

Abstract
Dystrophin-deficient muscles experience large reductions in expression of nitric oxide synthase (NOS), which suggests that NO deficiency may influence the dystrophic pathology. Because NO can function as an antiinflammatory and cytoprotective molecule, we propose that the loss of NOS from dystrophic muscle exacerbates muscle inflammation and fiber damage by inflammatory cells. Analysis of transgenic mdx mice that were null mutants for dystrophin, but expressed normal levels of NO in muscle, showed that the normalization of NO production caused large reductions in macrophage concentrations in the mdx muscle. Expression of the NOS transgene in mdx muscle also prevented the majority of muscle membrane injury that is detectable in vivo, and resulted in large decreases in serum creatine kinase concentrations. Furthermore, our data show that mdx muscle macrophages are cytolytic at concentrations that occur in dystrophic, NOS-deficient muscle, but are not cytolytic at concentrations that occur in dystrophic mice that express the NOS transgene in muscle. Finally, our data show that antibody depletions of macrophages from mdx mice cause significant reductions in muscle membrane injury. Together, these findings indicate that macrophages promote injury of dystrophin-deficient muscle, and the loss of normal levels of NO production by dystrophic muscle exacerbates inflammation and membrane injury in muscular dystrophy.
Keywords: muscle; muscular dystrophy; nitric oxide synthase; macrophages; inflammation
Introduction
Duchenne muscular dystrophy (DMD)* is one of the most prevalent and lethal inherited diseases of childhood. The deficient gene product, dystrophin (Hoffman et al., 1987), is a membrane-associated protein that provides a link in a chain of proteins between the actin cytoskeleton and the extracellular matrix (Ibraghimov-Beskrovnaya et al., 1992). The participation of dystrophin in this chain of proteins suggests that dystrophin serves a structural role in muscle cells. In addition, dystrophin deficiency in the mdx mouse model of DMD is associated with an increase in cell membrane lesions that can be detected by the presence of elevated creatine kinase (CK) levels in the serum, or by the presence of extracellular marker dyes within the muscle fibers (McArdle et al., 1994; Matsuda et al., 1995). These observations have provided the key evidence for the “mechanical defect hypothesis,” in which the loss of dystrophin from muscle is interpreted as causing muscle cell death by producing a cell membrane that more frequently experiences lethal, mechanical damage.
Part of the complexity in understanding the impact of dystrophin deficiency on muscle cell viability results from the secondary loss of the entire complex of proteins that interact with dystrophin at the muscle cell surface (Ervasti et al., 1990). Although the functions of most of these proteins remain obscure, neuronal nitric oxide synthase (nNOS) is a prominent member of the complex (Brenman et al., 1995) that has well-characterized functions in several tissues. In the absence of dystrophin, the concentration of nNOS at the cell membrane (Brenman et al., 1995) and in the cytoplasm (Chang et al., 1996) decreases tremendously, and the concentration of nNOS mRNA is also greatly diminished (Chang et al., 1996). Thus, it is feasible that the secondary loss of nNOS from muscle contributes significantly to the dystrophic pathology. Previous work has shown that mdx mice that are null mutants for nNOS show no detectable difference in muscle pathology compared with mdx mice which contain low levels of cytosolic nNOS (Chao et al., 1998; Crosbie et al., 1998). In addition, nNOS null mice that express dystrophin do not show muscular dystrophy (Chao et al., 1998; Crosbie et al., 1998), although that observation is not sufficient to conclude that nNOS deficiency is unimportant in dystrophinopathy. Null mutants for nNOS that express dystrophin and other members of the dystrophin complex would not have the primary defect of dystrophin deficiency that could then be exacerbated by the secondary loss of nNOS.
We propose that nNOS deficiency in dystrophic muscle could significantly promote muscle pathology if NO were to serve an antiinflammatory role in injured muscle. Dystrophinopathies are characterized by high concentrations of myeloid cells within the muscle (Arahata and Engel, 1984; McDouall et al., 1990; Gorospe et al., 1994; Cai et al., 2000). Although the possibility that these myeloid cells promote dystrophy has not been examined previously, they are known to be cytotoxic in other pathological conditions (Jansen et al., 1994; Jun et al., 1999). Furthermore, NO has been shown to inhibit the diapedesis, activation, and longevity of select myeloid cell populations (Clancy et al., 1992; Albina et al., 1993; Liu et al., 1998), so it is feasible that the loss of NO from dystrophic muscle would result in increased muscle inflammation. Finally, NO can scavenge cytolytic molecules that are generated by myeloid cells, so that a reduction in NO production could yield muscle that is more susceptible to lysis during inflammation.
Results
Normalization of NO production in mdx mice reduces muscle pathology in mdx mice
Western, Northern, and gene array analyses of NOS transgenic (Tg) muscles showed that there is an ∼250-fold increase in NOS mRNA and a 50-fold increase in NOS protein in muscles (Fig. 1, A and B). However, there is only an ∼0.2-fold increase in NO production by soleus muscle from NOS Tg mice compared with controls (Fig. 1 C). The relatively small increase in NO production relative to elevation of NOS protein in the NOS Tg muscles may reflect the increased expression of endogenous inhibitors of NOS. For example, gene array analysis and Western blots show a 10-fold increase in protein inhibitor of NOS in the NOS Tg muscles (unpublished data).
Figure 1.

Northern analysis of NOS Tg muscles. (A) Lanes 1–3 contained total RNA isolated from muscles that did not express the NOS transgene. Lane 4 was loaded with an equal mass of total RNA that was obtained from muscles of a NOS Tg mouse. The blot was probed with cDNA for full-length nNOS. The mass of the wild-type nNOS is greater than the transgenic nNOS, because the wild-type encodes an additional 34 amino acid nNOSμ peptide. (B) Western analysis of NOS Tg muscles. Muscle samples from littermates of NOS Tg mice that did not express the NOS transgene (three left lanes) and from NOS Tg mice (three right lanes). Densitometric analysis of the relative quantities of nNOS shows an ∼50-fold increase in nNOS in the NOS/Tg muscles. (C) NO release from control, NOS Tg, mdx, and NOS Tg/mdx. NO release from solei was measured for muscles obtained from C57 (white bar; n = 10), NOS Tg (black bar; n = 10), mdx (stippled bar; n = 8), and NOS Tg/mdx mice (striped; n = 8). Asterisk, differs from C57 at P < 0.05. Error bars indicate standard errors of the means in all figures.
