

Abstract
Caspase-3 is a cysteine protease located in both the cytoplasm and mitochondrial intermembrane space that is a central effector of many apoptotic pathways. In resting cells, a subset of caspase-3 zymogens is S-nitrosylated at the active site cysteine, inhibiting enzyme activity. During Fas-induced apoptosis, caspases are denitrosylated, allowing the catalytic site to function. In the current studies, we sought to identify the subpopulation of caspases that is regulated by S-nitrosylation. We report that the majority of mitochondrial, but not cytoplasmic, caspase-3 zymogens contain this inhibitory modification. In addition, the majority of mitochondrial caspase-9 is S-nitrosylated. These studies suggest that S-nitrosylation plays an important role in regulating mitochondrial caspase function and that the S-nitrosylation state of a given protein depends on its subcellular localization.
Keywords: nitric oxide; caspase-3; caspase-9; mitochondria; S-nitrosylation
Introduction
Apoptosis is a tightly regulated cell death program that removes excess or unwanted cells from organisms. Dysregulated apoptosis may be involved in the pathogenesis of diseases such as cancer, neurodegeneration, and autoimmunity (Thompson, 1995). The apoptotic program is executed by the caspase family of cysteine proteases that are expressed as inactive zymogens in cells and cleaved to form active tetrameric enzymes. Initiator caspases (e.g., caspases-8, -9, and -10) activate downstream effector caspases (e.g., caspases-3, -6, and -7), which in turn cleave specific cellular targets, resulting in cell death (Thornberry and Lazebnik, 1998).
Mitochondria serve as a central integrator of many apoptotic pathways. Several proapoptotic molecules are stored in the mitochondrial intermembrane space including apoptosis inhibitory factor (Susin et al., 1996, 1999b), cytochrome c (Li et al., 1997b), and a subset of caspase-2, -3, and -9 zymogens (Mancini et al., 1998; Krajewski et al., 1999; Susin et al., 1999a). When mitochondria receive an apoptotic signal, these proteins are released into the cytoplasm, triggering the cell suicide program. The percentage of caspase zymogens found in mitochondria is variable. In rat heart and brain, 90% of caspase-9 zymogens are mitochondrial (Krajewski et al., 1999), whereas only 10% of caspase-3 zymogens are found in mitochondria in HeLa cells (Mancini et al., 1998). Since caspases are activated in a cascade fashion, activation and release of a small pool of mitochondrial caspases may activate a much larger pool of cytoplasmic caspases. In addition, sequestering caspases in mitochondria may prevent inappropriate apoptosis by removing the proteases from cytoplasmic targets.
Apoptosis is also regulated by intracellular nitric oxide (NO)* production. NO can be either pro- or antiapoptotic. The proapoptotic effects of NO may be mediated by DNA damage, leading to p53 activation (Messmer and Brune, 1996), proteasome inhibition (Glockzin et al., 1999), and/or cytochrome c release from mitochondria, resulting from activation of the mitochondrial permeability transition pore (Messmer et al., 1996; Balakirev et al., 1997; Hortelano et al., 1997) or damage of mitochondrial membrane phospholipids (Ushmorov et al., 1999). NO is thought to exert its antiapoptotic effects through upregulation of protective proteins such as heat shock protein 70 (Kim et al., 1997a), heme oxygenase (Kim et al., 1995), and Bcl-2 (Genaro et al., 1995; Suschek et al., 1999): an increase in cGMP levels (Kim et al., 1997a,b), a decrease in ceramide levels (De Nadai et al., 2000), and/or S-nitrosylation of a critical cysteine residue expressed in the catalytic site of all caspase members (Dimmeler et al., 1997; Kim et al., 1997b, 2000; Li et al., 1997a; Mannick et al., 1999; Rossig et al., 1999).
We reported previously that a subset of caspase-3 zymogens is inhibited by S-nitrosylation of the catalytic site cysteine in unstimulated human lymphocyte cell lines. Upon activation of the Fas apoptotic pathway, the zymogens are denitrosylated, allowing the enzyme to function (Mannick et al., 1999). The studies did not identify the subpopulation of caspase-3 that is regulated by S-nitrosylation and did not analyze endogenous S-nitrosylation of other caspase zymogens. In the current studies, we determined whether mitochondrial caspase-3 is the subpopulation regulated by S-nitrosylation and whether caspase-9 zymogens also are endogenously S-nitrosylated.
Results and discussion
The majority of mitochondrial but not cytoplasmic caspase-3 is S-nitrosylated
Mitochondrial and cytoplasmic cellular fractions were isolated from a human B cell line (10C9) using differential centrifugation. The purity of the subcellular fractions was confirmed by superoxide dismutase (SOD1) (cytoplasm), cytochrome c (mitochondrial intermembrane space), and cytochrome oxidase (mitochondrial matrix) immunoblot analysis (Fig. 1) . Caspase-3 or control proteins were immunoprecipitated from the mitochondrial and cytoplasmic fractions using a caspase-3–specific monoclonal antibody or equal concentrations of an isotype-matched control antibody. Caspase-3 was immunoprecipitated efficiently with its specific antibody but not with control antibody (Fig. 2 A). Silver stains indicated that associated proteins did not significantly contaminate the caspase-3 immunoprecipitates (Fig. 2 A).
