

Abstract
Huntingtin-interacting protein 1 related (Hip1R) is a novel component of clathrin-coated pits and vesicles and is a mammalian homologue of Sla2p, an actin-binding protein important for both actin organization and endocytosis in yeast. Here, we demonstrate that Hip1R binds via its putative central coiled-coil domain to clathrin, and provide evidence that Hip1R and clathrin are associated in vivo at sites of endocytosis. First, real-time analysis of Hip1R–YFP and DsRed–clathrin light chain (LC) in live cells revealed that these proteins show almost identical temporal and spatial regulation at the cell cortex. Second, at the ultrastructure level, immunogold labeling of ‘unroofed’ cells showed that Hip1R localizes to clathrin-coated pits. Third, overexpression of Hip1R affected the subcellular distribution of clathrin LC. Consistent with a functional role for Hip1R in endocytosis, we also demonstrated that it promotes clathrin cage assembly in vitro. Finally, we showed that Hip1R is a rod-shaped apparent dimer with globular heads at either end, and that it can assemble clathrin-coated vesicles and F-actin into higher order structures. In total, Hip1R's properties suggest an early endocytic function at the interface between clathrin, F-actin, and lipids.
Keywords: endocytosis; clathrin; F-actin; coated pits; Huntington's disease
Introduction
Endocytosis is critically important for downregulation of growth factor signaling, nutrient uptake, antigen presentation, pathogen internalization, and maintenance of plasma membrane surface area. During receptor-mediated endocytosis, clathrin is recruited to the plasma membrane where it assembles on the lipid bilayer to form coated pits (for review see Marsh and McMahon, 1999; Brodin et al., 2000). Coated pits contain not only clathrin and receptors but also proteins that can promote clathrin polymerization and in some cases bind to the receptors (e.g., AP2 and AP180) (Ungewickell and Oestergaard, 1989; Keen, 1990). Coated pits subsequently invaginate and pinch off from the plasma membrane as clathrin-coated vesicles (CCVs)* in a dynamin-dependent manner (for review see Sever et al., 2000). In addition to clathrin, AP2, AP180/CALM, and dynamin, other proteins, including amphiphysin, synaptojanin, Eps15, epsin, endophilin, auxilin, cyclin G–associated kinase (GAK), syndapin, intersectin, and Abp1, all have been implicated in the early stages of endocytosis (for review see Brodin et al., 2000; Jarousse and Kelly, 2001; see also Kessels et al., 2001). Furthermore, evidence is emerging that both phospholipids and the actin cytoskeleton, which are integral parts of the cell cortex, are involved in endocytosis in mammalian cells (for review see Qualmann et al., 2000; Cremona and De Camilli, 2001).
A few molecular candidates that could function at the interface between actin and the endocytic machinery have been identified recently in mammalian cells (for review see Qualmann et al., 2000). One of these proteins is the huntingtin-interacting protein 1 related (Hip1R), a component of clathrin-coated pits and vesicles that binds to F-actin (Engqvist-Goldstein et al., 1999). Hip1R belongs to a conserved family of proteins that includes yeast Sla2p/End4p, a protein essential for both endocytosis and actin functions (Holtzman et al., 1993; Wesp et al., 1997; Yang et al., 1999), and Hip1, a protein implicated in the pathology of Huntington's disease (Kalchman et al., 1997; Wanker et al., 1997). Genetic and biochemical data have suggested that Sla2p and other cortical actin patch proteins function at an early stage of endocytosis in yeast (for review see Wendland et al., 1998).
Members of the Sla2/Hip1 family have a conserved domain structure. At the NH2 terminus, they contain a domain related to the epsin NH2-terminal homology domain (ENTH) found in several proteins implicated in endocytosis, including epsins and AP180/CALM, as well as their yeast homologues (Kay et al., 1999; McCann and Craig, 1999). Recently, it has been shown that the ENTH domains of CALM and epsin bind to phosphatidylinositol-4, 5-biphosphate (PI(4,5)P2) at the plasma membrane (Ford et al., 2001; Itoh et al., 2001). The NH2-terminal region of Hip1R is followed by a putative coiled-coil region of ∼300 amino acids, containing a short leucine zipper that in yeast Sla2p has been implicated in endocytosis and dimerization (Wesp et al., 1997; Yang et al., 1999). Finally, at the COOH terminus, these proteins contain two shorter predicted coiled-coils and a talin-like F-actin binding module (McCann and Craig, 1997, 1999; Engqvist-Goldstein et al., 1999; Yang et al., 1999).
Our previous data demonstrated that Hip1R is enriched in CCVs and colocalizes with markers for receptor-mediated endocytosis (Engqvist-Goldstein et al., 1999). We found that the NH2-terminal region plus the coiled-coil domain are sufficient for localizing Hip1R to the endocytic machinery. Since Hip1R binds to F-actin via its COOH-terminal talin-like domain, it could thereby recruit F-actin to the endocytic machinery, or vice versa. To learn more about the function of Hip1R in endocytosis and how it interacts with the endocytic machinery, we set out to identify proteins that bind to Hip1R.
Results
Hip1R associates with clathrin in vivo
To identify proteins that interact with Hip1R, we performed immunoprecipitation experiments and affinity chromatography. Fig. 1 a shows that clathrin coimmunoprecipitated with Hip1R from a microsomal fraction prepared from mouse brain extract (lane 4). This interaction appears specific since clathrin was not detected using beads alone (lane 2), preimmune guinea pig serum (unpublished data), or anti-Eps15 antibodies (lane 6). However, clathrin was detected in immunoprecipitations using antisera against the known clathrin-binding protein α-adaptin (lane 8). Similar results were obtained using affinity chromatography. Clathrin from brain extract was specifically retained on a column containing His6-tagged Hip1R (unpublished data).
Figure 1.
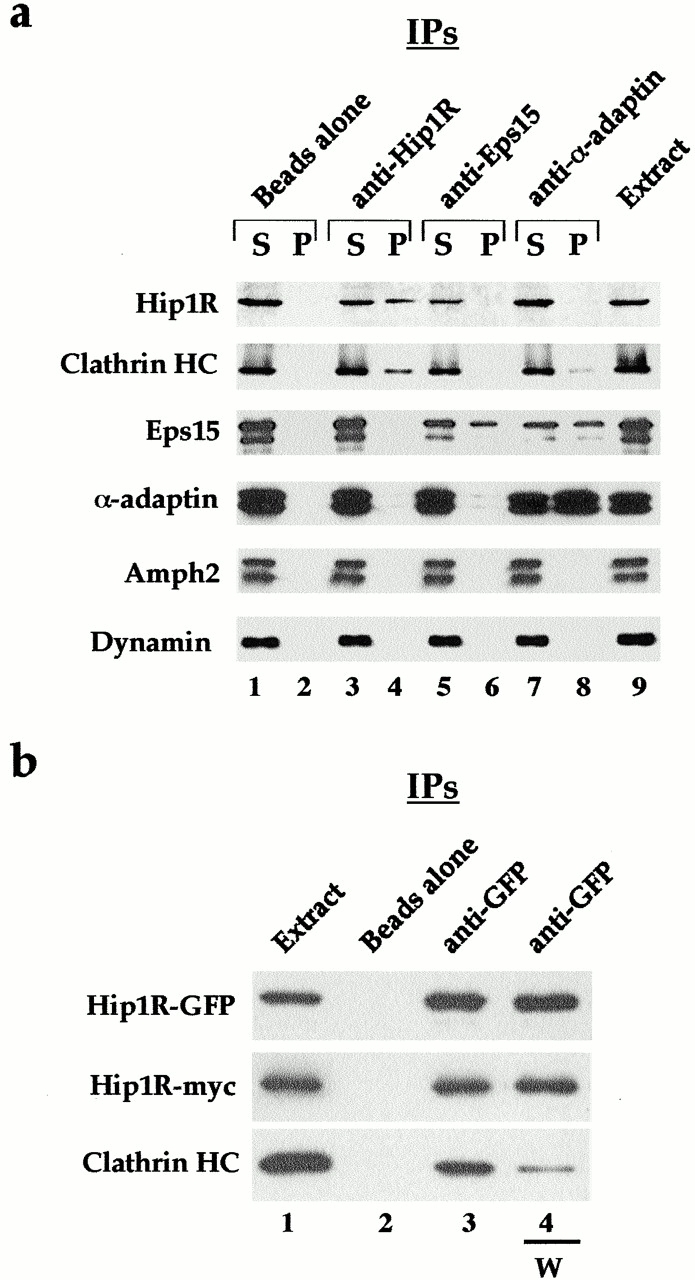
Coimmunoprecipitation of clathrin and Hip1R from an extract prepared from a mouse brain microsomal pellet (a) and from Cos-7 cell homogenate (b). Western blotting of different immunoprecipitation reactions. Antibodies used for immunoprecipitations are indicated at the top of each blot, while antibodies used to probe the blot are shown on the left side. (a) Lanes 1 and 2 show beads alone, lanes 3 and 4 show immunoprecipitation of Hip1R, lanes 5 and 6 show immunoprecipitation of Eps15, and lanes 7 and 8 show immunoprecipitation of α-adaptin. Lane 9 shows the amount of each protein in the extract. Equal portions of supernatant (S) and pellet (P) were loaded. (b) Lane 1 shows the extract and lanes 2–4 show pellets. Lane 2 shown beads alone, lane 3 shows immunoprecipitation of Hip1R–GFP, and lane 4 shows immunoprecipitation of Hip1R–GFP when beads where washed with 0.5 M Tris-HCl (W). The immunoprecipitations were performed three times with similar results.
When Hip1R–green fluorescent protein (GFP) and Hip1R–myc were coexpressed in Cos-7 cells, clathrin coimmunoprecipitated with both proteins, consistent with the results with endogenous brain proteins. Furthermore, Hip1R–myc coimmunoprecipitated with Hip1R–GFP (Fig. 1 b, lane 3), suggesting that Hip1R is a dimer (see below).
We had observed previously that buffer containing 0.5 M Tris-HCl dissociates Hip1R from CCVs (unpublished data). 0.5 M Tris-HCl is also known to disassemble clathrin cages into triskelions (Zaremba and Keen, 1983). To test whether 0.5 M Tris-HCl affects the interaction between clathrin and Hip1R, and/or the apparent Hip1R–Hip1R interaction, the immunoprecipitates were washed with a buffer containing 0.5 M Tris-HCl. As shown in Fig. 1 b, much less clathrin was detected in the immunoprecipitates that were washed with 0.5 M Tris-HCl (compare lanes 3 and 4). However, the interaction between Hip1R–GFP and Hip1R–myc was unaffected (compare lanes 3 and 4). In total, these results demonstrate that Hip1R is part of a complex with clathrin in vivo, and suggest that at least a pool of Hip1R exists in a complex with itself.