NOS Tg mice were crossed with mdx mice to generate NOS Tg/mdx mice. Measurements of NO production by age-matched solei from C57 controls, mdx, or NOS Tg/mdx muscles showed that the decrease in NOS in mdx muscle resulted in a reduction of NO production by ∼80% (Fig. 1 C). However, the NOS Tg returned the levels of muscle NO production to values that did not differ significantly from controls (Fig. 1 C). Normalization of NO production in NOS Tg/mdx muscles resulted in amelioration of all of the key indices of histopathology in dystrophin-deficient muscle (Fig. 2). For example, 4 wk mdx mouse muscle contains high concentrations of macrophages, central-nucleated muscle fibers, and high variability in muscle fiber diameter (Figs. 2–4). However, normalized NO production in dystrophin-deficient mice yields a 60% decrease in central-nucleated fibers in 4-wk-old mice (Fig. 3). In addition, 4-wk-old NOS Tg/mdx mice showed a 50% reduction in the concentration of macrophages and cells expressing major histocompatibility complex class II (MHC-II) (Fig. 3). NOS Tg/mdx mice also show an >50% reduction in the pathological variability in fiber size seen in mdx muscle (Fig. 4 A). Although it is feasible that the presence of the transgene could diminish pathology through an effect that was independent of normalized NO production, the finding that NOS inhibition reversed the ameliorative effects of the transgene argues strongly against that possibility. The reduction in macrophage concentration, MHC-II+ cell concentration, central-nucleated fibers, and fiber injury (Figs. 3 and 4) that were observed in NOS Tg/mdx muscles did not occur if the NOS Tg/mdx mice received L-NAME treatments.
Figure 2.
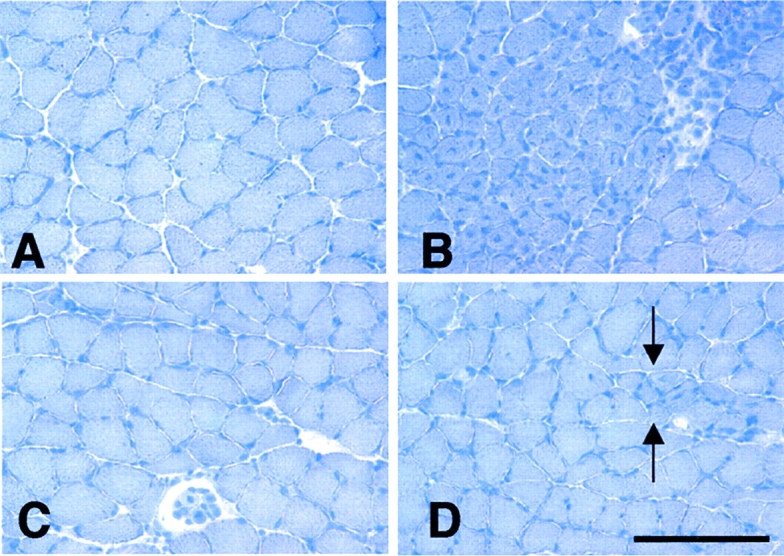
Sections of 4-wk-old soleus muscles stained by hematoxylin. (A) C57 muscle showing fibers of uniform diameter, no central nucleation, and no clusters of inflammatory cells between adjacent fibers. (B) Mdx muscle showing a typical focus of muscle pathology characterized by fiber populations of variable diameters and central nucleation. Dark staining nuclei of inflammatory cells appear between adjacent fibers. (C) NOS Tg/mdx muscle showing typical histology, where fiber diameter is more uniform than age-matched mdx muscle, and there is little inflammation or central nucleation. (D) NOS Tg/mdx muscle showing an example of the relatively small lesions that appear in NOS Tg/mdx muscle (between arrows) where there are small clusters of small-diameter, central-nucleated fibers. All micrographs are at the same magnification. Bar, 250 μm.
Figure 4.
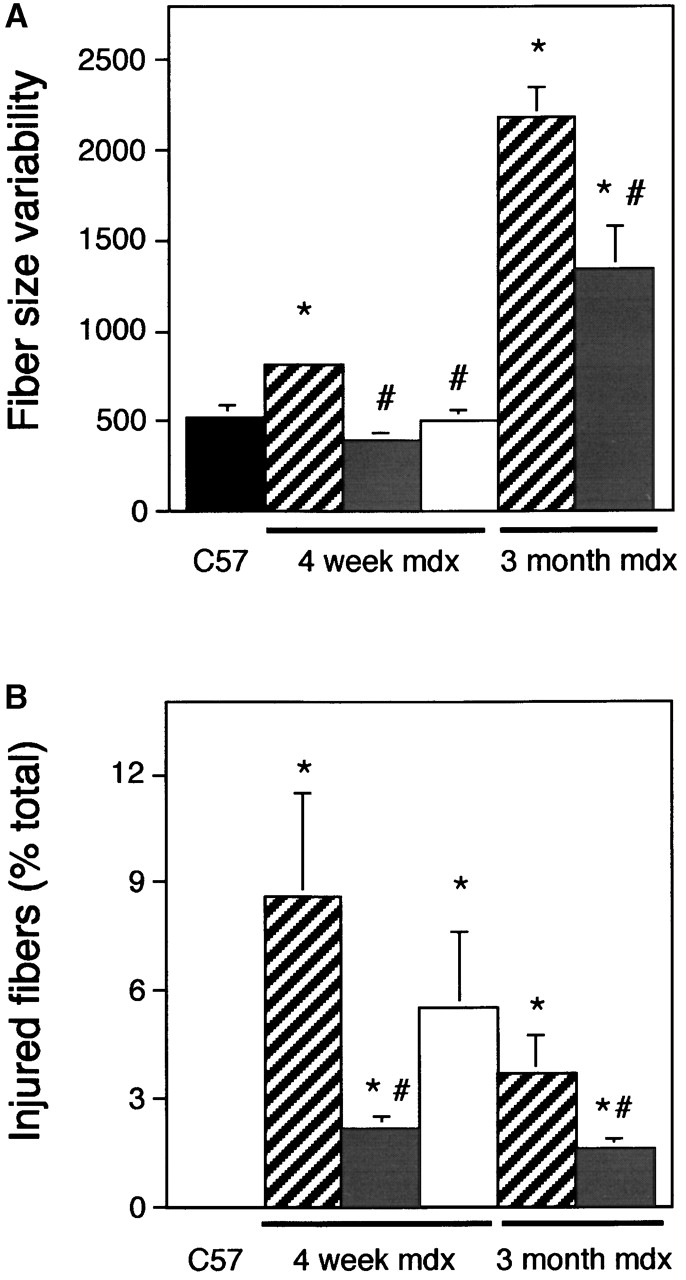
Expression of the NOS transgene reduces muscle fiber variability in size (A) and muscle fiber injury (B). Histogram shows variability in fiber diameter of 4-wk-old C57 (black bars; n = 10), 4-wk-old mdx (striped bars; n = 10), 4-wk-old NOS Tg/mdx (gray bars; n = 10), 4-wk-old, L-NAME–treated NOS Tg/mdx muscles (white bars; n = 6), 3-mo-old mdx (striped bars; n = 13), and 3-mo-old NOS Tg/mdx (grey bars; n = 5). (A) The data represent the mean of the standard deviation of fibers. (B) The data are the percentage of total fibers that contain procion red. Asterisk, differs from C57 (P < 0.05); pound sign, differs from age-matched mdx (P < 0.05).
