

Abstract
The β-catenin signaling pathway is deregulated in nearly all colon cancers. Nonhypercalcemic vitamin D3 (1α,25-dehydroxyvitamin D3) analogues are candidate drugs to treat this neoplasia. We show that these compounds promote the differentiation of human colon carcinoma SW480 cells expressing vitamin D receptors (VDRs) (SW480-ADH) but not that of a malignant subline (SW480-R) or metastasic derivative (SW620) cells lacking VDR. 1α,25(OH)2D3 induced the expression of E-cadherin and other adhesion proteins (occludin, Zonula occludens [ZO]-1, ZO-2, vinculin) and promoted the translocation of β-catenin, plakoglobin, and ZO-1 from the nucleus to the plasma membrane. Ligand-activated VDR competed with T cell transcription factor (TCF)-4 for β-catenin binding. Accordingly, 1α,25(OH)2D3 repressed β-catenin–TCF-4 transcriptional activity. Moreover, VDR activity was enhanced by ectopic β-catenin and reduced by TCF-4. Also, 1α,25(OH)2D3 inhibited expression of β-catenin–TCF-4-responsive genes, c-myc, peroxisome proliferator-activated receptor δ, Tcf-1, and CD44, whereas it induced expression of ZO-1. Our results show that 1α,25(OH)2D3 induces E-cadherin and modulates β-catenin–TCF-4 target genes in a manner opposite to that of β-catenin, promoting the differentiation of colon carcinoma cells.
Keywords: vitamin D; vitamin D receptor; β-catenin; E-cadherin; colon cancer
Introduction
Colon cancer is the third cause of cancer death in Western countries and a major health problem worldwide (Parker et al., 1997). Colon carcinogenesis is the result of the progressive transformation of colonic epithelial cells, which accumulate mutations that increase their proliferation and alter their phenotype.
E-cadherin is a transmembrane linker protein of the intercellular adheren junctions that play a key role in the maintenance of the adhesive and polarized phenotype of epithelial cells (Takeichi, 1995; Gumbiner, 1996). Loss of E-cadherin expression is a common event during the transition from adenoma to carcinoma, which involves the alteration of the normal epithelial phenotype and the acquisition of invasive capacity (Birchmeier and Behrens, 1994; Perl et al., 1998; Christofori and Semb, 1999). E-cadherin is regarded as a tumor suppressor gene and its loss as a predictor of poor prognosis (Vleminckx, et al., 1991; Takeichi, 1993; Mareel et al., 1994; Guilford et al., 1998). β-Catenin is a protooncogene encoding a cytoskeleton-associated protein, which in normal epithelial cells is bound mostly to the cytoplasmic tail of E-cadherin at the adherens junctions (for review see Morin, 1999). Free cytosolic β-catenin levels are strictly controlled by phosphorylation of the NH2-terminal region of the protein by glycogen synthase kinase (GSK)* 3β. This reaction requires association with axin/conductin and the product of the adenomatous polyposis coli (APC) tumor suppressor gene, and targets β-catenin for ubiquitin-mediated degradation by the proteasome (Rubinfeld et al., 1996; Behrens et al., 1998; Kishida et al., 1998). Interaction of Wnt ligands with their membrane receptors blocks GSK-3β, leading to the accumulation of free β-catenin (for reviews see Eastman and Grosschedl, 1999; Peifer and Polakis, 2000). In the cell nucleus, β-catenin binds members of the T cell transcription factor (TCF)/lymphoid enhancer-binding factor (LEF)-1 family and thus regulates gene expression (Behrens et al., 1996; Huber et al., 1996; Billin et al., 2000). Nearly all colon tumors present a deregulated β-catenin signaling pathway by mutation of either APC or β-catenin, which leads to the blockade of phosphorylation by GSK-3β, resulting in β-catenin stabilization (Inomata et al., 1996; Korinek et al., 1997; Morin et al., 1997), reduced APC-regulated nuclear export (Henderson, 2000; Rosin-Arbesfeld et al., 2000), and perhaps higher specific activity (Guger and Gumbiner, 2000). As a result, β-catenin accumulates in the nucleus, leading to both the activation of genes involved in the control of cell proliferation and invasiveness such as c-myc, cyclin D1, peroxisome proliferator-activated receptor (PPAR) δ, matrilysin, c-jun, fra1, uPA receptor, fibronectin, CD44, Tcf-1, Cdx-1 and gastrin, and the loss of expression of DRCTNNB1A and differentiated epithelial markers such as Zonula occludens (ZO)-1 (He et al., 1998, 1999; Crawford et al., 1999; Gradl et al., 1999; Mann et al., 1999; Tetsu and McCormick, 1999; Roose et al., 1999; Vera et al., 1999; Kawasoe et al., 2000; Koh et al., 2000; Lickert et al., 2000). Mutations in the TCF-4 gene may also contribute to this process (Duval et al., 2000). In addition, APC mutations may also be responsible at least in part for chromosomal instability in colon cancer cells (Fodde et al., 2001; Kaplan et al., 2001).
Epidemiological data suggest an inverse correlation between vitamin D dietary intake or sunlight exposure and human colorectal cancer (Garland et al., 1989; Newmark and Lipkin, 1992). Vitamin D, especially its most active metabolite 1α,25-dihydroxyvitamin D3 (1α,25[OH]2D3), not only contributes to calcium homeostasis but also regulates cell proliferation and differentiation (Saez et al., 1993; Xi and Feldman, 1993; Buras et al., 1994; Kane et al., 1996). 1α,25(OH)2D3 and several synthetic vitamin D derivatives (deltanoids), which show reduced calcemic activity such as EB1089, MC903, and KH1060, inhibit the growth of epithelial, melanoma, soft tissue sarcoma, and leukemic cells by inducing cell cycle arrest or apoptosis (Diaz et al., 2000; Park et al., 2000). Furthermore, they inhibit the invasive capacity in vitro, the synthesis of several invasion-associated proteins (Hansen et al., 1994; González-Sancho et al., 1998; Koli and Keski-Oja, 2000), and the tumor-induced angiogenesis (Majewski et al., 1993) of breast cancer cells, and they show a chemopreventive activity in animal models of colorectal and breast cancer (Akhter et al., 1997; van Weelden et al., 1998).
Vitamin D and its analogues regulate gene expression by binding to specific vitamin D receptors (VDRs) of the nuclear receptor superfamily, which are ligand-modulated transcription factors (for review see McDonald et al., 2001). Upon ligand activation, VDR binds specific nucleotide sequences (vitamin D response elements, VDREs) in target genes to activate or repress their expression through multiple but ill-defined interactions with coactivator complexes and components of the basal transcription machinery (for review see McDonald et al., 2001). Several vitamin D target genes have been characterized in several tumor cell types such as c-myc, p21Waf, p27Kip1, tenascin-C, fibronectin, laminin and its receptor, apolipoprotein D, insulin-like growth factor binding protein 3, cyclin C, and several members of the transforming growth factor family and their receptors (Freedman, 1999). Two studies have reported the regulation of epidermal growth factor receptor expression (Tong et al., 1998) or the cross-talk between vitamin D and tumor growth factor β signaling pathways (Yanagisawa et al., 1999), although the physiological relevance of these mechanisms remains unclear.
VDR is expressed in the normal colonic mucosa, slightly increases in the hyperplasic condition, and is clearly lower in late stages of carcinogenesis (Vandewalle et al., 1994). Accordingly, high VDR expression is associated with favorable prognosis in colorectal cancer patients, suggesting that these receptors are involved in this pathogenesis and their potential role as a predictive marker (Shabahang et al., 1993; Evans et al., 1998). Several clinical trials are underway to assess the activity of several vitamin D derivatives in patients with colorectal carcinoma and other neoplasias (Gross et al., 1998; Gulliford et al., 1998; Smith et al., 1999). However, their molecular and cellular mechanisms of action in colon carcinoma cells remain mostly unknown.
The SW480 cell line is one of the best characterized human colorectal cancer lines, and it has been widely used as a model system for the study of this neoplasia. It was established in 1976 from a primary human Duke's B colon adenocarcinoma of a 50-yr-old patient (Leibovitz et al., 1976). SW480 cells harbor most of the genetic abnormalities that characterize advanced colon cancers such as activation of K-ras oncogene, c-myc amplification, deletion of chromosome 18, and mutation of APC and p53 tumor suppressor genes (Tomita et al., 1992; Schwarte-Waldhoff et al., 1999). In addition, these cells are defective for E-cadherin and express high levels of nuclear β-catenin, transforming growth factor β, and epidermal growth factor receptors (Tomita et al., 1992). We used the SW480 cell line to examine the mechanism of action of 1α,25(OH)2D3 and several nonhypercalcemic analogues in colon cancer cells. Our results show that these compounds have a prodifferentiation phenotypic effect on VDR-positive SW480 cells parallel to the induction of E-cadherin, induce β-catenin nuclear export, and inhibit β-catenin gene regulatory activity. Moreover, 1α,25(OH)2D3 promotes a direct VDR–β-catenin interaction, which may decrease TCF-4–β-catenin complexes and may thus constitute another mechanism of inhibition of β-catenin signaling.
Results
1α,25(OH)2D3 induces the differentiation of a VDR-positive subpopulation of SW480 cells to an epithelial-like phenotype
To investigate its mechanism of action in human colon cancer cells, two cell lines from the same patient, SW480 cells established from a primary adenocarcinoma and SW620 from a lymph node metastasis, were treated with 1α,25(OH)2D3. Upon 1α,25(OH)2D3 addition, a proportion of SW480 cells changed in shape and properties to a more adhesive epithelial phenotype (Fig. 1 A, a and b), whereas the rest of the SW480 population and SW620 cells were unaffected (Fig. 1 A, a, b, g, and h). These two distinct responses in SW480 cultures correlated with two cell morphologies: flat, polygonal, and adherent to plastic dishes, which corresponded to 1α,25(OH)2D3-responsive cells, and rounded, refractile, and less adherent, which corresponded to nonresponsive cells (Fig. 1 A, a and b, arrows). This is consistent with previous reports of the existence of two populations in SW480 cell cultures (Tomita et al., 1992; Baulida et al., 1999) and led us to obtain clonal sublines of each cell type: SW480-ADH (adherent) and SW480-R (rounded). In agreement with the previous finding, the two established sublines retained their distinct morphology and hormonal response for >2 yr: upon addition of 1α,25(OH)2D3, the bulk of SW480-ADH cells acquired an epithelial-like phenotype, forming compact islands (Fig. 1 A, c and d), whereas SW480-R cells were unaffected (Fig. 1 A, e and f). Moreover, SW480-R cells were highly tumorigenic in athymic mice (eight of eight tumors were generated 9 wk after the subcutaneous injection of 106 cells), whereas in contrast SW480-ADH cells were weakly tumorigenic (3 of 24). The distinct behavior of these cell types was attributable to differences in the expression of VDR, which was checked in Northern blot assays (Fig. 1 B). VDR mRNA levels were high in the SW480-ADH subline and three times lower in parental SW480 cells. In contrast, SW620 cells expressed a very low level of VDR mRNA, which was undetectable in SW480-R cells. Further, 1α,25(OH)2D3 increased the expression of a transfected luciferase reporter gene cloned under the control of four copies of a direct repeat (DR) VDRE (4 × VDRE) in SW480-ADH and to a lesser extent in parental SW480 cells but had no effect in SW480-R or SW620 cells (Fig. 1 C). In addition, cotransfection of an exogenous VDR clearly induced the luciferase gene in all cell types (Fig. 1 C). Thus, the VDR expressed by SW480 and SW480-ADH cells was an active mediator of the gene regulatory activity of 1α,25(OH)2D3, whereas SW480-R and SW620 cells did not respond to this agent, owing to the lack of endogenous VDR expression. In agreement with these results and its effect in other cell types (Buras et al., 1994; Kane et al., 1996), 1α,25(OH)2D3 inhibited the proliferation of SW480-ADH cells and to a lesser extent that of SW480 cells, whereas SW480-R and SW620 cells were totally unresponsive (Fig. 1 D). We tested whether the reexpression of VDR reverted the malignant phenotype of SW480-R cells. However, no clones of SW480-R cells stably expressing VDR were obtained in any of three experiments carried out using various vectors that were functional in other cell systems, suggesting that VDR, perhaps by inhibiting cell growth, counterselects these cells and thus hampers clonal expansion.
Figure 1.
