

Abstract
ErbB2 is a receptor tyrosine kinase belonging to the family of epidermal growth factor (EGF) receptors which is generally involved in cell differentiation, proliferation, and tumor growth, and activated by heterodimerization with the other members of the family. We show here that type IV pilus–mediated adhesion of Neisseria meningitidis onto endothelial cells induces tyrosyl phosphorylation and massive recruitment of ErbB2 underneath the bacterial colonies. However, neither the phosphorylation status nor the cellular localization of the EGF receptors, ErbB3 or ErbB4, were affected in infected cells. ErbB2 phosphorylation induced by N. meningitidis provides docking sites for the kinase src and leads to its subsequent activation. Specific inhibition of either ErbB2 and/or src activity reduces bacterial internalization into endothelial cells without affecting bacteria-induced actin cytoskeleton reorganization or ErbB2 recruitment. Moreover, inhibition of both actin polymerization and the ErbB2/src pathway totally prevents bacterial entry. Altogether, our results provide new insight into ErbB2 function by bringing evidence of a bacteria-induced ErbB2 clustering leading to src kinase phosphorylation and activation. This pathway, in cooperation with the bacteria-induced reorganization of the actin cytoskeleton, is required for the efficient internalization of N. meningitidis into endothelial cells, an essential process enabling this pathogen to cross host cell barriers.
Keywords: ErbB2; homodimerization; src; Neisseria meningitidis; invasion
Introduction
The ErbB family of receptor tyrosine kinases (RTKs)* comprises four closely related members: EGF receptor (EGFR; also termed ErbB1), ErbB2/Neu, ErbB3, and ErbB4. These receptors are activated by numerous ErbB-specific ligands, each containing an EGF-like domain which confers binding specificity (for review see Olayioye et al., 2000). These ligands, which are produced as transmembrane precursors and are processed and released by proteolysis, bind the extracellular domain of ErbB receptors and lead to the formation of both homo- and heterodimers. No soluble ligand for ErbB2 has yet been discovered. However, increasing evidence suggests that the primary function of ErbB2 is as a coreceptor, since ErbB2 is the preferred heterodimerization partner for all other ErbB family members (Tzahar et al., 1996; Graus-Porta et al., 1997) and plays a role in the potentiation of ErbB receptor signaling (Beerli et al., 1995; Graus-Porta et al., 1995). Dimerization stimulates the intrinsic tyrosine kinase activity of the receptors and triggers autophosphorylation of specific tyrosine residues within the cytoplasmic domains. These phosphorylated residues provide docking sites for proteins which contain src homology 2 (SH2) or phosphotyrosine binding domains and which are involved in the regulation of intracellular signaling cascades. All ErbB family members couple via the adaptors Shc and/or Grb2 to the mitogen-activated protein kinase (MAPK) pathway. However, preferential modulation of specific pathways linked to specific homo- or heterodimers, or to specific cell surface sites, have been described (Olayioye et al., 2000; Carraway and Sweeney, 2001). Therefore, the signaling diversity associated with the ErbB family is dependent upon the repertoire of ErbB ligands, the combinatorial properties of induced receptor dimers, and receptor localization. Ultimately, downstream effects on gene expression determine the biological response to receptor activation. ErbB receptors are expressed in a variety of tissues of epithelial, mesenchymal, and neuronal origin, where they play fundamental roles in development, proliferation, and differentiation. Recently, an expression of ErbB receptors has also been described on endothelial cells, where they play a role in angiogenesis (Russell et al., 1999). Moreover, deregulated expression of ErbB receptors, in particular ErbB1 and ErbB2, has been implicated in the development and malignancy of numerous types of human cancers (Yarden and Sliwkowski, 2001). ErbB2 overexpression, observed in a significant proportion of breast and ovarian cancers, triggers ligand-independent activation of the kinase domain, apparently as a result of spontaneous dimer formation. A ligand-independent activation of ErbB RTKs can also be triggered by other classes of receptors, such as G protein–coupled receptors, cytokine receptors, or adhesion molecules (Carpenter, 1999; Gschwind et al., 2001). In the absence of ErbB kinase activity, phosphorylation of specific tyrosyl residues in the cytoplasmic tail of the receptor by nonreceptor kinases such as jak or src can provide docking sites for cytoplasmic signaling molecules and promote signaling cascades. In addition, a ligand-dependent transactivation of EGFR by G protein–coupled receptors can also occur by a proteolytic processing of the transmembrane pro-HB-EGF precursor followed by a paracrine activation of EGFR (Gschwind et al., 2001). Therefore, ErbB RTKs are central signal transducers which respond to the binding of specific ligands or via transactivation by distinct receptor systems.
Many bacterial pathogens exploit host cell signaling pathways in order to promote their adherence to or uptake by host cells (Dramsi and Cossart, 1998). For most invasive bacteria, a major consequence of these signaling events is a morphological change of the host cell surface resulting from a reorganization of the actin cytoskeleton. The nature and magnitude of such changes depend highly on the pathogen and its cellular host, although a common feature is the activation of host tyrosine phosphorylation leading to the activation of the small GTPases of the Rho family, key regulators of actin cytoskeleton dynamics. The best studied examples are those of Shigella or Salmonella which trigger the induction of large cellular membrane folds or ruffles around bacteria leading to their subsequent uptake by a macropinocytic process (Nhieu and Sansonetti, 1999; Galan and Zhou, 2000). These pathogens have evolved a complex type III secretion system allowing them to inject into host cells bacterial components that can directly initiate the activation of cellular signaling cascades. Alternatively, Listeria monocytogenes, which does not have a type III secretion system, promotes its entry by engaging cell adhesion receptors (E-cadherins) and the tyrosine kinase receptor Met (Mengaud et al., 1996; Shen et al., 2000).
Neisseria meningitidis (also referred to as meningococcus) is an extracellular, human-specific pathogen responsible for septicemia and meningitis. Despite the lack of type III secretion system, virulent capsulated N. meningitidis also initiates host cell signaling cascades leading to their subsequent uptake into host cells that are normally nonphagocytic (Nassif et al., 1999; Merz and So, 2000). This entry is likely to promote their transcytosis through cellular barriers by a mechanism that remains to be clearly identified, and therefore plays a major role in bacterial pathogenesis. Capsulated N. meningitidis interact with host cells in a multistep process (Pujol et al., 1997). Bacteria first adhere through their type IV pili, which are long filamentous protein structures that interact with an unidentified cellular receptor. Bacteria then proliferate, locally forming a colony at their site of attachment on the cell surface, a step referred to as localized adhesion. Subsequently, bacteria loose their pili, disperse from the colony, and spread over the cell surface to form a single monolayer of bacteria covering the cells, a step referred to as diffuse adhesion. The bacterial protein PilT, which is responsible for pilus retraction (Merz et al., 2000), is required for the transition from localized to diffuse adhesion (Pujol et al., 1999). Internalization of these bacteria occurs essentially during the first step of localized adhesion. During this interaction, N. meningitidis adhering to epithelial cells elicits the formation of specific molecular complexes underneath the bacterial colonies. These structures, called cortical plaques, result from the localized polymerization of cortical actin, recruitment of ezrin, a protein that links the cellular membrane to the actin cytoskeleton, and clustering of membrane integral proteins, such as the ezrin binding proteins CD44 or ICAM-1 as well as tyrosine phosphorylated proteins (Merz and So, 1997; Merz et al., 1999). These events are accompanied by the elongation of the epithelial cell microvilli towards the bacteria, leading to their engulfment and internalization into host cells. We recently showed that N. meningitidis interaction with endothelial cells also leads to cortical actin polymerization underneath the bacterial colonies and the massive recruitment of ezrin and ezrin-binding proteins (unpublished data). This recruitment promotes the formation of membrane projections, reminiscent of epithelial microvilli structures, which surround single bacteria and provoke their internalization within endothelial intracellular vacuoles. This has been demonstrated both in vitro and in vivo and suggests that this process is essential for the crossing of human endothelium via a transcytosis pathway. We further showed that N. meningitidis–induced actin polymerization requires the activation of the small GTPases of the Rho family RhoA and Cdc42, but not Rac1. Inhibition of Rho family GTPases prevented cortical actin polymerization, but only partially inhibited bacterial internalization, suggesting an alternative signaling pathway involved in N. meningitidis internalization. The process by which N. meningitidis interacts with the host cell and induces these signaling events has not been elucidated.