Figure 3.
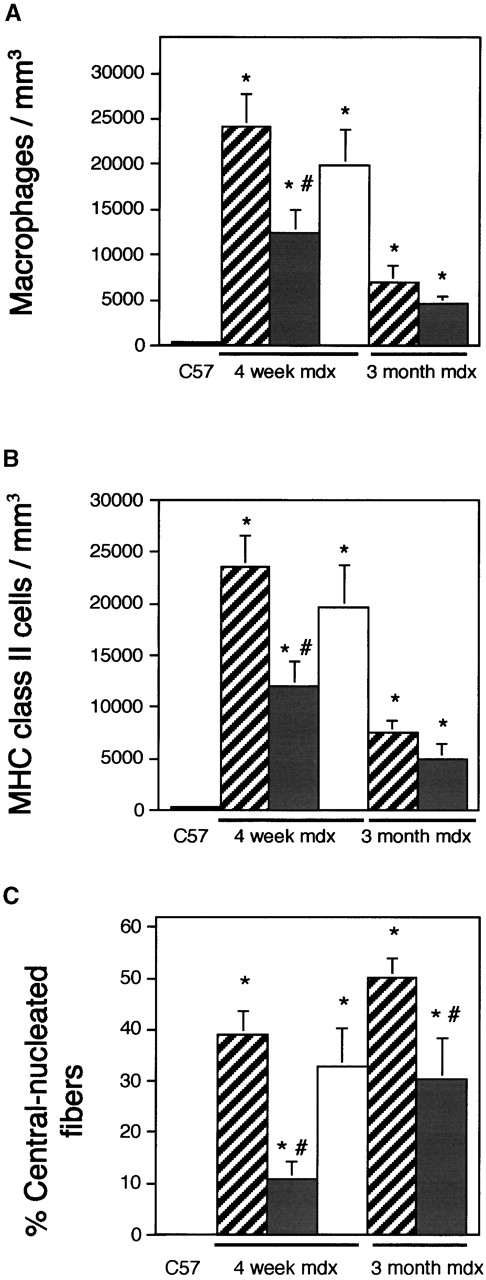
Expression of the NOS transgene reduces mdx muscle inflammation and regeneration. The concentrations of macrophages (A), MHC class II+ cells (B), and regenerative fibers (C) were measured in quadriceps from 4-wk-old C57 (black bars; n = 10), 4-wk-old mdx (striped bars; n = 10), 4-wk-old NOS Tg/mdx (gray bars; n = 10), 4-wk-old, L-NAME–treated NOS Tg/mdx muscles (white bars; n = 6), 3-mo-old mdx (striped bars; n = 13), and 3-mo-old, NOS Tg/mdx (grey bars; n = 5). Asterisk, differs from C57 (P < 0.05); pound sign, differs from age-matched mdx (P < 0.05).
We further tested whether the reduction of these indices of dystrophic pathology by the NOS transgene resulted from a delay in the onset of the pathology, rather than an ameliorization of the pathology, by evaluating soleus muscles from 3-mo-old NOS Tg/mdx mice and mdx mice that did not express the transgene. NOS Tg/mdx mice showed significantly less central nucleation at 3 mo of age than was observed in littermates that did not express the transgene (Fig. 3 C). This indicates that the cumulative muscle injury and regeneration experienced by the NOS Tg/mdx mice remains less than that which occurs in mdx mice, and also supports the interpretation that the transgene reduces the pathology rather than delays its onset.
Comparison of the concentrations of macrophages in 3-mo-old NOS Tg/mdx and in mdx mice showed that macrophages decline in concentration in both the transgenic and control mice between 1 and 3 mo of age (Fig. 3 A). A nearly identical change was seen in the concentrations of MHC-II–expressing cells (Fig. 3 B). These findings also indicate that expression of the transgene reduces dystrophinopathy, rather than delaying its onset.
Normalization of NO production in mdx mice reduces muscle membrane damage
NOS Tg/mdx mice showed a 70% decrease in the number of fibers possessing membrane lesions when compared with mdx mice (Figs. 4 B and 5). The proportion of injured fibers in NOS Tg/mdx mice remained significantly less than mdx mice at 3 mo of age, and fiber injury in NOS Tg/mdx muscles did not increase from 1 to 3 mo of age. Muscle membrane injury was also assessed by measuring serum CK concentration, which showed a similar 70% reduction in serum CK concentration in 4-wk-old NOS Tg/mdx compared with mdx (Fig. 6). However, both the NOS Tg/mdx and the mdx mice showed a trend for increased serum CK concentration at 3 mo of age and an increase in interanimal variability in serum CK concentration. The difference in CK concentration between the transgenic and control mdx was no longer significant at P < 0.05.
Figure 6.

Expression of the NOS transgene reduces the concentration of serum CK in mdx mice. 4-wk-old C57 (black bars; n = 5), 4-wk-old mdx (striped bars; n = 12), 4-wk-old NOS Tg/mdx (gray bars; n = 5), 4-wk-old, L-NAME–treated NOS Tg/mdx muscles (white bars; n = 6), 3-mo-old, mdx (striped bars; n = 13), and 3-mo-old NOS Tg/mdx (grey bars; n = 5). Asterisk, differs from C57 (P < 0.05); pound sign, differs from age-matched mdx (P < 0.05).