Hip1R binds to clathrin directly
To test whether Hip1R interacts directly with clathrin, full-length Hip1R was expressed in SF9 cells with an His6 tag at the NH2 terminus, purified, and tested for clathrin binding using a clathrin cage–pelleting assay. Hip1R pelleted with clathrin cages in a specific, saturable, and concentration-dependent manner (Fig. 2 a). This interaction is unlikely to be mediated through another protein since the purity of clathrin was very high with no detectable levels of AP2, as determined by analysis on Coomassie-stained gels and by Western blotting (Fig. 2 a, lane 9 and unpublished data). Furthermore, the purity of Hip1R was also high (Fig. 2 a, lane 1). Antibodies made against either the first 28 amino acids or the last 37 amino acids of Hip1R recognize a doublet at ∼115 kD (unpublished data). Therefore, the lower band is likely to be untagged Hip1R. Hip1R pelleting was not due to the His tag since His-LacI did not copellet with clathrin cages (Fig. 3 b). Since the data in Fig. 2 a suggested that in this experiment clathrin was saturated with Hip1R, we lowered the concentration of clathrin and Hip1R to estimate the dissociation constant (Kd) for this interaction. Clathrin cages (0–20 nM) were pelleted with a constant concentration of Hip1R (8 nM), and binding was detected by Western blotting. As shown in Fig. 2 b, Hip1R binds to clathrin cages with a Kd in the low nanomolar range.
Figure 2.
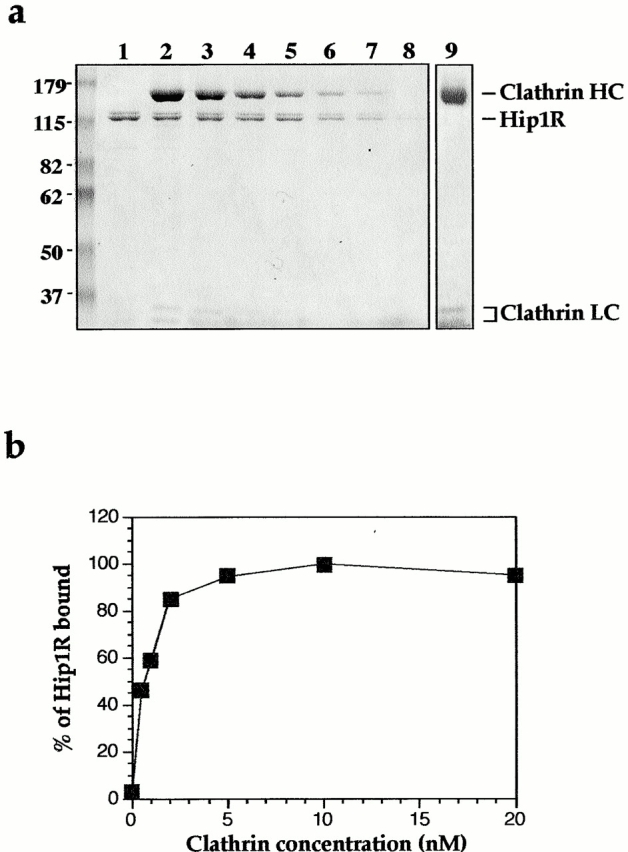
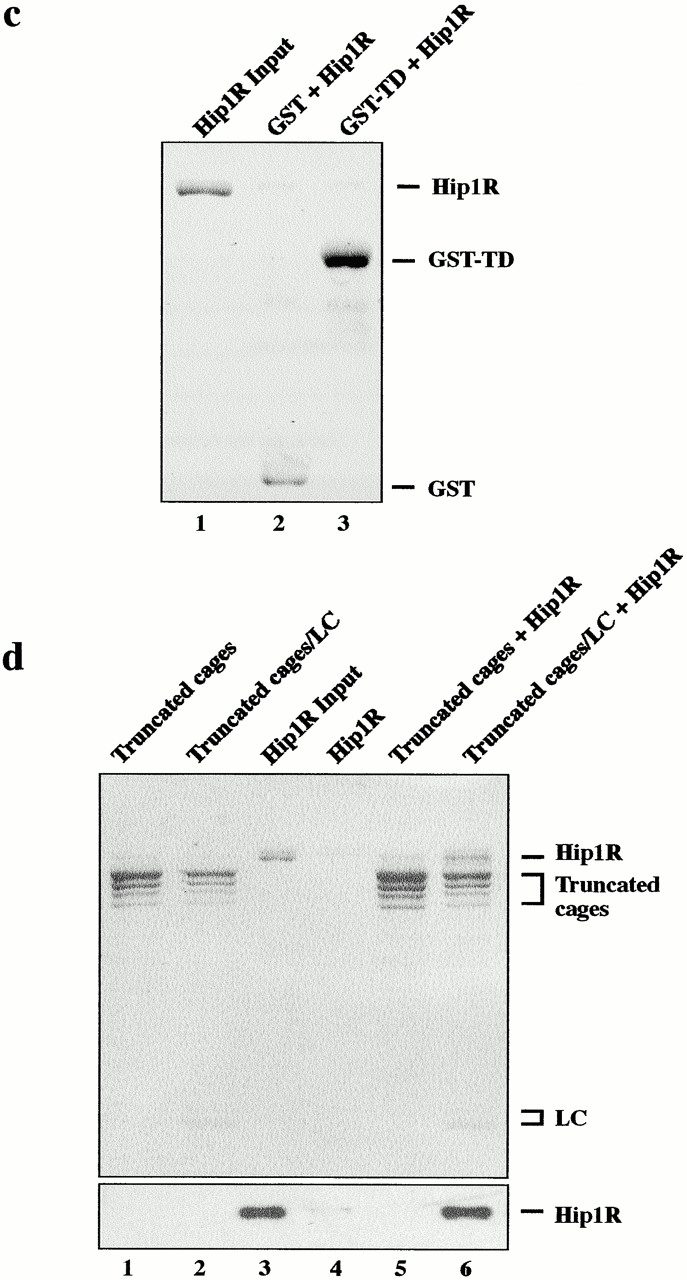
Hip1R binds directly to clathrin cages with high affinity. (a) Coomassie-stained gel of a clathrin cage–pelleting assay. Lane 1 shows total amount of recombinant His6–Hip1R (100 nM final). Lanes 2–8 show pellets from reactions containing 250, 150, 100, 50, 25, 12.5, and 0 nM of clathrin cages with a constant concentration of His6–Hip1R (100 nM). Lane 9 shows the purity of the purified clathrin used in this pelleting assay. (b) The graph shows the percentage of His6–Hip1R bound to clathrin cages as a function of clathrin concentration. In this experiment, 0–20 nM of clathrin cages was used with a constant concentration of Hip1R (8 nM). The bound Hip1R was detected by Western blotting and quantified by densitometry. The data shown are representative of three independent experiments. (c) Hip1R does not bind to GST-TD of clathrin HC (amino acids 1–579) immobilized on glutathione-agarose beads (Coomassie stained gel). Lane 1 shows the total amount of recombinant His6–Hip1R (100 nM final) added. Lane 2 shows a pellet of glutathione-agarose incubated with GST (500 nM) and His6–Hip1R (100 nM). Lane 3 shows a pellet of glutathione-agarose incubated with GST-TD (500 nM) and His6–Hip1R (100 nM). (d) Clathrin LC restores Hip1R binding to truncated cages. Coomassie-stained gel of a clathrin cage–pelleting assay (top). Western blot of Hip1R (bottom). Lanes 1 and 2 show truncated cages and truncated cages reconstituted with intact clathrin LCs, respectively. Lane 3 shows the total amount of recombinant His6–Hip1R (200 nM final) added to the cages. Lane 4 shows Hip1R alone (200 nM). Lane 5 shows truncated cages (∼400 nM) with His6–Hip1R (200 nM). Lane 6 shows truncated cages reconstituted with LC (∼400 nM) with His6–Hip1R (200 nM).
Figure 3.
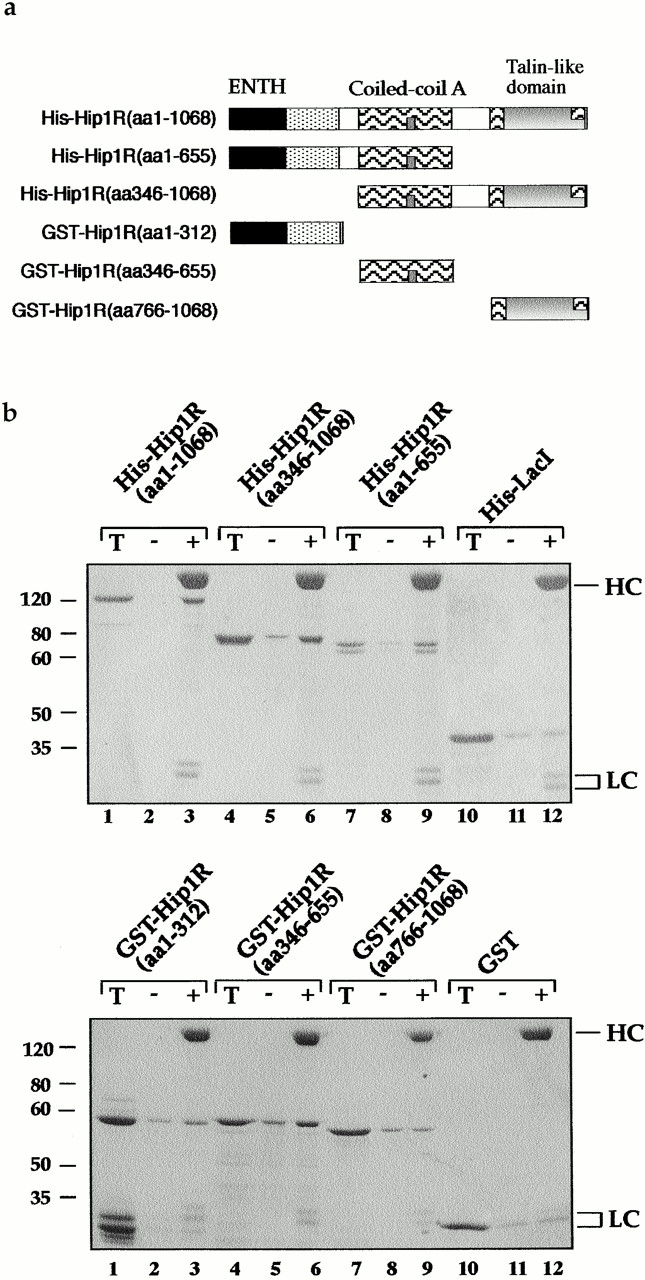


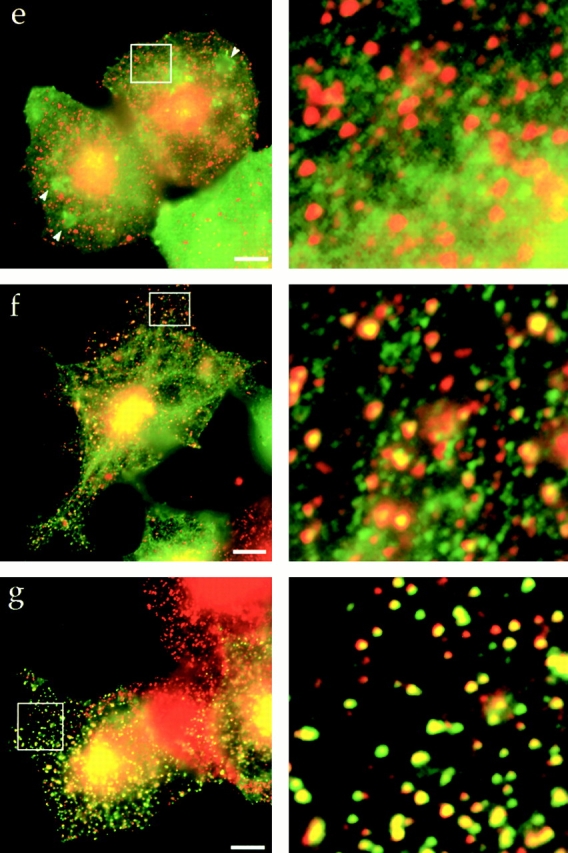
The central coiled-coil region of Hip1R binds to clathrin in vitro and in vivo. (a) Schematic diagrams of Hip1R domains expressed. (b) Coomassie-stained gel of the cage-pelleting assays. (T) shows the total amount of protein added in the assay, (−) represents pelleting without clathrin cages, and (+) represents pelleting with clathrin cages. For each assay, 600 nM of clathrin cages was used with ∼200 nM of HipR constructs. Because GST–Hip1R (amino acids 1–312) was partially degraded, a higher concentration of this construct was added to give ∼200 nM of nondegraded GST–Hip1R (amino acids 1–312). GST alone and His6LacI were used at ∼400 nM. (c) Graphical representation of results from cage-pelleting assays. The graph shows the percentage of Hip1R fragments pelleting with or without clathrin cages using data from three independent experiments. Error bars denote ±SE. (d) Endogenous clathrin specifically coimmunoprecipitated with Hip1R (amino acids 325–655)-6myc from Cos-7 cell extracts (lane 3). Clathrin was not detected using beads alone (lanes 2 and 6) or when Hip1R (amino acids 1–324)-6myc was immunoprecipitated (lane 7). The clathrin–Hip1R interaction was diminished when the beads were washed with buffer containing 0.5 M Tris-HCl (lane 4). Lanes 1 and 5 show extracts and lanes 2–4 and lanes 6 and 7 show pellets. (e–g and enlargements) Indirect immunofluorescence of endogenous clathrin HC (red) and myc-tagged Hip1R constructs (green) in Cos-7 cells. (e) Hip1R (amino acids 1–324)-6myc. (f) Hip1R (amino acids 325–655)-6myc. (g) Hip1R (amino acids 1–655)-6myc. Bar, 10 μm.
