

Abstract
Reactive oxygen species are believed to perform multiple roles during plant defense responses to microbial attack, acting in the initial defense and possibly as cellular signaling molecules. In animals, nitric oxide (NO) is an important redox-active signaling molecule. Here we show that infection of resistant, but not susceptible, tobacco with tobacco mosaic virus resulted in enhanced NO synthase (NOS) activity. Furthermore, administration of NO donors or recombinant mammalian NOS to tobacco plants or tobacco suspension cells triggered expression of the defense-related genes encoding pathogenesis-related 1 protein and phenylalanine ammonia lyase (PAL). These genes were also induced by cyclic GMP (cGMP) and cyclic ADP-ribose, two molecules that can serve as second messengers for NO signaling in mammals. Consistent with cGMP acting as a second messenger in tobacco, NO treatment induced dramatic and transient increases in endogenous cGMP levels. Furthermore, NO-induced activation of PAL was blocked by 6-anilino-5,8-quinolinedione and 1H-(1,2,4)-oxadiazole[4,3-a]quinoxalin-1-one, two inhibitors of guanylate cyclase. Although 6-anilino-5,8-quinolinedione fully blocked PAL activation, inhibition by 1H-(1,2,4)-oxadiazole[4,3-a]quinoxalin-1-one was not entirely complete, suggesting the existence of cGMP-independent, as well as cGMP-dependent, NO signaling. We conclude that several critical players of animal NO signaling are also operative in plants.
Keywords: reactive oxygen species/guanylate cyclase/pathogenesis-related proteins/calcium/plant signal transduction
Reactive oxygen species (ROS) have been proposed to serve as diffusible intercellular signals and/or second messengers for the activation of various defense genes in animals, bacteria and plants (1–5). In plants, ROS appear to induce genes involved in antioxidative responses as well as those associated with resistance to pathogens (3). In addition, ROS may contribute to programmed cell death as part of the hypersensitive response to pathogen attack (2, 4, 6). ROS-associated plant defenses have been compared with redox-regulated animal immune and inflammatory responses (5).
In the animal field, recent research has focused on nitric oxide (NO), which is involved in both physiological and pathophysiological conditions, such as inflammation, acute phase responses and programmed cell death (7). Because of its chemistry, which allows stability and reactivity at the same time, NO and its redox-activated forms are often regarded as the only ROS that can fulfill the requirements of a true intra- and intercellular signaling molecule (7, 8). NO may act through activation of guanylate cyclase, which produces the second messenger cyclic GMP (cGMP), or through S-nitrosylation of redox-sensitive transcription factors or ion channels (7, 9, 10).
Although NO is a byproduct of many cellular reactions and a natural constituent of all living cells, its diverse activities, including redox signaling, have been explored principally in animals (7). In plants, NO is produced nonenzymatically through light-mediated conversion of NO2 by carotenoids or enzymatically from NO2 by NADPH nitrate reductases (11). Mounting evidence points to NO as an effector for plant growth, development, and defense. NO can induce leaf expansion, root growth and phytoalexin production (12, 13). Recently, evidence was provided for the presence of a mammalian-type NO synthase (NOS) in plants (12, 14, 15).
Although suggestive, these findings provided only indirect evidence for the existence of a mammalian-type NO-mediated signal transduction pathway in plants. However, two important downstream components of NO signaling in mammals, cGMP and cyclic ADP-ribose (cADPR), are functional in plants. cGMP appears to be important in light-mediated signal transduction and ion channel regulation (16–18). In addition, cGMP can stimulate the induction of genes encoding chalcone synthase (CHS) and ferredoxin NADP+ oxidoreductase and can initiate anthocyanin biosynthesis in soybean (17). In animals, one mode of action of cGMP is to stimulate biosynthesis of cADPR, an agent that mobilizes Ca2+, and thereby serves as a further downstream messenger of NO (19). cADPR has been shown to play a physiological role in Ca2+ mobilization in Vicia faba, probably through modulation of vacuolar Ca2+ channels (20).
Here we have demonstrated that tobacco mosaic virus (TMV) infection of resistant, but not susceptible, tobacco results in elevated NOS activity. Furthermore, we have used NO as a tool to elucidate whether messengers of the NO-mediated pathway in animals are functional in plant defense responses. As markers, we used phenylalanine ammonia lyase (PAL) and pathogenesis-related 1 (PR-1) proteins. PAL is a key enzyme in the phenylpropanoid biosynthetic pathway and is probably involved in the biosynthesis of salicylic acid (SA) (21–23). PR-1 exhibits antifungal activity, and is considered an excellent marker of plant disease resistance against pathogens (24, 25). We have found that NO, cGMP, and cADPR can activate PAL and PR-1 gene expression. Furthermore, data are presented that suggest the presence of a NO-inducible guanylate cyclase in tobacco.
METHODS
Plant Material.
Tobacco plants [Nicotiana tabacum cv. Xanthi nc (NN) or Xanthi (nn)] and suspension culture cells were grown as described (26, 27). The TMV susceptible cultivar was used only in the experiments to determine NOS activity (Fig. 1). The transgenic NahG-10 was kindly provided by John Ryals.
Figure 1.

NOS activities in the extracts of mock-or TMV-infected tobacco leaves after temperature shift. (A) Xanthi nc (NN) and Xanthi (nn) plants were infected with TMV or buffer (mock) and defense responses were activated by shifting from 32°C to 22°C 48 hr later as described (32). Time is given in hr after shift to 22°C. NOS activity based on arginine to citrulline conversion is plotted against the y axis to the left. NOS activity assayed with the fluorometric method based on nitrite detection is plotted against the right axis. Data represent the mean of three experiments. (B) Inhibition of NOS activity by the NOS inhibitors NMMA and diphenyleneiodonium chloride (250 μM). Data represent the mean of duplicate measurements.
Treatment of Tobacco and Tobacco Cell Suspensions Cultures.