Normalization of NO production in mdx mice does not affect muscle vascularity
Vascularity of 4-wk-old NOS Tg/mdx mice and mdx mice that did not express the transgene was assessed by measuring capillary density and vascular volume fraction in soleus muscle. Both indices showed that there is no difference in the vascularity of mdx muscle that is caused by normalized NO production. Mdx mouse soleus muscles contained 6.96% of the total muscle volume in vascular luminal space (n = 10; SEM = 0.56), and 6.95% of NOS Tg/mdx soleus muscle volume consisted of vascular lumen (n = 10; SEM = 0.36). The capillary density in mdx soleus muscles was 110 capillaries/mm2 (n = 10; SEM = 10.8), and the capillary density in NOS Tg/mdx solei was 116 capillaries/mm2 (n = 10; SEM = 10.4).
Mdx muscle macrophages at pathophysiological concentrations lyse muscle cells
The observations reported above show that normalization of NO production in mdx muscle reduced muscle membrane lesions and inflammation. These reductions in inflammation and membrane damage did not occur in NOS Tg/mdx mice that were treated with the NOS inhibitor, L-NAME. This may indicate that NO functions as an antiinflammatory molecule, and that its normalization reduced macrophage-mediated damage to mdx fibers. Measurements show that macrophage concentrations can exceed 5 × 104 macrophages/mm3 muscle in 4 wk mdx soleus muscles (Fig. 7 A). We tested whether this concentration was sufficient to cause muscle membrane lysis in vitro by cytotoxicity assays using macrophages that were isolated from 4 wk mdx muscle. Greater than 40% of target myotubes were lysed within 16 h of coincubation with 5 × 104 mdx muscle macrophages/mm3, which indicates that these macrophages are potently cytolytic at this concentration (Fig. 7 B). However, 104 macrophages/mm3, which is the concentration observed in 4-wk-old NOS Tg/mdx muscles and in prenecrotic 2-wk-old mdx muscles, showed little cytotoxicity (Fig. 7 B).
Figure 7.
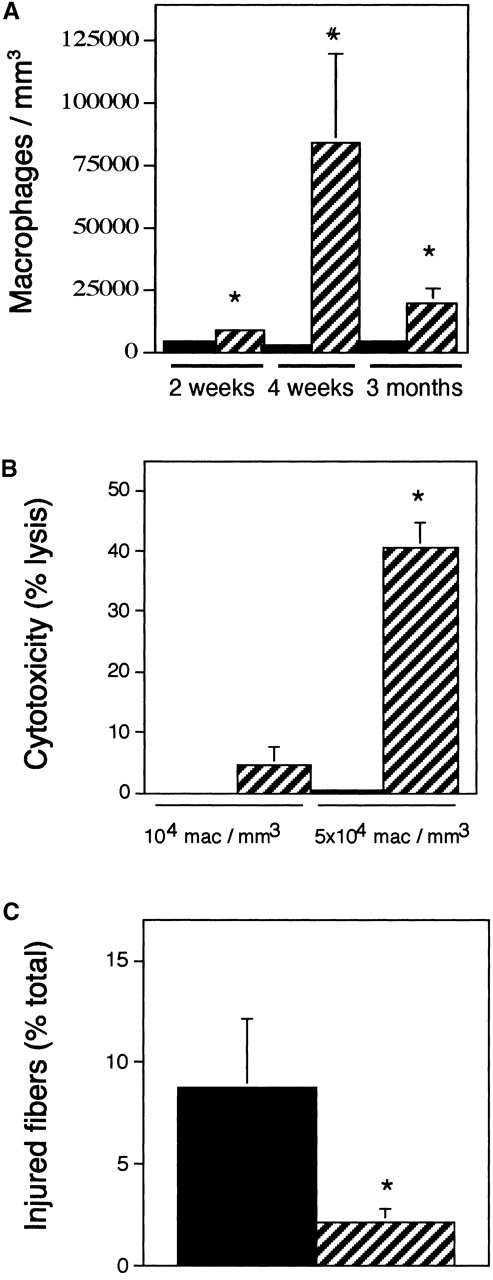
Macrophage concentration in mdx solei. (A) C57 (solid bars). Mdx (striped bars). Asterisk, differs from age-matched controls (P < 0.05). Pound sign, differs from all other data sets (P < 0.05). n = 14 for each data set. (B) Mdx muscle macrophages kill muscle cells in vitro. Values show the percentage lysis of C2C12 myotubes by macrophages isolated from 4-wk-old mdx muscles. Solid bars, nonactivated macrophages. Striped bars, PMA-activated macrophages. Asterisk, differs from C57 at P < 0.05. (C) Macrophage depletions reduce muscle membrane lesions. Data are the percentage of total muscle fibers in cross-sections of soleus muscle that contain intracellular procion red. Approximately 9% of the total fibers were injured and contained procion red in solei of mdx mice receiving intraperitoneal injections of PBS (black bar). Approximately 2% of the fibers of the fibers of mdx mice receiving intraperitoneal injections of anti-F4/80 showed intracellular procion red (striped bar). Asterisk, differs from PBS at P < 0.05.
We further tested whether mdx muscle macrophages lyse muscle membranes in vivo by depleting macrophages and assessing the occurrence of membrane lesions by entry of procion red into soleus muscle fibers. Muscles analyzed from macrophage-depleted mdx mice showed a >75% reduction in injured fiber concentration compared with nondepleted mdx mice (Figs. 7 C and 8).
Macrophage depletions from mdx muscles reduce the concentration of regenerative muscle fibers
We tested whether the reduction in muscle membrane injury in macrophage-depleted mdx mice resulted from increased muscle repair and regeneration by measuring the proportion of fibers in depleted and control mdx soleus muscles that contained central nuclei. PBS-injected mdx mice contained 31.2% central-nucleated fibers in soleus muscle (SE = 5.0; n = 6) and macrophage-depleted mice contained 9.9% (SE = 2.7; n = 8; differs from control at P < 0.002). This indicates that the cumulative muscle injury and regeneration experienced by macrophage-depleted mdx mice is less than control mdx mice, and that the proportion of central-nucleated fibers observed in 4-wk-old, macrophage-depleted mice is similar to the proportion in NOS Tg/mdx mice. This eliminates the possibility that either the normalization of NO production or macrophage depletion decreases mdx pathology by promoting regeneration.
Discussion
Mechanical damage to the cell membrane of dystrophin-deficient muscle may disrupt normal muscle homeostasis and lead to muscle cell pathology (Petrof et al., 1993). Membrane damage can also permit the unregulated transit of extracellular calcium into the cytoplasm that can increase activation of calcium-dependent proteases, which may promote muscle mass loss and muscle destruction (Spencer et al., 1995). The central importance of membrane lesions in the progression of dystrophinopathy has provided the foundation for studies directed toward increasing the expression of other proteins that may be able to recover membrane stability and prevent muscle damage in the absence of dystrophin. For example, elevating the expression of the dystrophin homologue utrophin (Tinsley et al., 1996) or dystrophin complex analogue α7-integrin (Burkin et al., 2001) have both been effective at preventing muscle membrane injury in dystrophin-deficient mice and reducing pathology.