Figure 1.

Isolation of mitochondrial and cytoplasmic cellular fractions. 10C9 cells were fractionated into mitochondrial (M) and cytoplasmic (C) fractions by differential centrifugation. Equal amounts of each fraction were electrophoresed, and the relative levels of cytochrome c (left), cytochrome oxidase subunit IV (COX; middle), and SOD1 (right) in each fraction were determined by immunoblotting. Molecular weights are indicated on the left. The results are representative of one of three separate experiments.
Figure 2.
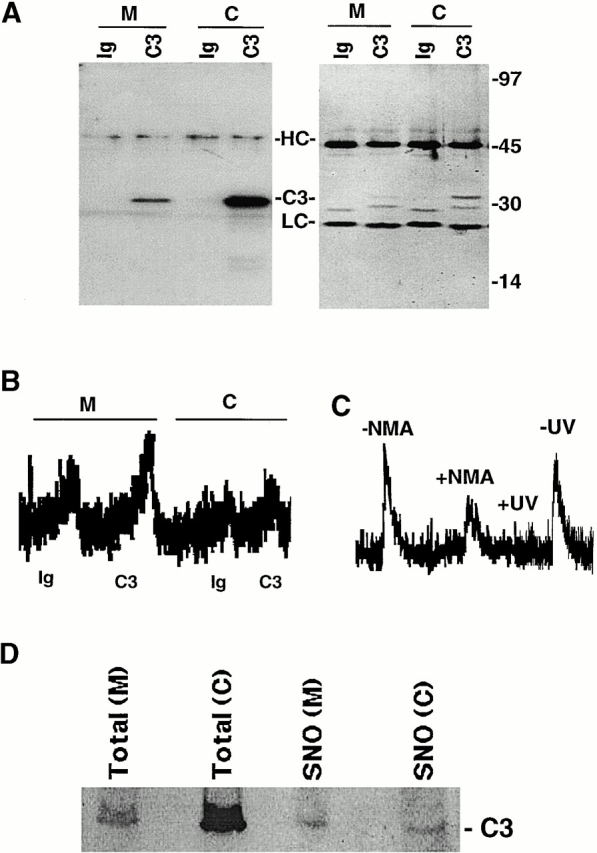
S-Nitrosylation of mitochondrial and cytoplasmic caspase-3. (A) Caspase-3 immunoprecipitation. Proteins were immunoprecipitated from mitochondrial (M) and cytoplasmic (C) cellular fractions using a caspase-3–specific monoclonal antibody (C3) or equal concentrations of an isotype-matched control antibody (Ig). Immunoprecipitated proteins were visualized on silver-stained gels (right) or caspase-3 Western blot analysis (left). Molecular weight markers, immunoglobulin heavy chain (HC), light chain (LC), and caspase-3 (C3) are shown. (B) S-Nitrosylation of caspase-3. The SNO-derived chemiluminescence signal of Ig control (Ig) and caspase-3 (C3) immunoprecipitations obtained from mitochondrial (M) and cytoplasmic (C) fractions of 10C9 cells are shown. NO chemiluminescence in arbitrary units is plotted on the y-axis, and time is plotted on the x-axis. The NO released from each sample is proportional to the area under the curve. The data are representative of 1 of 10 separate experiments. (C) Caspase-3 is S-nitrosylated endogenously. The SNO-derived chemiluminescence signal of mitochondrial caspase-3 immunoprecipitates from CEM cells after they had been grown for 48 h in the presence (+NMA) or absence (−NMA) of 4.5 mM L-NMA is shown. The data are representative of one of two separate experiments. Mitochondrial caspase-3 immunoprecipitates from control cells were divided into two samples, one of which was exposed to UV light for 10 min (+UV) and the other left untreated in the dark at room temperature for the same period (−UV). The SNO-derived chemiluminescence signal from UV-treated and untreated samples is shown. The data are representative of one of four separate experiments. (D) A higher percentage of mitochondrial than cytoplasmic caspase-3 is S-nitrosylated. S-nitrosylated proteins in cytoplasmic (C) and mitochondrial (M) fractions were selectively labeled with biotin and then purified over streptavidin-agarose as described previously (Jaffrey et al., 2001). The purified S-nitrosylated proteins were then analyzed by caspase-3 Western blot analysis. 1 out of 100 of the total protein in the mitochondrial or cytoplasmic starting sample (16 μg) was loaded in the lanes marked Total. Purified S-nitrosylated proteins obtained from each fraction were loaded in the lanes labeled SNO. Caspase-3 (C3) is indicated.