NOS (5 units) in 5 ml of 40 mM Hepes (pH 7.4), containing all cofactors and substrates of NOS (5 μM oxyhemoglobin/50 μM l-arginine/100 μM CaCl2/20 μg/ml calmodulin from spinach/120 μM NADPH/12 μM tetrahydro-l-biopterin/100 μM DTT; see Bredt and Snyder, ref. 28) or NO-releasing compounds (in 5 ml of water) were injected into the intercellular space of tobacco leaves. The injection technique was described (1, 6). Five units NOS in 5 ml generate an initial flux of 1 μM NO per min. After injection, plants were exposed to continuous light. At various times leaf discs (1 cm diameter) were harvested, quickly frozen and stored at −80°C until they were analyzed for SA and cGMP content, accumulation of PR-1 protein and mRNA and PAL mRNA.
For experiments with cADPR, tobacco leaf discs (1 cm diameter) were infiltrated for 2 min using a vacuum pump with 30 μM ruthenium red or water. After a 1-hr incubation at room temperature under light and with agitation (15 rpm), the leaf discs were vacuum infiltrated for a second time as described with 100 nM cADPR and/or 250 μM cGMP or 250 μM SA or water. The leaf discs were then floated in Petri dishes, agitated (15 rpm) at room temperature with continuous light for 6 or 14 hr and assayed for PAL or PR-1 transcripts by Northern hybridization.
Treatment of suspension cells with NO donors or cGMP was done in the dark. At various times, 10 ml of cells [0.4–0.8 g fresh weight (FW)] were harvested by filtration. The cells were quickly frozen in liquid nitrogen and stored at −80°C until further analysis for PAL and PR-1 mRNA, PAL activity and cGMP content.
RNA Extraction and Hybridization.
Northern hybridization followed standard protocols and was done as described (29). The tobacco cDNA clones used as probes for hybridization were acidic PR-1a and PAL (29).
Immunoblot Analyses of PR-1 Protein Levels.
Sample preparation, electrophoretic separation, and immunoblot analysis using a PR-1 specific mouse mAb (33G1) were done as described (1).
Quantitation of SA.
Free SA and SA β-glucoside were extracted and quantitated by HPLC according to a previously described protocol (30).
Quantitation of cGMP. Samples were treated and prepared as described by Brown and Newton (16). cGMP was estimated by using a 125I-based radioimmunoassay and a commercially available kit from Amersham. To increase the sensitivity, the samples were acetylated (16). cGMP was quantified by comparison to pure cGMP standards. For samples containing at least 50 fmol, recovery rates were >65%. Control experiments confirmed the specificity of the assay for cGMP (16). The 125I assay did not detect other nucleotides (cAMP, ATP, and GTP).
Assay for PAL Activity.
PAL enzyme activity was assayed as described (31). PAL activities in untreated cells were between 0.1 and 0.2 nmol/min per mg.
Assay for NOS Activity.
After infection with TMV, plants were maintained at 32°C for 48 hr before shifting back to the normal growth temperature of 22°C as described (32). Extracts from tobacco leaves harvested at various times postshift to 22°C and were prepared as described (14). NOS was measured by an arginine/citrulline assay (using l-[2,3-3H]arginine, 40 Ci/mmol; 1 Ci = 37 GBq; see ref. 28) as well as by a fluorometric assay based on nitrite detection via 2,3-diaminonaphthalene (33).
RESULTS
Infection of Tobacco with TMV Increases Endogenous NOS Activity.
Evidence is accumulating, that a mammalian-type NOS is activated during plant-microbe interactions (12, 14, 15, 34). To more readily follow changes in NOS activity in tobacco, advantage was taken of the reversible, high temperature inhibition of TMV-induced defense responses in these plants. At 32°C, TMV-infected Xanthi nc (NN) tobacco fail to (i) produce elevated levels of SA, (ii) synthesize PR proteins, (iii) restrict virus multiplication and spread, and (iv) develop necrotic lesions (hypersensitive response). Upon shifting these plants to lower temperatures (22°C), all of the above defense responses are rapidly and strongly induced in a more synchronous manner (32).
Within 2–3 hr after shifting TMV-infected Xanthi nc (NN) to 22°C, NOS activity rose 4–5 fold (Fig. 1A), as measured with two different assays (28, 33). In contrast, there was no increase in NOS activity after mock-infection or in TMV-infected susceptible Xanthi (nn) after shifting to the lower temperature. NOS activity was dependent on the NOS substrate arginine and cofactors NADPH, FAD, and FMN (data not shown). NOS activity was inhibited by the NOS inhibitors l-NG-monomethyl-l-arginine monoacetate (NMMA) (but not its inactive d-enantiomer) and diphenyleneiodonium chloride (Fig. 1B). However, tobacco NOS activity and neuronal rat NOS differed in their degree of sensitivity to their inhibitors. In sum, TMV infection of resistant, but not susceptible, tobacco led to elevated NOS activity.
NO Induces PR-1 Accumulation.
To determine whether plants have a mechanism to sense NO, we initially monitored PR-1 protein accumulation in tobacco containing artificially elevated NO levels. Injection of mammalian NOS into the intercellular spaces of tobacco leaves, a technique successfully used for other ROS-generating or scavenging systems (1, 6, 35), resulted in significant accumulation of PR-1 (Fig. 2A). By contrast, treatment of leaves with a substrate/cofactor mix lacking NOS failed to induce PR-1 accumulation. Confirmation that PR-1 accumulation was caused by NO came from the demonstration that the NOS inhibitor NMMA (1 mM) and the NO scavenger 2-phenyl-4,4,5,5-tetramethylimidazolinone-3-oxide-1-oxyl (PTIO; 150 μM) both suppressed NOS-induced PR-1 expression (Fig. 2B). The ability of NMMA and PTIO to inhibit PR-1 accumulation does not appear to be due to any toxic side effects, because coinjection of these compounds into leaves 3 hr after NOS infiltration did not prevent PR-1 accumulation. Furthermore, these chemicals did not affect SA-mediated induction of PR-1.