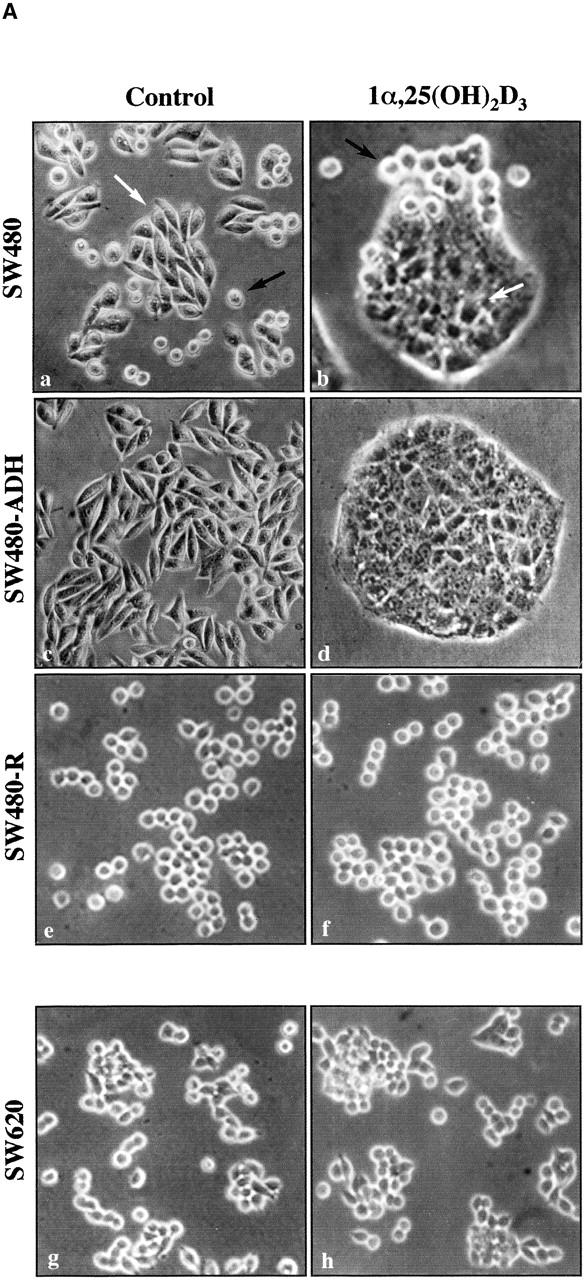
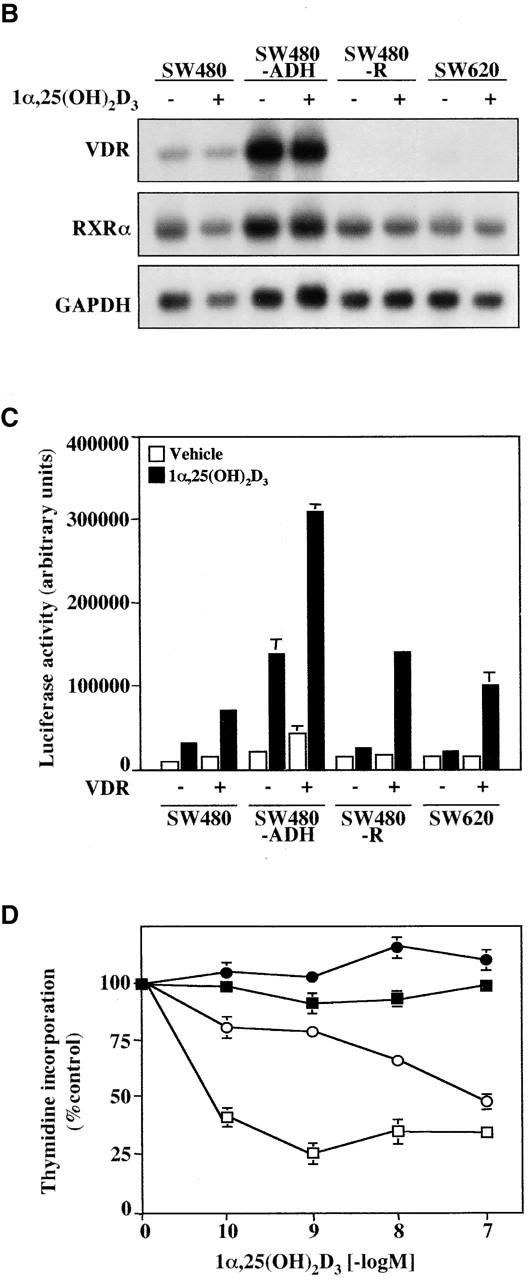
Effect of 1α,25(OH)2D3 on human colon carcinoma cells expressing variable levels of VDR. (A) Phase–contrast micrographs of various cell lines upon 48-h treatment with 1α,25(OH)2D3 (10−7 M) or vehicle (control): SW480 (a and b); SW480-ADH (c and d); SW480-R (e and f); SW620 (g and h). The two distinct cell types found in SW480 cultures are indicated in a and b: flat, polygonal, and 1α,25(OH)2D3-sensitive (white arrows), and 1α,25(OH)2D3-rounded 1α,25(OH)2D3-unresponsive (black arrows). (B) Northern blot analysis of VDR and RXRα expression in SW480, SW480-ADH, SW480-R, and SW620 cells untreated or treated with 10−7 M 1α,25(OH)2D3 for 48 h. 10 μg of poly(A)+ RNA was loaded per lane. GAPDH was used as internal control. (C) 1α,25(OH)2D3 transcriptional responsiveness of each cell line. Cells were transfected with the 4 × VDRE–DR3-tk-luc construct and a human VDR expression vector as indicated. After 48-h incubation in the presence or absence of 1α,25(OH)2D3 (10−7 M), luciferase activity in total cell extracts was measured as described in Materials and methods. A β-galactosidase expression vector was also transfected as internal control. Mean values and standard deviations of the mean obtained in three experiments using triplicates are shown. (D) Effect of 1α,25(OH)2D3 on DNA synthesis. Cells were untreated or treated with the indicated 1α,25(OH)2D3 concentrations for 48 h, and the level of DNA synthesis was measured by estimating the incorporation of labeled thymidine in TCA-precipitable material as described in Materials and methods. Mean values and standard deviations of the mean obtained in three experiments using duplicates are shown. ○, SW480 cells; •, SW480-R cells; □, SW480-ADH cells; ▪, SW620 cells.
1α,25(OH)2D3 induces E-cadherin transcription and regulates the expression and localization of other adhesion proteins
To examine the effect of 1α,25(OH)2D3 on the phenotype of SW480-ADH cells, we performed immunofluorescence and confocal laser microscopy analyses of several epithelial markers. Although SW480 cells have been described as E-cadherin–defective (Sadot et al., 1998; Baulida et al., 1999; Orsulic et al., 1999), SW480-ADH cells exhibited low but detectable membrane-bound expression of this intercellular adhesion protein that was drastically increased upon treatment with 1α,25(OH)2D3 (Fig. 2 A, a–c). The induction of E-cadherin expression correlated with the phenotypic change: it was observed as soon as 16 h after treatment, was especially high at 48 h, and lasted 7 d (Fig. 2 A, a–c). Like parental SW480 and other colon cancer lines defective for APC and E-cadherin, SW480-ADH cells showed high β-catenin levels in the nucleus and cytoplasm (Fig. 2 A, d). Remarkably, 1α,25(OH)2D3 severely altered the subcellular location of β-catenin, from predominantly nuclear to a nearly exclusive plasma membrane distribution (Fig. 2 A, d–f). This paralleled the induction of E-cadherin, which was maximal 48 h after 1α,25(OH)2D3 addition and agreed with the reported sequestration of β-catenin at the plasma membrane compartment after transient transfection of SW480 cells with E-cadherin (Sadot et al., 1998; Orsulic et al., 1999).
Figure 2.
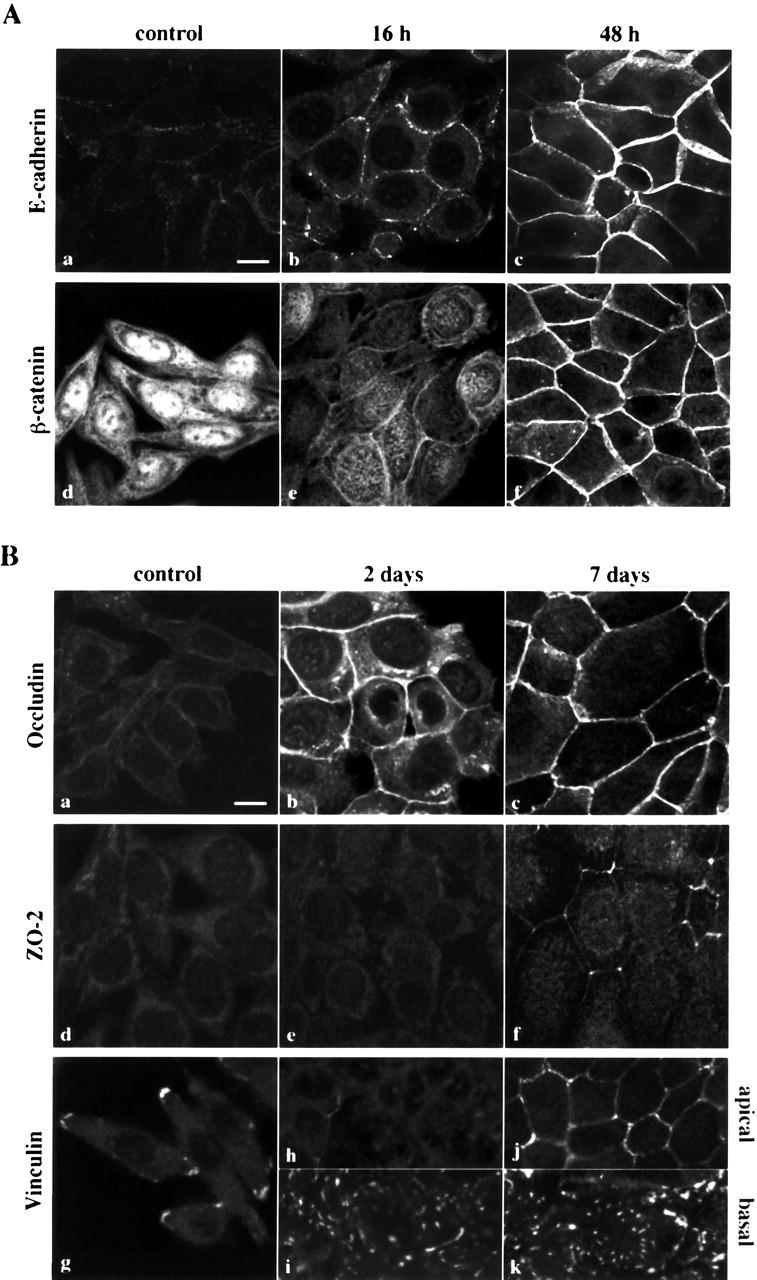
Induction of epithelial markers by 1α,25(OH)2D3 in SW480-ADH cells. (A) Analysis by immunofluorescence and confocal laser scanning microscopy of the expression of various adhesion proteins in cells treated with 10−7 M 1α,25(OH)2D3 for the indicated times or left untreated (control): E-cadherin (a–c); β-catenin (d–f). (B) Same as in A with longer treatments: a–c, occludin; d–f, ZO-2; g–k, vinculin. Vinculin expression was analyzed at two sections: basal (g, i, and k) and apical (h and j). Bars, 10 μm.
1α,25(OH)2D3 also progressively increased the amount of occludin and ZO-2, two components of the tight junctions that are located predominantly at the plasma membrane (Fig. 2 B, a–c and d–f, respectively). ZO-1, another component of these structures, was also enhanced (see Fig. 6) . We also studied the effects of 1α,25(OH)2D3 on the expression of vinculin, a protein located in tight junctions, adherens junctions, and focal adhesion plaques (Watabe-Uchida et al., 1998). Untreated SW480-ADH cells showed a punctate staining of vinculin at the cell periphery, which probably corresponded to focal adhesion plaques (Fig. 2 B, g). 2 d after 1α,25(OH)2D3 addition, an increased dotted basal, but not apical, vinculin staining was observed (Fig. 2 B, h–i), whereas at 7 d after treatment both basal and apical surfaces were strongly stained, revealing the relocalization of vinculin in intercellular junctional structures (Fig. 2 B, j–k). Altogether, these data indicated that 1α,25(OH)2D3 induces the expression of intercellular adhesion proteins in SW480-ADH cultures. The acquisition of the adhesive phenotype and the formation of compact islands increased with the duration of 1α,25(OH)2D3 treatment and depended on the presence of this agent in the medium, since its removal led to progressive cell disaggregation. Finally, 1α,25(OH)2D3 also relocated γ-catenin/plakoglobin from the nucleus and cytoplasm to the plasma membrane (unpublished data).
Figure 6.
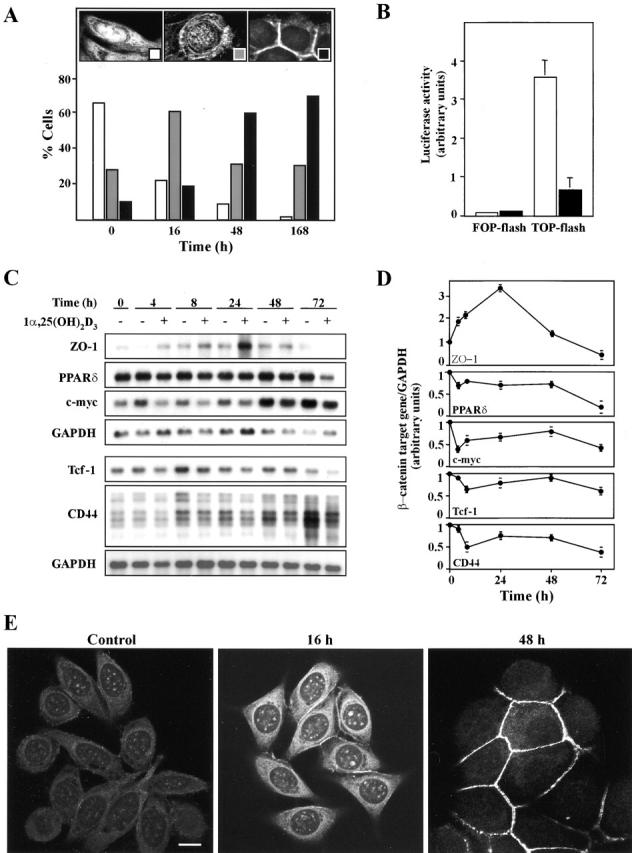
Inhibition of the β-catenin–TCF-4 signaling by 1α,25(OH)2D3. (A) 1α,25(OH)2D3 induces nuclear export of β-catenin. Quantification of the percentage of SW480-ADH cells showing predominant nuclear (left inset, white bars), mixed nuclear-cytoplasmic (middle inset, gray bars), or exclusively membranous (right inset, black bars) β-catenin localization after treatment with 1α,25(OH)2D3 (10−7 M) for the indicated times. 500 cells were analyzed at each time point. (B) Inhibition of β-catenin–TCF-4 transcriptional activity by 1α,25(OH)2D3. SW480-ADH cells were transfected with the wild-type (TOP-flash) or mutated (FOP-flash) β-catenin–TCF4/LEF-1–sensitive reporter plasmids and then left untreated (white bars) or treated (black bars) with 1α,25(OH)2D3 (10−7 M) for 48 h. Mean values and standard deviation of the mean of triplicated obtained in three experiments are shown. (C) Effects of 1α,25(OH)2D3 on the expression of β-catenin–TCF-4 target genes. Northern blots analysis of mRNA expression in SW480-ADH cells untreated or treated with 10−7 M 1α,25(OH)2D3 for the indicated times. Conditions were as above. (D) Quantification of the change in ZO-1, PPARδ, CD44, Tcf-1, and c-myc mRNA levels induced by 1α,25(OH)2D3. Mean values and error bars corresponding to triplicates obtained in three experiments are shown. (E) Induction and redistribution of ZO-1 protein by 1α,25(OH)2D3 treatment. Analysis by immunofluorescence and confocal laser microscopy of ZO-1 expression in SW480-ADH cells after addition of 1α,25(OH)2D3 (10−7 M). Bar, 10 μm.