To map the Hip1R binding domain of clathrin, we first tested whether Hip1R binds to the terminal domain (TD) of clathrin heavy chain (HC) (amino acids 1–579). As shown in Fig. 2 c, Hip1R did not copellet with GST-TD. We next tested whether Hip1R copellets with clathrin cages that have been subjected to limited tryptic digestion. This treatment hydrolyzes clathrin LCs and cleaves the 192-kD HCs, resulting in release of the globular 50-kD TD (Kirchhausen and Harrison, 1981). The remainder of the HCs, comprising the hub, proximal leg, and a portion of the distal leg (truncated cages), remain assembled and readily sedimentable (Murphy and Keen, 1992). As shown in Fig. 2 d, binding of Hip1R to truncated cages (lane 5) was undetectable. Reconstitution of truncated cages with intact clathrin LCs (Ungewickell and Oestergaard, 1982) restored binding of Hip1R to these cages (Fig. 2 d, lane 6).
The central coiled-coil domain of Hip1R binds to clathrin
To map the clathrin binding domain of Hip1R, several Hip1R truncation mutants were expressed in Escherichia coli or in Sf-9 cells (Fig. 3 a). Most of these mutants were expressed as stable proteins (Fig. 3 b, lanes labelled T), although GST–Hip1R (amino acids 1–312) was partially degraded to GST. His6–Hip1R (amino acids 1–1068), His6–Hip1R (amino acids 1–655), and His6–Hip1R (amino acids 346–1068) all pelleted with clathrin cages (Fig. 3, b, top, lanes 3, 6, and 9, and c), suggesting that amino acids 346–655 contain a clathrin binding domain. To verify that Hip1R (amino acids 346–655), which contains the putative coiled-coil domain, binds to clathrin, we also tested whether this domain alone can bind to clathrin. Fig. 3, b, bottom, lane 6, and c show that GST–Hip1R (amino acids 346–655) specifically copelleted with clathrin cages, whereas other Hip1R domains did not.
We next tested whether the putative coiled-coil domain of Hip1R interacts with clathrin in vivo. Hip1R (amino acids 325–655)-6myc and Hip1R (amino acids 1–324)-6myc were expressed in Cos-7 cells and immunoprecipitated using an anti-myc antibody. As shown in Fig. 3 d, endogenous clathrin coimmunoprecipitated with Hip1R (amino acids 325–655)-6myc (lane 3). This interaction appears to be specific since clathrin was not detected using beads alone (lane 2), or in immunoprecipitates of Hip1R (amino acids 1–324)-6myc (lane 7). As for full-length Hip1R, 0.5 M Tris-HCl disrupted the interaction between clathrin and the putative coiled-coil domain of Hip1R (lane 4). Since full-length Hip1R was not detected in immunoprecipitates with Hip1R (amino acids 325–655)-6myc, the interaction between clathrin and the coiled-coil domain is unlikely to be mediated through endogenous Hip1R (unpublished data).
We also tested whether the coiled-coil domain of Hip1R colocalizes with clathrin in Cos-7 cells. Although the NH2-terminal region that contains the ENTH-like domain followed by a conserved region (amino acids 1–324)-6myc showed no colocalization with clathrin (Fig. 3 e; Engqvist-Goldstein et al., 1999), the coiled-coil (amino acids 325–655)-6myc domain partially colocalized with clathrin in cells expressing this domain at intermediate to high levels (Fig. 3 f). At low expression levels, there was very little colocalization (unpublished data). However, Hip1R (amino acids 1–655)-6myc showed an almost perfect colocalization with clathrin, similar to full-length Hip1R (Fig. 3 g; Engqvist-Goldstein et al., 1999). We have reported previously that when the Hip1R NH2-terminal region (amino acids 1–324) is expressed in cells, it shows primarily cytosolic and nuclear localization (Engqvist-Goldstein et al., 1999). Upon closer examination, we also found that this construct localizes to patches at the cell cortex that are quite a bit larger than clathrin puncta (see arrowheads in Fig. 3 e). As indicated above, these patches do not colocalize with clathrin. In summary, the putative coiled-coil domain of Hip1R interacts with clathrin in vitro and in vivo.
Hip1R and clathrin show similar dynamics at the cortex in living cells
The dynamics of coated pit formation in live cells have been described (Gaidarov et al., 1999) by observing the behavior of GFP–clathrin LC isoform a (LCa) at the cell cortex. To gain further insight into the temporal and spatial regulation of Hip1R in comparison to clathrin during coated pit assembly, we observed the behavior of Hip1R–GFP or Hip1R–YFP and DsRed–clathrin LCa in live cells. Hip1R–GFP appears functional, since at low expression it shows similar localization to endogenous Hip1R (Engqvist-Goldstein et al., 1999), it coimmunoprecipitates with clathrin (Fig. 1 b), and does not appear to affect transferrin internalization (unpublished data). DsRed-clathrin LCa appears functional since it colocalizes with endogenous clathrin and has no apparent effects on endocytosis (unpublished data).
Hip1R and clathrin show an almost identical localization pattern in punctate structures on the cell cortex in live Cos-7 cells (Fig. 4 , a–c). Hip1R overlapped with clathrin in 98% of the puncta counted (n = 300). Similar results were obtained in other cell types, including HeLa and CHO cells (unpublished data). Approximately 85% of these structures are bona fide clathrin-coated pits, whereas the remaining structures are either CCVs or coated buds on endosomes (unpublished data). Since Hip1R colocalizes with internalized transferrin after a few minutes of uptake and is enriched in CCVs (Engqvist-Goldstein et al., 1999), it is likely that Hip1R also associates with clathrin in these compartments. We have shown previously that Hip1R–GFP exhibits similar behavior to that reported for puncta labeled with DsRed–clathrin-LCa at the cortex in Cos-7 cells (Engqvist-Goldstein et al., 1999; Gaidarov et al., 1999). That is, in both cases the majority of puncta remained relatively stationary, but their signal intensities changed dramatically, with puncta appearing and disappearing over time. The appearance of clathrin puncta at the cortex is likely to reflect assembly of coated pits and the subsequent disappearance may result when coated vesicles pinch off from the plasma membrane and are uncoated (Gaidarov et al., 1999). By observing clathrin and Hip1R in the same cell, we could now determine whether Hip1R appears and disappears together with clathrin. We observed individual puncta at the cell cortex at 2–4 s intervals for 60 frames. The lag in switching between Hip1R and clathrin fluorescence was between 1 and 2 s. At this time resolution, Hip1R and clathrin appear and disappear together (Fig. 4, d–f and Video 1, available at http://www.jcb.org/content/vol154/issue6). Out of 25 newly appearing clathrin puncta, Hip1R appeared when clathrin appeared 25 times. Out of 25 disappearing clathrin puncta, Hip1R disappeared when clathrin disappeared 25 times.
Figure 4.

Time-lapse video microscopy of Hip1R–YFP and DsRed–clathrin-LCa in live cells. These proteins display almost identical behavior at the cell cortex. Images were taken at 3 s intervals over 60 frames, where the lag between Hip1R and clathrin visualization is 1.5 s. (a–c) These images show a low magnification overview of a Cos-7 cell coexpressing Hip1R–YFP (green) and DsRed–clathrin-LCa (red). (a) Hip1R–YFP. (b) DsRed–clathrin-LCa. (c) Merge. (d–f) Selected images at the indicated times show that Hip1R–YFP and DsRed–clathrin-LCa appear (arrow) and disappear (arrowhead) at the same sites with indistinguishable timing. (d) Hip1R–YFP. (e) DsRed-clathrin-LCa. (f) Merge. A video containing images from this figure is available at http://www.jcb.org/content/vol154/issue6. Bar, 5 μm.
Hip1R localizes ultrastructurally to clathrin-coated pits in vivo, as revealed by immunogold labeling of unroofed cells, and appears to be an elongated dimer
The fact that Hip1R and clathrin show very similar temporal and spatial regulation in live cells suggested that Hip1R is recruited to the plasma membrane during formation of clathrin-coated pits. To investigate this possibility further, we determined the localization of endogenous Hip1R using an immunogold labeling technique in which clathrin-coated pits and vesicles are visualized at the plasma membrane of “unroofed cells” (Heuser, 2000b). Anti-Hip1R antibodies labeled clathrin-coated pits specifically in PC12 cells (Fig. 5 , a–j) and Cos-7 cells (Fig. 5, k–m). Fig. 5, b and k show Hip1R labeling of flat lattices, which presumably represent early stages of coat assembly, whereas Fig. 5, c–g, l, and m show Hip1R labeling of more deeply invaginated coated pits, which presumably represent later stages of coat remodeling, before the coated vesicles pinch off from the plasma membrane. In some cases, anti-Hip1R antibodies also labeled filamentous structures that appeared to connect the coated pits to adjacent actin filaments (Fig. 5, h–j, k, and m).