Figure 2.
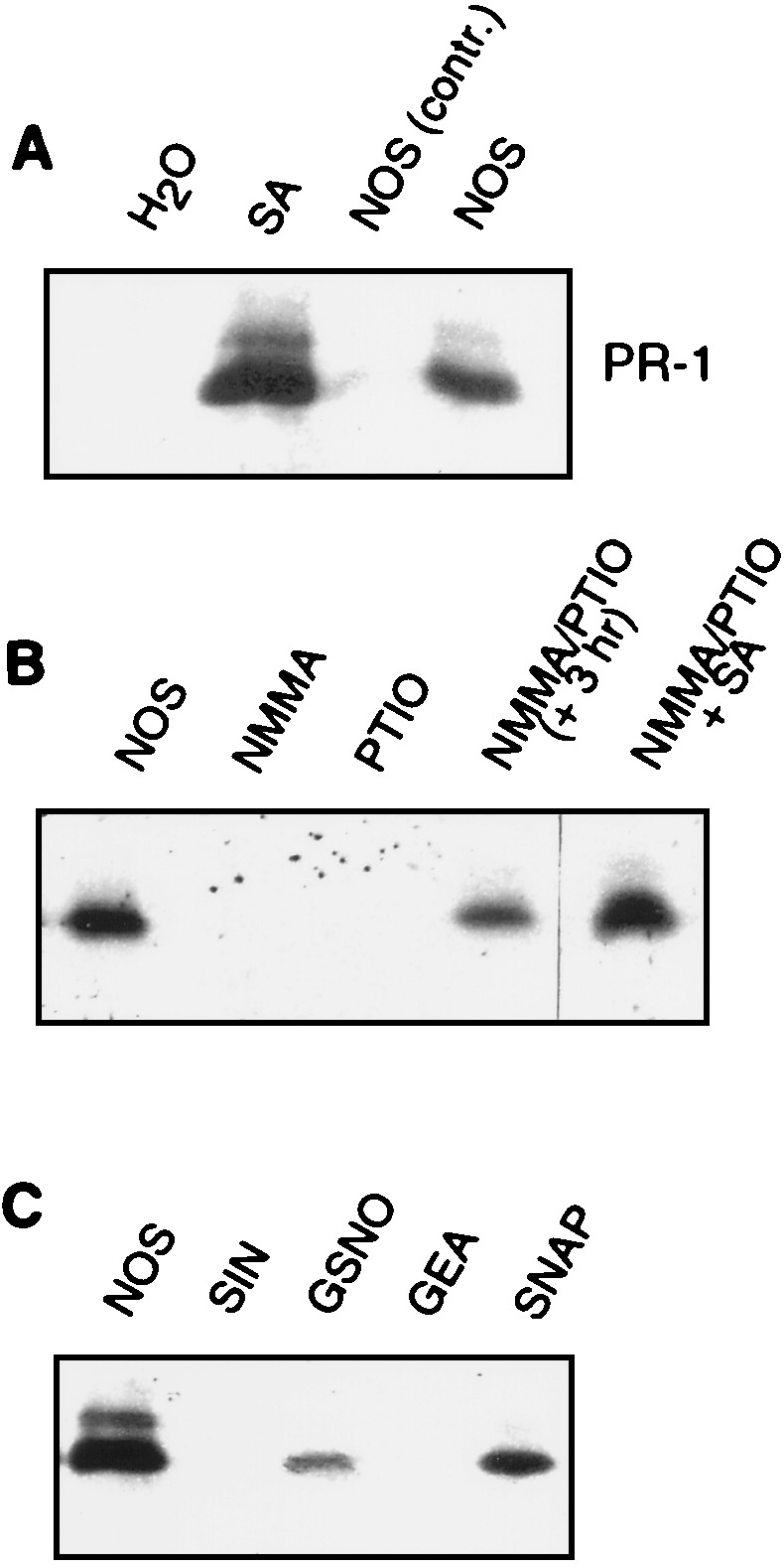
PR-1 protein accumulation in response to treatment with NOS or NO donors. Leaves were treated as indicated and PR-1 protein accumulation after 24 hr was determined by immunoblot analysis. (A) Leaves of Xanthi nc (NN) were infiltrated with SA (0.1 mM) or a NOS (5 units) plus a cofactor/substrate mix (see Materials and Methods). As a control, the cofactor/substrate mix without NOS was used (NOS control). (B) Inhibition of NOS-induced PR-1 accumulation by the NOS inhibitor NMMA (150 μM) or the NO scavenger PTIO (1 mM). The chemicals were added to the injection buffer containing NOS. Also shown are controls with infiltration of NMMA/PTIO delayed for 3 hr after addition of NOS (+ 3 hr), or coinfiltration of SA (in water) with these two chemicals. (C) Induction of PR-1 protein accumulation by NO donors: 3-morpholinosydnonimine-1 (SIN-1) (500 μM), GSNO (500 μM), 1,2,3,4-oxatriazolium-5-amino-3-(3-chloro-2-ethylphenyl)chloride 5024 (500 μM), and SNAP (150 μM).
The ability of NO generated enzymatically by NOS vs. that produced by biologically active NO donors to induce PR-1 accumulation was then compared. Using concentrations suggested for mammalian cells (150–500 μM), two of the most potent NO donors, S-nitroso-l-glutathione (GSNO), and S-nitroso-N-acetyl-dl-penicillamine (SNAP), effectively induced PR-1 accumulation. 3-Morpholinosydnonimine-1 (SIN-1), and 1,2,3,4-oxatriazolium-5-amino-3-(3-chloro-2-ethylphenyl) chloride (GEA) were effective only at concentrations of 1 mM or higher (data not shown). These results are consistent with the findings that the efficiency of the various NO donors depends on the biological system or cell type (9, 36). In sum, regardless of origin, NO led to a significant increase in PR-1 protein levels.