To determine whether the induction of occludin, ZO-1, and ZO-2 led to the formation of functional tight junctions, we measured the transepithelial electrical resistance (TER) of SW480-ADH cell monolayers grown on porous filters. As expected from their tumoral origin and spindle shape, untreated SW480-ADH cells showed no significant TER. In contrast, cells treated for 4 wk with 1α,25(OH)2D3 displayed low but detectable TER when seeded on filters (day 0), which progressively increased (fourfold) on the following days, revealing the formation of functional tight junctions (Table I). The requirement for long 1α,25(OH)2D3 treatments to induce TER (1–2 wk were insufficient) suggests that additional unknown proteins must be induced with slow kinetics and/or that the establishment of adequate molecular interactions is a slow process.
Table I.
Induction by 1α,25(OH)2D3 of TER in SW480-ADH cells
| TER
|
|||
|---|---|---|---|
| Time | Control | 1α,25(OH)2D3 | |
| d
|
Ωcm2
|
||
| 0 | 8.3 | 20.7 | |
| 1 | 8.3 | 33.2 | |
| 2 | 12.4 | 41.5 | |
| 3 | 12.4 | 58.1 | |
| 4 | 12.4 | 70.5 | |
| 5 | 12.4 | 66.4 | |
| 6 | 12.4 | 70.6 | |
| 7 | 12.4 | 91.3 | |
| 8 | 16.6 | 83.1 | |
Cells pretreated during 30 d with either 1α,25(OH)2D3 (10−7) or vehicle were then cultured on Transwells, and resistance measurements were performed as described in Materials and methods during the following 8 d under the same treatments. Mean values of triplicate cultures are shown.
Given that adherens junctions are essential to intercellular adhesion and epithelial polarity and that E-cadherin plays a role in these structures, we explored the mechanism by which 1α,25(OH)2D3 induces this protein. Northern blotting revealed that a 48-h treatment with 1α,25(OH)2D3 substantially increased (five- to sixfold) the level of E-cadherin 4.6 kb mRNA in SW480-ADH cells but not in cells lacking VDR (SW480-R and SW620) (Fig. 3 A). In contrast, the amount of β-catenin 3.4 kb mRNA was unaffected, and comparable levels, slightly lower in SW480-R, were detected in the three cell lines. In agreement with these results, a strong increase in the level of E-cadherin protein but no change in that of β-catenin were found upon 1α,25(OH)2D3 treatment (Fig. 3 B). Other hormones acting through members of the nuclear receptor superfamily such as dexamethasone, thyroid hormone, and all-trans retinoic acid failed to induce E-cadherin expression, showing the specificity of 1α,25(OH)2D3 (Fig. 3 C; unpublished data). Likewise, 9-cis retinoic acid, whose retinoid X receptor (RXR) is the common partner of VDR, was ineffective by itself, and it did not alter the induction of E-cadherin by 1α,25(OH)2D3 (Fig. 3 C; unpublished data). This strongly suggests that ligand activation of VDR is sufficient for E-cadherin induction by RXR-VDR heterodimers. We then studied the kinetics of the induction of E-cadherin mRNA in SW480-ADH cells by 1α,25(OH)2D3. The induction was abrupt with a rapid increase (10-fold) 4 h after addition and a peak at ∼8 h (12.5-fold) followed by a slow decay (Fig. 3, D and F). Accordingly, the cellular content of E-cadherin protein increased by 7-fold at 16 h and by ∼13-fold at 48 h after treatment (Fig. 3, E and F).
Figure 3.

Induction of E-cadherin expression by 1α,25(OH)2D3. (A) Northern blot analysis of E-cadherin and β-catenin mRNA expression in SW480-ADH, SW480-R, and SW620 cells untreated or treated with 10−7 M 1α,25(OH)2D3 for 48 h. 10 μg of poly(A)+ RNA was loaded per lane. GAPDH was used as an internal control. (B) Western blot analysis of E-cadherin and β-catenin protein expression in the same conditions. (C) Specificity of 1α,25(OH)2D3 action. Northern blot analysis of E-cadherin mRNA expression in SW480-ADH cells untreated or treated with 10−7 M of the indicated agent or the corresponding vehicles for 48 h. Conditions were as above. (D) Northern blot analysis of the kinetics of induction of E-cadherin mRNA by 1α,25(OH)2D3 in SW480-ADH cells. Times of treatment are indicated. Conditions were as above. (E) Western blot analysis of the kinetics of induction of E-cadherin protein by 1α,25(OH)2D3 in SW480-ADH cells. Times of treatment are indicated. (F) Quantification of the induction by 1α,25(OH)2D3 in SW480-ADH cells of E-cadherin mRNA (•) and protein (○) and of β-catenin mRNA (▪) and protein (□). Fold increase with respect to expression in untreated cells (time 0) is represented. Mean values of three experiments are shown. Quantifications were performed using NIH image software.
Transcription and translation inhibitors (actinomycin D and cycloheximide, respectively) were used to understand the E-cadherin induction. Actinomycin D completely blocked the effect of 1α,25(OH)2D3, whereas cycloheximide strongly inhibited it (Fig. 4 A). This indicates that 1α,25(OH)2D3 induces E-cadherin expression by a transcriptional mechanism that requires protein synthesis de novo. Since 1α,25(OH)2D3 interacts with the phosphoinositol 3-kinase and protein kinase C–signaling pathways in various cell types (Gniadecki et al., 1997; Hmama et al., 1999), we aimed to elucidate whether this also occurs in SW480-ADH cells. However, the induction of E-cadherin was unaffected by inhibitors of phosphoinositol 3-kinase (wortmannin, Ly294002), and it was only partially affected (30% reduction in RNA increase at 4 h) by a protein kinase C antagonist (bisindolylmaleimide), suggesting that these two signaling pathways are not relevant to the activity of 1α,25(OH)2D3 (unpublished data). The induction of E-cadherin transcription by 1α,25(OH)2D3 was tested in transient transfection experiments using two distinct constructs: either a short 270-bp (−178 to +92) or a long 1,072-bp (−987 to +92) fragment of the human gene promoter controlling the expression of the luciferase reporter gene. Consistently, a 1.8-fold induction of the large promoter construct but not of the small one was observed (Fig. 4 B). Further supporting the transcriptional effect of 1α,25(OH)2D3, no induction was detected when mutant forms of VDR (ΔAF2), lacking the COOH-terminal AF-2 domain responsible for transcriptional activation or harboring inactivating point mutations in this region (L417S and E420Q), were transfected (unpublished data). The discrepancy between the level of induction of the promoter construct used and the higher increase in E-cadherin mRNA content induced by 1α,25(OH)2D3 in SW480-ADH cells may be due to the lack of additional regulatory sequences in the available promoter region studied and/or effects on RNA stability. We ruled out the second hypothesis by analyzing whether 1α,25(OH)2D3 modulated the half-life of E-cadherin mRNA in SW480-ADH cells: the decay in E-cadherin mRNA content was the same in untreated and in 1α,25(OH)2D3-treated cells (Fig. 4 C).
Figure 4.
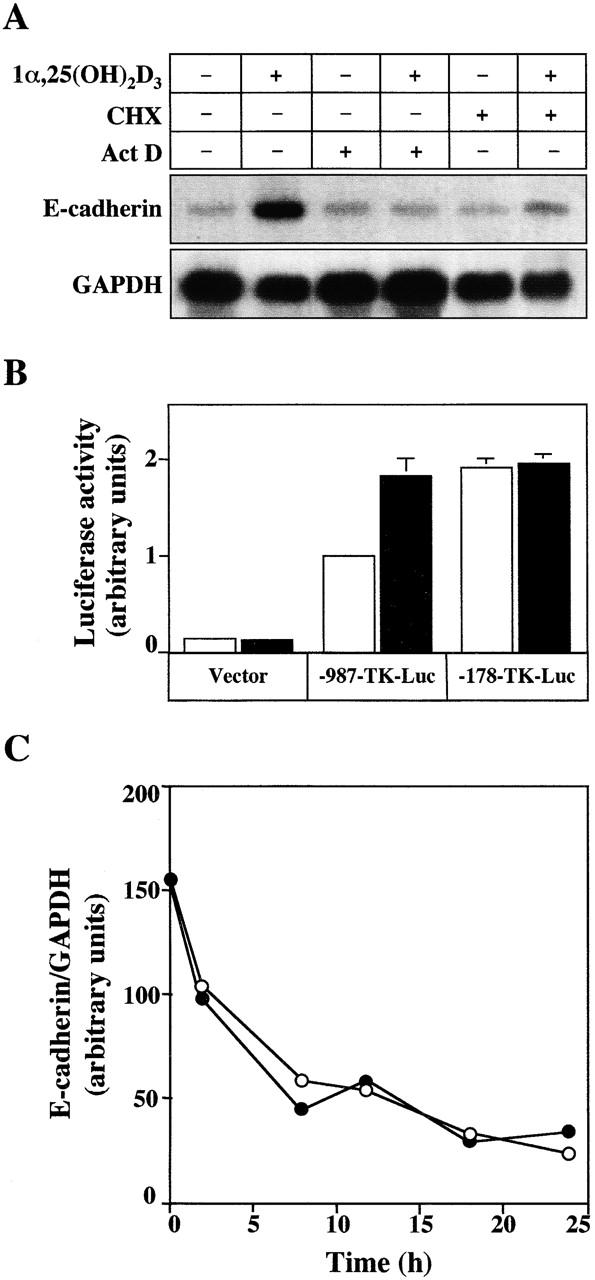
Mechanism of E-cadherin gene induction by 1α,25(OH)2D3. (A) Northern blot analysis of E-cadherin mRNA expression in SW480-ADH cells untreated or treated with 10−7 M 1α,25(OH)2D3 for 4 h. Where indicated, cells were pretreated with actinomycin D (Act D, 2 μg/ml) or cycloheximide (CHX, 8 μg/ml) 30 min before 1α,25(OH)2D3 addition. 10 μg of poly(A)+ RNA was loaded per lane. GAPDH was used as an internal control. (B) Activation of the human E-cadherin gene promoter by 1α,25(OH)2D3. SW480-ADH cells were transfected with either −987-TK-Luc plasmid, which contains the genomic sequence from +92 to −987 bp, or −178-TK-Luc containing the sequence from +92 to −178 bp of the human E-cadherin gene. The empty TK-Luc vector was used as control. Transfections were performed as described in Materials and methods. White bars, untreated cells; black bars, cells treated with 10−7 M 1α,25(OH)2D3 during 48 h after transfection. Mean values corresponding to five independent experiments done in triplicate are shown. (C) Lack of effect of 1α,25(OH)2D3 on E-cadherin mRNA stability. SW480-ADH cells were pretreated or not pretreated for 30 min with actinomycin D (2 μg/ml) and then incubated in the presence (•) or absence (○) of 1α,25(OH)2D3 (10−7 M), during the indicated times. Northern blot analysis of E-cadherin and GAPDH mRNA expression. Conditions were as above. Two independent experiments gave the same result.
1α,25(OH)2D3 causes β-catenin to translocate from the nucleus to E-cadherin complexes at the plasma membrane and inhibits its gene regulatory activity
Exogenous E-cadherin induces β-catenin relocation by nuclear export and the formation of E-cadherin–β-catenin complexes at the plasma membrane in SW480 and other cell lines lacking endogenous E-cadherin (Sadot et al., 1998; Orsulic et al., 1999). Since 1α,25(OH)2D3 induced E-cadherin expression in SW480-ADH cells, we determined whether it could promote a similar effect in these cells. 2-d treatments with 1α,25(OH)2D3 induced a nearly complete colocalization of both proteins at the region of the plasma membrane in contact with neighboring cells (Fig. 5 , top). An identical effect was observed when untreated SW480-ADH cells were transfected with an E-cadherin expression plasmid (Fig. 5, top). As controls, we used VDR-negative SW480-R cells, which did not show any change in the strong nuclear β-catenin staining upon 1α,25(OH)2D3 addition (Fig. 5, bottom). As expected, exogenous E-cadherin caused the same change in β-catenin localization as in SW480-ADH cells (Fig. 5, bottom). To correlate the change in β-catenin localization induced by 1α,25(OH)2D3 with the effects on β-catenin–TCF/LEF-1 target genes as a function of time, we studied the kinetics of β-catenin nuclear export. To this end, we analyzed the immunofluorescence of SW480-ADH cells stained with an anti–β-catenin antibody by confocal laser microscopy and counted the cells displaying predominantly nuclear, a mixed nuclear-cytosolic, or an exclusively membranous β-catenin localization (Fig. 6 A, insets). The number of nuclear-positive β-catenin cells clearly decreased (from 66 to 24%) as soon as 16 h after 1α,25(OH)2D3 addition, was further reduced (to 10%) at 48 h, and was extremely low (around 2%) 7 d later (Fig. 6 A, white bars). Conversely, the proportion of cells with predominant membrane-bound β-catenin increased progressively from 11% in nontreated cultures to 70% after 7 d of 1α,25(OH)2D3 treatment (Fig. 6 A, black bars).
Figure 5.