Our findings show that nonmechanical factors contribute importantly to membrane damage in dystrophinopathy. Normalization of NO production reduced the occurrence of many of the key indices of pathology that are characteristic of dystrophinopathy, such as central nucleation, inflammation, and fiber membrane damage. These observations are consistent with the view that muscle membrane lesions contribute to dystrophic muscle pathology. However, these findings also indicate that a large proportion of the lesions are the consequence of nNOS deficiency, rather than the direct result of mechanical damage.
Findings presented here support the conclusion that the protective effect of normalized NO production in dystrophin-deficient muscle is attributable to antiinflammatory actions of NO. Most importantly, the large reduction of inflammation of NOS Tg/mdx muscle supports this interpretation. Previous investigations conducted in vitro have shown potential mechanisms through which NO could inhibit inflammation. For example, NO can reduce diapedesis of myeloid cells by inhibition of P-selectin and ICAM expression (Liu et al., 1998), and NO can induce leukocyte apoptosis (Albina et al., 1993). Furthermore, NO can inhibit target cell cytolysis by myeloid cells by inhibiting the expression or activity of enzymes that generate cytolytic free radicals (Clancy et al., 1992; Abu-Soud and Hazen, 2000), and by scavenging free radicals that are generated by inflammatory cells (Miles et al., 1996). These antiinflammatory and free radical–scavenging functions of NO may be especially important in regulating the mdx pathology, because dystrophin-deficient muscle cells are more sensitive than control muscle to cytolysis that is induced by free radicals (Rando et al., 1998). The present findings, which show that macrophage depletion yields protection against muscle membrane lesions that is similar to that produced by normalization of NO production, supports the interpretation that normalized NO production protects dystrophic muscle from macrophage-mediated damage.
Inflammation in muscular dystrophy has been previously considered to be an inconsequential response to muscle injury, rather than a contributor to the pathological progression. However, the long-recognized benefits of some immunosuppressants in slowing the progress of dystrophinopathy have suggested that immune cells may contribute to the pathology (Drachman et al., 1974). Our findings, which show that macrophages that are isolated from mdx muscle are capable of lysing muscle cells in vitro, support the possibility that they may be directly cytotoxic in dystrophic muscle. The decrease in mdx muscle membrane damage after antibody depletions of macrophages is also consistent with a cytotoxic role of macrophages in mdx dystrophy. However, macrophages may also mediate cytotoxicity through less direct routes in mdx dystrophy if they were to activate other cytotoxic cells. Previous work has shown that depletion of CD8+ T cells (Spencer et al., 1997) or CD4+ T cells (Spencer et al., 2001) from mdx mice before the peak of pathology causes a reduction in histologically discernible pathology in dystrophic muscles. It is possible that mdx muscle macrophages activate autoaggressive T cells in mdx muscle, and thereby promote cytotoxicity indirectly.
Previous investigations have indicated that NOS deficiency in dystrophic muscle could exacerbate muscle pathology by disrupting normal regulation of blood flow, or by influencing angiogenesis. For example, mdx mice and DMD boys have an impaired ability to inhibit α-adrenergic–stimulated vasoconstriction (Thomas et al., 1998; Sander et al., 2000). However, both peripheral blood flow and vascular conductance in resting and contracting muscles were shown to be unaffected by the decreased concentration of nNOS in mdx muscles in vivo (Thomas et al., 1998). Those findings indicate that muscle nNOS would be important in modulating muscle blood flow, primarily in animals in which vasoconstriction was caused by increased sympathetic outflow. Whether nNOS-derived NO can induce vasodilation in vivo through mechanisms that do not involve inhibition of α-adrenergic–stimulated vasoconstriction has not been demonstrated. However, excised muscle subjected to electrical stimulation does experience vasodilation via the cGMP cascade (Lau et al., 2000; Grange et al., 2001). Although we were aware of no environmental stimuli that would have caused increased sympathetic outflow in the animals used in the present investigation, it is not possible to completely exclude that possibility.
Other reports have also shown that NO can either promote (Ziche et al., 1994; Montrucchio et al., 1997) or inhibit (Powell et al., 2000) angiogenesis, which could provide yet another mechanism through which normalization of NO production by dystrophic muscle could affect tissue pathology. However, our results show that the presence of the NOS transgene in mdx muscle did not affect capillary density or the volume fraction of the muscle that consisted of vascular structures. Thus, the effects of normalized NO production on dystrophinopathy are not attributable to an NO influence on angiogenesis.
Several previous observations have suggested the possibility that reductions in muscle NO production could affect muscle differentiation and regeneration (Lee et al., 1994; Anderson, 2000) and thereby influence the pathology of dystrophin-deficient muscle. For example, inhibition of muscle NOS activity significantly reduces the capacity of mononucleated muscle cells to fuse and differentiate into multinucleated myotubes in vitro (Lee et al., 1994). Thus, it is feasible that normalization of NO production in dystrophic muscle could reduce muscle pathology by promoting regeneration, rather than by decreasing damage. However, we have found that the NOS transgene greatly reduces the proportion of mdx muscle fibers that contain central nuclei, which are persistent indicators of fibers that have been damaged and experienced regeneration or repair. The reduction in regenerative fibers indicates that normalized NO production ameliorates the dystrophic pathology by reducing the initial damage to dystrophic muscle, rather than increasing regeneration.
We propose a model for the pathophysiology of dystrophin-deficient muscle in which nNOS deficiency and myeloid cells contribute significantly to the progression of the disease. In this model, initial muscle injury is a direct consequence of mechanical damage resulting from the absence of the membrane-stabilizing effects of dystrophin. However, muscle inflammation occurs as a secondary consequence of the mechanical injury, and inflammation may be increased by the reduction of NO-mediated inhibition of myeloid cell diapedesis. Activated myeloid cells, especially macrophages, then release cytolytic molecules from which healthy muscle can be protected by NO, but from which the cytoprotective effects of dystrophin-deficient muscle are diminished. This model suggests that further examination of the use of antiinflammatory agents to treat dystrophinopathies could yield productive, new therapeutic approaches.