Figure 5.
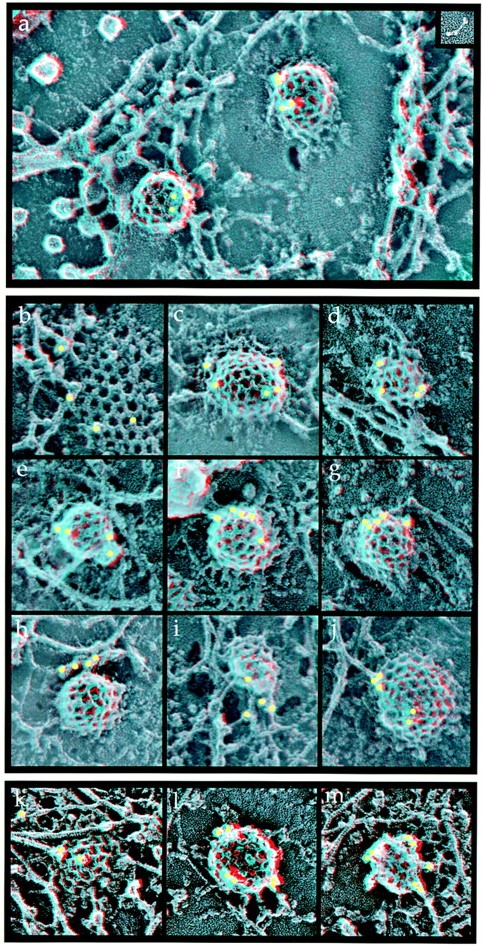
Hip1R localizes to clathrin-coated pits in vivo. Anaglyph stereo view of the inner surface of “unroofed” PC12 or Cos-7 cells that have been subjected to indirect EM immunocytochemistry with anti-Hip1R primary antibodies and 15 nm gold-tagged secondary antibodies. (a–j) PC12 cells. (k–m) Cos-7 cells. (a) A field showing the inner surface of the plasma membrane. Two clathrin-coated pits are labeled with gold (yellow dots). The inset shows a three-dimensional image of a Hip1R molecule at the same magnification absorbed on mica and visualized by quick-freeze, deep-etch electron microscopy (see also Fig. 6). (b and k) Hip1R labeling of a two-dimensional clathrin lattice. (c–g and l) Hip1R labeling of invaginated coated pits. (h–j, k, and m) In some cases, anti-Hip1R antibodies also labeled filamentous structures that appeared connected to the coated pits. Magnification, 75,000×.
To better understand Hip1R's structure and how this might relate to its function, as well as to determine what Hip1R might look like in in situ images like those shown in Fig. 5, a–m, we visualized the structure of highly purified Hip1R using the deep-etch technique, wherein macromolecules are adsorbed to mica in preparation for freeze drying and EM imaging (Heuser, 1983, 1989a). As shown in Fig. 6 , row 1–3, Hip1R appears to be very elongated and dimeric. Its size is clearly too large to be a single copy of a 120-kD protein. Its total length is ∼60 nm (±7 nm), which is comparable to that of the motor molecules kinesin (∼360 kD) (Fig. 6, row 4) and myosin II (∼450 kD) (Fig. 6, row 5). Indeed, when fully extended and adsorbed to mica, Hip1R is only slightly shorter than the diameter of a CCV (Fig. 5 a, inset). Of course, we do not yet know its actual conformation in situ. Its elongated central shaft measures ∼40 nm (±6 nm). This is presumably the central coiled-coil domain that binds to clathrin, since a 300 amino acid coiled-coil domain would be predicted to span ∼45 nm. At each end of the central shaft are found pairs of globular heads that look the same at each end, but different at the opposite ends, indicating that the molecule is a parallel dimer. At one end the heads tend to lie side by side and measure 7 to 8 nm in diameter, whereas on the other end they lie in more variable positions relative to each other and measure 8–10 nm in diameter. One set of these globular heads is presumably a pair of NH2-terminal regions containing the ENTH-like domain, whereas the other set is presumably a pair of the COOH-terminal regions containing the talin-like actin binding domain. However, we have not as yet determined which end is which. In addition to the elongated conformation, we also observed Hip1R molecules that appeared kinked (Fig. 6, row 6), a feature also shared with kinesin (Fig. 6, row 7; Hisanaga et al., 1989). Secondary structure predictions suggest a break in the central coiled-coil of Hip1R that may serve as a hinge region (Yang et al., 1999; Chopra et al., 2000).
Figure 6.
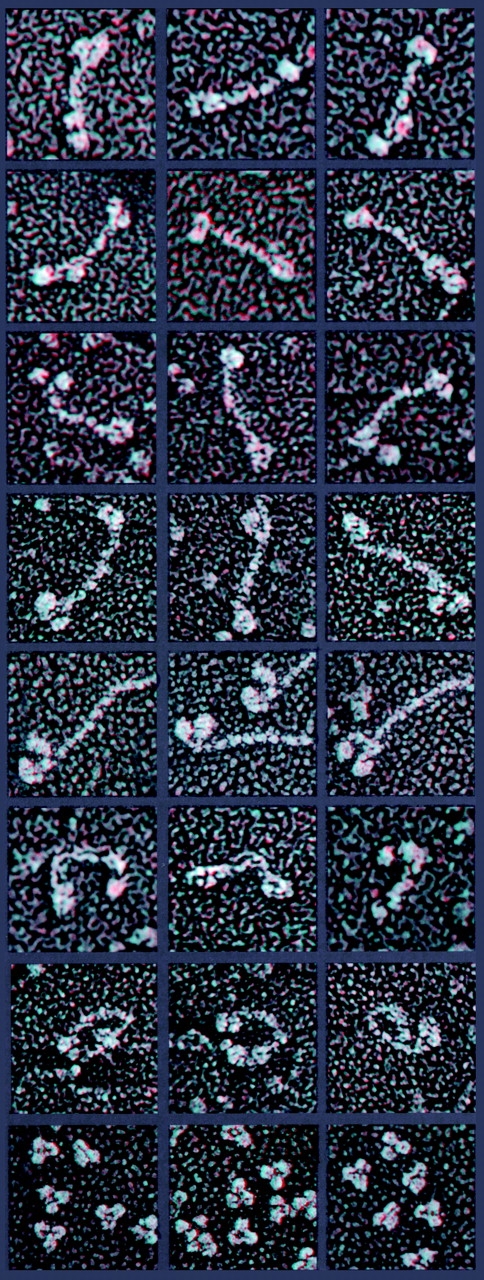
A gallery of anaglyph three-dimensional images. Images of Hip1R (rows 1–3, and 6), kinesin (rows 4 and 7), myosin II (row 5), and IgG (row 8) absorbed on mica and visualized by quick-freeze, deep-etch electron microscopy. The total length of the Hip1R molecule is ∼60 nm (±7 nm) with a central long shaft spanning 40 nm (±6 nm). At each end of the central shaft are pairs of 7–10-nm globular heads. We also observed Hip1R molecules that appeared kinked (row 6), a feature shared with kinesin (row 7). Magnification, 28,000×.
The fact that Hip1R appears by electron microscopy to be a dimer is supported by several previous observations. First, endogenous Hip1R stripped from CCVs with 0.5 M Tris-HCl migrated on a native gel filtration column close to AP2 (270 kD) (unpublished data), suggesting that Hip1R is a dimer (240 kD). Second, Hip1R coimmunoprecipitates with itself, even when clathrin is disassembled (Fig. 1 b). Moreover, we have recently shown that Hip1R and Hip1 can interact with each other in the yeast two-hybrid system (Chopra et al., 2000). Finally, studies on yeast Sla2p suggest that it also dimerizes (Wesp et al., 1997; Yang et al., 1999).
In summary, Hip1R appears in vitro to be a rod-shaped dimer some 60 nm in length, with globular heads at either end. Hip1R appears to localize in situ to the surfaces or circumferences of clathrin-coated pits, and it often appears to link coated pits with adjacent actin filaments.
Overexpression of Hip1R in Cos-7 cells causes redistribution of clathrin LC
We next investigated whether overexpression of Hip1R affects clathrin distribution. To test this possibility, we overexpressed Hip1R–myc in Cos-7 cells and looked at the distribution of endogenous clathrin. In cells expressing high levels of Hip1R–6myc, clathrin LC showed less punctate staining at the cell cortex and appeared more diffuse in the cytosol (Fig. 7 , a–c). This affect was dose dependent because at low expression levels, Hip1R–6myc and clathrin LC showed excellent colocalization (Fig. 7, d–f). Approximately 90% of cells expressing high levels of Hip1R showed clathrin LC delocalization (n = 100). However, the redistribution of clathrin LC was specific, since overexpression of Hip1R (amino acids 1–324)-6myc, which does not bind to clathrin in vitro and does not coimmunoprecipitate with clathrin, did not have this effect (Fig. 7, g–i). Only 5% of cells expressing high levels of Hip1R (amino acids 1–324)-6myc showed clathrin LC delocalization. Since clathrin LC is tightly associated with clathrin HC in vivo, we also determined whether overexpression of Hip1R affected the distribution of clathrin HC. As shown in Fig. 7, j–l, clathrin HC staining still appeared punctate in cells expressing Hip1R at high levels, although in some cells more of the clathrin HC appeared to be cytosolic.
Figure 7.
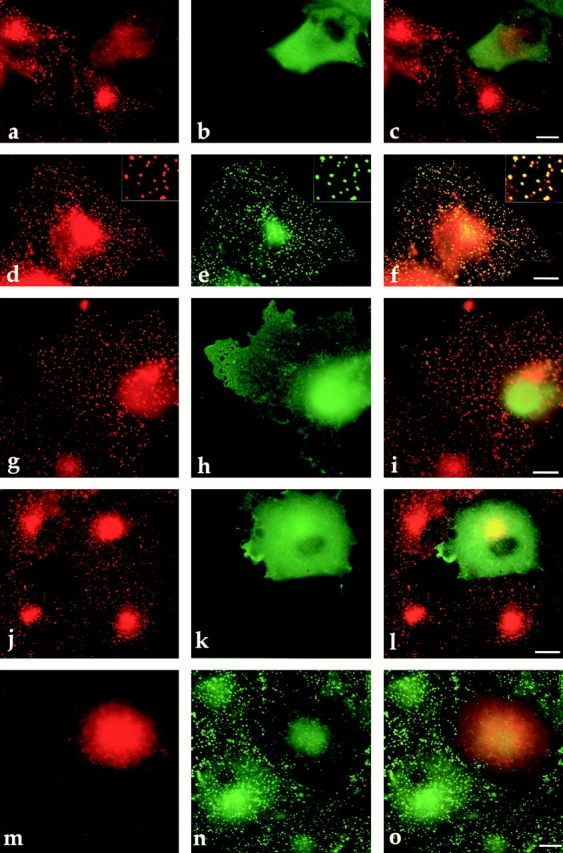
Overexpression of full-length Hip1R affects the distribution of clathrin LC. (a–l) Indirect immunofluorescence of endogenous clathrin LC (red) or clathrin HC (red) in Cos-7 cells expressing myc-tagged Hip1R constructs (green). (a–c) In cells expressing high levels of Hip1R (amino acids 1–1068)-6myc, clathrin LC is more diffuse and shows reduced staining at the cell cortex. (d–f) At low expression of Hip1R (amino acids 1–1068)-6myc, clathrin LC colocalizes with Hip1R. (g–i) High expression of Hip1R (amino acids 1–324)-6myc does not affect the distribution of clathrin LC. (j–l) Clathrin HC still shows a punctate staining pattern in cells expressing high levels of Hip1R (amino acids 1–1068). (m–o) Indirect immunofluorescence of endogenous Hip1R (green) in Cos-7 cells expressing high levels of DsRed–clathrin LCa (red). Merged images are shown in the right column. Bars, 10 μm.