Induction of colocalization of E-cadherin and β-catenin at the plasma membrane by 1α,25(OH)2D3. Analysis by immunofluorescence and confocal laser scanning microscopy of the expression of these two proteins in SW480-ADH or SW480-R cells at 48 h after either treatment with 1α,25(OH)2D3 (10−7 M) or transfection with an expression vector for human E-cadherin. Double immunofluorescence was performed using anti–E-cadherin and anti–β-catenin antibodies followed by the addition of the corresponding secondary TRICT-conjugated (E-cadherin, red) or FITC-conjugated (β-catenin, green) antibodies. The merge of both signals (yellow) indicates the areas of colocalization of both proteins. Bars, 10 μm.
Therefore, we aimed to examine the putative effects of 1α,25(OH)2D3 on β-catenin signaling. First, we checked whether TCF-4 RNA expression was modified by 1α,25 (OH)2D3 or not as in the case of β-catenin. No changes were detected by Northern and Western blotting (unpublished data). To measure β-catenin–mediated transcription, we used a reporter gene placed under the control of a promoter constructed by fusing three copies of the TCF/LEF-1 consensus responsive sequence and a c-fos minimal promoter (TOP-flash). A mutated form of this plasmid (FOP-flash) was used as negative control. 1α,25(OH)2D3 effectively inhibited β-catenin–TCF/LEF-1 transcriptional activity in transfected SW480-ADH cells (Fig. 6 B). Moreover, since these cells express a mutant APC protein and 1α,25 (OH)2D3 did not modify β-catenin levels this result shows that wild-type APC is not required by 1α,25(OH)2D3 to inhibit β-catenin signaling.
Next, we analyzed the regulation of endogenous β-catenin–TCF/LEF-1 target genes. 1α,25(OH)2D3 reduced the cellular RNA levels of c-myc, Tcf-1, CD44, and PPARδ genes induced by β-catenin–TCF/LEF-1 (He et al., 1998, 1999; Roose et al., 1999; Vera et al., 1999) (Fig. 6, C and D). They were downregulated with biphasic kinetics: a rapid (4–8 h) and slight (25–30%) reduction followed (after 48 h) by an additional diminution (to 50–80%) when β-catenin was no longer present into the nucleus. Though in untreated cells c-myc and CD44 RNA levels increased during incubation probably due to the proliferation status, 1α,25(OH)2D3 always had an inhibitory effect. In addition, 1α,25(OH)2D3 also rapidly induced the expression of ZO-1 (Fig. 6, C and D), which is repressed by β-catenin–TCF/LEF-1 (Mann et al., 1999). Like β-catenin, ZO-1 is involved in intercellular adhesion and perhaps signaling (Gottardi et al., 1996). This increase was confirmed at the protein level by immunofluorescence and confocal microscopy analyses (Fig. 6 E). In untreated SW480-ADH cells, ZO-1 showed a diffuse cytoplasmic and punctate nuclear localization. 16 h after 1α,25(OH)2D3 addition, the cellular content of ZO-1 clearly increased and was also located in cell-to-cell contact areas. At 48-h after treatment, the subcellular distribution of ZO-1 was drastically altered, since it was almost completely peripheral. Thus, 1α,25(OH)2D3 has a dual effect on ZO-1: it relieves the repression by β-catenin–TCF/LEF-1 at the transcriptional level, and it induces the redistribution of ZO-1 protein to the plasma membrane. This relocation was concomitant with that of β-catenin in accordance with the nuclear coexport of ZO-1 and catenins that takes place during the differentiation of MDCK epithelial cells (Rajasekaran et al., 1996). Although cyclin D1 has been proposed as a β-catenin–regulated gene (Tetsu and McCormick, 1999), neither cyclin D1 RNA nor protein expression were modified upon 1α,25(OH)2D3 addition to SW480-ADH cells (unpublished data). These data indicate that in SW480-ADH cells 1α,25(OH)2D3 downregulates β-catenin–TCF/LEF-1 target genes.
To examine how general the effects of 1α,25(OH)2D3 observed in SW480-ADH cells were, we extended the study to other human colon carcinoma cell lines such as Caco-2, SW1417, HT-29 M6, and LS-147T. As in SW480-ADH cells, 1α,25(OH)2D3 induced a parallel activation (two- to fourfold) of a VDRE–reporter construct and an inhibition of β-catenin–TCF-4 transcriptional activity (25–50%) (Fig. 7 A). SW480-R and SW620 cells were used as negative control. In addition, E-cadherin expression was increased (two- to threefold after 48 h) by 1α,25(OH)2D3 in these cell lines but in LS-174T, which were totally defective for this protein (Fig. 7 B).
Figure 7.
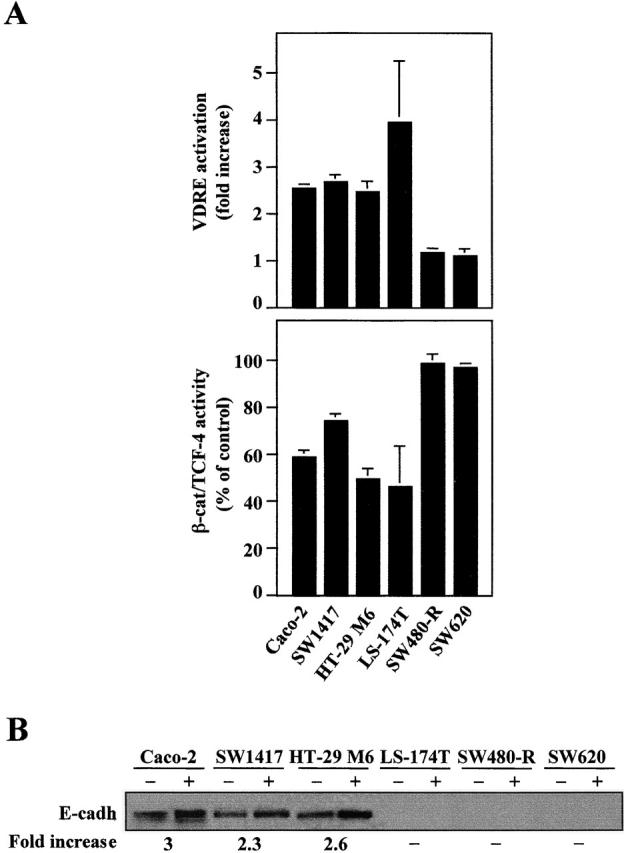
1α,25(OH)2D3 regulates VDR and β-catenin–TCF-4 transcriptional activity and E-cadherin expression in multiple human colon cancer cell lines. (A) Activation of VDR (top) and inhibition of β-catenin–TCF-4 transcriptional activity (bottom) by 1α,25(OH)2D3. Cells were transfected with either the 4 × VDRE–DR3-tk-luc construct (top) or the TOP-flash plasmid (bottom) and then treated or not with 1α,25(OH)2D3 (10−7 M) for 48 h in DME supplemented with 0.5% FCS. SW480-R and SW620 cells were used as negative control. Mean values and standard deviation of the mean of triplicated obtained in two or three experiments after normalization are shown. (B) Effect of the same 1α,25(OH)2D3 treatment on the expression of E-cadherin in the same cell lines. 20 μg of total cell protein extracts (with the exception of 60 μg in the case of LS-174T cells) were analyzed by Western blot. The result shown is representative of two or three experiments performed with each cell line. Quantification of the stimulation by 1α,25(OH)2D3 is shown below.
VDR and β-catenin interact in a ligand-regulated manner
The rapid changes in the RNA levels of target genes after 1α,25(OH)2D3 treatment, which occurred earlier than the induction of β-catenin nuclear export, suggested additional mechanisms of action for 1α,25(OH)2D3. We tested whether VDR and β-catenin interacted, thus affecting the formation of β-catenin–TCF-4 complexes. To this end, in vitro–translated VDR was incubated with either glutathione S-transferase (GST)–β-catenin or GST proteins in the presence or absence of 1α,25(OH)2D3. After precipitation with GSH–Sepharose beads, the presence of VDR in the precipitates was tested by immunoblotting using an anti-VDR antibody, revealing a direct mostly ligand-independent VDR–β-catenin interaction (Fig. 8 A). To determine whether VDR and β-catenin form complexes in vivo, extracts from cells incubated with 1α,25(OH)2D3 for various periods were immunoprecipitated with either anti–β-catenin or anti-VDR antibodies and then subjected to immunoblotting with the opposite antibody. 1α,25(OH)2D3 rapidly (4 h) enhanced the VDR–β-catenin interaction (Fig. 8 B). Conversely, 1α,25(OH)2D3 decreased the amount of TCF-4 present in β-catenin immunoprecipitates (Fig. 8 B), suggesting that VDR and TCF-4 compete for β-catenin binding. 48 h after treatment, the level of VDR–β-catenin interaction was similar to that of untreated cells, probably because of the strong induction of E-cadherin, which leads to β-catenin sequestration at the cell junctions. In fact, coimmunoprecipitation using anti–E-cadherin and anti–β-catenin antibodies revealed the formation of E-cadherin–β-catenin complexes, which replaced VDR–β-catenin complexes as early as 16 h after 1α,25(OH)2D3 treatment in the same extracts (Fig. 8, B–F). We would like to point out that the change in complex formation was concomitant with E-cadherin protein induction and phenotypic modulation by 1α,25(OH)2D3. Similar results were obtained when the presence of β-catenin was determined in VDR immunoprecipitates. Differences in the amount of β-catenin–VDR complexes immunoprecipitated with one or the other antibodies at 16 h after 1α,25(OH)2D3 addition must be attributed to differences in affinity and/or epitope recognition. As stated above, 1α,25(OH)2D3 did not modify the cellular content of VDR, whereas in the case of β-catenin and TCF-4 the apparent changes were due to the weak extracting conditions used to preserve protein–protein interactions in these assays (Fig. 8 B). However, no changes in total β-catenin, VDR, or TCF-4 levels were detected under strong solubilization conditions as mentioned above (unpublished data). As expected, no VDR–β-catenin complexes were detected in SW480-R cells expressing only residual amounts of VDR (Fig. 8 G) that were unable to mediate VDRE activation (Fig. 1 C and Fig. 7).
Figure 8.
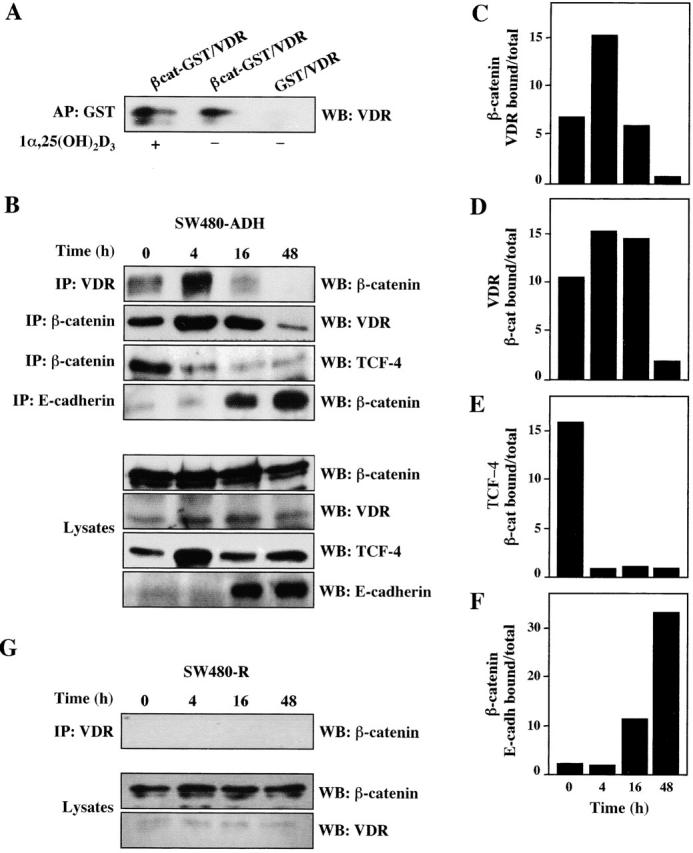
Induction of VDR–β-catenin interaction by 1α,25(OH)2D3. (A) Interaction in vitro. In vitro–translated human VDR and bacterially produced GST– β-catenin were incubated in the presence or absence of 1α,25(OH)2D3 as described in Materials and methods. Western blot (WB) analysis using a specific anti-VDR antibody of the material precipitated upon incubation with GSH–Sepharose beads. GST alone was used in parallel to rule out unspecific binding. (B) Interaction in vivo. Extracts of SW480-ADH cells untreated or treated with 1α,25(OH)2D3 (10−7 M) for the indicated times were subjected to immunoprecipitation (IP) with anti-VDR, or β-catenin, or anti-E-cadherin antibodies followed by Western blot with the antibodies indicated in each case. Western blot analysis showing the proportion of each protein present in the lysates. (C) Quantification of the amount of β-catenin bound to VDR after 1α,25(OH)2D3 addition. (D) Quantification of the amount of VDR bound to β-catenin after 1α,25(OH)2D3 addition. (E) Quantification of the amount of TCF-4 bound to β-catenin after 1α,25(OH)2D3 addition. (F) Quantification of the amount of β-catenin bound to E-cadherin after 1α,25(OH)2D3 addition. (G) Absence of VDR–β-catenin coimmunoprecipita- tion in SW480-R cells. Extracts of SW480-R cells untreated or treated with 1α,25(OH)2D3 (10−7 M) for the indicated times were subjected to immunoprecipitation with anti-VDR antibody followed by Western blotting using anti–β-catenin antibody. Western blot analysis of the lysates show the presence of only residual level of VDR.