S-nitrosothiol (SNO) bonds in immunoprecipitated proteins were measured by reduction/chemiluminescence as described (Fang et al., 1998). Immunoprecipitated caspase-3 zymogen was significantly more S-nitrosylated in mitochondria than in cytoplasm (Fig. 2, B and D, and Fig. 4) . Similar results were obtained using two additional human cell lines (CEM and U937). If caspases are S-nitrosylated on a single cysteine residue, as has been suggested by studies in vitro (Dimmeler et al., 1997; Kim et al., 1997b; Rossig et al., 1999), then the stoichiometry of SNO to caspase suggests that ∼85% of caspase-3 zymogens in mitochondria are S-nitrosylated compared with only 17% of zymogens in the cytoplasmic fraction (see Fig. 4). Treatment of cells with the NO synthase inhibitor N-G-monomethyl-l-arginine (L-NMA) for 48 h before immunoprecipitation or treatment of the immunoprecipitates with UV light for 10 min decreased the level of NO detected in the immunoprecipitates, suggesting that the NO signal is not an artifact of protein purification (Fig. 2 C). To confirm our chemiluminescence results, we analyzed caspase-3 S-nitrosylation using a recently described technique in which S-nitrosylated proteins are selectively biotinylated, purified over streptavidin-agarose, and then detected by immunoblot (Jaffrey et al., 2001). Although the concentration of caspase-3 was much higher in the cytoplasmic than in the mitochondrial lysates, similar levels of S-nitrosylated caspase-3 were detected in each fraction (Fig. 2 D). This data also suggests that a higher percentage of mitochondrial than cytoplasmic caspase-3 is S-nitrosylated.
Figure 4.

Caspase-3 and -9 are S-nitrosylated more extensively in mitochondria than cytoplasm. The SNO content (nM) of caspase-3 and -9 was determined by subtracting the SNO-derived chemiluminescence signal in control immunoprecipitates (background) from the signal in caspase-3 or -9 immunoprecipitates. Protein concentrations (nM) were determined by silver stain analysis using known concentrations of caspase-3 (BD PharMingen), caspase-9 (Medical and Biological Laboratories Co., Ltd.), and BSA standards. The SNO/caspase stoichiometry of mitochondrial and cytoplasmic caspase-3 and -9 are shown. The data represents the mean ± SEM of 10 separate experiments. *p < 0.02; † p < 0.04.
To rule out preferential denitrosylation of cytoplasmic caspase-3 during sample preparation, we spiked our cytoplasmic preparations with 60 nM of in vitro S-nitrosylated caspase-3 and then analyzed S-nitrosylated caspase-3 recovery in our immunoprecipitates. The SNO/protein stoichiometry of caspase-3 immunoprecipitates obtained from the spiked cytosolic preparations was 0.75, very similar to the stoichiometry (0.85) of mitochondrial caspase-3 immunoprecipitates. This data suggests that caspase-3 is not preferentially denitrosylated in our cytosolic preparations. In addition, treatment of cells with L-NMA (5 mM) throughout the time of cell lysis and immunoprecipitation had no effect on mitochondrial caspase-3 S-nitrosylation (unpublished data). Therefore, increased S-nitrosylation of mitochondrial caspase-3 is not likely to be due to an increased exposure to NO synthase (NOS) activity during sample preparation.
The possible sources of NO-related species that S-nitrosylate mitochondrial caspase-3 include a putative mitochondrial NOS (Kobzik et al., 1995; Ghafourifar and Richter, 1997; Tatoyan and Giulivi, 1998), NO produced by cytoplasmic NOS, which diffuses into the mitochondria, or transnitrosation from S-nitrosylated species within mitochondria such as S-nitrosoglutathione. To determine if increased S-nitrosylation of mitochondrial caspase-3 is due to increased NO production within mitochondria, we analyzed whether cytoplasmic caspase-3 S-nitrosylation is increased when cells are exposed to increasing concentrations of an NO donor. Exposure of cells to 0, 10, 100, or 1,000 μM of the NO donor S-nitrosopenicillamine for 24 h did not increase S-nitrosylation of immunoprecipitated cytoplasmic caspase-3 (unpublished data). Therefore, factors in the subcellular microenvironment of cytoplasmic and mitochondrial caspase-3 other than NO concentration are likely to be important determinants of S-nitrosylation.
Mitochondrial caspase-3 is denitrosylated after Fas stimulation
We have reported previously that S-nitrosylated caspase-3 zymogen obtained from whole cell lysates is denitrosylated after Fas stimulation (Mannick et al., 1999). To determine if mitochondrial caspase-3 is denitrosylated after Fas stimulation, we immunoprecipitated caspase-3 from mitochondrial fractions of cells obtained at 0 or 2–3 h after stimulation with Fas agonist antibody (Clone CH-11, 60 ng/ml; Upstate Biotechnology). Mitochondrial caspase-3 S-nitrosylation decreased 79 ± 12% (mean ± SEM, n = 4, p = 0.01, paired Student's t test) after Fas stimulation, indicating that mitochondrial caspase-3 is denitrosylated during Fas-induced apoptosis.