Materials and methods
Measurement of macrophage concentration
Mdx mice were killed at 2 wk, 4 wk, or 3 mo of age to represent prenecrotic, peak necrotic, and regenerative stages of the mdx pathology. Muscles were frozen, sectioned, and immunolabeled for macrophages (anti-F4/80) or MHC-II (PharMingen). The concentrations of F4/80+ cells and MHC-II+ cells were determined by histomorphometry (Spencer et al., 2001).
Cytotoxicity assays
Muscles from 4-wk-old mdx mice were minced into 2.5 mg/ml collagenase (Sigma-Aldrich) in DME and incubated at 37°C for 40 min. The tissue was transferred to 0.1% trypsin for 30 min, centrifuged, and the cells were resuspended in 10% FBS in DME. Digested tissue was filtered though a 70-μm filter and centrifuged. The cells were resuspended in HBSS, overlaid on Histopaque 1077 (Sigma-Aldrich), and then centrifuged at 400 g for 45 min at 4°C. Macrophages were collected at the HBSS–Histopaque interface. Some cells were then adhered to slides by centrifugation with a Cytospin (Shandon), and stained with anti-F4/80 to confirm that they were macrophages. The remaining cells were used for cytotoxicity assays.
C2C12 cells that were used for cytotoxicity assays were plated in 96-well plates in 10% FBS in DME until confluent, and then placed in serum-free media overnight to promote fusion. The cells were returned to 10% FBS for 2 d, and then subjected to serum withdrawal overnight to further promote fusion. They were then returned to complete media for ∼4 d before use in cytotoxicity assays.
Myotubes were incubated in HBSS containing 51Cr and 0.25% FBS for 2 h and washed in HBSS before use in cytotoxicity assays. Myotubes were cocultured with macrophages in HBSS containing 0.25% FBS, 400 μM arginine, and 0.6 μM PMA for 16 h, after which the media was assayed for 51Cr release by scintillation counting. Macrophages were added at concentrations that occur in 4 wk mdx soleus muscles, as determined above. Cytotoxicity was expressed as a percentage of total lysis by setting 0% as 51Cr released spontaneously by myotubes incubated identically, except without macrophages. 100% cytotoxicity was set at the 51Cr release into the media by myotubes incubated with 0.1% Triton X-100 in HBSS.
Macrophage depletions from mdx mice
Circulating macrophage populations were depleted from mdx mice by repeated intraperitoneal injections of anti-F4/80. Antibody from F4/80 hybridoma culture supernatant was isolated by ammonium sulfate precipitation. Precipitated IgG was then resuspended in sterile PBS, and IgG concentration determined by ELISA assay. Mice undergoing macrophage depletion received 65 μg IgG by intraperitoneal injection in five daily injections per week, beginning at 1 wk of age and continuing to 4 wk of age. Control animals received injections of the same volume of PBS on the same schedule. For some animals, serum was assayed for macrophage concentration in cytospin preparations. Macrophage-depleted animals showed at least a 90% reduction in circulating macrophage concentrations. Soleus muscles were removed from 4-wk-old animals and used to assess the presence of membrane lesions or muscle fiber regeneration, as described below.
Production of NOS Tg mice
Rat brain nNOS in pCMV5 (provided by Dr. James T. Stull, University of Texas, Southwestern, Dallas, TX) was cloned downstream of the 2.2-kb human skeletal actin promoter and the vp1 intron (provided by Dr. Jeffrey Chamberlain, University of Washington, Seattle, WA). The NOS cDNA was followed by the SV-40 large T poly-A site. The ClaI-DraI–digested DNA was injected to the pronucleus of F2 hybrid zygotes of C57 Bl6 C3H parents. Positive transgenic mice were identified by Southern and PCR analysis for human skeletal actin promoter. Overexpression of the transgene was confirmed by Northern and Western blotting and by gene array analysis.
Production of NOS Tg/mdx mice
NOS Tg males and mdx females were paired to produce females that were heterozygous for the dystrophin mutation and males that were hemizygous for dystrophin deficiency. Thus, all male offspring were dystrophic. Dystrophin null mutation was confirmed by ARMS PCR (Amalfitano and Chamberlain, 1996). Male offspring that expressed the transgene were identified by isolating genomic DNA from tail tissue that was digested with proteinase K at 55°C overnight. F1 males that did not express the transgene were used as dystrophic controls.
Inhibition of NOS in vivo
We assessed whether the effects of the presence of the NOS transgene on mdx muscle pathology were attributable to normalized NO production by administering NOS inhibitor to NOS Tg/mdx mice. NOS Tg/mdx mice were supplied with 0.5 mg/ml of N-nitro-L-arginine methyl ester (L-NAME) in their drinking water from 3 to 4 wk of age. Muscles and sera were collected from the mice at 4 wk of age for analysis of muscle fiber damage, muscle inflammation, and muscle fiber regeneration, as described below.
Assay of NO release
NO release by unstimulated soleus muscles was measured as described previously (Tidball et al., 1998).
Assessment of muscle membrane damage
The occurrence of muscle membrane lesions was assessed by testing for the presence of an extracellular marker dye in the cytoplasm of muscle fibers. Soleus muscles were held at rest length and incubated in 0.2% procion red in Kreb's Ringer solution for 1 h and then washed, frozen, cross-sectioned, and viewed by fluorescent microscopy to identify injured fibers. Muscle injury was also tested by measuring the concentration of muscle CK in serum collected by cardiac puncture. CK assays were performed as described by the manufacturer of the assay kit (Sigma-Aldrich).
Measurement of muscle fiber size variability
Variability of muscle fiber cross-sectional area was evaluated because high variability of fiber size is a pathological feature of the mdx phenotype (Bulfield et al., 1984). Fiber cross-sectional area was measured for every fiber in each section using a digitized imaging system (Bioquant). Variability in cross-sectional areas between samples was expressed as the mean of the standard deviations for each population.
Assessment of muscle fiber regeneration
Previous observations have indicated that NO may affect muscle differentiation and regeneration (Lee et al., 1994; Anderson, 2000). We examined the effect of the NOS transgene on muscle fiber regeneration by determining the proportion of soleus muscle fibers that were central-nucleated. Muscle fibers that have experienced injury, repair, and regeneration contain centrally located myonuclei which provide a persistent morphological indicator of fibers that have experienced damage (Carnwath and Shotton, 1987). The percentage of the total muscle fibers containing central nuclei that were present in complete cross-sections of entire soleus muscles was determined microscopically in 4-wk-old and 3-mo-old NOS Tg/mdx and control mice.