Functional interplay between VDR, β-catenin, and TCF-4
Given that β-catenin is a transcriptional coactivator and interacts with VDR, we examined whether it enhances the activation of a VDRE by 1α,25(OH)2D3. To this end, we used a plasmid encoding a stable form of β-catenin resistant to APC-induced degradation in which the GSK-3β target Ser37 residue is mutated to Ala (S37A) (Easwaran et al., 1999). Exogenous β-catenin S37A expression did not modify VDRE activation in SW480-ADH or SW480-R cells, probably because of the high level of endogenous nuclear β-catenin (Fig. 9 A). To circumvent this problem, we transfected human MCF-7 mammary epithelial cells containing extremely low amounts of nuclear β-catenin. In these cells, β-catenin S37A fostered the induction by 1α,25(OH)2D3 of a consensus VDRE from 6- to 49-fold (Fig. 9 B). In contrast, β-catenin S37A did not affect the basal VDRE levels in untreated cells (unpublished data). No such effect was observed on thyroid or glucocorticoid hormone response elements or on a κB-dependent construct (unpublished data). In contrast to β-catenin, exogenous TCF-4 caused a twofold reduction in VDRE activation in SW480-ADH (over endogenous VDR) and SW480-R (over exogenous VDR) cells (Fig. 9 A), further supporting the hypothesis that VDR and TCF-4 compete for β-catenin binding. A dose-response assay of transfection of increasing amounts of TCF-4 expression plasmid in SW480-ADH cells confirmed that TCF-4 hinders VDR activity (Fig. 9 C). In contrast, a mutant TCF-4 lacking the NH2-terminal region required for β-catenin binding (ΔΝ-TCF-4) was unable to inhibit VDR activity (Fig. 9 C). Additionally, the inhibition of VDRE activation by TCF-4 was completely reversed by cotransfection of β-catenin S37A (Fig. 9 C).
Figure 9.
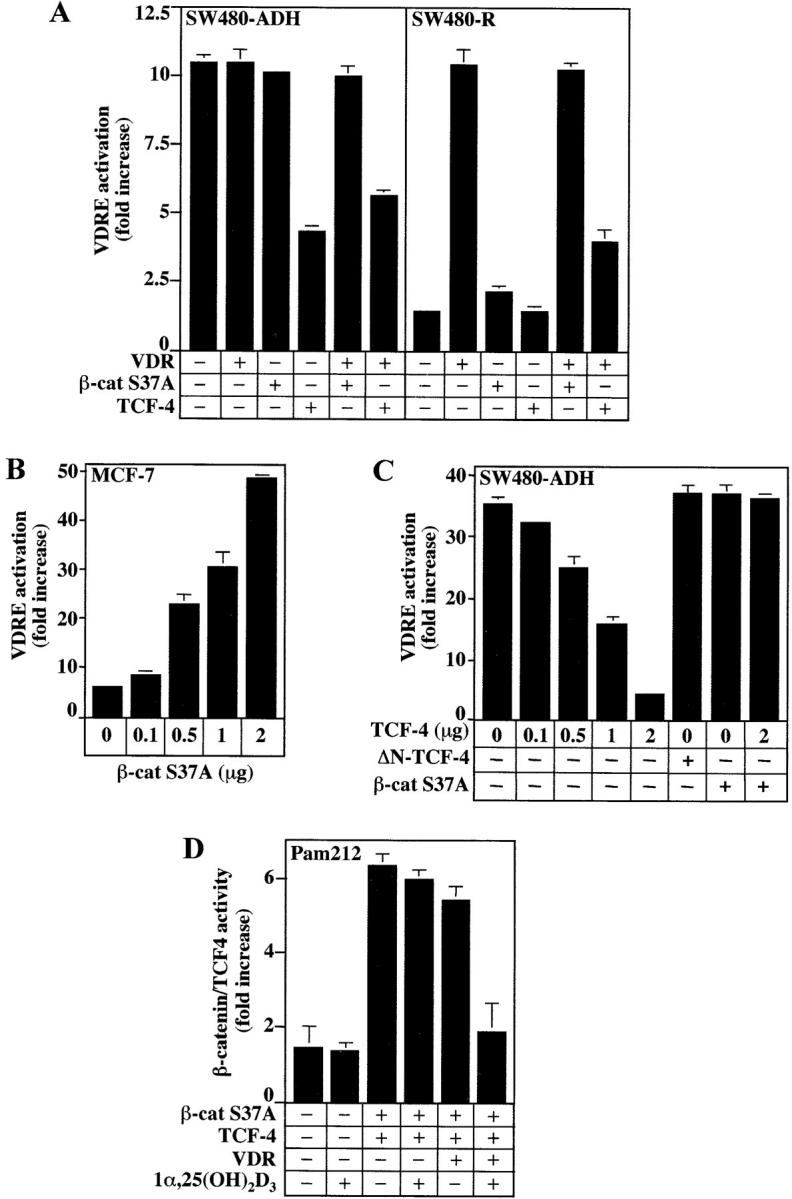
Modulation of the transcriptional activity of VDR by β-catenin and TCF-4. (A) SW480-ADH and SW480-R cells were transfected with the 4 × VDRE–DR3-Tk-Luc construct in combination with expression vectors for VDR, β-catenin S37A, or TCF-4 as indicated. Luciferase activity was measured in extracts of cells untreated or treated with 1α,25(OH)2D3 (10−7 M) for 48 h. (B) β-Catenin enhances VDRE activation in MCF-7 cells. Cells were cotransfected with 4 × VDRE–DR3-Tk-Luc and increasing amounts of β-catenin S37A expression vector. (C) TCF-4 inhibits VDRE activation in SW480-ADH cells. Cells were cotransfected with 4 × VDRE–DR3-Tk-Luc construct and variable amounts of expression vectors for wild-type TCF-4, the mutant ΔN-TCF-4, or β-catenin S37A as indicated. Conditions and measurements were as above. In A–C, VDRE activation is represented as fold increase in treated versus untreated cells. (D) 1α,25(OH)2D3 inhibits β-catenin–TCF-4 transcriptional activity in Pam212 cells. Cells were co-transfected with TOP-flash or FOP-flash reporter constructs and with expression vectors for β-catenin, TCF-4, and VDR as indicated. 1α,25(OH)2D3 (10−7 M) was added 24 h after transfection, and cell extracts were prepared 24 h later. Fold increase values of luciferase activity (TOP/FOP) after normalization were calculated. In A–D, mean values and standard deviation of the mean obtained in duplicates of three independent experiments are shown.
Next, we examined whether ligand-activated VDR inhibits the transcriptional activity of β-catenin–TCF/LEF-1 complexes regardless of E-cadherin induction. To this end, we transfected β-catenin and TCF-4 in Pam212, a keratinocyte murine cell line expressing high amounts of E-cadherin and lacking nuclear β-catenin. In these cells, which express only residual levels of VDR, the activation of the TOP-flash reporter by β-catenin–TCF-4 was inhibited (greater than threefold) by 1α,25(OH)2D3 upon ectopic expression of VDR (Fig. 8 D) without concomitant induction of E-cadherin as assessed by Western blotting (unpublished data). This result is in line with those obtained in LS-174T cells in which 1α,25(OH)2D3 induces VDRE activation and inhibits β-catenin–TCF-4 transcriptional activity in spite of a total lack of E-cadherin induction (Fig. 7 B).
Nonhypercalcemic vitamin D3 analogues are potent inhibitors of the β-catenin signaling pathway
Three synthetic vitamin D analogues with low calcemic properties, EB1089, KH1060, and MC903, used currently in clinical trials to treat several neoplasias (see Introduction for references) were studied for their effects on β-catenin signaling in SW480-ADH cells. Like 1α,25(OH)2D3, all three analogues displayed antiproliferative activity (unpublished data). We then studied whether these compounds induce the expression of E-cadherin mRNA and protein by Northern and Western blotting. As 1α,25(OH)2D3, doses in the 10−11–10−7 M range of each compound induced E-cadherin expression (Fig. 10 A). All three compounds reduced the expression of a β-catenin–TCF-4 reporter construct in SW480-ADH cells (Fig. 10 B). The effects observed in the assays show that EB1089 and KH1060 are more potent than 1α,25(OH)2D3, whereas MC903 is less active. In agreement with our previous results, all three analogues promoted the morphological differentiation and nuclear export of β-catenin in SW480-ADH cells but had no effect in VDR-defective SW480-R or SW620 cells as revealed by immunofluorescence (Fig. 10 C).
Figure 10.



Nonhypercalcemic 1α,25(OH)2D3 derivatives induce E-cadherin and inhibit β-catenin–TCF-4 transcriptional activity in SW480-ADH cells, causing β-catenin nuclear export and morphological differentiation. (A) Northern and Western blot analyses of E-cadherin expression in cells treated for 24 h and 48 h, respectively, with various doses of 1α,25(OH)2D3, MC903, KH1060, or EB1089. Conditions were as above. (B) β-catenin–TCF-4 transcriptional activity in cells transfected with the TOP-flash and FOP-flash constructs and treated 48 h with 10−7 M of the indicated compound. Mean values and standard deviation of the mean obtained in duplicates of two independent experiments are shown. (C) Induction of differentiation and nuclear export of β-catenin. Immunofluorescence and confocal laser microscopy analysis of β-catenin expression was done as before. SW480-R and SW620 cells were used as negative control.
Discussion
Our results show that 1α,25(OH)2D3 and several nonhypercalcemic analogues used currently in clinical trials as potential anticancer drugs induce the differentiation of human colon carcinoma cells. They promote a phenotypic change involving the induction of E-cadherin expression and the blockade of the β-catenin gene regulatory activity in SW480-ADH cells.
1α,25(OH)2D3 inhibits the transcriptional activity of β-catenin by two mechanisms. On the one hand, it rapidly increases the amount of VDR bound to β-catenin, blocking the interaction of this catenin to TCF-4. Therefore, 1α,25(OH)2D3 modulates TCF/LEF-1 target genes in a manner opposite to β-catenin. In some cells, such as Pam212 and LS-174T, this effect is independent of changes in E-cadherin expression. Secondly, in SW480-ADH cells changes in β-catenin transcriptional activity caused by 1α,25(OH)2D3 are accompanied by the nuclear export of β-catenin and its relocalization to the plasma membrane that happens concomitantly to E-cadherin protein expression. 1α,25(OH)2D3 might enhance the sequestration of β-catenin in the plasma membrane compartment by E-cadherin or alternatively might stimulate β-catenin nuclear export, a process mediated by APC (Henderson, 2000; Rosin-Arbesfeld et al., 2000). Alternatively, 1α,25(OH)2D3 may stimulate the APC-independent β-catenin export described recently (Eleftheriou et al., 2001). Although an estimation of the respective contribution of these activities to the inhibition of β-catenin signaling is very difficult, their respective timing supports the hypothesis that the primary effect is due to the formation of VDR–β-catenin complexes. By favoring this, 1α,25(OH)2D3 may indirectly regulate the transcription of β-catenin–TCF/LEF-1 target genes such as Tcf-1, CD44, PPARδ, and ZO-1. The changes in RNA content for these genes after 1α,25(OH)2D3 treatment occur earlier than those in β-catenin localization, which suggests that they are initiated as a result of the VDR–TCF-4 competition for β-catenin and later strengthened by the nuclear export of β-catenin.
The physical interaction of VDR with β-catenin adds to that reported previously for retinoic acid receptor (RAR) (Easwaran et al., 1999). However, the interaction of β-catenin with these two nuclear receptors differs. RAR strictly depends on ligand binding, whereas a certain amount of VDR–β-catenin complexes were found in vitro in the absence of 1α,25(OH)2D3. In contrast, this basal interaction augments in the presence of 1α,25(OH)2D3 in vivo, suggesting the participation of ligand-dependent nuclear mediator(s) or mechanism(s). However, given the high activity of 1α,25 (OH)2D3 in our cell system, low amounts of metabolically active vitamin D derivatives in the culture medium may be sufficient to activate VDR in the absence of added agent. Likewise, the stimulation by β-catenin of the effect of 1α,25 (OH)2D3 on a VDRE-dependent promoter agrees with that reported for RAR-responsive promoters (Easwaran et al., 1999). In vivo, the interaction between VDR and β-catenin may ameliorate β-catenin–TCF-4 signaling. Upon β-catenin stabilization due to its mutation or that of APC, binding to VDR may buffer its stimulatory action on TCF-4 target genes, a protective effect which can be lost along with VDR expression during malignant progression. Additionally, our data suggest that nuclear β-catenin might transiently potentiate VDR transcriptional activity before β-catenin moves out of the nucleus and/or VDR is extinguished.
The β-catenin homologue γ-catenin/plakoglobin is also regulated by APC and functions as an oncogene (Kolligs et al., 2000). Like β-catenin, it activates c-myc expression in APC-mutated cells, which together with mutations in its NH2-region is thought to be critical for its oncogenic activity (Kolligs et al., 2000). γ-Catenin/plakoglobin indirectly activates TCF/LEF-1–regulated genes by increasing the levels of β-catenin and by inducing its nuclear translocation (Zhurinsky et al., 2000). These data show similarities but also differences in the mechanism of action of β-catenin and γ-catenin/plakoglobin. 1α,25(OH)2D3 induces the nuclear export of γ-catenin/plakoglobin to the plasma membrane, which may also contribute to the phenotypic change described in this study.
Our results show the competition between VDR, TCF-4, and E-cadherin for binding to β-catenin. Since the E-cadherin gene has been proposed to be down-regulated by β-catenin upon binding of β-catenin–TCF/LEF-1 to its promoter (Huber et al., 1996) and 1α,25(OH)2D3 induces a concomitant increase in VDR–β-catenin binding and E-cadherin mRNA, an effect on the E-cadherin promoter is plausible. The formation of VDR–β-catenin complexes may reduce the β-catenin binding to TCF-4 and so lead to a relief of promoter repression and/or have a direct activating effect. In disagreement with this hypothesis, VDR overexpression in SW480-ADH cells did not increase the basal activation of the 1.1-kb E-cadherin promoter construct, although potential regulatory sequences outside this region cannot be ruled out. The moderate induction by 1α,25(OH)2D3 of this E-cadherin gene promoter construct does not unambiguously demonstrate that E-cadherin gene is regulated transcriptionally by this agent. However, this effect is consistently observed, and together with the inhibitory action of actinomycin and the lack of regulation of E-cadherin RNA half-life suggests an effect at the transcription level.