In this study, we therefore analyzed the tyrosine phosphorylation events occurring in human endothelial cells during adhesion of N. meningitidis. We provide evidence that pilus-mediated adhesion of capsulated N. meningitidis induces the clustering and tyrosyl phosphorylation of the host cell tyrosine kinase receptor ErbB2, but not of the other ErbBs. ErbB2 activation leads to downstream activation of the protein tyrosine kinase src and the tyrosine phosphorylation of the src-substrate cortactin. Furthermore, we show that ErbB2/src signaling pathway is not involved in the formation of cortical plaques per se, but plays a crucial role in facilitating bacterial entry into endothelial cells. By analyzing the respective contributions to bacterial entry of both ErbB2/src and Rho GTPases–dependent signaling pathways, we show that ErbB2/src pathway regulates N. meningitidis entry through signaling events distinct from those leading to bacteria-induced actin cytoskeleton reorganization. Moreover, both pathways cooperate in bacterial entry. Therefore, our results provide evidence for a new mode of activation of ErbB2 by bacteria-induced homodimerization, and for a novel biological function of ErbB2 in supporting bacterial internalization into host cells.
Results
Adhesion of N. meningitidis to human endothelial cells induces tyrosine phosphorylation of cellular proteins, which can be prevented by AG1478, an ErbB family inhibitor
We investigated the tyrosine phosphorylation events occurring in a human endothelial cell line (HBMEC) during interaction with N. meningitidis. For this purpose, HBMECs were infected with a piliated capsulated strain of N. meningitidis (wild-type 2C43), and two derivative mutants of that strain: a highly piliated and adhesive mutant (PilT−) and a nonpiliated (nonadherent) PilE− mutant. As shown in Fig. 1 A, infection with the wild-type strain 2C43 for 4 h induced the major tyrosyl phosphorylation of proteins of ∼80/85 and 180 kD. These phosphorylation events were markedly increased when cells were infected with the PilT− mutant of 2C43, whereas they were not observed in cells in contact with the nonadherent PilE− mutant. Kinetics analysis showed that these signals were detectable as soon as 30 min after infection, maximal at 4 h (during the pilus-mediated localized adhesion). By contrast, they were no longer detected at 8 h with the wild-type strain (diffuse adhesion), whereas the signal remained persistent in cells infected with the PilT− mutant, which is unable to retract pili and to proceed to the diffuse adhesion step (unpublished data). Therefore, these events occur during the localized pilus-mediated adhesion of N. meningitidis to human endothelial cells. In parallel, experiments were performed with an isolate of a highly virulent strain, designated as ROU, obtained from the cerebrospinal fluid of a patient with fulminant meningococcal infection (Fig. 1 B). This strain was highly efficient in inducing tyrosyl phosphorylation of the p80/85 and p180, suggesting a correlation between these cellular events and the infectious process of N. meningitidis. Moreover, similar data were observed with a primary culture of human umbilical vein endothelial cells (unpublished data), suggesting that these phosphorylation events might be a general response of endothelial cells to N. meningitidis adhesion.
Figure 1.
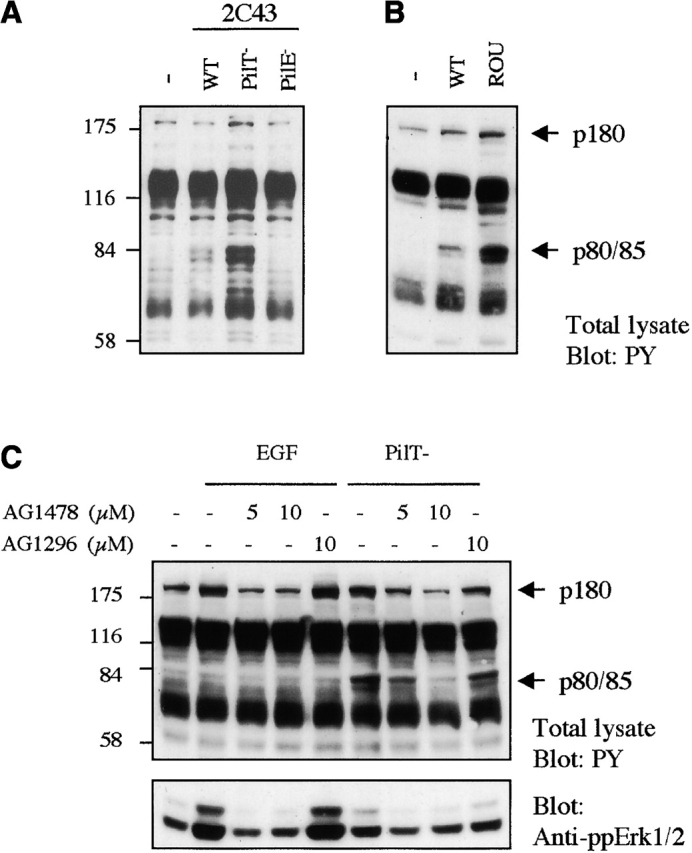
Localized adhesion of N. meningitidis induces tyrosine phosphorylation events that are prevented by AG1478 treatment. (A) HBMECs (starved for 18 h) were either uninfected (−) or infected with the wild-type strain (WT), the PilT-defective mutant (PilT−), or the PilE-defective mutant (PilE−) of the 2C43 strain of N. meningitidis for 4 h before lysis. (B) HBMECs starved for 18 h were either uninfected (−) or infected with the 2C43 wild-type strain (WT) or ROU strain (ROU). (C) HBMECs (starved for 18 h) were pretreated for 2 h with 5 or 10 μM AG1478, 10 μM AG1296, or left untreated (−). Cells were then infected for 4 h with the PilT-defective mutant strain in the presence of these inhibitors before lysis. As control, cells pretreated the same way were stimulated with 10 ng/ml EGF for 3 min before lysis. (A–C) Samples of whole cell extracts were analyzed by immunoblotting with an antiphosphotyrosine antibody (4G10). The electrophoretic mobilities of proteins of 80/85 and 180 kD (arrows) and of molecular weight standards (175, 116, 84, and 58 kD) are indicated. (C) Bottom, blot was reprobed with an antiphosphoErK1/2 antibody, which detects Erk1/2 when activated by phosphorylation.
Since ErbB receptors are ∼180 kD, compatible with that of p180, we hypothesized that tyrosine kinase receptors of the EGFR family might be involved in the cellular response to bacterial adhesion. We tested whether these phosphorylation events would be affected by AG1478, a very potent and selective inhibitor of both EGFR and ErbB2 kinases (Kurokawa et al., 2000). As shown in Fig. 1 C, cell treatment with 5 or 10 μM AG1478 totally prevented the increase of tyrosyl phosphorylation of p180 protein induced by either EGF or infection for 4 h with the PilT− mutant, whereas cell treatment with 10 μM AG1296, a selective inhibitor of the PDGF receptor kinase, had no effect. Similarly to EGF, N. meningitidis induced the activation of the MAPK Erk1/2 that was also prevented by treatment with AG1478, but not by AG1296. Altogether, these data strongly suggest that the infection by N. meningitidis leads to the activation of member(s) of the ErbB family. Interestingly, AG1478 treatment also drastically reduced the phosphorylation of p80/85 induced by N. meningitidis, suggesting these proteins as downstream targets of ErbB signaling cascade. However, phosphorylation of p80/85 was not observed in response to EGF stimulation.
Adhesion of N. meningitidis to human endothelial cells induces tyrosine phosphorylation and clustering of ErbB2, but not of the other ErbB members
Therefore, we analyzed the tyrosyl phosphorylation state of members of the ErbB family in cells infected for 4 h with either the 2C43 wild-type, the PilT− mutant, or the ROU strain in the presence or in the absence of 5 μM AG1478. Stimulation of HBMECs with EGF at 10 ng/ml for 3 min highly stimulated EGFR tyrosine phosphorylation as expected, whereas N. meningitidis adhesion had no effect on tyrosine phosphorylation of the EGFR (Fig. 2 A). Analysis of not only EGFR, but also ErbB3 or ErbB4 phosphorylation over several hours of infection by N. meningitidis failed to reveal any modification (unpublished data). By contrast, bacterial infection induced tyrosyl phosphorylation of ErbB2, which could be prevented by AG1478 treatment (Fig. 2), indicating that N. meningitidis leads to the selective activation of ErbB2, whereas EGF stimulated both EGFR and ErbB2 phosphorylation, consistent with EGFR/ErbB2 heterodimerization (Fig. 2 C).
Figure 2.