The majority of mitochondrial but not cytoplasmic caspase-9 is S-nitrosylated
Caspase-3 is the only caspase zymogen which has been shown to be S-nitrosylated by endogenous NOS activity in cells. To determine if other caspase zymogens are endogenously S-nitrosylated, S-nitrosylation of mitochondrial and cytoplasmic caspase-9 zymogens was analyzed. Caspase-9 was immunoprecipitated from mitochondrial and cytoplasmic fractions of two human B cell lines, BJAB and10C9. These cell lines were chosen because they express relatively high levels of mitochondrial caspase-9, allowing sufficient protein to be immunoprecipitated to assess the presence of SNO bonds. Caspase-9 was immunoprecipitated with its specific antibody but not with equal concentrations of control antibody (Fig. 3 A). Silver stain analysis revealed that caspase-9 was the only protein brought down in detectable amounts by the caspase-9 antiserum but not by control antiserum (Fig. 3 A). Chemiluminescence analysis of the immunoprecipitates indicated that S-nitrosylated caspase-9 was located predominantly in the mitochondrial fraction of cells (Fig. 3 A and Fig. 4). Treatment of the immunoprecipitates with UV light eliminated the NO signal in the immunoprecipitates, again suggesting that the signal is not derived from contaminating nitrite (unpublished data). If caspase-9 is S-nitrosylated on a single cysteine residue, then the stoichiometry of SNO to caspase suggests that ∼68% of caspase-9 zymogens in mitochondria are S-nitrosylated compared with only 11% of zymogens in the cytoplasmic fraction (Fig. 4). Thus, the mitochondrial subpopulations of both caspase-3 and -9 zymogens are the primary targets of S-nitrosylation in cells.
Figure 3.
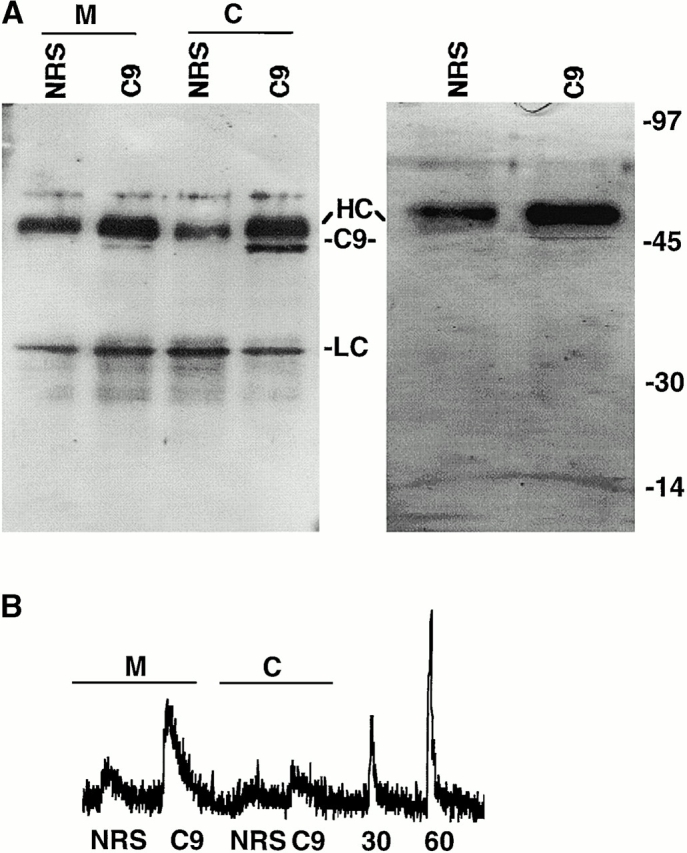
S-Nitrosylation of cytoplasmic and mitochondrial caspase-9. Caspase-9 immunoprecipitation. Proteins were immunoprecipitated from mitochondrial (M) or cytoplasmic (C) cellular fractions using a caspase-9–specific rabbit polyclonal antiserum (C9) or equal concentration of normal rabbit serum (NRS). Immunoprecipitated proteins were visualized on silver-stained gels (right) or caspase-9 Western blot analysis (left). In the silver-stained gel, immunoprecipitates from whole cell lysates are shown. Molecular weight markers, immunoglobulin heavy chain (HC), light chain (LC), and caspase-9 (C9) are shown. (B) S-Nitrosylation of caspase-9. The SNO-derived chemiluminescence signal of control normal rabbit serum (NRS) or caspase-9 (C9) immunoprecipitates obtained from mitochondrial (M) and cytoplasmic (C) fractions of BJAB cells and from 30 nM (30) and 60 nM (60) S-nitrosoglutathione standards are shown. The data are representative of 1 of 10 separate experiments.
S-Nitrosylation is not required to target caspase-3 to mitochondria
It is possible that S-nitrosylation, like phosphorylation, localizes proteins to specific subcellular compartments (Teruel and Meyer, 2000). To determine if S-nitrosylation targets caspase-3 to mitochondria, cells were grown for 48 h in the presence of NOS inhibitors. NOS inhibition decreased caspase-3 S-nitrosylation (Fig. 2 C) but had no affect on mitochondrial caspase-3 levels (Fig. 5 A). In addition, mutation of the catalytic site cysteine of caspase-3 to an alanine did not alter mitochondrial caspase-3 levels (Fig. 5 B), despite inhibiting caspase S-nitrosylation (Mannick et al., 1999). These data suggest that S-nitrosylation is not required to target caspase-3 to mitochondria.
Figure 5.