Total SA Levels Increase in NO-Treated Tobacco.
In many cases, the accumulation of PR-1 is associated with elevated SA levels. SA is known to be an effective inducer of PR-1 genes (24, 25). To determine whether NO treatment activates PR-1 accumulation through an SA-dependent signaling pathway, SA levels were monitored in NO-treated plants. Indeed, treatment of tobacco leaves with NOS as described above induced a significant increase in total SA (free SA plus conjugated SA forms), reaching about 2 μg/g FW by 18 hr after treatment (Fig. 3A). It should be noted, however, that most of the SA was in the conjugated form, with free SA comprising <5% of total SA. Similar results have been reported for tobacco resisting pathogen attack or after treatment with H2O2 (23, 37). Further demonstration that SA is required for the NO-mediated induction of PR-1 genes came from studies on transgenic plants expressing the nahG gene, which encodes a bacterial salicylate hydroxylase. These transgenic plants accumulate little, if any, SA and, as a consequence, show reduced PR-1 accumulation upon SA treatment or pathogen infection (24, 25). As expected, SA infiltration did not cause PR-1 protein (Fig. 3B) or mRNA (Fig. 3D) accumulation in NahG plants. Similarly, NOS treatment failed to raise PR-1 protein or mRNA levels in these NahG plants (Fig. 3D). Thus, NO originating from injected NO donors or recombinant NOS appears to activate PR-1 gene expression through SA.
Figure 3.
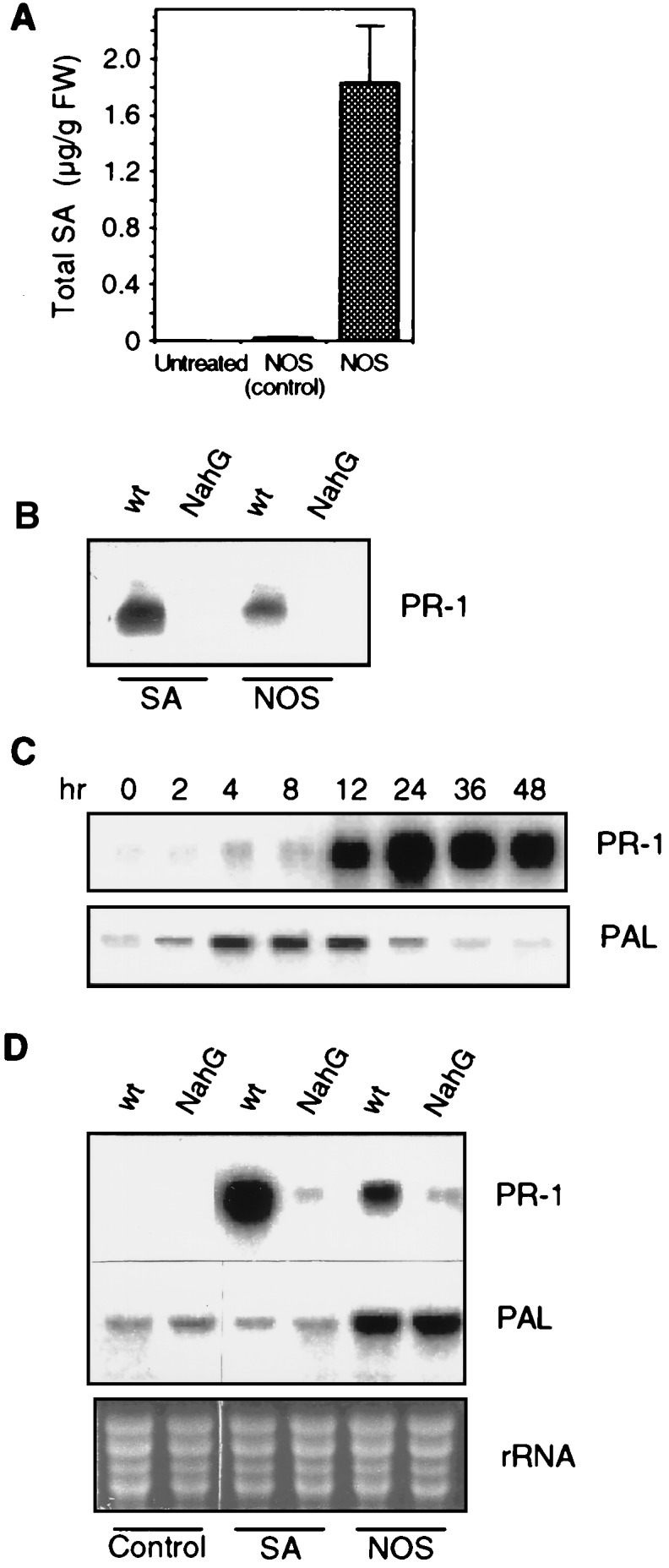
Effects of NOS on SA accumulation and PR-1 and PAL gene expression. (A) NOS treatment induces SA accumulation. Levels of total SA (free SA plus conjugated forms) were determined in either untreated leaves or 18 hr after infiltration with NOS or a substrate/cofactor mix lacking NOS (control). In untreated plants, SA was below the detection limit of 0.01 μg/g FW. (B) PR-1 protein accumulation in wild-type (wt) and NahG tobacco in response to treatment with SA or NOS. Treatment of leaves and PR-1 immunoblot analysis were done as described for Fig. 2. (C) Kinetics of PR-1 and PAL mRNA accumulation in response to treatment with NOS. (D) PR-1 and PAL mRNA accumulation in wild-type and NahG tobacco in response to treatment with SA or NOS. Treatment of leaves was as described for Fig. 2. Samples were taken after 8 and 18 hr for PAL and PR-1 mRNA. Ethidium bromide-stained rRNAs are shown as a control for gel loading.
NO Induces PAL via a SA-Independent Pathway.