Since overexpression of Hip1R affected the subcellular distribution of clathrin LC, we also tested whether overexpression of clathrin LC affected localization of endogenous Hip1R. In cells expressing high levels of DsRed–clathrin LCa, Hip1R showed reduced punctate staining at the cell cortex (Fig. 7, m–o).
Hip1R induces clathrin cage assembly in vitro
The data so far indicate that Hip1R is tightly associated with clathrin at the plasma membrane, suggesting that Hip1R might function during CCV formation. To address this possibility, we tested whether Hip1R affects clathrin assembly in vitro. Clathrin triskelia, alone and in the presence of Hip1R, were dialyzed against a buffer that supports assembly of pure clathrin only when assembly proteins are present (e.g., AP2, AP180/CALM, auxilin, and GAK) (Ungewickell and Oestergaard, 1989; Ahle and Ungewickell, 1990; Keen, 1990). Fig. 8 b, lane 6, and c, show that when His6–Hip1R was added to the assembly reaction, 40–50% of the clathrin was shifted from the high speed supernatant to the high-speed pellet, suggesting that Hip1R promoted clathrin assembly. This effect is similar to what has been reported previously for AP2 (Goodman et al., 1997) and for the auxilin-like domain of GAK (Fig. 8 a; Ahle and Ungewickell, 1990) using similar buffer conditions. The assembly of clathrin was not caused by the His6 tag since another His6-tagged protein (His6–GAK kinase domain) did not have this activity (Fig. 8 b, lane 7–9). Even a fourfold molar excess of His6–GAK kinase domain to clathrin did not shift clathrin to the high-speed pellet (unpublished data).
Figure 8.
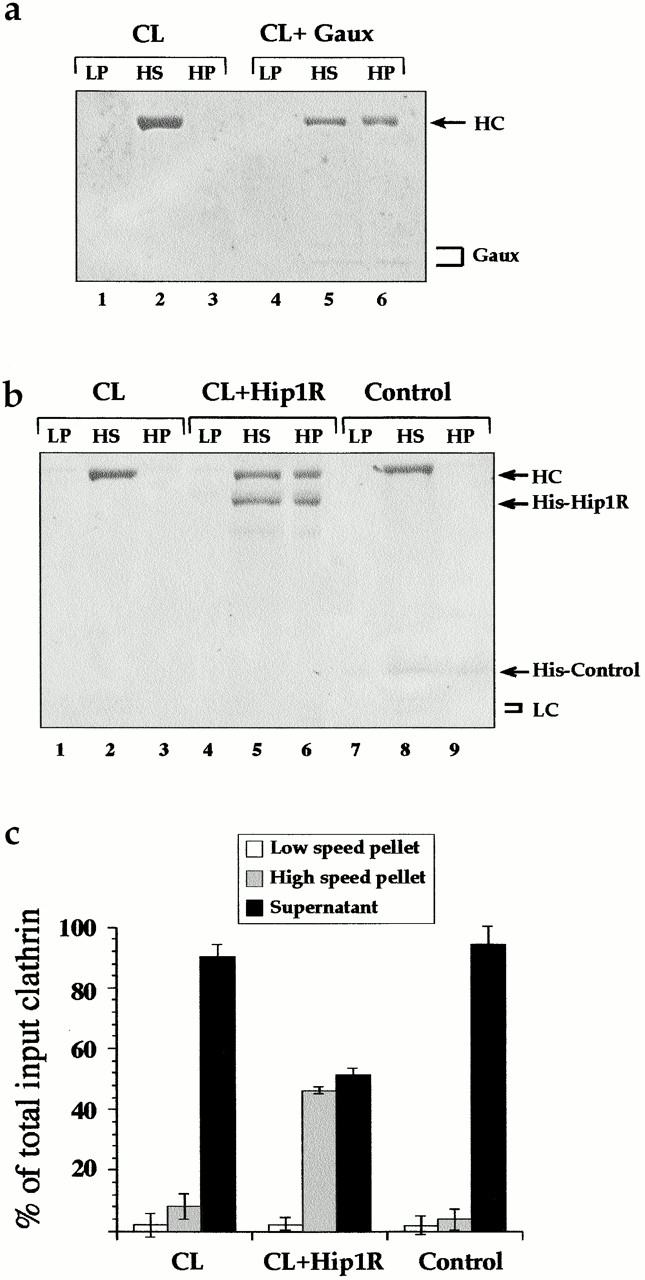

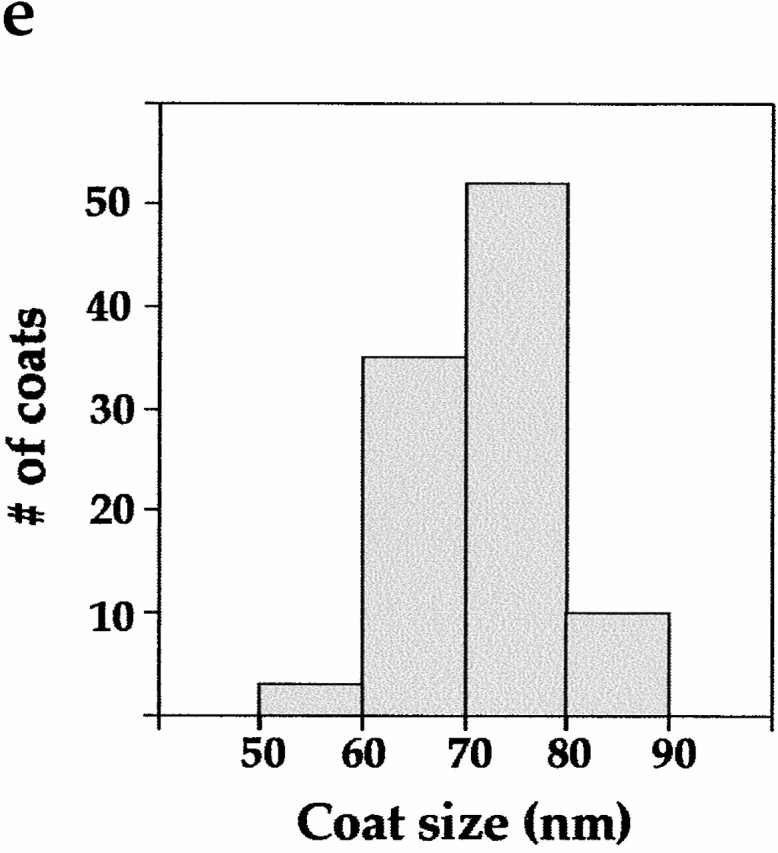
Hip1R promotes clathrin cage assembly in vitro. (a) Clathrin (400 nM) was dialyzed overnight in the absence (CL) or presence of an equimolar quantity of His6–GAK auxilin-like domain (Gaux). Equal proportions of the low speed pellet (LP), high speed supernatant (HS), and high speed pellet (HP) were run on an SDS-PAGE gel which was then stained with Coomassie blue. (b) Clathrin (200 nM) was dialyzed overnight in the absence (CL) or presence of an equimolar quantity of His6–Hip1R or His6–GAK kinase domain (control) as indicated. Equal proportions of the low speed pellet (LP), high speed supernatant (HS), and high speed pellet (HP) were run on an SDS-PAGE gel and stained with Coomassie blue. (c) The graph shows the relative amount of clathrin HC in low speed pellet, high speed supernatant, and high speed pellet using data from three independent experiments. Clathrin HC was quantified by densitometry. Values are expressed as the fraction of total input clathrin. Error bars denote ±SE. (d) Electron microscopy of negatively stained clathrin coat structures formed in the presence of His6–Hip1R. (e) Histogram of coat sizes formed in the presence of His6–Hip1R (n = 100). Bar, 100 nm.
Electron microscopy of negatively stained aliquots of the dialysate confirmed that Hip1R promoted clathrin cage assembly (Fig. 8 d). The size distribution of these cages ranged from 50–90 nm. 85% of the cages were 60–79 nm (Fig. 8 e). This is similar to what has been reported for other clathrin assembly factors (e.g., AP2 and AP180) (Ahle and Ungewickell, 1986; Keen, 1987).
Hip1R can physically link F-actin to clathrin cages in vitro
We have shown previously that the talin-like domain of Hip1R binds to F-actin in vitro and colocalizes with F-actin in vivo (Engqvist-Goldstein et al., 1999). Our biochemical and ultrastructural findings suggesting that Hip1R is a parallel dimer raise the possibility that Hip1R could cross-link clathrin and actin filaments to themselves and to each other. To test this hypothesis, low speed pelleting of mixtures of F-actin, Hip1R, and CCVs was performed. We first tested whether full-length Hip1R binds to F-actin. As shown in Fig. 9 a, lanes 15 and 16, mixtures of Hip1R and F-actin copelleted at low speed, which suggests that Hip1R can crosslink F-actin. We then mixed CCVs together with Hip1R, which caused a shift of clathrin and Hip1R to the low speed pellet (Fig. 9 a, lanes 17 and 18). Other components of CCVs, AP2 and Eps15, also shifted to the low speed pellet as determined by Western blotting (unpublished data). Finally, we added CCVs together with F-actin and Hip1R, which caused a shift of clathrin, Hip1R, and F-actin to the low speed pellet (Fig. 9 a, lanes 19 and 20). Thus, CCVs and actin do not appear to compete for binding to Hip1R, raising the possibility (tested below) that Hip1R might crosslink actin filaments and CCVs to each other. The Hip1R pelleting of CCVs appeared specific, since another actin crosslinking protein, coronin (Goode et al., 1999), did not cause a shift in clathrin pelleting (lanes 9–12), although it did crosslink actin filaments (lanes 7 and 8, and 11 and 12).
Figure 9.