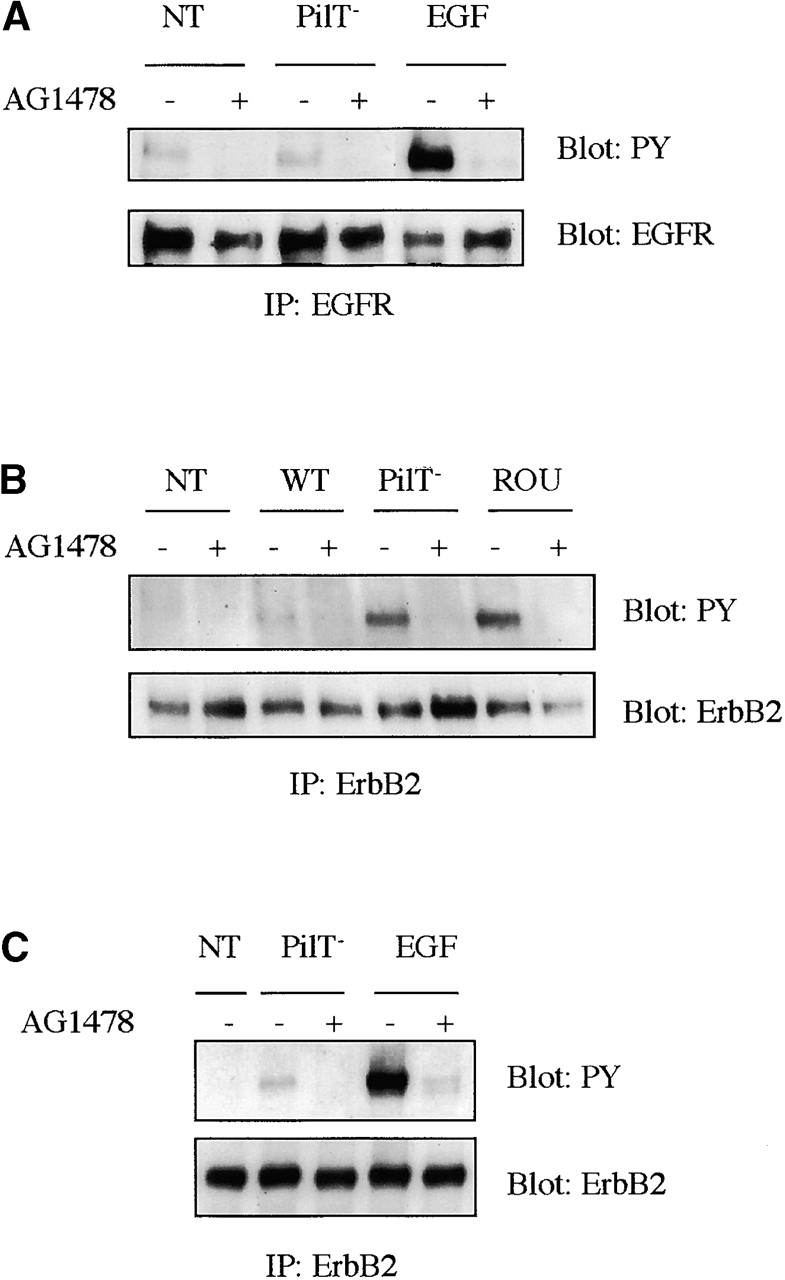
Localized adhesion of N. meningitidis induces the tyrosyl phosphorylation of ErbB2, but not of the other ErbB members. HBMECs (starved for 24 h) were pretreated for 2 h with 5 μM AG1478 (+) or left untreated (−). Cells were either noninfected (NT) or infected with the wild-type (WT), the PilT-defective mutant (PilT−) of the 2C43 strain, or the ROU strain of N. meningitidis, as indicated, for 4 h before lysis. AG1478 treatment was maintained during infection. As control, cells pretreated the same way were stimulated with 10 ng/ml EGF for 3 min before lysis. (A) EGF receptor was immunoprecipitated and immunoblotted with an antiphosphotyrosine antibody (4G10) (top). Blot was reprobed with an anti-EGFR antibody (bottom) to show that similar levels of receptors were immunoprecipitated. (B and C) ErbB2 receptor was immunoprecipitated and immunoblotted with an antiphosphotyrosine antibody (4G10) (top). Blots were reprobed with an anti-ErbB2 antibody (bottom). Three independent experiments were carried out with similar results and representative results from one experiment are shown.
Therefore, cellular localization of the ErbB receptors was analyzed in cells infected for 4 h with the 2C43 wild-type strain. As shown in Fig. 3, although ErbB2 displayed a punctuated staining in uninfected cells, it was highly recruited underneath the bacterial colonies in infected cells, whereas EGFR, ErbB3, and ErbB4 staining was not affected. Altogether, these data show that adhesion of N. meningitidis to human endothelial cells induces tyrosine phosphorylation and clustering of ErbB2, but not of the other ErbB members, suggesting that most likely N. meningitidis induces ErbB2 activation by promoting the formation of ErbB2 homodimers.
Figure 3.
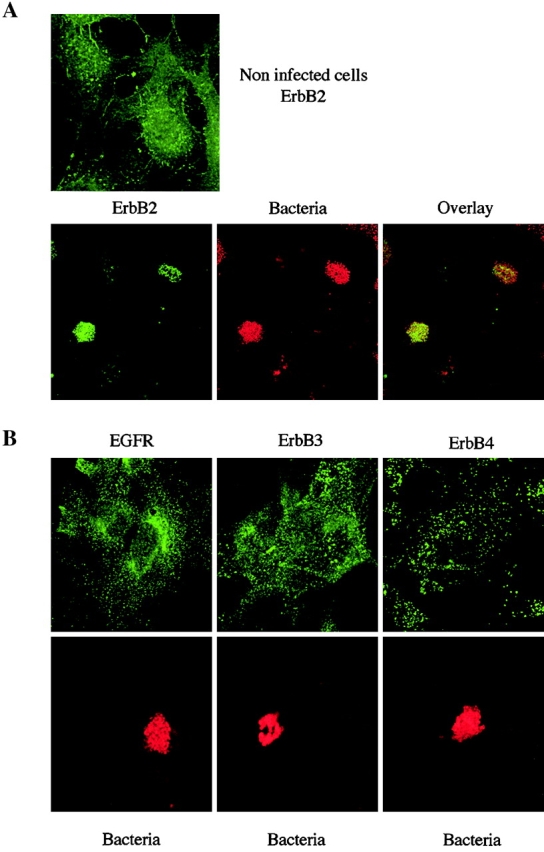
Localized adhesion of N. meningitidis induces the clustering of ErbB2, but not of the other ErbB members. (A) Top, uninfected cells were stained for ErbB2; bottom, cells infected for 3 h with the 2C43 wild-type strain of N. meningitidis were double-stained for ErbB2 (left) and bacterial colonies (middle). Merged images (overlay) of the same fields are presented on the right. (B) Cells infected for 3 h with the 2C43 wild-type strain of N. meningitidis were double-stained for either EGFR, ErbB3, or ErbB4 (top, as indicated) and bacterial colonies (bottom). Panels show receptor and bacterial staining in the same fields.
ErbB2 activation is not required for cortical plaque formation induced by N. meningitidis
We have reported previously that N. meningitidis induces actin cytoskeleton rearrangements and cortical plaque formation in human endothelial cells (unpublished data). Since stimulation of ErbBs may lead to Rho GTPase activation and actin cytoskeleton rearrangements, we investigated the contribution of ErbB2 activation in the HBMEC response to N. meningitidis. Immunofluorescence analysis of ezrin, actin, and ErbB2 was performed in cells infected for 4 h with the 2C43 wild-type strain of N. meningitidis in the presence of 5 μM AG1478. As shown in Fig. 4 A, AG1478 did not prevent the massive recruitment of ezrin and cortical actin polymerization underneath the bacterial colonies that we described previously in the absence of inhibitor. This observation indicates that ErbB2 activation is not required for cortical plaque formation induced by N. meningitidis. Moreover, we observed that inhibition of ErbB2 activity did not affect its own massive recruitment underneath the bacterial colonies, thus suggesting that N. meningitidis–induced ErbB2 clustering may precede receptor activation.
Figure 4.