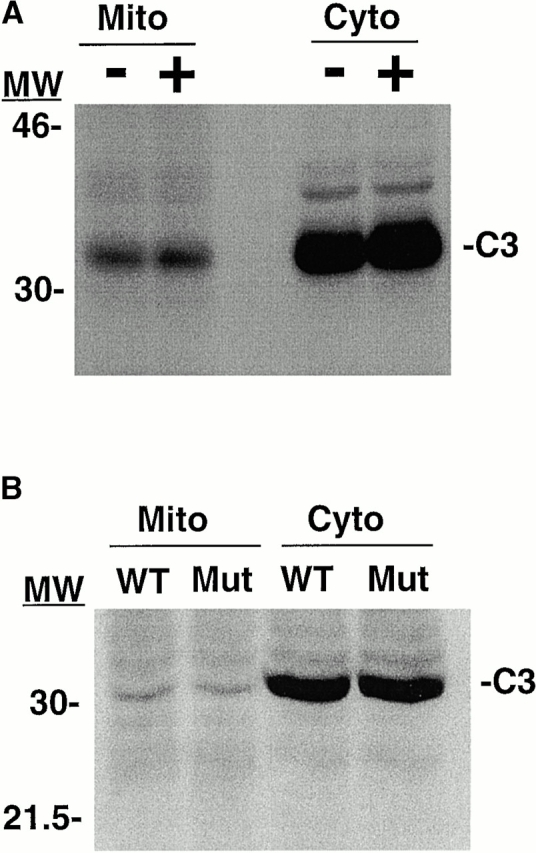
S-Nitrosylation does not target caspase-3 to mitochondria. (A) NOS inhibition does not decrease mitochondrial caspase-3 levels. CEM cells were grown in the presence (+) or absence (−) of the NOS inhibitor N-G-nitro-l-arginine (5 mM). After 48 h, a caspase-3 (C3) Western blot of equal protein concentrations from mitochondrial (Mito) and cytoplasmic (Cyto) fractions were performed. The blot is representative of one of three separate experiments. Similar results were obtained using the NOS inhibitor L-NMA. Molecular weight markers are indicated on the left. (B) Mutation of the catalytic site cysteine does not decrease mitochondrial caspase-3 levels. MCF-7 cells, which do not express caspase-3, were stably transfected with plasmids expressing wild-type procaspase-3 (WT) or procaspase-3 in which the catalytic site cysteine was mutated to an alanine (Mut) (Mannick et al., 1999). A caspase-3 Western blot of equal concentrations of mitochondrial and cytoplasmic fractions was performed. The data is representative of one of three separate experiments.
In summary, the results demonstrate that the majority of mitochondrial, but not cytoplasmic, caspase-3 zymogens are S-nitrosylated in resting cells. Since caspase-3 is S-nitrosylated on its catalytic site cysteine (Mannick et al., 1999) and this inhibits enzyme activity (Dimmeler et al., 1997; Kim et al., 1997a,b, 2000; Li et al., 1997a; Mannick et al., 1999; Rossig et al., 1999), S-nitrosylation is likely to play a particularly important role in regulating mitochondrial caspase-3 function. We have also shown that mitochondrial caspase-9 is S-nitrosylated, raising the possibility that S-nitrosylation is a general mechanism by which mitochondrial caspase activity is controlled. Although the S-nitrosylated cysteine residue(s) of caspase-9 has not been identified, S-nitrosylation of the active site thiol of caspase-3, and perhaps caspase-9, may prevent inappropriate caspase autoactivation in mitochondria when zymogens are brought into relatively close proximity in the intermembrane space.
S-Nitrosylation of proteins has been suggested to play a role comparable to phosphorylation in regulating signaling pathways (Stamler et al., 1992). Phosphorylation involves the reversible covalent attachment of a phosphate group to serine, threonine, or tyrosine residues on proteins, whereas nitrosylation involves the reversible covalent attachment of an NO group to cysteine residues. One of the differences between phosphorylation and S-nitrosylation is that the SNO bond is easily reduced and therefore less stable than the protein–phosphate bond. This has made characterization of SNO proteins difficult and led to questions regarding their intracellular stability. The factors that influence intracellular SNO protein formation and stability include pO2 (Eu et al., 2000), pH, redox environment (Stamler, 1994; Stamler et al., 1997), the intracellular location of NOS, the different chemistries of the various NO species that react with thiols (Stamler, 1994), the reactivity of the target thiol itself, which can vary sixfold (Stamler et al., 1997), and the possibility of enzymes comparable to kinases and phosphatases that catalyze the attachment or removal of NO groups from thiols (Stamler et al., 1997; Gaston, 1999; Fang et al., 2000). Of note, the rate constant for formation of the inorganic S-nitrosylating intermediate nitrogen dioxide from NO is substantially higher in lipid membranes (which are abundant in mitochondria) than in aqueous solution (Liu et al., 1998).