Our data show that 1α,25(OH)2D3 increases E-cadherin gene transcription by a mechanism that is dependent on the AF-2 domain of VDR and mediated by the novo synthesis of short-lived proteins, although additional posttranscriptional routes cannot be discarded. E-cadherin expression is inhibited by the product of Snail gene, which is a transcriptional repressor on the E-boxes located near the transcription start site (Batlle et al., 2000; Cano et al., 2000). We show that 1α,25(OH)2D3 regulates the human E-cadherin promoter when a region of ∼1.1 kb upstream of the +92 site is studied, but it has no effect on the activity of the proximal promoter region that contains the E-boxes bound by Snail. These data suggest that 1α,25(OH)2D3 and Snail have opposite effects on distinct sequences of the E-cadherin promoter. Though no changes in Snail mRNA expression were detected upon 1α,25(OH)2D3 treatment, further research is required to determine a putative interplay between these agents.
Whether the increase observed in the cellular content of other intercellular adhesion proteins such as occludin, ZO-1, ZO-2, and vinculin is a direct effect of 1α,25(OH)2D3 or a corollary of its primary effect on E-cadherin and/or β-catenin is unknown. Since the ZO-1 gene is repressed by β-catenin–TCF-4 (Mann et al., 1999) and ZO-1 mRNA increases before E-cadherin protein levels are significantly elevated, that is, when β-catenin is abundant in the nucleus, the effect of 1α,25(OH)2D3 on this gene is probably due to the rapid formation of VDR–β-catenin complexes and the subsequent reduction of β-catenin–TCF-4 complexes. Additionally, ZO-1 protein forms complexes with α-, β- and γ-catenins (Rajasekaran et al., 1996), and like β-catenin it can locate in the cell nucleus where it may function as a signaling molecule (Gottardi et al., 1996). In untreated SW480-ADH cells, ZO-1 is distributed diffusely in the cytoplasm and in intranuclear foci. Upon 1α,25(OH)2D3 treatment, ZO-1 is first increased and then redistributes to the cell surface as in MDCK cells after E-cadherin expression (Rajasekaran et al., 1996). The relevance of the effects of 1α,25(OH)2D3 on ZO-1 reported here is supported by a recent report describing the regulation of c-ErbB2/Neu expression by ZO-1 (Balda and Matter, 2000). It is also significant that other genes regulated by 1α,25(OH)2D3 in SW480-ADH cells play important roles: occludin expression has been associated with tumor differentiation (Kimura et al., 1997), whereas vinculin is involved in the organization of tight junctions (Watabe-Uchida et al., 1998). These changes are consistent with the key role of E-cadherin in the maintenance of epithelial characteristics and account for the drastic change induced by 1α,25(OH)2D3 in the differentiation status of SW480-ADH cells.
The rapid inhibitory effect of 1α,25(OH)2D3 on c-myc expression, another TCF/LEF-1–β-catenin target gene, probably results from the combined effects of activities at various levels, which take place before nuclear export of β-catenin. First, 1α,25(OH)2D3 may induce the binding of regulatory proteins to the first intron of the gene (Pan and Simpson, 1999). Second, the formation of VDR–β-catenin complexes may inhibit the activation by β-catenin–TCF/LEF-1, as may the alteration of γ-catenin/plakoglobin localization (Kolligs et al., 2000).
The effects of 1α,25(OH)2D3 in SW480-ADH cells are transient and depend on its nuclear receptors. VDR content is low in normal colon epithelial cells, increases at the early stages of tumor progression, and is almost absent in the more malignant carcinoma cells (Vandewalle et al., 1994). This agrees with the finding that 1α,25(OH)2D3 inhibits proliferation in human rectal mucosa (Thomas et al., 1992) and with its activity in the weakly tumorigenic SW480-ADH cells and its inefficacy in the highly tumorigenic SW480-R cells. Our data are consistent with the proposed protective role of dietary vitamin D or sunlight exposure and with the predictive use of VDR expression in colon cancer biopsies. Our results suggest that liganded VDR may hinder the loss of differentiation and the increase in proliferation at the early stages of carcinogenesis. 1α,25(OH)2D3 has a pleiotropic biological activity with complex cell-specific antitumor properties that include induction of apoptosis, growth arrest, inhibition of invasiveness, and stimulation of differentiation. The pattern of expression of the coactivators (SRC-1, CBP, GRIP-1/TIF-2, and others) and corepressors (NCoR, SMRT, Alien) (Polly et al., 2000; for review see McDonald et al., 2001) that interact with VDR and the activation of other signaling pathways such as that of TGF-β (Yanagisawa et al., 1999) may be responsible for the effects of 1α,25(OH)2D3 in a particular cell type. Our data reveal a complex network of interactions between cell junction proteins with signaling abilities such as β-catenin, γ-catenin/plakoglobin, and ZO-1 and nuclear hormone receptors such as VDR, which together with other signaling mediators regulate gene expression and the phenotype of epithelial cells in a combinatorial fashion.
In summary, we report here on a novel activity of 1α,25(OH)2D3 in human colon carcinoma cells, consisting of the induction of E-cadherin and the inhibition of β-catenin signaling, which has an antitumor effect in vivo. 1α,25(OH)2D3 exerts these protective effects in SW480-ADH cells carrying a panel of mutations in critical genes such as p53, ras, and APC and expressing negligible amounts of E-cadherin but overexpressing c-myc and in other human colon cell lines expressing functional VDR. These results point to the key role of VDR expression in colon carcinogenesis and support the use of nonhypercalcemic vitamin D derivatives for the treatment of this neoplasia.
Materials and methods
Cell culture
SW480, SW620, Caco-2, HT-29 M6, SW1417, LS-147T, MCF-7, and Pam212 cells were grown in DME supplemented with 10% FCS. SW480-ADH and SW480-R lines were obtained from SW480 by limit dilution and cultured in the same medium. All experiments using 1α,25(OH)2D3, MC903, KH1060, and EB1089 (a gift from Dr. Lise Binderup, Leo Pharmaceutical Products, Copenhagen, Denmark) were performed in DME supplemented with charcoal-treated FCS to remove liposoluble hormones. 1α,25(OH)2D3 and deltanoids were dissolved in isopropanol, and dexamethasone, all-trans-retinoic acid, and 9-cis-retinoic acid (all from Sigma-Aldrich) were dissolved in ethanol.
Cloning of the human E-cadherin gene promoter
A −178/+92 human E-cadherin promoter fragment was amplified by PCR using Pfu DNA polymerase (Stratagene) and 5′-GACTACGCGTACTCCAGGCTAGAGGGTCAC-3′ and 5′-GATCGATATCCGGGTGCGGTC-GGGTCGGGCCGGGCA-3′ as sense and antisense oligonucleotides, respectively. The −987/+92 E-cadherin promoter fragment was amplified using the same antisense oligonucleotide and 5′-AGTCGGTACCGA-GAGTGCAGTGGCTCACGC-3′ as sense oligonucleotide. Fragments were digested with MluI/EcoRV or SacI/NcoI restriction enzymes and inserted in pGL3 reporter vector (Promega). Cloned fragments were sequenced in order to rule out differences with respect to the published sequence (sequence data available from GenBank/EMBL/DDBJ under accession no. L34545).
Antibodies
The following antibodies were used: rabbit polyclonal anti-VDR (sc-1008; Santa Cruz Biotechnology, Inc.), mouse monoclonal anti–β-catenin (C19220; Transduction Laboratories), rat monoclonal anti–mouse E-cadherin (ECCD-2; a gift from Dr. M. Takeichi, Kyoto University, Kyoto, Japan), rabbit polyclonal antioccludin (71-1500; Zymed Laboratories), rabbit polyclonal anti-ZO1 (61-7300; Zymed Laboratories), rabbit polyclonal anti–ZO-2 (71-1400; Zymed Laboratories), rabbit polyclonal antivinculin IgG (sc-7649; Santa Cruz Biotechnology, Inc.), goat anti–TCF-4 (sc-8632; Santa Cruz Biotechnology, Inc.), FITC-conjugated goat anti–mouse IgG (115-095-003; Jackson ImmunoResearch Laboratories), TRICT-conjugated anti–rat IgG (112-025-003; Jackson ImmunoResearch Laboratories), TRICT-conjugated anti–rabbit IgG (Jackson ImmunoResearch Laboratories), goat anti–rabbit IgG (H + L) HRP-conjugated (67437; ICN Biomedicals), goat anti–mouse IgG (H + L) HRP-conjugated (67428; ICN Biomedicals), goat anti–rat IgG (H + L) HRP-conjugated (31472; Pierce Chemical Co.), and rabbit anti–goat IgG (612762; ICN Biomedicals).
Immunostaining
Cells were rinsed four times in PBS, fixed in cold methanol for 30 s at −20°C and rinsed in PBS. The nonspecific sites were blocked by incubation with PBS containing 1% BSA for 1 h at room temperature. Cells were then washed four times in PBS and incubated with the primary antibodies diluted in PBS containing 1% BSA for 1 h at room temperature or overnight at 4°C. After four washes with PBS, cells were incubated with secondary antibodies for 45 min at room temperature, washed, and mounted in VectaShield (Vector Laboratories). Confocal microscopy was performed with a Bio-Rad Laboratories MRC-1024 laser scanning microscope equipped with an Axiovert 100 invert microscope (ZEISS) at excitation wavelengths of 488 nm (for FITC) and 543 nm (for TRICT). Each channel was recorded independently, and pseudocolor images were generated and superimposed. Images were processed by the Adobe Photoshop® 5.0 software (Adobe Systems, Inc.).
RNA preparation and Northern analysis
Purification of poly(A)+ RNA was carried out as reported elsewhere (Vennström and Bishop, 1982). Northern blots were performed on nylon membranes (Nytran; Schleicher & Schuell) following standard protocols (Sambrook et al., 1989). All probes were labeled by the random priming method (Feinberg and Vogelstein, 1983). Hybridizations were carried out overnight at 65°C in 7% SDS, 500 mM sodium phosphate buffer, pH 7.2, and 1 mM EDTA as described by Church and Gilbert (1984). Filters were washed twice for 30 min each in 1% SDS and 40 mM sodium phosphate buffer, pH 7.2, at 65°C. The sizes of respective mRNAs were calculated using RNA ladder markers (Bio-Rad Laboratories). Membranes were exposed to Hyperfilm™ MP films (Amersham Pharmacia Biotech). The following probes were used: for VDR and RXRα, the complete human cDNA; for E-cadherin, a fragment (nucleotides 2209–2649) of human cDNA; for β-catenin, the whole mouse cDNA; for c-myc, the third exon of the human cDNA (donated by Dr. J. León, Facultad de Medicina, University of Cantabria, Santander, Spain); for CD44, the full-length human cDNA; for Tcf-1, the full-length ΦTcf-1 (van de Wetering et al., 1991) (both donated by Drs. E. Sancho and H. Clevers, University Medical Center, Utrecht, Netherlands); for ZO-1, a 558-pb fragment of the human cDNA obtained by reverse transcriptase-PCR with the 5′-CTTAACTATGCCCAGTGG-3′ and 5′-CTTGTGGTGAGTAAGGAG-3′ oligonucleotides as sense and antisense primers, respectively; for PPARδ, a 407-bp fragment of the human cDNA obtained by reverse transcriptase-PCR with the 5′-CTACGGTGTTCATGCATGTGAG-3′ and 5′-CACAATGTCTCGATGTCGTGG-3′ oligonucleotides as sense and antisense primers, respectively; and for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), the complete human cDNA.
Immunoprecipitation and Western blotting
Immunoprecipitation of whole cell extracts with specific antibodies was carried out as described elsewhere (Lozano and Cano, 1998). Whole cell extracts were prepared by washing the monolayers twice in PBS, and the cells were lysed by incubation in RIPA buffer (150 mM NaCl, 1.5 mM MgCl2, 10 mM NaF, 10% glycerol, 4 mM EDTA, 1% Triton X-100, 0.1% SDS, 1% deoxycholate, 50 mM Hepes, pH 7.4, plus phosphatase- and protease-inhibitor mixture [PPIM: 25 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin]) for 15 min on ice followed by centrifugation at 13,000 rpm for 10 min at 4°C. Immunoprecipitated proteins were analyzed in 7.5% or 12% SDS-PAGE gels. Immunoblotting of cell lysates or immunoprecipitates was performed by protein transfer to Immobilon-P membranes (Millipore Corp.) and incubation with the appropriate specific antibody. Blots were developed using the ECL detection system (Amersham Pharmacia Biotech).
In vitro protein–protein interaction
100 ng each of bacterially produced GST–β-catenin protein and human VDR translated in vitro in rabbit reticulocyte lysates (TNT® T7 Quick Kit; Promega) were mixed with 500 μl of immunoprecipitation buffer (IB: 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, 1% NP-40, 1% Triton X-100) and incubated for 1 h at 4°C. GST–β-catenin complexes were collected by addition of glutathione–Sepharose 4B (Amersham Pharmacia Biotech), washed twice with IB, and resuspended in Laemmli sample buffer. Proteins in the complexes were analyzed in 7.5% SDS-PAGE gels, which was followed by immunoblotting with the indicated antibodies.