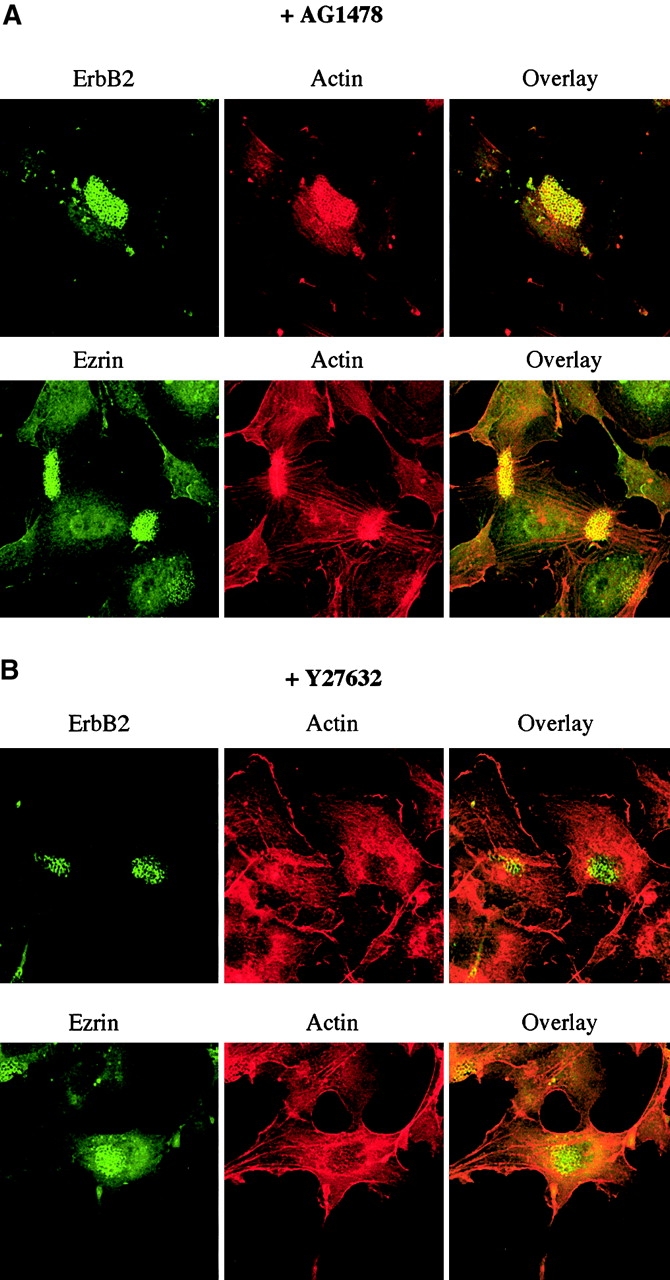
ErbB2 activation is not required for cortical plaque formation induced by N. meningitidis. HBMECs were pretreated for 2 h with 5 μM AG1478 (A) or with 30 μM Y27632 (B) before infection for 3 h with the 2C43 wild-type strain of N. meningitidis in the presence of the inhibitors. Cells were double-stained for ErbB2 and bacterial colonies (top) or ezrin and actin (bottom). Merged images (overlay) of the same fields are presented on the right.
Cellular localization of ErbB2 was then analyzed in the presence of 30 μM Y27632, a selective inhibitor of the Rho effector Rho-kinase (Ishizaki et al., 2000), which prevents N. meningitidis–induced actin cytoskeleton rearrangement, but does not affect ezrin recruitment underneath the bacterial colonies (Fig. 4 B). Interestingly, Y27632 did not affect either ErbB2 clustering (Fig. 4 B) or phosphorylation (unpublished data) induced by bacterial interaction. Therefore, the Rho-GTPase–dependent cortical actin polymerization induced by N. meningitidis is not required for ErbB2 recruitment underneath the bacterial colonies or ErbB2 activation.
ErbB2 activation induced by N. meningitidis leads to the activation of the tyrosine kinase src and the subsequent phosphorylation of the src substrate cortactin
To define the role of ErbB2 in the interaction process of N. meningitidis with human endothelial cells, we attempted to identify the tyrosyl-phosphorylated proteins p80/85 affected by AG1478 treatment described in Fig. 1. The migration pattern of p80/85 was suggestive of a few candidate proteins, including the actin-binding protein cortactin, ezrin, and the p85α subunit of the PI-3 kinase. Indeed, as shown in Fig. 5 A, infection for 4 h with the 2C43 wild-type strain of N. meningitidis induced the tyrosyl phosphorylation of cortactin, whereas ezrin or the p85α subunit of the PI-3 kinase were not tyrosine phosphorylated under the same conditions (unpublished data). In agreement with the fact that cortactin is a known substrate of the tyrosine kinase src, an autophosphorylation assay demonstrated that src was indeed activated in cells infected for 4 h with the 2C43 wild-type (Fig. 5 B). As expected, no src activity nor tyrosyl phosphorylation of cortactin was detected in cells in contact with the 2C43 PilE− mutant, whereas the PilT− mutant was more efficient in inducing both src activity and cortactin phosphorylation. Src activation induced by either 2C43 wild-type or ROU strains of N. meningitidis was largely reduced by treatment with 5 μM AG1478 (Fig. 5 C). Moreover, cortactin phosphorylation was fully inhibited by a treatment with 5 μM PP2, a selective inhibitor of src family kinases (Fig. 5 D), and was also largely inhibited by treatment with 5 μM AG1478 (Fig. 5 E), whereas EGF stimulation did not stimulate cortactin phosphorylation. Together, these results demonstrate that N. meningitidis induces src activation and subsequent cortactin phosphorylation and point to src as a downstream signaling effector of ErbB2 in this pathway. In addition, since AG1478 does not fully inhibit src activity and cortactin phosphorylation, whereas it completely prevents ErbB2 phosphorylation, an alternative pathway, independent of ErbB2, may also contribute to the activation of src induced by N. meningitidis.
Figure 5.

ErbB2 activation induced by N. meningitidis leads to src activation and the phosphorylation of cortactin. (A and B) HBMECs (starved for 18 h) were either uninfected (−) or infected with the wild-type (WT), the PilT-defective mutant (PilT−), or the PilE-defective mutant (PilE−) of the 2C43 strain of N. meningitidis for 4 h before lysis. (C–E) HBMECs starved for 18 h were pretreated for 2 h with either 5 μM PP2 or 5 μM AG1478 (+) or left untreated (−). Cells were either noninfected (NT) or infected with the wild-type 2C43 or the ROU strains as indicated for 4 h before lysis. PP2 and AG1478 treatments were maintained during infection. As control, cells pretreated the same way were stimulated with 10 ng/ml EGF for 3 min before lysis. (A, D, and E) Cortactin was immunoprecipitated and immunoblotted with an antiphosphotyrosine antibody (4G10) (top). Blots were reprobed with an anticortactin antibody (bottom) to show that similar protein levels were immunoprecipitated. (B and C) Src was immunoprecipitated and subjected to either an autophosphorylation assay (B) or an in vitro kinase assay using acid-denaturated enolase as a substrate (C). Three independent experiments were carried out with similar results and representative results from one experiment are shown.
Therefore, we examined whether src was associated with phosphorylated ErbB2 in infected cells. As shown in Fig. 6 A, an increased amount of src was detected in ErbB2 immunoprecipitates in cells infected for 4 h with either the 2C43 wild-type or PilT− mutant. These interactions were largely prevented by treatment with 5 μM AG1478, thus confirming that ErbB2 phosphorylation induced by N. meningitidis results in the recruitment and activation of src. To further confirm that bacteria- or EGF-induced ErbB2 phosphorylation can provide a docking site for the SH2 domain of src, we performed a pull-down assay with a recombinant gluthatione S-transferase (GST)–src SH2 fusion protein (Fig. 6, B and C). Analysis of the pattern of tyrosine-phosphorylated proteins precipitated with the fusion protein showed that both N. meningitidis and EGF induced the interaction of src–SH2 with a 185-kD phosphoprotein identified by Western blotting as ErbB2, whereas only EGF stimulation induced the interaction of src-SH2 with a prominent tyrosine-phosphorylated band of 175 kD identified as EGFR. These interactions were dependent on the intrinsic kinase activity of ErbB2 or EGFR, since it was prevented by AG1478 treatment. These observations confirmed our conclusion that, although EGF induced EGFR/ErbB2 heterodimerization, bacteria induced the clustering of ErbB2 as homodimers.
Figure 6.

Localized adhesion of N. meningitidis leads to src association with phosphorylated ErbB2. HBMECs (starved for 18 h) were either uninfected (−) or infected with the wild-type (WT), or the PilT-defective mutant (PilT−) of the 2C43 strain of N. meningitidis for 4 h before lysis. As control, cells pretreated the same way were stimulated with 10 ng/ml EGF for 3 min before lysis. (A) ErbB2 was immunoprecipitated and immunoblotted with an anti-src antibody (top). Blot was reprobed with an anti-ErbB2 antibody (bottom) to show that similar protein levels were immunoprecipitated. (B and C) Whole cell extracts were incubated with 5 μg of a GST-tagged peptide containing the SH2 domain of src and coupled to glutathione-Sepharose. The complexes were immunoblotted with an antiphosphotyrosine antibody (4G10) (top). Blots were reprobed with an anti-ErbB2 antibody (middle) and anti-EGFR antibody (bottom). Three independent experiments were carried out with similar results and representative results from one experiment are shown.