Our findings are the first to suggest the S-nitrosylation state of a given protein may depend on its subcellular location. Since many of the factors listed above that influence the formation and stability of S-nitrosylated proteins are likely to vary between subcellular compartments, it is possible that certain cellular microenvironments, such as the mitochondrial intermembrane space, are privileged sites favoring SNO protein stability. During apoptosis, mitochondrial caspase-3 is released into the cytoplasm, and caspase-3 is denitrosylated (Mancini et al., 1998; Mannick et al., 1999; Samali et al., 1999). We speculate that movement of proteins out of privileged sites into environments that do not favor SNO stability may be a mechanism involved in protein activation by denitrosylation.
Materials and methods
Materials
RPMI 1640, FCS, penicillin, streptomycin, and l-glutamine were purchased from Cellgro/Mediatech. Caspase-3, caspase-9, cytochrome c, and SOD1 antibodies were purchased from BD Transduction Laboratories. Cytochrome oxidase antibody (subunit 1V) was purchased from Molecular Probes. Control normal rabbit serum was obtained from the laboratory of Dr. William Marshall (University of Massachusetts Medical School, Worcester, MA). All other chemicals were purchased from Sigma-Aldrich.
Cell lines
10C9 and BJAB are human Burkitt's lymphoma B cell lines. CEM is a human T cell line, and U937 is a human monomyelocytic leukemia cell line. BJAB was obtained from the laboratory of Dr. Elliott Kieff (Harvard Medical School, Boston, MA), CEM and U937 were obtained from the laboratory of Dr. Robert Finberg (University of Massachusetts Medical School), and 10C9 was purchased from American Type Culture Collection. MCF-7 cells stably transfected with WT and mutant caspase-3 were generated as described previously (Mannick et al., 1999). Cells were grown at 37°C, 5% CO2, in RPMI 1640 supplemented with 10% heat-inactivated FCS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Subcellular fractionation
0.7 × 108 cells were washed in PBS and then resuspended in 1 ml buffer A (150 mM NaCl, 50 mM Tris, pH 8, containing 100 μM EDTA and the following protease inhibitors: 10 μg/ml leupeptin, 5 μg/ml aprotinin, and 0.5 mM phenylmethyl-sulfonyl fluoride). The cells were then homogenized (Kontes) until >80% of the cells were stained with trypan blue. Nuclei and unbroken cells were removed during two 10-min centrifugations at 1,000 g. The postnuclear supernatant was centrifuged at 10,000 g for 30 min to obtain a pellet highly enriched in mitochondria. The mitochondrial pellet was resuspended in a volume of buffer A equal to that of the postmitochondrial supernatant. In some experiments, the postmitochondrial supernatant (S10) was further centrifuged at 100,000 g to obtain an S100 cytosolic fraction.
Immunoprecipitation
Mitochondrial or cytosolic fractions were lysed for 30 min at 4°C in high salt buffer (1% NP-40, 500 mM NaCl, 50 mM Tris, pH 8, 100 μM EDTA, and protease inhibitors). Insoluble material was pelleted for 10 min at 10,000 g at 4°C. The supernatant was precleared as described previously (Mannick et al., 1999). Cellular proteins were immunoprecipitated in the dark with 5 μg of an anti–caspase-3 IgG2a monoclonal antibody, 5 μg of a control IgG2a antibody (Sigma-Aldrich), 1 μl of a 1:5 dilution of rabbit anti–caspase-9 polyclonal antiserum or equal concentrations of control normal rabbit serum. The antigen–antibody complexes were isolated with protein A–Sepharose beads for 2 h at 4°C. The beads were then washed five times in high salt buffer to which 1 mM n-ethyl-maleimide was added to block free thiols and thereby prevent artifactual S-nitrosylation. Antigen–antibody complexes were removed from the protein A–sepharose beads by three 10-min incubations in 70 μl of 100 mM glycine (pH 3) at 4°C before NO measurements.
S-Nitrosylation measurements
Caspase S-nitrosylation was detected by reduction/chemiluminescence as described previously (Fang et al., 1998) with minor modifications. Immunoprecipitated samples (after separation from beads) were injected into 5 ml of a solution containing 100 μM CuCl, 1 mM cysteine, and 0.01% antifoam (pH 3.5, 50°C) purged continuously with argon or helium (grade 5; BOC gases). NO evolved was measured by chemiluminescence (Sievers). Data were interpreted as raw photoelectric output (integrated using Sievers software) and as absolute NO evolved (using NO standards generated by S-nitrosoglutathione).
Detection of S-nitrosylated caspase-3 on gels
S-nitrosylated proteins in mitochondrial and cytoplasmic fractions were selectively biotinylated and purified as described previously (Jaffrey et al., 2001). The purified proteins were then analyzed by caspase-3 Western blot analysis.
In vitro S-nitrosylation of caspase-3
Immunoprecipitated caspase-3 zymogen eluted from protein A–Sepharose beads in 100 mM glycine (pH 3) was reduced with 20 mM DTT for 30 min on ice. The DTT was removed by passage over a Sephadex G-25 column (Amersham Pharmacia Biotech) preequilibrated with buffer B (100 mM Hepes, pH 7.4, 140 mM NaCl, 100 μM EDTA, and protease inhibitors). The protein was then incubated with 200 μM S-nitrosocysteine for 1 h on ice and purified by passage over a Sephadex G-25 column. The extent of caspase-3 S-nitrosylation was determined by chemiluminescence as described above.