Measurements of muscle vascularity
NO has been shown previously to affect vascularization of tissues (Ziche et al., 1994; Montrucchio et al., 1997), which could feasibly affect the time course or severity of the dystrophic pathology. We measured the capillary density in soleus muscles of 4-wk-old NOS Tg/mdx mice and in their littermates that did not express the transgene using techniques described previously (Carry et al., 1986). Because measurements of capillary density indicates the number of capillaries, but does not reflect possible differences in the size of blood vessels, we also measured the percent volume of the muscles that were comprised of vascular structures. Vascular percent volume was measured in frozen cross-sections of entire soleus muscles that were immunolabeled with rat anti–mouse CD31 (PharMingen). CD31 is expressed constitutively by vascular endothelial cells, so that vascular structures could be identified unambiguously by anti-CD31 binding. The luminal area of all vascular structures in each muscle cross-section was measured microscopically (Bioquant) and the total cross-sectional area of each section was determined so that vascular percent volume for each muscle could be calculated.
Gene array analysis
All hindlimb muscles from three NOS Tg mice and three non-Tg littermates were collected and pooled into NOS Tg and control samples. RNA was isolated as described previously (Chomczynski and Sacchi, 1987). Total RNA was reverse transcribed using a SuperScript system (GIBCO BRL) with an oligo-dT primer. The cDNA was then used for in vitro transcription to generate biotinylated cRNA using an ENZO bioarray RNA transcript labeling kit (Affymetrix). 15 μg of cRNA from each sample was subjected to random cleavage, after which cRNA samples were hybridized for 16 h at 45°C to an Affymetrix microarray (EukGE-WS2 high density array; murine U74A gene chip). The microarray was washed with buffer before reacting biotinylated bound cRNA with streptavidin-conjugated phycoerythrin. The fluorescent signal corresponding to cRNA bound to each known cDNA in the microarray was measured using a gene array scanner.
Figure 5.

Expression of the NOS transgene reduces muscle membrane lesions. Solei of C57 mice showed procion red almost exclusively located extracellularly (A), indicating infrequent occurrence of membrane lesions. Mdx muscles (B) showed that ∼8% of the fibers were injured and contained procion red. NOS Tg/mdx muscles (C and D) showed a frequency of procion red–containing injured fibers that was reduced to levels nearing C57 controls. Bar, 100 μm.
Figure 8.

Macrophage depletions reduce mdx muscle fiber injury. Soleus muscles from 4-wk-old mdx mice showed a frequent occurrence of fibers with intracellular procion red, indicating the presence of membrane lesions (A). Depletion of circulating macrophages by anti-F4/80 injections decreased the occurrence of membrane lesions (B). Bar, 100 μm.
Acknowledgments
This investigation was supported by Michael's Miracle Makers, the Muscular Dystrophy Association, and the National Institutes of Health.
Footnotes
Abbreviations used in this paper: CK, creatine kinase; DMD, Duchenne muscular dystrophy; MHC-II, major histocompatibility complex class II; nNOS, neuronal NOS; NO, nitric oxide; NOS, NO synthase; Tg, transgenic.
References
- Abu-Soud, H.M., and S.L. Hazen. 2000. Nitric oxide modulates the catalytic activity of myeloperoxidase. J. Biol. Chem. 275:5425–5430. [DOI] [PubMed] [Google Scholar]
- Albina, J.E., S. Cui, R.B. Mateo, and J.S. Reichner. 1993. Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J. Immunol. 150:5080–5085. [PubMed] [Google Scholar]
- Amalfitano, A., and J.S. Chamberlain. 1996. The mdx-amplification-resistant mutation system assay, a simple and rapid polymerase chain reaction-based detection of the allele. Muscle Nerve. 19:1549–1553. [DOI] [PubMed] [Google Scholar]
- Anderson, J.E. 2000. A role for nitric oxide in muscle repair: nitric oxide-mediated activation of muscle satellite cells. Mol. Biol. Cell. 11:1859–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arahata, K., and A.G. Engel. 1984. Monoclonal antibody analysis of mononuclear cells in myopathies. I: Quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann. Neurol. 16:193–208. [DOI] [PubMed] [Google Scholar]
- Brenman, J.E., D.S. Chao, H. Xia, K. Aldape, and D.S. Bredt. 1995. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 82:743–752. [DOI] [PubMed] [Google Scholar]
- Bulfield, G., W.G. Siller, P.A.L. Wight, and K.J. Moore. 1984. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. USA. 81:1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin, D.J., G.Q. Wallace, K.J. Nicol, D.J. Kaufman, and S.J. Kaufman. 2001. Enhanced expression of the α7β1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol. 152:1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, B., M.J. Spencer, G. Nakamura, L. Tseng-Ong, and J.G. Tidball. 2000. Eosinophilia of dystrophin-deficient muscle is promoted by perforin-mediated cytotoxicity by T cell effectors. Am. J. Pathol. 156:1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnwath, J.W., and D.M. Shotton. 1987. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci. 80:39–54. [DOI] [PubMed] [Google Scholar]
- Carry, M.R., S.P. Ringel, and J.M. Starcevich. 1986. Distribution of capillaries in normal and diseased human skeletal muscle. Muscle Nerve. 9:445–454. [DOI] [PubMed] [Google Scholar]
- Chao, D.S., F. Silvagno, and D.S. Bredt. 1998. Muscular dystrophy in mdx mice despite lack of neuronal nitric oxide synthase. J. Neurochem. 71:784–789. [DOI] [PubMed] [Google Scholar]
- Chang, W.J., S.T. Iannaccone, K.S. Lau, B.S. Masters, T.J. McCabe, K. McMillan, R.C. Padre, M.J. Spencer, J.G. Tidball, and J.T. Stull. 1996. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA. 93:9142–9147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform-extraction. Anal. Biochem. 162:156–159. [DOI] [PubMed] [Google Scholar]