Transactivation assays
Nearly confluent cells were transfected in triplicate P-60 dishes using LipofectAMINE™ reagent (Life Technologies) following the manufacturer guidelines. The 4 × VDRE–DR3-Tk-Luc construct containing four copies in tandem of a consensus DR3 response element for vitamin D cloned upstream of the herpes virus simplex thymidine kinase gene promoter and the luciferase reporter gene was provided by Dr. C. Carlberg (Heinrich-Heine-Universität, Düsseldorf, Germany). To study β-catenin–TCF/LEF-1 transcriptional activity, we transfected either TOP-flash and FOP-flash containing multimerized wild-type (CCTTTGATC) or mutated (CCTTTGGCC) TCF/LEF-1–binding sites upstream of a minimal c-fos promoter driving luciferase gene expression (Korinek et al., 1997) (a gift from Dr. H. Clevers). 1 μg of human VDR cDNA (cloned in pSG5) or human wild-type TCF-4 (cloned in pcDNA3.1) or mutant TCF-4 (ΔN-TCF-4 lacking the NH2-terminal β-catenin–interacting region in pcDNA3) (Baulida et al., 1999) and β-catenin S37A (cloned in pCGN; a gift from Dr. A. Ben-ZeÄev, The Weizman Institute, Rehovot, Israel) were used for exogenous expression together with 0.25 μg of the reporter luciferase plasmids. For exogenous E-cadherin expression, we used the complete murine cDNA (cloned in the pBATEM2 vector; a gift from Dr. M. Takeichi, Kyoto University). The expression vectors for the truncated VDR lacking the 11 COOH-terminal amino acids (ΔAF2) (Jiménez-Lara and Aranda, 1999) and for other point-mutated VDR (L417S and E420Q) (cloned in pSG5) were donated by Dr. Ana Aranda (Instituto de Investigaciones Biomédicas). As internal control of the transfection efficiency, 0.5 μg of the pRSV-LacZ containing a β-galactosidase reporter gene was used. Luciferase and β-galactosidase activities were measured 48 h after transfection.
Transepithelial electrical resistance
The functional integrity of tight junctions was assayed by measuring the electrical resistance towards ion flux of epithelial cell layers cultured on porous tissue culture inserts (3090; Falcon). We used the Millicell electrical resistance system (Millipore Corp.) connected to the electrode system Endohm-24 (World Precision Instruments) following the manufacturer guidelines. TERs were calculated after subtracting the background given by a blank culture insert.
Proliferation assays
50,000 cells were seeded in 24-well dishes (Falcon) and incubated in normal growth medium in the presence or absence of the indicated concentrations of 1α,25(OH)2D3. 48-h later, cells were pulsed with 1 μCi/ml 3H-thymidine for 3 h. At the end of the labeling period, the medium was removed and the cells were rinsed twice with PBS and fixed with chilled 10% trichloroacetic acid for 10 min. Trichloroacetic acid was then removed and the monolayers were air-dried at room temperature for 20 min. Thereafter, precipitated cellular macromolecules were dissolved in 500 μl of 0.1 N NaOH-0.1% SDS, and 450 μl of each sample was diluted in 5 ml of scintillation solution OptiPhase HighSafe (Wallac Scintillation Products). Radioactivity was measured using a 1209 RackBeta counter (LKB Wallac).
Acknowledgments
We thank Dr. Lise Binderup and Christina Hansen (Leo Pharmaceuticals Products) for the generous gift of 1α,25(OH)2D3, EB1089, KH1060, and MC903, and Dr. Francisco Real for isolating SW480-R cells. We are also indebted to those who appear in Materials and methods for providing us with plasmids and antibodies, A. Castillo and D. Gómez for their help in the transfection assays, and M. González and T. Martínez for technical assistance.
H.G. Pálmer and J.M. González-Sancho were recipients of fellowships from the Comunidad Autónoma de Madrid. This work was supported by a grant from the Plan Nacional de Investigación y Desarrollo (SAF98-0060), Ministerio de Ciencia y Tecnología of Spain.
H.G. Pálmer and J.M. González-Sancho contributed equally to this work.
Footnotes
Abbreviations used in this paper: APC, adenomatous polyposis coli; DR, direct repeat; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GSK, glycogen synthase kinase; GST, glutathione S-transferase; LEF, lymphoid enhancer-binding factor; 1α,25(OH2)D3, 1α,25-dihydroxyvitamin D3; PPAR, peroxisome proliferator-activated receptor; RAR, retinoic acid receptor; RXR, retinoid X receptor; TCF, T cell transcription factor; TER, transepithelial electrical resistance; VDR, vitamin D receptor; VDRE, vitamin D response element; ZO, Zonula occludens.
References
- Akhter, J., X. Chen, P. Bowrey, E.J. Bolton, and D.L. Morris. 1997. Vitamin D3 analog, EB1089, inhibits growth of subcutaneous xenographs of the human colon cancer cell line, LoVo, in nude mouse model. Dis. Colon Rec. 40:317–321. [DOI] [PubMed] [Google Scholar]
- Balda, M.S., and K. Matter. 2000. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J. 19:2024–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle, E., E. Sancho, C. Francí, D. Domínguez, M. Monfar, J. Baulida, and A. García de Herreros. 2000. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumor cells. Nat. Cell Biol. 2:84–89. [DOI] [PubMed] [Google Scholar]
- Baulida, J., E. Batlle, and A. García de Herreros. 1999. Adenomatous polyposis coli protein (APC)-independent regulation of β-catenin/Tcf4 mediated transcription in intestinal cells. Biochem. J. 344:565–570. [PMC free article] [PubMed] [Google Scholar]
- Behrens, J., J.P. von Kries, M. Külh, L. Bruhn, D. Wedlich, R. Grosschedl, and W. Birchmeier. 1996. Functional interaction of β-catenin with the transcriptional factor LEF-1. Nature. 382:638–642. [DOI] [PubMed] [Google Scholar]
- Behrens, J., B.A. Jerchow, M. Würtele, J. Grimm, C. Asbrand, R. Wirtz, M. Kühl, D. Wedlich, and W. Birchmeier. 1998. Functional interaction of an axin homolog, conductin, with β-catenin, APC, and GSK3β. Science. 280:596–599. [DOI] [PubMed] [Google Scholar]
- Billin, A.N., H. Thirlwell, and D.E. Ayer. 2000. β-Catenin-histone deacetylase interactions regulate the transition of LEF-1 from a transcriptional repressor to an activator. Mol. Cell. Biol. 20:6882–6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier, W., and J. Behrens. 1994. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochem. Biophys. Acta. 1198:11–26. [DOI] [PubMed] [Google Scholar]
- Buras, R.R., L.M. Schumaker, F. Davoodi, R.V. Brenner, M. Shabahang, R.J. Nauta, and S.R.T. Evans. 1994. Vitamin D receptors in breast cancer cells. Breast Cancer Res. Treat. 31:191–202. [DOI] [PubMed] [Google Scholar]
- Cano, A., M.A. Pérez-Moreno, I. Rodrigo, A. Locascio, M.J. Blanco, M.G. del Barrio, F. Portillo, and M.A. Nieto. 2000. The transcription factor Snail controls the epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2:76–83. [DOI] [PubMed] [Google Scholar]
- Christofori, G., and H. Semb. 1999. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. TIBS. 24:73–76. [DOI] [PubMed] [Google Scholar]
- Church, G.M., and W. Gilbert. 1984. Genome sequencing. Proc. Natl. Acad. Sci. USA. 81:1991–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford, H.C., B.M. Fingleton, L.A. Rudolph-Owen, K.J. Heppner-Goss, B. Rubinfeld, P. Polakis, and L.M. Matrisian. 1999. The metalloproteinase matrilysin is a target of β-catenin transactivation in intestinal tumors. Oncogene. 18:2883–2891. [DOI] [PubMed] [Google Scholar]
- Diaz, G.D., C. Paraskeva, M.G. Thomas, L. Binderup, and A. Hague. 2000. Apoptosis is induced by the active metabolite of vitamin D3 and its analogue EB1089 in colorectal adenoma and carcinoma cells: possible implication for prevention and therapy. Cancer Res. 60:2304–2312. [PubMed] [Google Scholar]
- Duval, A., S. Rolland, E. Tubacher, H. Bui, G. Thomas, and R. Hamelin. 2000. The human T-cell transcription factor-4 gene: structure, extensive characterization of alternative splicings, and mutational analysis in colorectal cancer cell lines. Cancer Res. 60:3872–3879. [PubMed] [Google Scholar]
- Eastman, Q., and R. Grosschedl. 1999. Regulation of LEF-1/TCF transcription factors by Wnt and other signals. Curr. Opin. Cell Biol. 11:233–240. [DOI] [PubMed] [Google Scholar]
- Easwaran, V., M. Pishvaian, Salimuddin, and S. Byers. 1999. Cross-regulation of β-catenin-LEF/TCF and retinoid signaling pathways. Curr. Biol. 9:1415–1418. [DOI] [PubMed] [Google Scholar]
- Eleftheriou, A., M. Yoshida, and B.R. Henderson. 2001. Nuclear export of human β-catenin can occur independently of CRM1 and APC. J. Biol. Chem. M102656200, in press. [DOI] [PubMed] [Google Scholar]
- Evans, S.R.T., J. Nolla, J. Hanfelt, M. Shabahang, R.J. Nauta, and I.B. Shchepotin. 1998. Vitamin D receptor expression as a predictive marker of biological behavior in human colorectal cancer. Clin. Cancer Res. 4:1591–1595. [PubMed] [Google Scholar]
- Feinberg, P.A., and B.A. Vogelstein. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 137:266–267. [DOI] [PubMed] [Google Scholar]
- Fodde, R., J. Kuipers, C. Rosenberg, R. Smits, M. Kielman, C. Gaspar, J.H. van Es, C. Breukel, J. Wiegant, R.H. Giles, et al. 2001. Mutations in the APC tumor suppressor gene cause chromosomal instability. Nat. Cell Biol. 3:433–438. [DOI] [PubMed] [Google Scholar]
- Freedman, L.P. 1999. Transcriptional targets of the vitamin D3 receptor-mediating cell cycle arrest and differentiation. J. Nutr. 129:581S–586S. [DOI] [PubMed] [Google Scholar]
- Garland, C.F., F.C. Garland, E.K. Shaw, G.W. Comstock, K.J. Helsing, and E.D. Gorham. 1989. Serum 25-hydroxyvitamin D and colon cancer: eight-year prospective study. Lancet. 2:1176–1178. [DOI] [PubMed] [Google Scholar]
- Guilford, P., J. Hopkins, J. Harraway, M. McLeod, N. McLeod, P. Harawira, H. Taite, R. Scoular, A. Miller, and A.E. Reeve. 1998. E-cadherin germline mutations in familial gastric cancer. Nature. 392:402–405. [DOI] [PubMed] [Google Scholar]
- Gniadecki, R., B. Gajkowska, and M. Hansen. 1997. 1,25-dihydroxyvitamin D3 stimulates the assembly of adherens junctions in keratinocytes: involvement of protein kinase C. Endocrinology. 138:2241–2248. [DOI] [PubMed] [Google Scholar]
- González-Sancho, J.M., M. Alvarez-Dolado, and A. Muñoz. 1998. 1,25-dihydroxyvitamin D3 inhibits tenascin-C expression in mammary epithelial cells. FEBS Lett. 426:225–228. [DOI] [PubMed] [Google Scholar]
- Gottardi, C.J., M. Arpin, A.S. Fanning, and D. Louvard. 1996. The junctions-associated protein, zonula occludens-1, localizes to the nucleus before the maturation and during the remodeling of cell-cell contacts. Proc. Natl. Acad. Sci. USA. 93:10779–10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradl, D., M. Kühl, and D. Wedlich. 1999. The Wnt/Wg signal transducer β-catenin controls fibronectin expression. Mol. Cell. Biol. 19:5576–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C., T. Stamey, S. Hancock, and D. Feldman. 1998. Treatment of early recurrent prostate cancer with 1α,25-dihydroxyvitamin D3. J. Urol. 159:2035–2040. [DOI] [PubMed] [Google Scholar]
- Guger, K.A., and B.M. Gumbiner. 2000. A mode of regulation of β-catenin signaling activity in Xenopus embryos independent of its levels. Dev. Biol. 223:441–448 [DOI] [PubMed] [Google Scholar]
- Gulliford, T., J. English, K.W. Colston, P. Menday, S. Moller, and R.C. Coombes. 1998. A phase I study of the vitamin D analogue EB1089 in patients with advanced breast and colorectal cancer. British J. Cancer. 78:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbiner, B.M. 1996. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 84:345–357. [DOI] [PubMed] [Google Scholar]
- Hansen, C.M., T.L. Frandsen, N. Brunner, and L. Binderup. 1994. 1α,25-dihydroxyvitamin D3 inhibits the invasive potential of human breast cancer cells in vitro. Clin. Exp. Metastasis. 12:195–202. [DOI] [PubMed] [Google Scholar]
- He, T.C., A.B. Sparks, C. Rago, H. Hermeking, L. Zawel, L.T. da Costa, P.J. Morin, B. Vogelstein, and K.W. Kinzler. 1998. Identification of c-myc as a target of the APC pathway. Science. 281:1509–1512. [DOI] [PubMed] [Google Scholar]
- He, T.C., T.A. Chan, B. Vogelstein, and K.W. Kinzler. 1999. PPARδ is an APC-regulated target of nonsteroideal anti-inflammatory drugs. Cell. 99:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson, B.R. 2000. Nuclear-cytoplasmatic shuttling of APC regulates β-catenin subcellular localization and turnover. Nat. Cell Biol. 2:653–660. [DOI] [PubMed] [Google Scholar]