Since cortactin is known to interact with actin, we assessed whether cortactin was recruited at the site of actin polymerization. Although cortactin displayed a generalized cytoplasmic staining with some enrichment in membrane ruffles in uninfected cells, it was recruited in cortical plaques in cells infected for 4 h with a the 2C43 wild-type strain of N. meningitidis (Fig. 7). AG1478 and PP2 only slightly affected cortactin recruitment into cortical plaques, which was instead totally prevented by Y27632 treatment. Thus, ErbB2 activation promotes cortactin phosphorylation, but this event is not required for cortactin recruitment within cortical plaques, a process dependent on cortactin interaction with the actin cytoskeleton.
Figure 7.
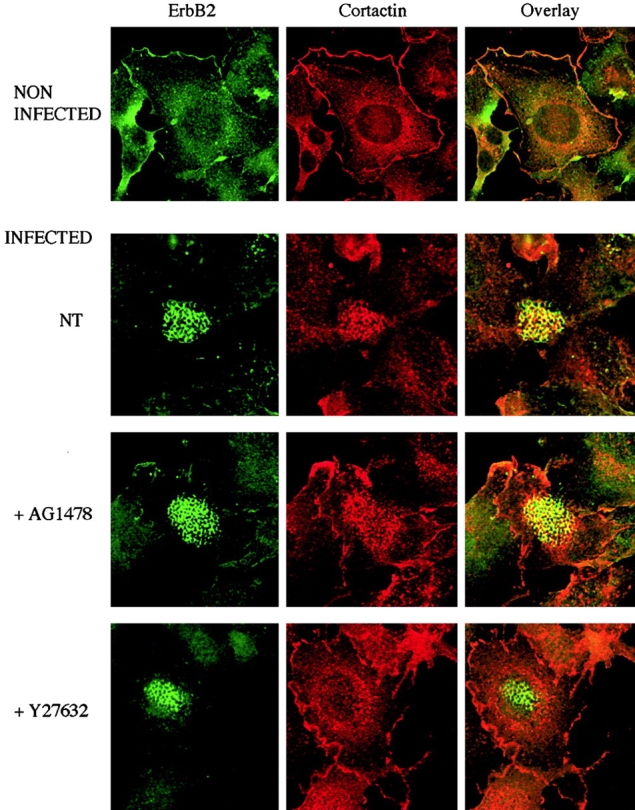
ErbB2 activation is not required for cortactin recruitment within cortical plaques. Cells were pretreated for 2 h with either 5 μM AG1478 or 30 μM Y27632. Cells noninfected or infected for 3 h with the 2C43 wild-type strain of N. meningitidis were double-stained for ErbB2 (left) and cortactin (middle). Merged images (overlay) of the same fields are presented on the right.
ErbB2/Src signaling pathway activation is involved in bacterial internalization by human endothelial cells
The internalization of N. meningitidis in endothelial cells, as shown previously, is maximal during localized adhesion, i.e., when the ErbB2/src pathway is fully activated, as shown here. The potential role of the ErbB2/src signaling pathway on bacterial internalization into endothelial cells was therefore investigated. We observed that the 2C43 PilT− mutant and the ROU strain, which activate the ErbB2/src signaling pathway more efficiently than the 2C43 wild-type strain, were also more effectively internalized (2.5 and 10×, respectively) (Fig. 8 A). As expected, no internalization of the nonadherent PilE− mutant was observed. Furthermore, treatment with 5 μM AG1478 or 5 μM PP2 decreased bacterial internalization by ∼50 or 70%, respectively (Fig. 8 B). In agreement with our conclusion that src is a downstream signaling effector of ErbB2, the addition of both inhibitors did not further reduce bacterial entry. By contrast, AG1296, the specific inhibitor of the PDGF receptor which failed to affect the tyrosyl phosphorylation induced by N. meningitidis (Fig. 1), had no effect on bacterial entry. Thus, these experiments indicate that the ErbB2/src pathway is involved in N. meningitidis internalization into endothelial cells.
Figure 8.
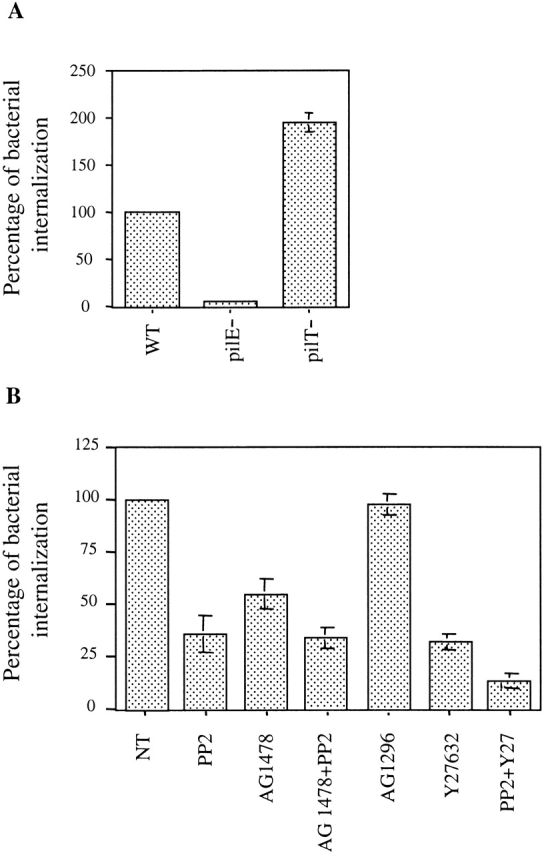
(A) Entry levels of the different strains of N. meningitidis in HBMECs. HBMECs were infected with the wild-type (WT), the PilT-defective mutant (PilT−), or the PilE-defective mutant (PilE−) of the 2C43 strain of N. meningitidis. After 4 h, the number of internalized bacteria in relation to the number of adherent bacteria was determined. (B) Effects of PP2, AG1478, AG1296, and Y27632 on N. meningitidis entry in HBMECs. Cells were either left untreated (NT) or pretreated for 2 h with 5 μM PP2, 5 μM AG1478, 5 μM AG1296, 30 μM Y27632, or with both inhibitors PP2+AG1478 or PP2+Y27632, as indicated. Cells were infected with the wild-type strain of N. meningitidis for 4 h in the presence of the inhibitors. The percentage of internalized bacteria in relation to the entry or adhesion of the wild-type strain in absence of any activator or inhibitor was determined, and average values (±SEM) from four independent experiments are presented.
Since we recently showed that the small Rho GTPases were involved in cortical plaque formation and subsequent N. meningitidis internalization into endothelial cells (unpublished data), we analyzed the respective contributions of both ErbB2/src and Rho GTPase–dependent signaling pathways on bacterial entry into endothelial cells. As shown in Fig. 8 B, treatment with PP2 in the presence of Y27632 almost totally prevented bacterial entry (90% inhibition), whereas Y27632 alone inhibited bacterial entry by 60%. None of these treatments affected bacterial adhesion to endothelial cells (unpublished data). Together, these data demonstrate that N. meningitidis promotes two distinct signaling pathways, the ErbB2/src and Rho-GTPase pathways, which cooperate to promote N. meningitidis internalization into endothelial cells.
Discussion
We have investigated here the tyrosine phosphorylation events occurring during the interaction of N. meningitidis with human endothelial cells. We demonstrate that the activation of the ErbB2 receptor tyrosine kinase is directly involved in host signal transduction promoted by N. meningitidis interaction and subsequent bacterial internalization.
N. meningitidis induces a massive recruitment and tyrosine phosphorylation of ErbB2. Our data suggest that the interaction of N. meningitidis with human endothelial cells leads to the activation of ErbB2, most likely via formation of ErbB2 homodimers, since no recruitment nor tyrosine phosphorylation of any other known ErbB family members was detected. We believe that these results constitute an important finding since, although no soluble ligand for ErbB2 has been identified so far, this receptor appears to be the preferred heterodimerization partner of all ErbB proteins (Tzahar et al., 1996; Graus-Porta et al., 1997). Although the formation of most heterodimers is mediated by ErbB ligands, ErbB2-containing heterodimers may also be triggered by other classes of receptors through ligand-independent pathways (Qiu et al., 1998). ErbB2 homodimerization was previously described only in a few cases: a ligand-independent activation of the kinase domain of ErbB2 is observed in a significant proportion of breast and ovarian cancers overexpressing ErbB2, apparently as a result of spontaneous dimer formation (Yarden and Sliwkowski, 2001). Homodimerization of this receptor can also be achieved by mutating a single amino acid residue in the transmembrane domain, leading to constitutive ErbB2 dimerization and activation (Bargmann et al., 1986; Olayioye et al., 1998). Alternatively, antibody binding to the extracellular domain can also promote ErbB2 homodimerization and its subsequent activation (Sarup et al., 1991; Olayioye et al., 1998). Thus, our results constitute the first example of ErbB2 activation by homodimerization induced in nontumor cells (expressing physiological levels of ErbB2) through a ligand-independent pathway.