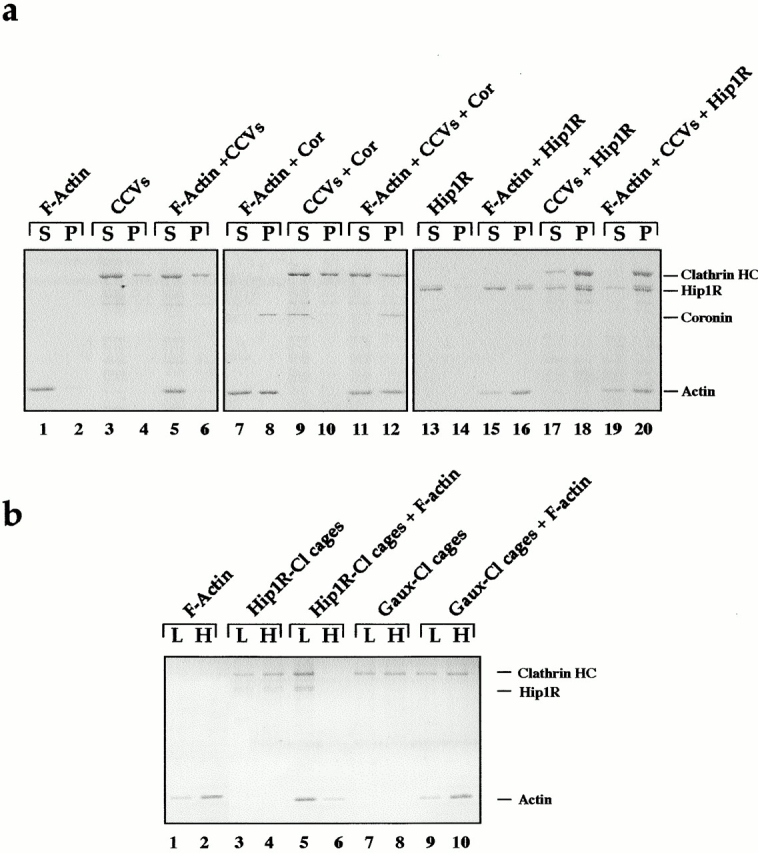
Hip1R can physically link F-actin and clathrin in vitro. (a) Hip1R crosslinks F-actin and CCVs. SDS-PAGE of a low speed F-actin cosedimentation assay. Equal proportions of the low speed pellet (P) and low speed supernatant (S) were run on an SDS-PAGE gel which was then stained with Coomassie blue. Different components (2 μM F-actin, 0.5 μM Hip1R, 0.5 μM coronin (cor), and 0.2 mg/ml CCVs) were included in the assay as indicated at the top of the gel. (b) Clathrin cages assembled with Hip1R copellet with F-actin at low speed. Clathrin (200–400 nM) was dialyzed overnight in the presence of an equimolar quantity of either His6–Hip1R (200 nM) or His6–GAK auxilin-like domain (400 nM). The Hip1R–clathrin cages and the auxilin–clathrin cages were then tested in an F-actin copelleting assay. Equal proportions of the low speed pellet (L) and the high speed pellet (H) were run on an SDS-PAGE gel which was then stained with silver. Lanes 1 and 2 show F-actin alone. Lanes 3 and 4 show clathrin cages assembled with Hip1R. Lanes 5 and 6 show clathrin cages assembled with Hip1R plus F-actin. Lanes 7 and 8 show clathrin cages assembled with the auxilin-like domain of GAK. Lanes 9 and 10 show clathrin cages assembled with the auxilin-like domain of GAK plus F-actin. The data shown are representative of three independent experiments.
Since Hip1R alone caused a shift of CCVs to the low speed pellet, we could not determine in the above experiment whether Hip1R could link clathrin and F-actin physically. To address this question, we performed F-actin pelleting assays using Hip1R-assembled clathrin cages instead of CCVs. Under buffer conditions used for F-actin pelleting, 30–40% of clathrin cages assembled with Hip1R pelleted at low speed (Fig. 9 b, lanes 3 and 4). Addition of F-actin resulted in 100% of the Hip1R-assembled clathrin cages pelleting at low speed and a shift of F-actin from the high speed pellet to the low speed pellet (Fig. 9 b, lanes 5 and 6). This effect appeared specific since GAK (auxilin-like domain)-assembled clathrin cages did not shift to the low speed pellet upon adding F-actin (Fig. 9 b, lanes 9 and 10). Furthermore, GAK (auxilin-like domain)-assembled clathrin cages did not cause a shift of F-actin to the low speed pellet (Fig. 9 b, lanes 9 and 10). Taken together, these results demonstrate that Hip1R can physically link F-actin and clathrin cages in vitro.
Discussion
We have shown previously that Hip1R is a component of clathrin-coated pits and vesicles that may link the endocytic machinery to the actin cytoskeleton (Engqvist-Goldstein et al., 1999). To date, very little is known about how the members of Sla2/Hip1 family interact with the endocytic machinery. Here, we showed that Hip1R binds directly to clathrin cages with high affinity and coimmunoprecipitates with clathrin from cell and tissue extracts. Furthermore, Hip1R and clathrin show a remarkably similar temporal and spatial regulation at the cell cortex in live cells. Not only do these proteins show a very similar localization pattern, but they appear and disappear at the cortex together, within the time resolution of these experiments. The appearance of clathrin puncta at the cortex is likely to reflect assembly of clathrin-coated pits (Gaidarov et al., 1999). These data suggest that Hip1R is recruited to the cell cortex during early stages of endocytosis. Further support for this conclusion comes from data showing that Hip1R localizes to coated pits at the ultrastructural level. Anti-Hip1R antibodies specifically labeled two-dimensional clathrin lattices and invaginated clathrin-coated pits in “unroofed” cells.
Similar to other endocytic proteins present in clathrin-coated pits (e.g., AP2, AP180) (Ungewickell and Oestergaard, 1989; Keen, 1990), Hip1R promotes assembly of clathrin in vitro. Although we currently do not know whether the clathrin assembly promoting activity of Hip1R is important in vivo, our data so far clearly demonstrate that Hip1R associates with assembled clathrin in vivo. AP2 and AP180 are both major components of the clathrin coat, involved in recruiting clathrin to the plasma membrane through interactions with PI(4,5)P2, and, in the case of AP2, through interactions with receptors (for review see Marsh and McMahon, 1999; Brodin et al., 2000). Recent efforts to reconstitute the early stages of endocytosis in vitro showed that the minimal requirements for the initial stage of coated pit invagination are clathrin, AP2, AP180, and PIP2-containing membranes (Ford et al., 2001). However, with these components the pits did not invaginate completely, suggesting that additional components may be required for formation of coated pits (Ford et al., 2001). Hip1R may be required to form coated pits with proper morphology and might affect the kinetics of pit formation. In addition, since the actin cytoskeleton is an integral part of the cell cortex and has been implicated in early stages of endocytosis, the actin-binding properties of Hip1R might affect coated pit formation in vivo.
The putative central coiled-coil domain of Hip1R interacts with clathrin. The coiled-coil domain copelleted with clathrin cages in vitro and coimmunoprecipitated from extracts with clathrin. Furthermore, the Hip1R coiled-coil domain partially colocalized with clathrin. The Hip1R coiled-coil domain does not contain the clathrin binding motifs L(L,I)(D,E,N)(L,F)(D,E), referred to as the “clathrin box,” or the shorter DLL motif found in many proteins that interact with the TD of clathrin HC (Dell'Angelica et al., 1998; Morgan et al., 2000; ter Haar et al., 2000). Consistent with these observations, we did not detect any binding of Hip1R to the TD of clathrin. Instead, our in vitro binding studies suggest that LCs contribute to the binding of Hip1R to clathrin. Truncated cages reconstituted with LCs restored Hip1R binding to clathrin. Furthermore, overexpression of Hip1R affected the subcellular distribution of endogenous clathrin LC in vivo. In cells expressing high levels of Hip1R, clathrin LC appeared more diffuse and there were fewer large puncta at the cortex. This may result if Hip1R binds to LC and sequesters LC in the cytosol. An alternative possibility that we considered is that Hip1R blocks the access of clathrin LC antibody by binding tightly to LC. However, we favor the former possibility because clathrin LC antibody can immunoprecipitate clathrin cages saturated with Hip1R in vitro (unpublished data).
Although the localization of clathrin LC was affected by Hip1R overexpression, clathrin HC localization appeared essentially normal. However, we cannot exclude the possibility that there was a partial redistribution of the HC that would be difficult to detect by light microscopy. In light of the redistribution of clathrin LC, it is surprising that there was no detectable reduction in internalization of transferrin in cells overexpressing Hip1R (unpublished data). One possibility is that Hip1R only partially competes and/or sequesters clathrin LC from coated pits and that the remaining LC can maintain clathrin function. It is interesting to note that clathrin LCs are thought to inhibit HC assembly, so that lattice formation does not spontaneously occur under physiological conditions (Ungewickell and Ungewickell, 1991; Ybe et al., 1998). Since Hip1R binding to truncated clathrin cages depends on LC, Hip1R might promote clathrin assembly via interactions with LC.
A Hip1R fragment containing the NH2-terminal region plus the coiled-coil domain colocalized with clathrin in cells much better than the coiled-coil domain alone. This suggests the coiled-coil domain alone is not sufficient for proper association of Hip1R with clathrin-coated pits and vesicles. However, the NH2-terminal region alone did not colocalize with clathrin, although it localizes to patches at the cell cortex. The NH2-terminal region contains an ENTH-like domain and a region of ∼150 amino acids that is conserved within the Sla2/Hip1 family. The ENTH-like domain of the Sla2/Hip1 family members is similar to domains of AP180/CALM, which have been shown recently to interact with PI(4,5)P2 at the plasma membrane (Ford et al., 2001). Because all of the CALM residues implicated in PI(4,5)P2 binding are conserved in Hip1R, it is highly likely that Hip1R also associates with PI(4,5)P2 (Ford et al., 2001). This activity may increase the affinity of Hip1R for coated pits, as has been shown for other endocytic proteins (e.g., AP2) that bind to PIP2 (Gaidarov and Keen, 1999). In yeast, it has also been shown that both the NH2-terminal region and the coiled-coil domain of Sla2p are important for endocytosis (Wesp et al., 1997).
Studies on clathrin dynamics in vivo have suggested that the actin cytoskeleton might provide a scaffold at the plasma membrane for the endocytic machinery. Depolymerization of F-actin caused an increase in the lateral movement of clathrin-coated pits (Gaidarov et al., 1999). There is also evidence that in some cell types, depolymerization of F-actin traps CCVs before detachment from the plasma membrane (Lamaze et al., 1997). Our previous data suggested that Hip1R may provide a link between the endocytic machinery and F-actin (Engqvist-Goldstein et al., 1999). We now demonstrate that full-length Hip1R can directly link F-actin and clathrin in vitro. Clathrin cages assembled with Hip1R shifted from the high speed pellet to the low speed pellet when F-actin was included in the reaction. Furthermore, Hip1R copelleted with both F-actin and CCVs at low speed, demonstrating that Hip1R promotes assembly of clathrin and F-actin into higher order structures in vitro. Immunolocalization of Hip1R to the interface between clathrin-coated pits and actin filaments in “unroofed” cells provides support for the in vivo relevance of this activity.