One important defense gene whose induction is known to be among the earliest plant responses to a variety of stimuli such as UV, light, wounding, oxidative stress, and pathogen attack is PAL (21). PAL is expressed much earlier than PR-1 during plant defense responses, and its expression is probably not induced by SA (23). Thus, we examined whether NO treatment of tobacco leaves would result in the accumulation of PAL transcripts. NOS injection into leaves resulted in transient expression of PAL, which preceded PR-1 expression (Fig. 3C). SA infiltration did not induce the PAL gene (Fig. 3D). Moreover, the NOS-mediated activation of PAL expression was not affected in NahG plants. Thus, induction of the PAL gene by NO appears to be independent of SA.
PAL Expression Is Mediated by Guanylate Cyclase.
To delineate the mechanism(s) by which artificially generated NO might act, PAL gene expression was monitored in tobacco suspension cells rather than leaves. The synchronized treatment of many cells greatly facilitates the analysis of transiently expressed genes such as PAL. In addition, dark-grown heterotrophic cells exhibit only low basal expression of genes involved in photosynthesis or phenylpropanoid biosynthesis such as PAL or CHS (17). To confirm that tobacco suspension cells and leaves respond similarly to NO, the induction of PR-1 and PAL by the NO donors GSNO (200 μM) and SNAP (100 μM) was examined (Fig. 4). Both NO donors induced PR-1, as well as PAL, expression in suspension cells. Similar concentrations have been used in experiments with animal cells or bacteria (9, 36). As was observed in leaves, PTIO (500 μM) suppressed the action of GSNO and SNAP in suspension cells (data not shown). In addition, SA treatment induced the accumulation of PR-1, but not PAL transcripts in suspension cells (Fig. 4).
Figure 4.

NO donors induce PAL and PR-1 gene expression in tobacco suspension cells. (A) PR-1 protein and mRNA accumulation after treatment with SA (100 μM), GSNO (200 μM), or SNAP (100 μM). Total RNA was isolated from cells 9 hr posttreatment and subjected to Northern hybridization. Protein samples were taken after 24 hr. (B) Accumulation of PAL mRNA in suspension cells 5 hr posttreatment. Ethidium bromide-stained rRNAs are shown as a control for gel loading.
In animals, one action of NO is to activate guanylate cyclase, which leads to a transient increase in the second messenger cGMP (7). To elucidate whether guanylate cyclase is present in tobacco, NO-induced activation of PAL was studied in detail. In tobacco suspension cells, the NO donor GSNO (350 μM) transiently activated the PAL gene at the mRNA level, with full induction occurring between 2.5 and 5 hr posttreatment (Fig. 5A). PAL activity in GSNO-treated cells showed an 8- to 10-fold increase with respect to the untreated control (Fig. 5B). The rise in enzymatic activity paralleled the appearance of PAL transcript, and PAL enzyme activity remained at high levels for several hours. Strikingly, PAL expression and PAL activity were also activated by the NO downstream messenger cGMP (Fig. 5 A and B), which was administered as 8-Br-cGMP (100 μM), a cGMP derivative that is known to be biologically effective when applied to plant cells (17, 18). Moreover, the guanylate cyclase inhibitors 6-anilino-5,8-quinolinedione (LY 83583; 200 μM) and 1H-(1,2,4)-oxadiazole[4,3-a]quinoxalin-1-one (ODQ; 200 μM) suppressed the action of GSNO. 8-Br-cGMP was able to reverse the effect of LY 83583 on GSNO-induced PAL gene activation.
Figure 5.

Guanylate cyclase participates in the NO-induced activation of the tobacco PAL gene. (A) Accumulation of PAL mRNA in suspension cells after treatment with either the NO donor GSNO or cGMP in the absence or presence of the guanylate cyclase inhibitors LY 83583 and ODQ. Total RNA was isolated from cells after incubation for the indicated times. Treatments are indicated. Concentrations were 350 μM GSNO, 200 μM LY 83583, 200 μM ODQ, and 100 μM 8-Br-cGMP. The control was untreated cells. Equal loading of RNA was confirmed by ethidium bromide staining for rRNA (data not shown). (B) PAL enzyme activity in cells treated as described for A. At the indicated times, extracts of suspension cells were prepared and PAL activity in each was determined. Data represent the mean of duplicate measurements. Standard errors are indicated.
cGMP Levels Increase in NO-Treated Tobacco.
If NO’s mechanism of action for PAL induction is through the activation of guanylate cyclase, then treatment with NO donors or NOS should alter cGMP levels. Using a radioimmunoassay (16, 18, 38), cGMP levels were measured in tobacco leaves and suspension cells treated with NO-generating systems. Control levels were ≈5 pmol/g FW for suspension cells and about 10 pmol/g FW for leaves (Fig. 6). cGMP contents in various plants have been shown to vary significantly, ranging from less than 0.1 pmol/g FW in barley aleurone to more than 1 μmol/g FW in spruce needles (18, 39). Infiltration of leaves with NOS or treatment of cells with GSNO resulted in a 10–15-fold increase in cGMP. This increase was rapid and transient, consistent with its potential role in cell signaling and PAL induction. In contrast, cGMP was not produced by suspension cells treated with GSNO and LY 83583 (data not shown).
Figure 6.

cGMP levels in NO-treated tobacco leaves and suspension cells. (A) Leaves were infiltrated with NOS as described for Fig. 2, or (B) cells were treated with 350 μM GSNO as described for Fig. 4 (arrows indicate start of treatment). At the indicated times, samples were taken as described (Materials and Methods) and analyzed for cGMP content by a radioimmunoassay. Data represent the mean of two independent experiments, with each time point assayed in triplicate. Standard errors are indicated.
cADPR Induces Defense Gene Expression in Tobacco.