ErbB2 activation by N. meningitidis infection apparently results from bacteria-induced clustering of this receptor, since ErbB2 tyrosine phosphorylation is dependent on its intrinsic kinase activity. In contrast, ErbB2 recruitment underneath the bacterial colonies appears to be activation-independent, inasmuch as it was not affected by the ErbB2 kinase inhibitor AG1478. Clustering of ErbB2 may result in the spontaneous homodimer formation due to an increased local concentration of ErbB2 molecules, in a similar way to what has been described in tumor cells overexpressing ErbB2. Indeed, previous studies performed with antibody-induced clustering of ErbB2 showed that the activation state of ErbB2 correlates with the size of ErbB2 clusters (Nagy et al., 1999). However, how N. meningitidis interaction leads to ErbB2 clustering is still unclear. No activation of this signaling pathway is observed in cells in contact with the nonpiliated strain of N. meningitidis (PilE−), thus indicating that these events are not due to the presence of any bacterial-secreted molecule in the infection medium. Moreover, this activation is maximal during the localized adhesion of N. meningitidis and decreases when bacteria loose their pili and disperse at the cell surface, whereas it remains persistent in cells infected with the PilT− mutant. This mutant, defective in pili retraction, is highly piliated and cannot disperse at the cell surface (Pujol et al., 1999; Merz and So, 2000). Thus, these data provide evidence that ErbB2 activation requires a pilus-dependent interaction of the bacteria with the host cells. Type IV pili initiate the interaction of virulent, capsulated N. meningitidis with human cells by interacting with a cellular receptor that remains to be clearly identified. The membrane protein MCP/CD46 was suggested as a receptor for N. meningitidis pili (Kallstrom et al., 1997). However the weak affinity of the pili–CD46 interaction suggests the existence of an alternative human cell receptor for N. meningitidis. Thus, ErbB2 clustering may result from a direct interaction with bacterial pili. Alternatively, ErbB2 might be recruited underneath the bacterial colonies by interacting in a multimer complex with other cellular components recruited after interaction of bacteria with an unknown cellular receptor. Indeed, the evidence provided suggests that an interaction between ErbB2 and other transmembrane glycoproteins may potentiate ErbB2 activation. In normal and transformed mammary epithelial cells, the transmembrane subunit of sialomucin complex ASGP-2 was shown to form a stable complex through its EGF-like domain with ErbB2, but not EGFR, ErbB3, or ErbB4, and to potentiate neuregulin-induced ErbB2–ErbB3 heterodimer activation (Carraway et al., 1999). Moreover, a direct interaction between CD44 and ErbB2 has been described, leading to a potentiation of the constitutive tyrosine phosphorylation of ErbB2 in an ovarian carcinoma cell line overexpressing ErbB2 (Bourguignon et al., 1997), or to a significant increase in neuregulin-induced ErbB2–ErbB3 heterodimerization and ErbB2 phosphorylation in Schwann cells (Sherman et al., 2000). Interestingly, we show here that ErbB2 was recruited together with ezrin and CD44. Further studies are required to assess whether such an interaction of ErbB2 with other transmembrane glycoproteins may occur in endothelial cells in response to N. meningitidis adhesion and may be critical for bacteria-induced ErbB2 clustering and/or activation.
How does the activation of ErbB2 lead to the entry of N. meningitidis into host cells that are normally nonphagocytic? We showed that ErbB2 phosphorylation results in the recruitment and activation of the tyrosine kinase src, which are dependent on the intrinsic kinase activity of ErbB2. A selective inhibition of both ErbB2 and src revealed a major role of the ErbB2/src signaling in N. meningitidis internalization. Interestingly, the ErbB2/src pathway is not required for the activation of GTPase-dependent cytoskeletal modifications that we have observed recently in response to the adhesion of N. meningitidis (unpublished data). Inhibition of ErbB2 or src activity did not affect the cortical actin reorganization induced underneath the bacterial colonies. Inversely, inhibition of bacteria-induced cortical actin polymerization did not prevent the activation of ErbB2/src pathway. Therefore, N. meningitidis promotes the activation of two distinct pathways, both of which contribute to an efficient internalization of bacteria. Interestingly, the implication in bacterial internalization of host cell src family tyrosine kinases and Rho family GTPases has been reported in several cases of invasive pathogens (Dramsi and Cossart, 1998). For example, Opa52-mediated phagocytosis of N. gonorrhoeae by human neutrophils requires, for an efficient uptake, the activity of the related Rho family member Rac, which appears to be controlled by an src-like tyrosine kinase (Fgr or Hck) (Hauck et al., 1998). In the same way, src, together with Rho family GTPases, is involved in the internalization of Shigella (Dehio et al., 1995; Adam et al., 1996) and is required for triggering the formation of actin polymerization foci induced by Shigella (Dumenil et al., 1998). By contrast, invasion by Salmonella, which requires the activation of Cdc42, probably does not involve tyrosine phosphorylation (Galan and Zhou, 2000). When required, tyrosine kinase activity is then directly involved in the activation of small GTPases and controls bacterial entry by eliciting cytoskeleton changes. N. meningitidis seems to have developed an alternative pathway leading to its subsequent uptake into endothelial cells, since we observed that src tyrosine kinase activity controls N. meningitidis entry at a different level than Rho family GTPases. We showed that src activation by N. meningitidis leads to the tyrosyl phosphorylation of a major susbstrate (p80/85), which we identified as cortactin. Cortactin is an actin-binding protein, which can promote in vitro F-actin crosslinking. We consistently observed that cortactin was highly recruited underneath the bacterial colonies, and that this recruitment was not correlated with tyrosyl phosphorylation of cortactin, but was instead dependent on cortical actin polymerization induced by N. meningitidis. Interestingly, ErbB2 activation induced by EGF, while activating src (unpublished data), did not lead to cortactin phosphorylation. This apparent discrepancy may result from the activation of specific signaling pathways activated by ErbB2 homodimers (in response to N. meningitidis) or ErbB2/EGFR heterodimers (in response to EGF), including the activation of alternative kinases involved in cortactin phosphorylation, such as Fer (Kim and Wong, 1998). Alternatively, cortactin phosphorylation induced in infected cells may result from the local concentration of signaling molecules in microdomains underneath the bacterial colonies. Further studies are required to investigate the consequences of the tyrosyl phosphorylation of cortactin and to characterize the mechanism by which the src signaling pathway facilitates N. meningitidis internalization into endothelial cells.
In summary, these results, summarized in Fig. 9, provide evidence for a novel mode of activation and an unsuspected involvement of ErbB2 in host pathogen interaction. We believe that they will help unravel the pathophysiological mechanism of meningitis associated with N. meningitidis infection.
Figure 9.

Schematic representation of the signaling pathways activated by N. meningitidis and involved in bacterial entry into endothelial cells. (1) Type IV pili initiate the interaction of virulent, capsulated N. meningitidis with human endothelial cells by interacting with a cellular receptor that remains to be clearly identified. Bacteria then proliferate, locally forming a colony at their site of attachment on the cell surface. (2) This pili- dependent adhesion induces, through a Rho GTPase-independent pathway, the recruitment of ezrin and moesin, two proteins that link the cellular membrane to the actin cytoskeleton, and the clustering of several transmembrane proteins: ErbB2 and the ezrin binding proteins CD44 and ICAM-1. (3) The activation of both Rho and Cdc42 GTPases induces a localized polymerization of cortical actin, leading to the formation of membrane projections reminiscent of epithelial microvilli structures, which surround single bacteria and provoke their internalization within endothelial intracellular vacuoles. Downstream of bacteria- induced ErbB2 activation are the ras/MAPK signaling pathway, and the src tyrosine kinase which phosphorylates cortactin, and is involved in bacterial entry.