Our biochemical data are consistent with the structural analysis of Hip1R, showing that it is a rod-shaped apparent dimer with globular heads at either end, spanning 60 nm in length. These globular heads are presumably the NH2-terminal region, which contains the ENTH-like domain, and the COOH-terminal region, which contains the talin-like actin binding domain. Therefore, a Hip1R dimer may bind to at least two actin filaments and two clathrin molecules, thereby creating higher order complexes which would associate with the plasma membrane via interactions of the ENTH-like domain with PIP2. Since Hip1R can also promote assembly of clathrin cages in vitro, we speculate that Hip1R could assemble clathrin on the lipid bilayer. In total, Hip1R's properties suggest that it may act as a scaffolding protein at the interface between clathrin, F-actin, and lipids during endocytosis.
Materials and methods
DNA constructs
Full-length Hip1R (amino acids 1–1068), Hip1R (amino acids 1–655), Hip1R (amino acids 346–1068), and the auxilin and kinase domains of GAK were expressed with an His6 tag at the NH2 terminus in Sf9 cells. Hip1R (amino acids 1–1068), Hip1R (amino acids 1–655), and Hip1R (amino acids 346–1068) were amplified by PCR using primers that generate Sal1 and Not1 sites at the 5′ and 3′ ends, respectively. The PCR products were then ligated into pFASTBAC Hta (GIBCO BRL). Recombinant baculoviruses were generated according to the manufacturer (GIBCO BRL).
For in vitro clathrin cage binding assays, Hip1R fragments were expressed as GST fusion proteins in E. coli. Hip1R (amino acids 1–312) was amplified by PCR using primers that generate BamHI and NsiI sites at the 5′ and 3′ ends, respectively. Hip1R (amino acids 346–655) was amplified using primers that generate XhoI and HindIII sites at the 5′ and 3′ ends, respectively. The PCR products were cut with the above restriction enzymes and ligated into the pGAT2 as described in (Engqvist-Goldstein et al., 1999).
Construction of GST-TD of clathrin (amino acids 1–579) was described in Goodman et al. (1997).
For expression of Hip1R constructs in mammalian cells, full-length protein (amino acids 1–1068) and fragments of Hip1R were expressed as GFP fusions, or were expressed with an myc tag at the COOH terminus. Hip1R (amino acids 325–655)-6myc was constructed as described in Engqvist-Goldstein et al. (1999). Construction of Hip1R (amino acids 1–1068)-GFP, Hip1R (amino acids 1–1068)-6myc, Hip1R (amino acids 1–324)-6myc, Hip1R (amino acids 1–655)-6myc, Hip1R (amino acids 346–1068)-6myc, and Hip1R (amino acids 766–1068)-6myc were described in Engqvist-Goldstein et al. (1999). All constructs were sequenced to ensure that no mutations were introduced during PCR.
Expression and purification of recombinant proteins
Expression in insect cells (Sf9) and purification of His6-tagged full-length Hip1R (amino acids 1–1068), Hip1R (amino acids 1–655), Hip1R (amino acids 346–1068), GAK (auxilin-like domain), and GAK (kinase domain), were done according to the manufacturer (GIBCO BRL). Pooled fractions containing pure proteins were dialyzed overnight in 1 L of HEKG buffer (20 mM K-Hepes, pH 7.4, 1 mM EDTA, 50 mM KCl, 5% glycerol) containing 0.4 mM PMSF with one exchange of buffer, snap frozen in liquid N2 and stored at −80°C.
GST–Hip1R (amino acids 1–312), GST–Hip1R (amino acids 346–655), GST–Hip1R (amino acids 766–1068), GST-TD of clathrin HC (amino acids 1–579) and GST alone were expressed in E. coli and purified as described in Engqvist-Goldstein et al. (1999).
Immunoprecipitations
For immunoprecipitation from brain extracts, five mouse brains (Pel-Freez Biologicals) were minced using a razor blade and homogenized at 4°C using a dounce glass homogenizer (size AA; ∼20 strokes). For homogenization, 2 ml/g tissue homogenization buffer (10 mM Hepes, pH 7.4, 1 mM EDTA) containing protease inhibitors cocktail A (10 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitors, and 1 mM PMSF) was used. The samples were centrifuged at 4°C for 50 min at 7,500 rpm in a SA-600 rotor (Beckman Coulter). The resulting supernatant fraction was then centrifuged at 4°C for 50 min at 55,000 rpm in a TLA 100.3 rotor (Beckman Coulter). The pellet was resuspended in 1.2 mL buffer A (20 mM Hepes, pH 7.4, 1 mM EDTA, 5 mM MgCl, 50 mM KCl, 1% NP-40) containing the protease inhibitor cocktail A and homogenized as described above. The extract was then spun at 4°C for 15 min at 15,000 rpm in a microcentrifuge. Antibodies were then added to 200 μL of extract and incubated at 4°C on a rotating platform. After 2 h, swollen protein A or protein G beads were added to the extract/antibody mixture (30 μL of 50% slurry) and incubated an additional 1.5 h at 4°C on a rotating platform. The supernatant (200 μL) was collected and mixed with 100 μL 3× SDS sample buffer and boiled. The beads were washed three times with 1 mL of buffer A and then resuspended in 300 uL of SDS sample buffer and boiled. Equal portions of homogenate, supernatant (S), and pellet (P) were loaded on SDS-PAGE gels and analyzed by immunoblotting.
For immunoprecipitation from cell extracts, Cos-7 cells were plated at 1.5 × 106 cells per T-75 flask. Transfections were performed using the lipofectamine method according to the manufacturer (GIBCO BRL). 36 h after transfection, cells were washed once with PBS, pH 7.4, and incubated on ice. To lyse the cells, 0.2 ml of cold lysis buffer (20 mM Hepes, pH 7.4, 1 mM EDTA, 50 mM KCl, 1% NP-40) containing protease cocktail A was added directly to the plates and cells were scraped off and homogenized by passaging through a 27-gauge needle. Immunoprecipitations were performed as described above. The beads were resuspended in 20 μL of SDS sample buffer and boiled. For treatment with 0.5 M Tris-HCl, beads were washed an additional three times with the same buffer with or without 0.5 M Tris-HCl. Equal volumes of homogenate and beads were loaded on SDS-PAGE gels and analyzed by immunoblotting.
The following antibodies were used for immunoprecipitation experiments. Polyclonal anti-Hip1R (GP#8; Engqvist-Goldstein et al., 1999) was used at 1:20; monoclonal anti–α-adaptin antibody (AP.6; Affinity BioReagents, Inc.) was used at 1:100; polyclonal anti-Eps15 antibody (Transduction Laboratories) was used at 1:100; polyclonal anti-myc antibody (Santa Cruz Biotechnology, Inc.) was used at 1:100; and polyclonal anti-GFP antibody (provided by Dr. Pam Silver and Dana Farber, Harvard Medical School, Cambridge, MA) was used at 1:500. The following antibodies were used for Western blotting. Polyclonal anti-Hip1R antibodies (GP#8 and GP#6) were used at 1:1,000; monoclonal anti–α-adaptin antibody (AC1-M11; Affinity BioReagents, Inc.) was used at 1:1,000; polyclonal anti-Eps15 antibody was used at 1:2,000; monoclonal anticlathrin antibody (Transduction Laboratories) was used at 1:1,000; monoclonal antidynamin antibody (hudy 1; Upstate Biotechnology) was used at 1:2,000; monoclonal anti-myc antibody (9E10; Santa Cruz Biotechnology, Inc.) was used at 1:1,000; polyclonal anti-GFP antibody was used at 1:20,000; and polyclonal amphiphysin II antibody (provided by Harvey T. McMahon, MRC Laboratories, Cambridge, UK) was used at 1:2,000.
Clathrin cage binding assays
To obtain pure clathrin for binding assays, clathrin triskelia were purified from bovine brain. First, CCVs were purified as described in Engqvist-Goldstein et al. (1999). Then clathrin was purified from CCVs as described (Zaremba and Keen, 1983). Fractions containing only clathrin heavy chain (HC) and light chain (LC), as judged by Coomassie-stained gels, and no detectable levels of AP2, as judged by immunoblotting, were used for assembly of clathrin cages. Preparation of intact and truncated clathrin cages reconstituted with clathrin LCs has been described elsewhere (Keen et al., 1991). Hip1R binding to cages was performed as described in Goodman et al. (1997). In brief, full-length Hip1R or Hip1R domains were incubated in the presence of clathrin cages in buffer B (0.1 M Mes, pH 6.5, 1 mm EGTA, 0.5 mm MgCl2) for 10 min at room temperature. Complexes were centrifuged through a 50 μL 0.2 M sucrose cushion prepared in buffer B using a TLA100 rotor (Beckman Coulter) at 75,000 rpm for 5 min at 4°C and analyzed on 8.5% SDS-PAGE gels by Coomassie staining or immunoblotting. Hip1R bands were quantified by densitometry.
GST-TD binding assay
GST and GST-TD of clathrin (0.5 μM) in a 15-μl bed volume of glutathione-agarose and His6–Hip1R (0.1 μM) were incubated in 100 μl 20 mM Hepes, pH 7.4, 1 mM EDTA, 50 mM KCl for 1 h at 4°C on a rotator. The beads were then centrifuged and washed three times with 1 ml of the same buffer containing 1% Triton X-100 and 100 mM KCl and eluted with SDS-PAGE sample buffer and analyzed on 8.5% SDS-PAGE gels by Coomassie staining.
Transfection of COS-7 cells with cDNA encoding Hip1R and clathrin fusion proteins
Transfections were performed using the lipofectamine method according to the manufacturer (GIBCO BRL). Typically, 1 μg of DNA per T-25 flask was used. Transfections were performed for 5 h at 37°C. The solution was then replaced by complete medium (DME containing 10% FBS). After 3 h, cells were trypsinized and replated on coverslips at 20–30% confluency. Approximately 36 h later, cells were processed for immunofluorescence or for imaging of Hip1R–YFP and DsRed–clathrin-LCa in living cells or for transferrin uptake assay.
Cell culture and indirect immunofluorescence
Cell culture and immunofluorescence were performed as described in (Engqvist-Goldstein et al., 1999). The following primary antibodies were used: polyclonal anti-Hip1R antibody (GP#8) was used at 1:20; monoclonal anti–α-adaptin antibody (AP.6) and monoclonal anti-clathrin antibody (X22; Affinity Bioreagents, Inc.) were used at 1:150; monoclonal anticlathrin LC antibody (CON.1; Covance Research Products) was used at 1:200; monoclonal anti-myc antibody (9E10) was used at 1:4,000; and polyclonal anti-myc antibody (Santa Cruz Biotechnology, Inc.) was used at 1:400. The following secondary antibodies were used. FITC goat anti–guinea pig (ICN Biomedicals); FITC donkey anti–mouse (Jackson ImmunoResearch Laboratories); rhodamine donkey anti–mouse (Jackson ImmunoResearch Laboratories); FITC donkey anti–rabbit (Jackson ImmunoResearch Laboratories); rhodamine donkey anti–rabbit (Jackson ImmunoResearch Laboratories). The coverslips were viewed using an inverted fluorescence microscope (Eclipse TE300; Nikon). Images were captured using a Hamamatsu ORCA cooled CCD camera. Images were recorded digitally (Phase3 Imaging Systems; ImageProPlus). All images were prepared for publication using Adobe Photoshop® software.