Western blot and silver stain analysis
Immunoprecipitated proteins or control BSA standards were separated on 12% SDS-PAGE. The gels were either silver stained as per the manufacturer's instructions (Sigma-Aldrich), or proteins were transferred to nitrocellulose and Western blots were performed as described previously (Mannick et al., 1999).
Acknowledgments
We thank Qian Miao for excellent technical assistance.
This work was supported by National Institutes of Health grants GM57601 (to J.B. Mannick), HL59337 (to B. Gaston), a Leukemia and Lymphoma Society Translational Research award (to J.B. Mannick), an American Cancer Society Research Project grant (to J.B. Mannick), and Bundesministerium für Bildung und Forschung (to M. Szibor).
M. Szibor is currently on leave from the Institute of Pathophysiology, Martin-Luther-University, Halle, Germany.
Footnotes
Abbreviations used in this paper: L-NMA, N-G-monomethyl-l-arginine; NO, nitric oxide; NOS, nitric oxide synthase; SNO, S-nitrosothiol; SOD1, superoxide dismutase.
References
- Balakirev, M., V.V. Khramtsov, and G. Zimmer. 1997. Modulation of the mitochondrial permeability transition by nitric oxide. Eur. J. Biochem. 246:710–718. [DOI] [PubMed] [Google Scholar]
- De Nadai, C., P. Sestili, O. Cantoni, J.P. Lievremont, C. Sciorati, R. Barsacchi, S. Moncada, J. Meldolesi, and E. Clementi. 2000. Nitric oxide inhibits tumor necrosis factor-alpha-induced apoptosis by reducing the generation of ceramide. Proc. Natl. Acad. Sci. USA. 97:5480–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler, S., J. Haendeler, M. Nehls, and A.M. Zeiher. 1997. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J. Exp. Med. 185:601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eu, J.P., J. Sun, L. Xu, J.S. Stamler, and G. Meissner. 2000. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 102:499–509. [DOI] [PubMed] [Google Scholar]
- Fang, K., N.V. Ragsdale, R.M. Carey, T. MacDonald, and B. Gaston. 1998. Reductive assays for S-nitrosothiols: implications for measurements in biological systems. Biochem. Biophys. Res. Commun. 252:535–540. [DOI] [PubMed] [Google Scholar]
- Fang, K., R. Johns, T. Macdonald, M. Kinter, and B. Gaston. 2000. S-nitrosoglutathione breakdown prevents airway smooth muscle relaxation in the guinea pig. Am. J. Physiol. Lung Cell Mol. Physiol. 279:L716–L721. [DOI] [PubMed] [Google Scholar]
- Gaston, B. 1999. Nitric oxide and thiol groups. Biochim. Biophys. Acta. 1411:323–333. [DOI] [PubMed] [Google Scholar]
- Genaro, A.M., S. Hortelano, A. Alvarez, C. Martinez, and L. Bosca. 1995. Splenic B lymphocyte programmed cell death is prevented by nitric oxide release through mechanisms involving sustained Bcl-2 levels. J. Clin. Invest. 95:1884–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafourifar, P., and C. Richter. 1997. Nitric oxide synthase activity in mitochondria. FEBS Lett. 418:291–296. [DOI] [PubMed] [Google Scholar]
- Glockzin, S., A. von Knethen, M. Scheffner, and B. Brune. 1999. Activation of the cell death program by nitric oxide involves inhibition of the proteasome. J. Biol. Chem. 274:19581–19586. [DOI] [PubMed] [Google Scholar]
- Hortelano, S., B. Dallaporta, N. Zamzami, T. Hirsch, S.A. Susin, I. Marzo, L. Bosca, and G. Kroemer. 1997. Nitric oxide induces apoptosis via triggering mitochondrial permeability transition. FEBS Lett. 410:373–377. [DOI] [PubMed] [Google Scholar]
- Jaffrey, S.R., H. Erdjument-Bromage, C.D. Ferris, P. Tempst, and S.H. Snyder. 2001. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3:193–197. [DOI] [PubMed] [Google Scholar]
- Kim, Y.M., H. Bergonia, and J.R. Lancaster, Jr. 1995. Nitrogen oxide-induced autoprotection in isolated rat hepatocytes. FEBS Lett. 374:228–232. [DOI] [PubMed] [Google Scholar]
- Kim, Y.M., M.E. de Vera, S.C. Watkins, and T.R. Billiar. 1997. a. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-alpha-induced apoptosis by inducing heat shock protein 70 expression. J. Biol. Chem. 272:1402–1411. [DOI] [PubMed] [Google Scholar]
- Kim, Y.M., R.V. Talanian, and T.R. Billiar. 1997. b. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J. Biol. Chem. 272:31138–31148. [DOI] [PubMed] [Google Scholar]
- Kim, Y.M., T.H. Kim, H.T. Chung, R.V. Talanian, X.M. Yin, and T.R. Billiar. 2000. Nitric oxide prevents tumor necrosis factor alpha-induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology. 32:770–778. [DOI] [PubMed] [Google Scholar]
- Kobzik, L., B. Stringer, J.L. Balligand, M.B. Reid, and J.S. Stamler. 1995. Endothelial type nitric oxide synthase in skeletal muscle fibers: mitochondrial relationships. Biochem. Biophys. Res. Commun. 211:375–381. [DOI] [PubMed] [Google Scholar]