In animal cells, NO-induced cGMP can stimulate the synthesis of the NAD+ metabolite cADPR (19). cADPR is a potent Ca2+ mobilizing agent that binds to intracellular Ca2+ channels and activates Ca2+ release. To test the possibility that cADPR might be a second messenger in our system, tobacco leaves were treated with cADPR and changes in PAL and PR-1 transcript levels were monitored 6 or 14 hr later, respectively. cADPR induced both PAL and PR-1 expression (Fig. 7A). Moreover, the cADPR-induced expression of PAL and PR-1 was blocked by 30 μM ruthenium red, which inhibits Ca2+ release from internal stores in animal and Vicia faba cells (20, 40). Thus, cADPR appears to activate PAL and PR-1 expression through a Ca2+ release mechanism, which is sensitive to ruthenium red. When similar experiments were done in NahG plants, cADPR-activated PAL expression was not affected (Fig. 7B). In contrast, only a low accumulation of PR-1 mRNA after cADPR treatment was seen in NahG plants compared with wild-type plants. In sum, cADPR appears to act through a SA-dependent pathway for PR-1 activation but a SA-independent pathway for PAL induction. When cGMP was applied in combination with cADPR, induction of both genes was further enhanced (Fig. 7C), suggesting a coordinate action of cGMP and cADPR. The synergistic effect of cGMP and cADPR on PAL and PR-1 expression was suppressed by ruthenium red.
Figure 7.

PAL and PR-1 mRNA accumulation in response to treatment with cADPR. (A) Leaf discs were first vacuum infiltrated with 30 μM ruthenium red (RR) or water and 1 hr later with either cADPR (100 nM), SA (250 μM), or water. The leaf discs were assayed for PAL or PR-1 mRNA 6 or 14 hr after the second infiltration, respectively. (B) PAL and PR-1 mRNA accumulation in wild-type and NahG tobacco after cADPR treatment. Experimental procedures were the same as described in A. (C) Combined effects of cGMP and cADPR on PAL and PR-1 transcript accumulation. Experiments were done as described in A except that the second vacuum infiltration was done with 100 nM cADPR and/or 250 μM cGMP or water.
DISCUSSION
In animals, NO is known to act as a redox transmitter in the regulation of a diverse array of physiological processes, and NOS plays a central role in host responses to pathogen infection (7, 36). By contrast, whether NO and NOS play any role in activating various plant processes has been less clear. A growing body of evidence, however, suggests that portions of the innate immune system are highly conserved among vertebrates, invertebrates and plants (41, 42). In particular, two key enzymes involved in mammalian macrophage action, NADPH oxidase and NOS, appear to be activated during plant-microbe interactions (12, 14, 15, 34, 43). Moreover, in an elegant set of experiments Delledonne et al. (44) recently demonstrated the involvement of NO in disease resistance in both soybean suspension cells and Arabidopsis. Our study complements these findings by demonstrating that NOS activity increases in tobacco after TMV infection of resistant, but not susceptible, cultivars. In addition, we provide the first demonstration that one of the action(s) of NO in plants is to mediate defense gene expression via the second messenger cGMP.
The main goal of this study was to elucidate how and by which mechanism plants sense NO. Thus, we used artificially generated NO to address whether key components of the NO-signaling pathway in animals, namely cGMP and cADPR, can induce plant defense gene expression. It should be emphasized, however, that NO’s action in animals affects many types of cellular responses. Moreover, NO not only acts through second messengers, but also via S-nitrosylation of critical transcription factors and direct regulation of ion channels (7, 10).
To test whether plants can respond to NO, PR-1 gene expression was monitored in tobacco treated with NOS or NO donors. Increases in PR-1 expression are a reliable indicator for various stress situations including exposure to pathogen attack, UV light, ozone, or imbalances in cellular homeostasis and metabolism (25, 45). Micromolar concentrations of NO were found to induce PR-1 protein and mRNA accumulation effectively (Figs. 2–4). In contrast, H2O2, which has been hypothesized to play an important role in the induction of defense genes such as PR-1 (1, 2), must be administered in the range of 1 mM to 1 M to obtain substantial PR activation (1, 37, 46). It should be noted that many signaling pathways in animals originally thought to be activated by H2O2 (e.g., signaling cascades involving NF-κB, AP-1, oxyR, or soxRS) may, in fact, be regulated by other cellular metabolites including NO (7). Interestingly, PR-1 activation by NO is mediated by SA. NO treatment resulted in elevated levels of SA and its conjugates (Fig. 3A) and PR-1 induction by NO was blocked in NahG transgenic plants that are unable to accumulate SA (Fig. 3 B and D).
NO also induced the expression of another defense gene, PAL, whose expression during the defense response generally precedes that of PR-1. PAL provides precursors for phenylpropanoid (and hence SA) biosynthesis (21). Like PR-1, PAL is also activated by a variety of other stresses. Treatment with NOS activated PAL expression in NahG transgenic as well as wt plants (Fig. 3D). Thus, in contrast to PR-1, PAL induction by NO appears to be SA independent. In tobacco suspension cells, the NO donors GSNO and SNAP, as well as the membrane permeable analog of the NO second messenger cGMP, 8-Br-cGMP, were found to induce PAL expression (Figs. 4 and 5). The rapid and transient nature of NO- or cGMP-mediated PAL expression resembles the cGMP-induced activation of the soybean CHS gene, which encodes another enzyme in the phenylpropanoid pathway (17).