Materials and methods
Antibodies and reagents
Antibodies to cortactin (p80/85) and pp60c-src were purchased from Upstate Biotechnology; antiphosphotyrosine antibody (4G10) was from Meditech; polyclonal antibodies directed against EGFR, ErbB2, ErbB3, ErbB4 were purchased from Santa Cruz Biotechnology, Inc. Antiphospho Erk1/2 (Thr202/Tyr204) was from Cell Signaling Technology. Rhodamine phalloidin and recombinant human EGF were from Sigma-Aldrich. GST fusion protein containing src-SH2 domain was provided by Dr. S. Fisher (Institut Cochin de Génétique Moléculaire, Paris, France). The selective inhibitors AG1478, AG1296, and PP2 were from Calbiochem, and Y27632 was provided by Yoshitomi Pharmaceutical.
Bacterial strains and growth conditions
2C43 (formerly clone 12), a piliated capsulated Opa− variant of the serogroup C meningococcal strain 8013, the nonpiliated derivative strain pilE::Km, and the PilT− derivative strain pilT::Erm were described previously (Nassif et al., 1993; Pujol et al., 1999). The strain designated ROU (group W135, ET37), is a piliated, capsulated isolate obtained from the cerebrospinal fluid of a 2-mo-old infant and has been described previously (Pron et al., 1997). Before experiments, N. meningitidis was grown overnight on GCB solid medium (4% GC medium base; Difco), 1% agar, 0.4% glucose, 0.2 mg/ml thiamine, 0.0005% Fe(NO3)39H2O, 0.01% l-glutamine at 37°C under 5% CO2. Several colonies were then selected and grown in DME supplemented with 0.1% BSA for 2 h and were finally diluted to ∼107 bacteria/ml suspension for cell infection.
Cell culture, infection, and quantification of N. meningitidis adhesion or entry into endothelial cells
HBMECs (Schweitzer et al., 1997), provided by Dr. B. Weksler (Weill Medical College of Cornell University, Ithaca, NY), were cultured in DME Glutamax (Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum, 7.5 μg/ml endothelial cell growth supplement (Sigma-Aldrich), 7 UI heparin (Sigma-Aldrich) and 10 mM Hepes.
Infections were performed as follows: confluent monolayers of HBMECs were starved for 18 h before infection in DME supplemented with 0.1% BSA (starvation medium) to lower the basal level of tyrosine phosphorylations. HBMECs were then overlaid for 30 min with ∼107 bacteria in starvation medium. Cells were then washed three times with starvation medium to remove nonadherent bacteria and infection was allowed to proceed for various periods of time. To avoid reinfection of the monolayers, cells were washed once every hour with starvation medium. When indicated, cells were pretreated for 2 h with AG1478, AG1296, PP2, or Y27632 before infection. Cells were then infected as above and the infection was allowed to proceed for 4 h in the presence of the inhibitors. For adhesion and internalization assays, cells grown to confluency in 6-well plates were infected in starvation medium, washed three times, and the infection was allowed to proceed for 4 h. To quantify adherent bacteria, the infected cell layers were extensively washed, resuspended by scraping into GCB liquid medium, serially diluted, and spread onto GCB plates. Bacteria were grown overnight and colony-forming units were counted. To quantify internalized bacteria, the infected cell layers were extensively washed, incubated with 150 μg/ml gentamicin at 37°C for 1 h, and washed extensively before scraping and plating dilutions onto GCB plates. Each condition was performed in triplicate and each experiment was carried out four times independently.
Immunoprecipitations and immunoblotting
Cells were washed once with cold PBS and lysed in a NP-40–based buffer (0.1% NP-40, 50 mM Tris-HCl, pH 7.4, 250 mM NaCl, 5 mM EDTA, 2 mM NaVO4, 5 mM NaF, 1 mM PMSF, 10 μg/ml each of aprotinin/leupeptin/pepstatin). For the coimmunoprecipitation assay, cells were lysed in a Triton X-100–containing buffer (1% Triton X-100, 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM MgCl2, 1 mM CaCl2, 2 mM NaVO4, 5 mM NaF, 1 mM PMSF, 10 μg/ml each of aprotinin/leupeptin/pepstatin). The insoluble fraction was removed by centrifugation and the cleared lysates were used for immunoprecipitation with specific antibodies or pull down experiment with the GST fusion protein. Precipitated proteins or fractions of the whole cell lysate were separated on a 7.5% SDS-PAGE gel and transferred to nitrocellulose (Schleicher & Schuell). After blocking for 1 h in PBS/1% BSA/0.05% Tween 20, filters were probed overnight with specific antibodies. Proteins were visualized with peroxidase-coupled secondary antibody using the ECL system (Amersham Pharmacia Biotech).
Src autophosphorylation/kinase assays
Immunoprecipitation of src was carried out as described in the previous section, using anti-p60c-src antibody and protein G–Sepharose beads. Beads were washed three times in an autophosphorylation buffer (20 mM Pipes, pH 7, 10 mM MnCl2, 1 mM PMSF, 0.1 mM NaVO4, 2 μg/ml aprotinin/leupeptin/pepstatin). Beads were resuspended in autophosphorylation buffer and incubated for 5 min at 30°C with 1 μM ATP and 25 μCi 32P ATP. Alternatively, beads were washed three times in an enolase buffer (100 mM Pipes, pH 7, 10 mM MnCl2, 1 mM PMSF, 0.1 mM NaVO4, 2 μg/ml aprotinin/leupeptin/pepstatin) and incubated for 5 min at 30°C with 3 μM ATP, 25 μCi 32P ATP, and 5 μg of denatured enolase (5 min at 30°C in 25 mM acetate buffer, pH 3.3). Reactions were stopped with Laemmli's buffer. Samples were heated for 10 min at 100°C, separated by electrophoresis on a 12% SDS-polyacrylamide gel, and transferred to nitrocellulose membranes. Incorporations of radioactive ATP by src ro enolase were detected by autoradiography.
Confocal immunofluorescence microscopy
HBMECs were grown to confluence on Permanox chamber slides (Lab-Tek). After infection, cells were fixed in 4% paraformaldehyde for 10 min, washed three times with PBS, and permeabilized with 0.2% Triton X-100 in PBS for 10 min. Cells were blocked for 30 min with 3% BSA in PBS and were incubated for 2 h with the primary antibodies. After three washes with PBS, cells were incubated for 1 h with CY3-conjugated anti–mouse IgG and CY2-conjugated anti–rabbit IgG (Jackson ImmunoResearch Laboratories). Actin staining was performed with rhodamine-labeled phalloidin (Sigma-Aldrich) and bacteria staining with 0.5 μg/ml ethidium bromide. Labeled preparations were mounted in mowiol and analyzed with a confocal microscope (Bio-Rad Laboratories).
Acknowledgments
We thank Dr. B.B. Weksler (Cornell University, Ithaca, New York) for providing HBMECs and for helpful comments on the manuscript, Dr. Josiane Pierre (INSERM U461, Chatenay Malabry, France) for providing us with specific antibodies to ErbB family members, Yoshitomi Pharmaceutical (Osaka, Japan) for the generous gift of Y27632, and Dr. Sylvie Cazaubon and Dr. Simon Louis for critical reading of the manuscript.
This work was supported by grants from the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, and the Ministère de la Recherche et de la Technologie.
Footnotes
Abbreviations used in this paper: EGFR, EGF receptor; GST, glutathione S-transferase; HBMEC, human bone marrow endothelial cell; MAPK, mitogen-activated protein kinase; RTK, receptor tyrosine kinase; SH2, src homology 2.