- Krajewski, S., M. Krajewska, L.M. Ellerby, K. Welsh, Z. Xie, Q.L. Deveraux, G.S. Salvesen, D.E. Bredesen, R.E. Rosenthal, and G. Fiskum. 1999. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA. 96:5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J., T.R. Billiar, R.V. Talanian, and Y.M. Kim. 1997. a. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem. Biophys. Res. Commun. 240:419–424. [DOI] [PubMed] [Google Scholar]
- Li, P., D. Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri, and X. Wang. 1997. b. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91:479–489. [DOI] [PubMed] [Google Scholar]
- Liu, X., M.J. Miller, M.S. Joshi, D.D. Thomas, and J.R. Lancaster. 1998. Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc. Natl. Acad. Sci. USA. 95:2175–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini, M., D.W. Nicholson, S. Roy, N.A. Thornberry, E.P. Peterson, L.A. Casciola-Rosen, and A. Rosen. 1998. The caspase-3 precursor has a cytosolic and mitochondrial distribution: implications for apoptotic signaling. J. Cell Biol. 140:1485–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannick, J.B., A. Hausladen, L. Liu, D.T. Hess, M. Zeng, Q.X. Miao, L.S. Kane, A.J. Gow, and J.S. Stamler. 1999. Fas-induced caspase denitrosylation. Science. 284:651–654. [DOI] [PubMed] [Google Scholar]
- Messmer, U.K., and B. Brune. 1996. Nitric oxide-induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochem. J. 319:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messmer, U.K., U.K. Reed, and B. Brune. 1996. Bcl-2 protects macrophages from nitric oxide-induced apoptosis. J. Biol. Chem. 271:20192–20197. [DOI] [PubMed] [Google Scholar]
- Rossig, L., B. Fichtlscherer, K. Breitschopf, J. Haendeler, A.M. Zeiher, A. Mulsch, and S. Dimmeler. 1999. Nitric oxide inhibits caspase-3 by S-nitrosation in vivo. J. Biol. Chem. 274:6823–6826. [DOI] [PubMed] [Google Scholar]
- Samali, A., J. Cai, B. Zhivotovsky, D.P. Jones, and S. Orrenius. 1999. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 18:2040–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler, J.S. 1994. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 78:931–936. [DOI] [PubMed] [Google Scholar]
- Stamler, J.S., D.I. Simon, J.A. Osborne, M.E. Mullins, O. Jaraki, T. Michel, D.J. Singel, and J. Loscalzo. 1992. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA. 89:444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler, J.S., E.J. Toone, S.A. Lipton, and N.J. Sucher. 1997. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 18:691–696. [DOI] [PubMed] [Google Scholar]
- Suschek, C.V., V. Krischel, D. Bruch-Gerharz, D. Berendji, J. Krutmann, K.D. Kroncke, and V. Kolb-Bachofen. 1999. Nitric oxide fully protects against UVA-induced apoptosis in tight correlation with Bcl-2 up-regulation. J. Biol. Chem. 274:6130–6137. [DOI] [PubMed] [Google Scholar]
- Susin, S.A., N. Zamzami, M. Castedo, T. Hirsch, P. Marchetti, A. Macho, E. Daugas, M. Geuskens, and G. Kroemer. 1996. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 184:1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin, S.A., H.K. Lorenzo, N. Zamzami, I. Marzo, C. Brenner, N. Larochette, M.C. Prevost, P.M. Alzari, and G. Kroemer. 1999. a. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med. 189:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin, S.A., H.K. Lorenzo, N. Zamzami, I. Marzo, B.E. Snow, G.M. Brothers, J. Mangion, E. Jacotot, P. Costantini, M. Loeffler, et al. 1999. b. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 397:441–446. [DOI] [PubMed] [Google Scholar]
- Tatoyan, A., and C. Giulivi. 1998. Purification and characterization of a nitric-oxide synthase from rat liver mitochondria. J. Biol. Chem. 273:11044–11048. [DOI] [PubMed] [Google Scholar]
- Teruel, M.N., and T. Meyer. 2000. Translocation and reversible localization of signaling proteins: a dynamic future for signal transduction. Cell. 103:181–184. [DOI] [PubMed] [Google Scholar]
- Thompson, C.B. 1995. Apoptosis in the pathogenesis and treatment of disease. Science. 267:1456–1462. [DOI] [PubMed] [Google Scholar]
- Thornberry, N.A., and Y. Lazebnik. 1998. Caspases: enemies within. Science. 281:1312–1316. [DOI] [PubMed] [Google Scholar]
- Ushmorov, A., F. Ratter, V. Lehmann, W. Droge, V. Schirrmacher, and V. Umansky. 1999. Nitric-oxide-induced apoptosis in human leukemic lines requires mitochondrial lipid degradation and cytochrome C release. Blood. 93:2342–2352. [PubMed] [Google Scholar]