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz, C., L.G. Payne, J. Gubser, and R. Wittek. 1991. A mutation in the gene encoding the vaccinia virus 37,000-Mr protein confers resistance to an inhibitor of virus envelopment and release. J. Virol. 65:3435–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodeik, B. 2000. Mechanisms of viral transport in the cytoplasm. Trends Microbiol. 8:465–472. [DOI] [PubMed] [Google Scholar]
- Sodeik, B., M.W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes, G.V. 1976. High-voltage electron microscope study of the release of vaccinia virus from whole cells. J. Virol. 18:636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storrie, B., J. White, S. Rottger, E.H. Stelzer, T. Suganuma, and T. Nilsson. 1998. Recycling of Golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J. Cell Biol. 143:1505–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen, M., M. Nakano, S. Keller, K. Boucke, R.P. Stidwill, and U.F. Greber. 1999. Microtubule-dependent plus and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 144:657–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze, J., M. Hollinshead, B. Reis, K. Radsak, and H. Kern. 1993. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur. J. Cell Biol. 60:163–178. [PubMed] [Google Scholar]
- Trinczek, B., A. Ebneth, E.M. Mandelkov, and E. Mandelkov. 1999. Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J. Cell Sci. 112:2355–2367. [DOI] [PubMed] [Google Scholar]
- van Eijl, H., M. Hollinshead, and G.L. Smith. 2000. The vaccinia virus A36R protein is a type Ib membrane protein present on intracellular but not extracellular enveloped particles. Virology. 271:26–36. [DOI] [PubMed] [Google Scholar]
- Wolffe, E.J., S.N. Isaacs, and B. Moss. 1993. Deletion of the vaccinia virus B5R gene encoding a 42-kiloDalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J. Virol. 67:4732–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe, E.J., E. Katz, A. Weisberg, and B. Moss. 1997. The A34R glycoprotein gene is required for induction of specialized actin-containing microvilli and efficient cell-to-cell transmission of vaccinia virus. J. Virol. 71:3904–3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe, E.J., A.S. Weisberg, and B. Moss. 1998. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell virus spread. Virology. 244:20–26. [DOI] [PubMed] [Google Scholar]
- Zhang, W.-H., D. Wilcock, and G.L. Smith. 2000. The vaccinia virus F12L protein is required for actin tail formation, normal plaque size and virulence. J. Virol. 74:11663–11670. [DOI] [PMC free article] [PubMed] [Google Scholar]