The occurrence of cGMP in plants has been unambiguously demonstrated by various mass spectrometry techniques (16, 17, 47). Furthermore, it has been shown that various stimuli, such as gibberellic acid treatment of barley aleurone, light stimulation of bean cells or NO treatment of spruce needles, can cause transient increases in cGMP levels (16, 18, 39). In animals cGMP is produced by guanylate cyclase. There are two major families of guanylate cyclases—the transmembrane receptor class, which contains the guanylate cyclase domain within the intracellular portion of the protein, and the soluble form, which is activated by NO (48). NO activates soluble guanylate cyclase either by binding to the heme-iron or through S-nitrosylation at critical cysteine residues (7). The following observations strongly support the existence of an NO-inducible guanylate cyclase in our tobacco system: (i) NO induction of PAL was suppressed by LY 83583 and ODQ (Fig. 5A), two inhibitors of NO-inducible mammalian guanylate cyclase, (ii) this suppression was reversed by 8-Br-cGMP (Fig. 5A), and (iii) NO treatment resulted in an increase of cGMP (Fig. 6). Interestingly, the increase in tobacco cGMP was similar in magnitude to that reported during NO-induced smooth muscle relaxation in animals (7). However, we cannot exclude other NO-based pathways for PAL activation. Indeed, the incomplete inhibition of PAL induction by ODQ, which is a more specific inhibitor of guanylate cyclase than LY 83583, may suggest the presence of a cGMP-independent mechanism, in addition to a cGMP-dependent pathway.
Current evidence suggests that one of several actions of cGMP is to regulate the second messenger Ca2+, either via cyclic nucleotide-gated ion channels (18) or via cADPR. cADPR is known to be involved in the abscisic acid signaling pathway in tomato and Arabidopsis (49, 50). In addition, cADPR has been found to elicit Ca2+ release from beet vacuoles (20). This release showed similarities to cADPR-gated Ca2+ flux in animal cells and could be inhibited by ruthenium red. Similarly, we found that cADPR induced PAL and PR-1 expression (Fig. 7). Ruthenium red suppressed this induction, arguing that Ca2+ participates in the signal transduction pathway. Interestingly, expression of these defense genes was amplified when cGMP and cADPR were added simultaneously (Fig. 7C). Thus, cGMP and cADPR appear to act synergistically to increase gene expression, just as reported for mammalian cells or sea urchin eggs (19, 51). However, it should be noted, that, at least in case of PAL, cGMP alone was able to induce significant gene expression. Thus, PAL expression appears to be enhanced by, but not dependent on cADPR. Moreover, we have not established that NO or cGMP act through cADPR to activate PR-1 and PAL. For example, NO may directly regulate Ca2+ channels via S-nitrosylation (10), which would be independent of cGMP and cADPR.
In summary, we have provided evidence for a mammalian-type NO-responsive, defense gene activation pathway in tobacco, which involves cGMP-dependent components. However, plants may have many ways to transduce a NO signal. The most efficient NO donors in our system, GSNO and SNAP, are also known to mediate S-nitrosylation in animal and bacteria cells (9, 36). Therefore, further investigations will be necessary to determine the complexity of NO’s action in plants.
Acknowledgments
We thank Frank Tsui for SA analyses and D’Maris Dempsey for critical reading of the manuscript. This work was supported by grants MCB 9723952 and MCB 9514239 from the National Science Foundation.
ABBREVIATIONS
- cADPR
cyclic ADP-ribose
- cGMP
cyclic GMP
- GSNO
S-nitroso-l-glutathione
- LY-83583
6-anilino-5,8-quinolinedione
- NMMA
NG-monomethyl-l-arginine monoacetate
- NOS
NO synthase
- PAL
phenylalanine ammonia lyase
- PR
pathogenesis-related
- PTIO
2-phenyl-4,4,5,5-tetramethylimidazolinone-3-oxide-1-oxyl
- ODQ
1H-(1,2,4)-oxadiazole[4,3-a]quinoxalin-1-one
- ROS
reactive oxygen species
- SA
salicylic acid
- SNAP
S-nitroso-N-acetyl-dl-penicillamine
- TMV
tobacco mosaic virus
- FW
fresh weight
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Chen Z, Silva H, Klessig D F. Science. 1993;262:1883–1886. doi: 10.1126/science.8266079. [DOI] [PubMed] [Google Scholar]
- 2.Levine A, Tenhaken R, Dixon R, Lamb C. Cell. 1994;79:583–595. doi: 10.1016/0092-8674(94)90544-4. [DOI] [PubMed] [Google Scholar]
- 3.Mehdy M C, Sharma Y K, Sathasivan K, Bays N W. Physiol Plant. 1996;98:365–374. [Google Scholar]
- 4.Baker C J, Orlandi E W. Annu Rev Phytopathol. 1995;33:299–321. doi: 10.1146/annurev.py.33.090195.001503. [DOI] [PubMed] [Google Scholar]
- 5.Khan A K, Wilson T. Chem Biol. 1995;2:437–445. doi: 10.1016/1074-5521(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 6.Jabs T, Dietrich R A, Dangl J L. Science. 1996;273:1853–1856. doi: 10.1126/science.273.5283.1853. [DOI] [PubMed] [Google Scholar]
- 7.Stamler J S. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 8.Stamler J S, Singel D L, Loscalzo J. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 9.Melino G, Bernassola F, Knight R A, Corasaniti M T, Nistico G, Finazzi-Agro A. Nature (London) 1997;388:432–433. doi: 10.1038/41237. [DOI] [PubMed] [Google Scholar]
- 10.Xu L, Eu J P, Meissner G, Stamler J S. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 11.Millar A H, Day D A. Trends Plant Sci. 1997;2:289–290. [Google Scholar]