- Hmama, Z., D. Nandan, L. Sly, K.L. Knutson, P. Herrera-Velit, and N.E. Reiner. 1999. 1α,25-dihydroxyvitamin D3-induced myeloid cell differentiation is regulated by a vitamin D receptor-phosphatidylinositol 3-kinase signaling complex. J. Exp. Med. 190:1583–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, O., R. Korn, J. McLaughlin, M. Ohsugi, B.G. Herrmann, and R. Kemler. 1996. Nuclear localization of β-catenin by interaction with transcription factor LEF-1. Mech. Dev. 59:3–10. [DOI] [PubMed] [Google Scholar]
- Inomata, M., A. Ochiai, S. Akimoto, S. Kitano, and S. Hirohashi. 1996. Alteration of β-catenin expression in colonic epithelial cells of familial adenomatous polyposis patiens. Cancer Res. 56:2213–2217. [PubMed] [Google Scholar]
- Jiménez-Lara, A.M., and A. Aranda. 1999. Vitamin D represses retinoic acid-dependent transactivation of the retinoic acid receptor-β2 promoter: the AF-2 domain of the vitamin D receptor is required for transrepression. Endocrinology. 140:2898–2907. [DOI] [PubMed] [Google Scholar]
- Kane, K.F., M.J.S. Langman, and G.R. Williams. 1996. Antiproliferative responses of two human colon cancer cell lines to vitamin D3 are differentially modified by 9-cis-retinoic acid. Cancer Res. 56:623–632. [PubMed] [Google Scholar]
- Kaplan, K.B., A.A. Burds, J.R. Swedlow, S.S. Bekir, P.K. Sorger, and I.S. Näthke. 2001. A role for the Adenomatous Polyposis coli protein in chromosome segregation. Nat. Cell Biol. 3:429–432. [DOI] [PubMed] [Google Scholar]
- Kawasoe, T., Y. Furukawa, Y. Daigo, T. Nishiwaki, H. Ishiguro, M. Fujita, S. Satoh, N. Miwa, Y. Nagasawa, Y. Miyoshi, et al. 2000. Isolation and characterization of a novel human gene, DRCTNNB1A, the expression of which is down-regulated by β-catenin. Cancer Res. 60:3354–3358. [PubMed] [Google Scholar]
- Kimura, Y., H. Shiozaki, M. Hirao, Y. Maeno, Y. Doki, M. Inoue, T. Monden, Y. Ando-Akatsuka, M. Furuse, S. Tsukita, et al. 1997. Expression of occludin, tight-junction-associated protein, in human digestive tract. Am. J. Pathol. 151:45–54. [PMC free article] [PubMed] [Google Scholar]
- Kishida, S., H. Yamamoto, S. Ikeda, M. Kishida, I. Sakamoto, S. Koyama, and A. Kikuchi. 1998. Axin, a negative regulator of the wnt signaling pathway, directly interacts with the adenomatous polyposis coli and regulates the stabilization of the β-catenin. J. Biol. Chem. 273:10823–10826. [DOI] [PubMed] [Google Scholar]
- Koh, T.J., C.J. Bulitta, J.V. Fleming, G.J. Dockray, A. Varro, and T.C. Wang. 2000. Gastrin is a target of the β-catenin/TCF4 growth-signaling pathway in a model of intestinal polyposis. J. Clin. Invest. 106:533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koli, K., and J. Keski-Oja. 2000. 1α,25-dihydroxyvitamin D3 and its analogues down-regulate cell invasion-associated proteases in cultured malignant cells. Cell Growth Differ. 11:221–229. [PubMed] [Google Scholar]
- Kolligs, F.T., B. Kolligs, K.M. Hajra, G. Hu, M. Tani, K.R. Cho, and E.R. Fearon. 2000. γ-Catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of β-catenin. Genes Dev. 14:1319–1331. [PMC free article] [PubMed] [Google Scholar]
- Korinek, V., N. Barker, P.J. Morin, D. van Wichen, R. de Weger, K.W. Kinzler, B. Vogelstein, and H. Clevers. 1997. Constitutive transcriptional activation by β-catenin-Tcf complex in APC−/− colon carcinoma. Science. 275:1784–1787. [DOI] [PubMed] [Google Scholar]
- Leibovitz, A., J.C. Stinton, W.B. McCombs, C.E. McCoy, K.C. Mazur, and N.D. Mabry. 1976. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 36:4562–4569. [PubMed] [Google Scholar]
- Lickert, H., C. Domon, G. Huls, C. Wehrle, I. Duluc, H. Clevers, B. Meyer, J.N. Freund, and R. Kemler. 2000. Wnt/β-catenin signaling regulates the expression of the homeobox gene Cdx1 in embryonic tissue. Development. 127:3805–3813. [DOI] [PubMed] [Google Scholar]
- Lozano, E., and A. Cano. 1998. Cadherin/catenin complexes in murine epidermal keratinocytes: E-cadherin complexes containing either β-catenin or plakoglobin contribute to stable cell-cell contacts. Cell Adh. Commun. 6:51–67. [DOI] [PubMed] [Google Scholar]
- Majewski, S., A. Szmurlo, M. Marczak, S. Jablonska, and W. Bollag. 1993. Inhibition of tumor cell-induced angiogenesis by retinoids, 1α,25-dihydroxyvitamin D3 and their combination. Cancer Lett. 75:35–39. [DOI] [PubMed] [Google Scholar]
- Mann, B., M. Gelos, A. Siedow, M.L. Hanski, A. Gratchev, M. Ilyas, W.F. Bodmer, M.P. Moyer, E.O. Riecken, H.J. Buhr, et al. 1999. Target genes of β-catenin-T cell factor/lynphoid-enhancer-factor signaling in human colorectal carcinomas. Proc. Natl. Acad. Sci. USA. 96:1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareel, M., M. Brake, and F. van Roy. 1994. Invasion promoter versus invasion suppressor molecules: the paradigm of E-cadherin. Mol. Biol. Rep. 19:45–67. [DOI] [PubMed] [Google Scholar]
- McDonald, P.N., D.M. Kraichely, and A.J. Brown. 2001. The vitamin D receptor. Nuclear Receptors and Genetic Disease. T.P. Burris and E.R.B. McCabe, editors. Academic Press, Inc., Orlando, FL. 197–243.
- Morin, J.P. 1999. β-Catenin signaling in cancer. Bioessays. 21:1021–1030. [DOI] [PubMed] [Google Scholar]
- Morin, J.P., A.B. Sparks, V. Korinek, N. Barker, H. Clevers, B. Vogelstein, and K.W. Kinzler. 1997. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 275:1787–1790. [DOI] [PubMed] [Google Scholar]
- Newmark, H.L., and M. Lipkin. 1992. Calcium, vitamin D, and colon cancer. Cancer Res. 52:2067–2070. [PubMed] [Google Scholar]
- Orsulic, S., O. Huber, H. Aberle, S. Arnold, and R. Kemler. 1999. E-cadherin binding prevents β-catenin nuclear localization and β-catenin/LEF-1-mediated transactivation. J. Cell Sci. 112:1237–1245. [DOI] [PubMed] [Google Scholar]
- Pan, Q., and R.U. Simpson. 1999. c-myc intron element-binding proteins are required for 1,25-dihydroxyvitamin D3 regulation of c-myc during HL-60 cell differentiation and the involvement of HOXB4. J. Biol. Chem. 274:8437–8444. [DOI] [PubMed] [Google Scholar]
- Park, W.H., J.G. Seol, E.S. Kim, C.W. Jung, C.C. Lee, L. Binderup, H.P. Koeffler, B.K. Kim, and Y.Y. Lee. 2000. Cell cycle arrest induced by the vitamin D (3) analog EB1089 in NCI-H929 myeloma cells is associated with induction of the cyclin-dependent kinase inhibitor p27. Exp. Cell Res. 254:279–286 [DOI] [PubMed] [Google Scholar]
- Parker, S.L., T. Tong, S. Bolden, and P.A. Wingo. 1997. Cancer statistics. CA Cancer J. Clin. 47:5–27. [DOI] [PubMed] [Google Scholar]
- Peifer, M., and P. Polakis. 2000. Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science. 287:1606–1609. [DOI] [PubMed] [Google Scholar]
- Perl, A.-K., P. Wilgenbus, U. Dahl, H. Semb, and G. Christofori. 1998. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 392:190–193. [DOI] [PubMed] [Google Scholar]
- Polly, P., M. Herdick, U. Moehren, A. Baniahmad, T. Heinzel, and C. Carlberg. 2000. VDR-Alien: a novel, DNA-selective vitamin D3 receptor corepressor partnership. FASEB J. 14:1455–1463. [DOI] [PubMed] [Google Scholar]
- Rajasekaran, A.K., M. Hojo, T. Huima, and E. Rodriguez-Boulan. 1996. Catenins and zonula occludens-1 form a complex during early stages in the assembly of tight junctions. J. Cell Biol. 132:451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roose, J., G. Huls, M. van Beest, P. Moerer, K. van der Horn, R. Goldschmeding, T. Logtenberg, and H. Clevers. 1999. Synergy between tumor suppressor APC and the β-catenin-TCF4 target Tcf1. Science. 285:1923–1926. [DOI] [PubMed] [Google Scholar]
- Rosin-Arbesfeld, R., F. Townsley, and M. Bienz. 2000. The APC tumour suppressor has a nuclear export function. Nature. 406:1009–1012. [DOI] [PubMed] [Google Scholar]
- Rubinfeld, B., I. Albert, E. Porfiri, C. Fiol, S. Munemitsu, and P. Polakis. 1996. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science. 272:1023–1026. [DOI] [PubMed] [Google Scholar]
- Sadot, E., I. Simcha, M. Shtuman, A. Ben-ZeÄev, and B. Geiger. 1998. Inhibition of β-catenin-mediated transactivation by cadherin derivatives. Proc. Natl. Acad. Sci. USA. 95:15339–15344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez, S., N. Falette, C. Guillot, F. Meggouh, M.F. Lefebvre, and M. Crepin. 1993. 1α,25(OH)2D3 modulation of mammary tumor cell growth in vitro and in vivo. Breast Cancer Res. Treat. 27:69–81. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schwarte-Waldhoff, I., S. Klein, S. Blass-Kampmann, A. Hintelmann, C. Eilert, S. Dreschers, H. Kalthoff, S.A. Hahn, and W. Schmiegel. 1999. DPC4/SMAD4 mediated tumor suppression of colon carcinoma cells is associated with reduced urokinase expression. Oncogene. 18:3152–3158. [DOI] [PubMed] [Google Scholar]
- Shabahang, M., R.R. Buras, F. Davoodi, L.M. Schumaker, R.J. Nauta, and S.R.T. Evans. 1993. 1,25-dihydroxyvitamin D3 receptor as a marker of human colon carcinoma cell line differentiation and growth inhibition. Cancer Res. 53:3712–3718. [PubMed] [Google Scholar]
- Smith, D.C., C.S. Johnson, C.C. Freeman, J. Muindi, J.W. Wilson, and D.L. Trump. 1999. A phase I trial of calcitriol (1α,25-dihydroxycholecalciferol) in patients with advanced malignancy. Clin. Cancer Res. 5:1339–1345. [PubMed] [Google Scholar]
- Takeichi, M. 1993. Cadherins in cancer: implications for invasion and metastasis. Curr. Opin. Cell Biol. 5:806–811. [DOI] [PubMed] [Google Scholar]
- Takeichi, M. 1995. Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 7:619–627. [DOI] [PubMed] [Google Scholar]
- Tetsu, O., and F. McCormick. 1999. β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 398:422–426. [DOI] [PubMed] [Google Scholar]
- Thomas, M.G., S. Tebbutt, and R.C. Williamson. 1992. Vitamin D and its metabolites inhibit cell proliferation in human rectal mucosa and a colon cancer cell line. Gut. 33:1660–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita, N., W. Jiang, H. Hibshoosh, D. Warburton, S.M. Kahn, and I.B. Weinstein. 1992. Isolation and characterization of a highly malignant variant of the SW480 human colon cancer cell lines. Cancer Res. 52:6840–6847. [PubMed] [Google Scholar]
- Tong, W.M., E. Kallay, H. Hofer, W. Hulla, T. Manhardt, M. Peterlik, and H.S. Cross. 1998. Growth regulation of human colon cancer cells by epidermal growth factor and 1α,25-dihydroxyvitamin D3 is mediated by mutual modulation of receptor expression. Eur. J. Cancer. 34:2119–2125. [DOI] [PubMed] [Google Scholar]
- Vandewalle, B., A. Adenis, L. Hornez, F. Revillion, and J. Lefebvre. 1994. 1,25-dihydroxyvitamin D3 receptors in normal and malignant human colorectal tissues. Cancer Lett. 86:67–73. [DOI] [PubMed] [Google Scholar]
- van de Wetering, M., M. Oosterwegel, D. Dooijes, and H. Clevers. 1991. Identification and cloning of TCF-1, a T lymphocyte-specific transcription factor containing a sequence-specific HMG box. EMBO J. 10:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weelden, K., L. Flanagan, L. Binderup, M. Tenniswood, and J. Welsh. 1998. Apoptotic regression of MCF-7 xenografts in nude mice treated with the vitamin D3 analog, EB1089. Endocrinology. 139:2102–2110. [DOI] [PubMed] [Google Scholar]
- Vennström, B., and J.M. Bishop. 1982. Isolation and characterization of chicken DNA homologous to the two putative oncogenes of avian erythroblastosis virus. Cell. 28:135–143. [DOI] [PubMed] [Google Scholar]
- Vera, J., M. Wielenga, R. Smits, V. Korinek, C. Smit, M. Kielman, R. Fodde, H. Clevers, and S.T. Pals. 1999. Expression of CD44 in APC and Tcf1 mutant mice implies regulation by the WNT pathway. Am. J. Pathol. 154:515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vleminckx, K., L. Vakaet, M. Mareel, W. Fiers, and F. van Roy. 1991. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 66:107–119. [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida, M., N. Uchida, Y. Imamura, A. Nagafuchi, K. Fujimoto, T. Uemura, S. Vermeulen, F. van Roy, E.D. Adamson, and M. Takeichi. 1998. α-Catenin–vinculin interaction functions to organize the apical junctional complex in epithelial cells. J. Cell Biol. 142:847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi, Z., and D. Feldman. 1993. Regulation of vitamin D receptor abundance and responsiveness during differentiation of HT-29 colon cancer cells. Endocrinology. 132:1808–1814. [DOI] [PubMed] [Google Scholar]
- Yanagisawa, J., Y. Yanagi, Y. Masuhiro, M. Suzawa, M. Watanabe, K. Kashiwagi, T. Toriyabe, M. Kawabata, K. Miyazono, and S. Kato. 1999. Convergence of transforming growth factor-β and vitamin D signaling pathways on SMAD transcriptional coactivators. Science. 283:1377–1321. [DOI] [PubMed] [Google Scholar]
- Zhurinsky, J., M. Shtutman, and A. Ben-Ze'ev. 2000. Differential mechanisms of LEF/TCF family-dependent transcriptional activation by β-catenin and plakoglobin. Mol. Cell. Biol. 20:4238–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]