- Clancy, R.M., J. Leszczynska-Piziak, and S.B. Abramson. 1992. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J. Clin. Invest. 90:1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosbie, R.H., V. Straub, H.Y. Young, J.C. Lee, J.A. Rafael, J.S. Chamberlain, V.L. Dawson, T.M. Dawson, and K.P. Campbell. 1998. Mdx muscle pathology is independent of nNOS perturbation. Hum. Mol. Genet. 7:823–829. [DOI] [PubMed] [Google Scholar]
- Drachman, D.B., K.V. Toyka, and E. Myer. 1974. Prednisone in Duchenne muscular dystrophy. Lancet. 2:1409–1412. [DOI] [PubMed] [Google Scholar]
- Ervasti, J.M., K. Ohlendieck, S.D. Kahl, M.G. Gaver, and K.P. Campbell. 1990. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 345:315–319. [DOI] [PubMed] [Google Scholar]
- Gorospe, J.R.M., M.D. Tharp, J. Hinckley, J.N. Kornegay, and E.P. Hoffman. 1994. A role for mast cells in the progression of Duchenne muscular dystrophy? J. Neurol. Sci. 122:44–56. [DOI] [PubMed] [Google Scholar]
- Grange, R.W., E. Isotani, K.S. Lau, K.E. Kamm, P.L. Huang, and J.T. Stull. 2001. Nitric oxide contributes to vascular smooth muscle relaxation in contracting fast-twitch muscles Physiol Genomics. 5:35–44. [DOI] [PubMed] [Google Scholar]
- Hoffman, E.P., R.H. Brown, and L.M. Kunkel. 1987. Dystrophin, the protein product of the Duchenne muscular dystrophy locus. Cell. 51:919–928. [DOI] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya, O., J.M. Ervasti, C.J. Leveille, C.A. Slaughter, S.W. Sernett, and K.P. Campbell. 1992. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 355:696–702. [DOI] [PubMed] [Google Scholar]
- Jansen, A., F. Homo-Delarche, H. Hooijkaas, P.J. Leenen, M. Dardenne, and H.A. Drexhage. 1994. Immunohistochemical characterization of monocyte-macrophages and dendritic cells involved in the initiation of insulitis and beta-cell destruction in NOD mice. Diabetes. 43:667–675. [DOI] [PubMed] [Google Scholar]
- Jun, H., C.S. Yoon, L. Zbytnuik, N. van Rooijen, and J.W. Yoon. 1999. The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Exp. Med. 189:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, K.S. R.W. Grange, E. Isotani, I.H. Sarelius, K.E. Kamm, P.L. Huang, and J.T. Stull. 2000. nNOS and eNOS modulate cGMP formation and vascular response in contracting fast-twitch skeletal muscle. Physiol. Genomics. 2:21–27. [DOI] [PubMed] [Google Scholar]
- Lee, K.H., M.Y. Baek, K.Y. Moon, W.K. Song, C.H. Chung, D.B. Ha, and M.S. Sung. 1994. Nitric oxide as a messenger molecule for myoblast fusion. J. Biol. Chem. 269:14371–14374. [PubMed] [Google Scholar]
- Liu, P., B. Xu, C.E. Hock, R. Nagele, F.F. Sun, and P.Y. Wong. 1998. NO modulates P-selectin and ICAM-1 mRNA expression and hemodynamic alterations in hepatic I/R. Am. J. Physiol. 275:H2191–H2198. [DOI] [PubMed] [Google Scholar]
- Matsuda, R., A. Nishikawa, and H. Tanaka. 1995. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J. Biochem. 118:959–964. [DOI] [PubMed] [Google Scholar]
- McArdle, A., R.H.T. Edwards, and M.J. Jackson. 1994. Time course of changes in plasma membrane permeability in the dystrophin-deficient mdx mouse. Muscle Nerve. 17:1378–1384. [DOI] [PubMed] [Google Scholar]
- McDouall, R.M., M.J. Dunn, and V. Dubowitz. 1990. Nature of the mononuclear infiltrate and the mechanism of muscle damage in juvenile dermatomyositis and Duchenne muscular dystrophy. J. Neurol. Sci. 99:199–207. [DOI] [PubMed] [Google Scholar]
- Miles, A.M., D.S. Bohle, P.A. Glassbrenner, B. Hansert, D.A. Wink, and M.B. Grisham. 1996. Modulation of superoxide-dependent oxidation and hydroxylation reactions by nitric oxide. J. Biol. Chem. 271:40–47. [DOI] [PubMed] [Google Scholar]
- Montrucchio, G., E. Lupia, A. de Martino, E. Battaglia, M. Arese, A. Tizzani, F. Bussolino, and B. Camussi. 1997. Nitric oxide mediates angiogenesis induced in vivo by platelet-activating factor and tumor necrosis factor-alpha. Am. J. Pathol. 151:557–563. [PMC free article] [PubMed] [Google Scholar]
- Petrof, B.J., J.B. Shrager, H.H. Stedman, A.M. Kelly, and H.L. Sweeney. 1993. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA. 90:3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell, J.A., S.N. Mohamed, J.S. Kerr, and S.A. Mousa. 2000. Antiangiogenesis efficacy of nitric oxide donors. J. Cell. Biochem. 80:104–114. [DOI] [PubMed] [Google Scholar]
- Rando, T.A., M.H. Disatnik, Y. Yu, and A. Franco. 1998. Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul. Disord. 8:14–21. [DOI] [PubMed] [Google Scholar]
- Sander, M., B. Chavoshan, S.A. Harris, S.T. Iannoccone, J.T. Stull, G.D. Thomas, and R.G. Victor. 2000. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA. 97:13818–13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, M.J., D.E. Croall, and J.G. Tidball. 1995. Calpains are activated in necrotic fibers from mdx dystrophic mice. J. Biol. Chem. 270:10909–10914. [DOI] [PubMed] [Google Scholar]
- Spencer, M.J., C.M. Walsh, K.A. Dorshkind, E.M. Rodriguez, and J.G. Tidball. 1997. Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J. Clin. Invest. 99:2745–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer, M.J., E. Montecino-Rodriguez, K. Dorshkind, and J.G. Tidball. 2001. Helper (CD4+) and cytotoxic (CD8+) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol. 98:235–243. [DOI] [PubMed] [Google Scholar]
- Thomas, G.D., M. Sander, K.S. Lau, P.L. Huang, J.T. Stull, and R.G. Victor. 1998. Impaired metabolic modulation of α-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc. Natl. Acad. Sci. USA. 95:15090–15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball, J.G., E. Lavergne, K.S. Lau, M.J. Spencer, J.T. Stull, and M. Wehling. 1998. Mechanical loading regulates NOS expression and activity in developing and adult skeletal muscle. Am. J. Physiol. 275:C260–C266. [DOI] [PubMed] [Google Scholar]
- Tinsley, J.M., A.C. Potter, S.R. Phelps, R. Fisher, J.I. Trickett, and K.E. Davies. 1996. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 384:349–353. [DOI] [PubMed] [Google Scholar]
- Ziche, M., L. Morbidelli, E. Masini, S. Amerini, H.J. Granger, C.A. Maggi, P. Geppetti, and F. Ledda. 1994. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration invitro promoted by substance P. J. Clin. Invest. 94:2036–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]