- 12.Leshem Y Y. Plant Growth Regul. 1996;18:155–159. [Google Scholar]
- 13.Noritake T, Kawakita K, Doke N. Plant Cell Physiol. 1996;37:113–116. doi: 10.1093/oxfordjournals.pcp.a028952. [DOI] [PubMed] [Google Scholar]
- 14.Ninnemann H, Maier J. Photochem Photobiol. 1996;64:393–398. doi: 10.1111/j.1751-1097.1996.tb02477.x. [DOI] [PubMed] [Google Scholar]
- 15.Cueto M, Hernandéz-Perera O, Martin R, Bentura M L, Rodrigo J, Lamas S, Golvano M P. FEBS Lett. 1996;398:159–164. doi: 10.1016/s0014-5793(96)01232-x. [DOI] [PubMed] [Google Scholar]
- 16.Brown E G, Newton R P. Phytochem Anal. 1992;3:1–13. [Google Scholar]
- 17.Bowler C, Neuhaus G, Yamagata H, Chua N-H. Cell. 1994;77:73–81. doi: 10.1016/0092-8674(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 18.Penson S P, Schuurink R C, Fath A, Gubler F, Jacobsen J V, Jones R L. Plant Cell. 1996;8:2325–2333. doi: 10.1105/tpc.8.12.2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galione A, White A. Trends Cell Biol. 1994;4:431–436. doi: 10.1016/0962-8924(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 20.Allen G J, Muir S R, Sanders D. Science. 1995;268:735–737. doi: 10.1126/science.7732384. [DOI] [PubMed] [Google Scholar]
- 21.Dixon R A, Paiva N L. Plant Cell. 1995;7:1085–1097. doi: 10.1105/tpc.7.7.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pallas A P, Paiva N P, Lamb C, Dixon R A. Plant J. 1997;10:281–293. [Google Scholar]
- 23.Mauch-Mani B, Slusarenko A J. Plant Cell. 1996;8:203–212. doi: 10.1105/tpc.8.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryals J A, Neuenschwander U H, Willits M G, Molina A, Steiner H-Y, Hunt M D. Plant Cell. 1996;8:1809–1819. doi: 10.1105/tpc.8.10.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durner J, Shah J, Klessig D F. Trends Plant Sci. 1997;2:266–274. [Google Scholar]
- 26.Durner J, Klessig D F. J Biol Chem. 1996;271:28492–28501. doi: 10.1074/jbc.271.45.28492. [DOI] [PubMed] [Google Scholar]
- 27.Conrath U, Chen Z, Ricigliano J R, Klessig D F. Proc Natl Acad Sci USA. 1995;92:7143–7147. doi: 10.1073/pnas.92.16.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bredt D S, Snyder S H. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wendehenne D, Durner J, Chen Z, Klessig D F. Phytochemistry. 1997;47:651–657. [Google Scholar]
- 30.Bowling S A, Guo A, Cao H, Gordon A S, Klessig D F, Dong X. Plant Cell. 1994;6:1845–1857. doi: 10.1105/tpc.6.12.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rösler J, Krekel F, Amrhein N, Schmid J. Plant Physiol. 1997;113:175–179. doi: 10.1104/pp.113.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malamy J, Hennig J, Klessig D F. Plant Cell. 1992;4:359–365. doi: 10.1105/tpc.4.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Misko T P, Schilling R J, Salvemini D, Moore W M, Currie M G. Anal Biochem. 1993;214:11–16. doi: 10.1006/abio.1993.1449. [DOI] [PubMed] [Google Scholar]
- 34.Allen A C, Fluhr R. Plant Cell. 1997;9:1559–1572. doi: 10.1105/tpc.9.9.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willekens H, Chamnongpol S, Davey M, Schraudner M, Langebartels C, Van Montagu M, Inzé D, Van Camp W. EMBO J. 1997;16:4806–4816. doi: 10.1093/emboj/16.16.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hausladen A, Privalle C T, Keng T, DeAngelo J, Stamler J S. Cell. 1996;86:719–729. doi: 10.1016/s0092-8674(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 37.Neuenschwander U, Vernooij B, Friedrich L, Uknes S, Kessmann H, Ryals J. Plant J. 1995;8:227–233. doi: 10.1073/pnas.92.10.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janistyn B. Planta. 1983;159:382–385. doi: 10.1007/BF00393178. [DOI] [PubMed] [Google Scholar]
- 39.Pfeiffer S, Janystin B, Jessner G, Pichorner H, Ebermann R. Phytochemistry. 1994;36:259–262. [Google Scholar]
- 40.Galione A, McDougal A, Busa W B, Willmott N, Gillot I, Whitaker M. Science. 1993;261:348–352. doi: 10.1126/science.8392748. [DOI] [PubMed] [Google Scholar]
- 41.Medzhitov R, Preston-Hurlburt P, Janeway C A., Jr Nature (London) 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 42.Yang Y, Shah J, Klessig D F. Genes Dev. 1997;11:1621–1639. doi: 10.1101/gad.11.13.1621. [DOI] [PubMed] [Google Scholar]
- 43.Groom Q J, Torres M A, Fordham-Skelton A P, Hammond-Kosack K E, Robinson N J, Jones J D G. Plant J. 1996;10:515–522. doi: 10.1046/j.1365-313x.1996.10030515.x. [DOI] [PubMed] [Google Scholar]
- 44.Delledonne, M., Xia, Y., Dixon, R. A. & Lamb, C. (1998) Nature (London), in press. [DOI] [PubMed]
- 45.Green R, Fluhr R. Plant Cell. 1995;7:203–212. doi: 10.1105/tpc.7.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bi Y M, Kenton P, Mur L, Darby R, Draper J. Plant J. 1995;8:235–245. doi: 10.1046/j.1365-313x.1995.08020235.x. [DOI] [PubMed] [Google Scholar]
- 47.Chiatante D, Newton R P, Crignola S, Levi M, Brown E G. Phytochemistry. 1990;29:2815–2820. [Google Scholar]
- 48.McDonald L J, Murad F. Adv Pharmacol. 1995;34:263–276. doi: 10.1016/s1054-3589(08)61091-1. [DOI] [PubMed] [Google Scholar]
- 49.Leckie, C. P., McAinsh, M. R. & Hetherington, A. M. (1997) Plant Physiol. 114, Suppl., 272.
- 50.Wu Y, Kuzma J, Marechal E, Graeff R, Lee H C, Foster R, Chua N-H. Science. 1997;278:2126–2130. doi: 10.1126/science.278.5346.2126. [DOI] [PubMed] [Google Scholar]
- 51.Graeff R, Franco L, De Flora A, Lee H C. J Biol Chem. 1998;273:118–125. doi: 10.1074/jbc.273.1.118. [DOI] [PubMed] [Google Scholar]