References
- Adam, T., M. Giry, P. Boquet, and P. Sansonetti. 1996. Rho-dependent membrane folding causes Shigella entry into epithelial cells. EMBO J. 15:3315–3321. [PMC free article] [PubMed] [Google Scholar]
- Bargmann, C.I., M.C. Hung, and R.A. Weinberg. 1986. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 45:649–657. [DOI] [PubMed] [Google Scholar]
- Beerli, R.R., D. Graus-Porta, K. Woods-Cook, X. Chen, Y. Yarden, and N.E. Hynes. 1995. Neu differentiation factor activation of ErbB-3 and ErbB-4 is cell specific and displays a differential requirement for ErbB-2. Mol. Cell. Biol. 15:6496–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguignon, L.Y., H. Zhu, A. Chu, N. Iida, L. Zhang, and M.C. Hung. 1997. Interaction between the adhesion receptor, CD44, and the oncogene product, p185HER2, promotes human ovarian tumor cell activation. J. Biol. Chem. 272:27913–27918. [DOI] [PubMed] [Google Scholar]
- Carpenter, G. 1999. Employment of the epidermal growth factor receptor in growth factor–independent signaling pathways. J. Cell Biol. 146:697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway, K.L., III, and C. Sweeney. 2001. Localization and modulation of ErbB receptor tyrosine kinases. Curr. Opin. Cell Biol. 13:125–130. [DOI] [PubMed] [Google Scholar]
- Carraway, K.L., III, E.A. Rossi, M. Komatsu, S.A. Price-Schiavi, D. Huang, P.M. Guy, M.E. Carvajal, N. Fregien, C.A. Carraway, and K.L. Carraway. 1999. An intramembrane modulator of the ErbB2 receptor tyrosine kinase that potentiates neuregulin signaling. J. Biol. Chem. 274:5263–5266. [DOI] [PubMed] [Google Scholar]
- Dehio, C., M.C. Prevost, and P.J. Sansonetti. 1995. Invasion of epithelial cells by Shigella flexneri induces tyrosine phosphorylation of cortactin by a pp60c-src-mediated signalling pathway. EMBO J. 14:2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi, S., and P. Cossart. 1998. Intracellular pathogens and the actin cytoskeleton. Annu. Rev. Cell Dev. Biol. 14:137–166. [DOI] [PubMed] [Google Scholar]
- Dumenil, G., J.C. Olivo, S. Pellegrini, M. Fellous, P.J. Sansonetti, and G.T. Nhieu. 1998. Interferon α inhibits a Src-mediated pathway necessary for Shigella-induced cytoskeletal rearrangements in epithelial cells. J. Cell Biol. 143:1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan, J.E., and D. Zhou. 2000. Striking a balance: modulation of the actin cytoskeleton by Salmonella. Proc. Natl. Acad. Sci. USA. 97:8754–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta, D., R.R. Beerli, and N.E. Hynes. 1995. Single-chain antibody-mediated intracellular retention of ErbB-2 impairs Neu differentiation factor and epidermal growth factor signaling. Mol. Cell. Biol. 15:1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta, D., R.R. Beerli, J.M. Daly, and N.E. Hynes. 1997. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 16:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwind, A., E. Zwick, N. Prenzel, M. Leserer, and A. Ullrich. 2001. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 20:1594–1600. [DOI] [PubMed] [Google Scholar]
- Hauck, C.R., T.F. Meyer, F. Lang, and E. Gulbins. 1998. CD66-mediated phagocytosis of Opa52 Neisseria gonorrhoeae requires a Src-like tyrosine kinase- and Rac1-dependent signalling pathway. EMBO J. 17:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki, T., M. Uehata, I. Tamechika, J. Keel, K. Nonomura, M. Maekawa, and S. Narumiya. 2000. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 57:976–983. [PubMed] [Google Scholar]
- Kallstrom, H., M.K. Liszewski, J.P. Atkinson, and A.B. Jonsson. 1997. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol. Microbiol. 25:639–647. [DOI] [PubMed] [Google Scholar]
- Kim, L., and T.W. Wong. 1998. Growth factor-dependent phosphorylation of the actin-binding protein cortactin is mediated by the cytoplasmic tyrosine kinase FER. J. Biol. Chem. 273:23542–23548. [DOI] [PubMed] [Google Scholar]
- Kurokawa, H., A.E. Lenferink, J.F. Simpson, P.I. Pisacane, M.X. Sliwkowski, J.T. Forbes, and C.L. Arteaga. 2000. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 60:5887–5894. [PubMed] [Google Scholar]
- Mengaud, J., H. Ohayon, P. Gounon, R.M. Mege, and P. Cossart. 1996. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell. 84:923–932. [DOI] [PubMed] [Google Scholar]
- Merz, A.J., and M. So. 1997. Attachment of piliated, Opa- and Opc-gonococci and meningococci to epithelial cells elicits cortical actin rearrangements and clustering of tyrosine-phosphorylated proteins. Infect. Immun. 65:4341–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz, A.J., and M. So. 2000. Interactions of pathogenic Neisseriae with epithelial cell membranes. Annu. Rev. Cell Dev. Biol. 16:423–457. [DOI] [PubMed] [Google Scholar]
- Merz, A.J., C.A. Enns, and M. So. 1999. Type IV pili of pathogenic Neisseriae elicit cortical plaque formation in epithelial cells. Mol. Microbiol. 32:1316–1332. [DOI] [PubMed] [Google Scholar]
- Merz, A.J., M. So, and M.P. Sheetz. 2000. Pilus retraction powers bacterial twitching motility. Nature. 407:98–102. [DOI] [PubMed] [Google Scholar]
- Nagy, P., A. Jenei, A.K. Kirsch, J. Szollosi, S. Damjanovich, and T.M. Jovin. 1999. Activation-dependent clustering of the erbB2 receptor tyrosine kinase detected by scanning near-field optical microscopy. J. Cell Sci. 112:1733–1741. [DOI] [PubMed] [Google Scholar]
- Nassif, X., J. Lowy, P. Stenberg, P. O'Gaora, A. Ganji, and M. So. 1993. Antigenic variation of pilin regulates adhesion of Neisseria meningitidis to human epithelial cells. Mol. Microbiol. 8:719–725. [DOI] [PubMed] [Google Scholar]
- Nassif, X., C. Pujol, P. Morand, and E. Eugene. 1999. Interactions of pathogenic Neisseria with host cells. Is it possible to assemble the puzzle? Mol. Microbiol. 32:1124–1132. [DOI] [PubMed] [Google Scholar]
- Nhieu, G.T., and P.J. Sansonetti. 1999. Mechanism of Shigella entry into epithelial cells. Curr. Opin. Microbiol. 2:51–55. [DOI] [PubMed] [Google Scholar]
- Olayioye, M.A., D. Graus-Porta, R.R. Beerli, J. Rohrer, B. Gay, and N.E. Hynes. 1998. ErbB-1 and ErbB-2 acquire distinct signaling properties dependent upon their dimerization partner. Mol. Cell. Biol. 18:5042–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olayioye, M.A., R.M. Neve, H.A. Lane, and N.E. Hynes. 2000. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 19:3159–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pron, B., M.K. Taha, C. Rambaud, J.C. Fournet, N. Pattey, J.P. Monnet, M. Musilek, J.L. Beretti, and X. Nassif. 1997. Interaction of Neisseria meningitidis with the components of the blood-brain barrier correlates with an increased expression of PilC. J. Infect. Dis. 176:1285–1292. [DOI] [PubMed] [Google Scholar]
- Pujol, C., E. Eugene, L. de Saint Martin, and X. Nassif. 1997. Interaction of Neisseria meningitidis with a polarized monolayer of epithelial cells. Infect. Immun. 65:4836–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol, C., E. Eugene, M. Marceau, and X. Nassif. 1999. The meningococcal PilT protein is required for induction of intimate attachment to epithelial cells following pilus-mediated adhesion. Proc. Natl. Acad. Sci. USA. 96:4017–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, Y., L. Ravi, and H.J. Kung. 1998. Requirement of ErbB2 for signalling by interleukin-6 in prostate carcinoma cells. Nature. 393:83–85. [DOI] [PubMed] [Google Scholar]
- Russell, K.S., D.F. Stern, P.J. Polverini, and J.R. Bender. 1999. Neuregulin activation of ErbB receptors in vascular endothelium leads to angiogenesis. Am. J. Physiol. 277:H2205–H2211. [DOI] [PubMed] [Google Scholar]
- Sarup, J.C., R.M. Johnson, K.L. King, B.M. Fendly, M.T. Lipari, M.A. Napier, A. Ullrich, and H.M. Shepard. 1991. Characterization of an anti-p185HER2 monoclonal antibody that stimulates receptor function and inhibits tumor cell growth. Growth Regul. 1:72–82. [PubMed] [Google Scholar]
- Schweitzer, K.M., P. Vicart, C. Delouis, D. Paulin, A.M. Drager, M.M. Langenhuijsen, and B.B. Weksler. 1997. Characterization of a newly established human bone marrow endothelial cell line: distinct adhesive properties for hematopoietic progenitors compared with human umbilical vein endothelial cells. Lab Invest. 76:25–36. [PubMed] [Google Scholar]
- Shen, Y., M. Naujokas, M. Park, and K. Ireton. 2000. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell. 103:501–510. [DOI] [PubMed] [Google Scholar]
- Sherman, L.S., T.A. Rizvi, S. Karyala, and N. Ratner. 2000. CD44 enhances neuregulin signaling by Schwann cells. J. Cell Biol. 150:1071–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzahar, E., H. Waterman, X. Chen, G. Levkowitz, D. Karunagaran, S. Lavi, B.J. Ratzkin, and Y. Yarden. 1996. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell Biol. 16:5276–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden, Y., and M.X. Sliwkowski. 2001. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2:127–137. [DOI] [PubMed] [Google Scholar]