Imaging of Hip1R–YFP and DsRed–clathrin-LCa in living cells
Cos-7, HeLa, and CHO cells were transfected with Hip1R–GFP or Hip1R–YFP and DsRed–clathrin-LCa as described above. Cells were grown on uncoated 35-mm coverslip dishes (MatTek Corporation), and transferred to medium containing 25 mM Hepes, pH 7.4, before viewing. The microscope stage was fitted with an open perfusion microincubator attached to a temperature controller (Harvard Apparatus) maintained at 37°C for the duration of the experiment, unless stated otherwise. Temperature of the viewing objective (Plan-Apo 1.40 NA 63× oil immersion lens; ZEISS) was monitored separately with a YSI Precision 4000A Thermometer and regulated with an external heater fan. Images were collected on a Quantix cooled, CCD camera containing either a Kodak 1400 chip, or the newer Kodak 1401E chip (Photometrics), yielding a pixel size of 108 nm for all images. GFP and YFP were visualized using specific individual filter sets for each fluorophore (Chroma). To image YFP and DsRed1, excitation/emission filter sets were switched between FITC and Texas red (TxRd), using a common FITC/TxRd double dichroic mirror (Chroma). Excitation light originated from a 100-W mercury lamp, and was attenuated to 32% with a neutral density filter. Filter wheels (dual or single 6-position wheels with shutter; Ludl Electronic Products) were mounted on the excitation and emission ports of the microscope and were controlled with IPLab software (Scanalytics). All images were processed for analysis with IPLab and Photoshop® (Adobe).
To collect high quality images with good sensitivity, cells were photographed by rapidly alternating excitation/emission filters (YFP/DsRed1), and exposing for 0.4–1.0 s to capture each fluorescent marker individually. The resulting data consisted of two image stacks (one for each marker), where frame 1 of stack 1 was the first image taken, frame 1 of stack 2 was the second image, frame 2 of stack 1 was third, and so on. For analysis, each grayscale frame from stack 1 (green) was color merged with the same frame number in stack 2 (red), to create a pseudosimultaneous color stack.
Immunogold labeling of “unroofed” cells
Labeling of “unroofed” cells with anti-Hip1R antibodies was performed as described in (Heuser, 1989b, 2000b). In brief, PC12 and Cos-7 cells were grown on carbon-coated glass and “unroofed” by brief exposure to an ultrasonic burst in “KHMgE medium” (70 mM KCl, 30 mM Hepes buffer, pH 7.2, 5 mM MgCl2, and 3 mM EGTA). Immediately after “unroofing,” the cells were fixed for 30 min in 2% formaldehyde freshly dissolved from paraformaldehyde into KHMgE medium, “quenched” for 10 min with 50 mM lysine and 50 mM glycine in KHMgE, and labeled for 1 h each with anti-Hip1R antibody (GP#8) at 1:10 dilution followed by a commercial 15-nm gold-tagged secondary antibody. Samples were then processed by quick-freezing, freeze-drying, platinum replication in preparation for viewing in the electron microscope. For stereomicroscopy, the replica was mounted in the eucentric side-entry goniometer stage of a JEOL 200CX electron microscope, imaged at 20,000–70,000×, and photographed at ±10° of tilt off the vertical axis. For preparation of digital “anaglyph” three-dimensional images from the resultant stereo pairs of EM negatives, see Heuser (2000a)(c).
Deep-etch technique for macromolecules adsorbed to mica flakes
Purified His6–Hip1R (∼10 μg/ml) was briefly adsorbed to freshly cleaved mica and then quick-frozen, freeze-fractured, and deep-etched as described previously (Heuser, 1983, 1989a). Replicas were viewed in a JEOL 200CX transmission electron microscope operated at 100 kV, and stereo images were obtained using ±10° of tilt with an eucentric side-entry goniometer stage. Final digital “anaglyph” three-dimensional images were generated by making digital copies of stereo pairs of standard EM negatives and merging them together in separate colors with Adobe Photoshop®, as described in Heuser (2000a).
Clathrin assembly assay
Clathrin assembly assays have been described (Keen, 1987; Goodman et al., 1997). In brief, clathrin triskelia (100–400 nM) was dialyzed overnight in 0.1 M Na-MES, pH 6.8, containing 0.1 mM PMSF in the presence of 0–400 nM His6–Hip1R (amino acids 1–1068) and/or 0–800 nM of His6–GAK (kinase domain) and/or ∼400 nM of His6–GAK (auxilin-like domain) using a Slide-A-Lyzer (Pierce Chemical Co.). The dialyzates were microcentrifuged at 10,000 g for 2 min (generating low speed pellet and supernatant fractions) and then at 75,000 rpm for 5 min in a TL100 rotor (generating high speed pellet and supernatant fractions). Equal portions of the fractions were analyzed on 8.5% SDS-PAGE, and clathrin HC was quantified by densitometry of Coomassie blue staining. Aliquots of the dialyzate were also stained with 1% uranyl acetate and viewed in a FEI Tecnai 12 (120 kV) transmission electron microscope.
Assay for F-actin pelleting with CCVs
Low speed F-actin cosedimentation assays were performed as described in Goode et al. (1999) with minor alterations. In brief, 2 μM rabbit skeletal muscle actin (Cytoskeleton, Inc.) was incubated with 0.2 mg/mL CCVs (purified from mouse brain), and/or 0.5 μM Hip1R and/or 0.5 μM yeast coronin in actin polymerization buffer (5 mM Tris-HCl, pH 7.5, 0.2 mM DTT, 0.2 mM CaCl2, 0.7 mM ATP, 150 mM KCl, 2 mM MgCl2) for 30 min at room temperature in a final volume of 30 μl. The samples were then spun for 5 min at 14,000 rpm in a tabletop centrifuge. The supernatant (30 μL) was collected and mixed with 15 μL 3× SDS sample buffer and boiled. The pellets were resuspended in 45 μL 1× SDS sample buffer and boiled. Equal portions of supernatant (S) and pellet (P) samples were loaded on SDS-PAGE gels.
Assay for F-actin pelleting with clathrin cages
Clathrin cages were assembled with Hip1R or with the auxilin-like domain of GAK as described above (see clathrin assembly assay). The dialyzates were then centrifuged at 75,000 rpm for 5 min in a TL100 rotor and resuspended in buffer B (0.1 M MES, 1.0 mM EGTA, 0.5 mM MgCl2, pH 6.5). To remove any aggregates, the cages were spun for 5 min at 14,000 rpm in a tabletop centrifuge. F-actin (1.25 μM) was polymerized in a final volume of 30 μl for 30 min as described above (see F-actin pelleting assays with CCVs). 10 μL of the assembled clathrin cages, containing either Hip1R or the auxilin-like domain of GAK, were added to the polymerized actin and incubated for an additional 10 min at room temperature. The samples were then centrifuged through a 50 μl 0.2 M sucrose cushion prepared in buffer B at 10,000 g for 5 min (generating low speed pellet and supernatant fractions) and then at 75,000 rpm for 5 min in a TL100 rotor (generating high speed pellet and supernatant fractions). Equal portions of the fractions were analyzed on 8.5% SDS-PAGE gels by Coomassie staining.
Online supplemental material
The video shows time-lapse analysis of Hip1R–YFP (green) and DsRed–clathrin-LCa (red) coexpressed in a Cos-7 cell. We observed individual puncta at the cell cortex at 3 s intervals over 60 frames, where the lag between Hip1R and clathrin visualization is 1.5 s. At this time resolution, Hip1R and clathrin appear (arrow) and disappear (arrowhead) together. The first frame of this video is shown in Fig. 4 c. Additional frames of a selected area of this cell are shown in Fig. 4 f. The video was accelerated 15 times in relation to real time. The video is available at http://www.jcb.org/content/vol154/issue6.
Supplemental Material
Acknowledgments
We thank Ann Fischer for her advice on cell culture, Kent McDonald and Reena Zalpuri for advice on electron microscopy, Claire X. Zhang for providing GAK constructs, Kimberly L. Cunningham for technical help, Bruce L. Goode for providing purified yeast coronin, and Dr. Pam Silver for anti-GFP antibodies, and Harvey T. McMahon for antiamphiphysin II antibody. We are also grateful to Frances M. Brodsky and Claire X. Zhang for stimulating discussions and helpful comments on the manuscript.
This work was supported by a grant from the National Institutes of Health to D.G. Drubin (DK32094).
The online version of this article contains supplemental material.
Footnotes
Abbreviations used in this paper: CCV, clathrin-coated vesicles; ENTH, epsin NH2-terminal homology domain; GAK, cyclin G–associated kinase; GFP, green fluorescent protein; GST, glutathione S-transferase; HC, heavy chain; Hip1R, huntingtin-interacting protein 1 related; LC, light chain; PI(4,5)P2, phosphatidylinositol-4, 5-biphosphate; TD, terminal domain.
References
- Ahle, S., and E. Ungewickell. 1986. Purification and properties of a new clathrin assembly protein. EMBO J. 5:3143–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahle, S., and E. Ungewickell. 1990. Auxilin, a newly identified clathrin-associated protein in coated vesicles from bovine brain. J. Cell Biol. 111:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin, L., P. Low, and O. Shupliakov. 2000. Sequential steps in clathrin-mediated synaptic vesicle endocytosis. Curr. Opin. Neurobiol. 10:312–320. [DOI] [PubMed] [Google Scholar]
- Chopra, V.S., M. Metzler, D.M. Rasper, A.E.Y. Engqvist-Goldstein, R. Singaraja, L. Gan, K.M. Fichter, K. McCutcheon, D. Drubin, D.W. Nicholson, and M.R. Hayden. 2000. HIP12 is a non-proapoptotic member of a gene family including HIP1, an interacting protein with huntingtin. Mamm. Genome. 11:1006–1015. [DOI] [PubMed] [Google Scholar]
- Cremona, O., and P. De Camilli. 2001. Phosphoinositides in membrane traffic at the synapse. J. Cell Sci. 114:1041–1052. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica, E.C., J. Klumperman, W. Stoorvogel, and J.S. Bonifacino. 1998. Association of the AP-3 adaptor complex with clathrin. Science. 280:431–434. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein, A.E.Y., M.M. Kessels, V.S. Chopra, M.R. Hayden, and D.G. Drubin. 1999. An actin-binding protein of the Sla2/huntingtin interacting protein 1 family is a novel component of clathrin-coated pits and vesicles. J. Cell Biol. 147:1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, M.G.J., B.M.F. Pearse, M.K. Higgins, Y. Vallis, D.J. Owen, A. Gibson, C.R. Hopkins, P.R. Evans, and H.T. McMahon. 2001. Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science. 291:1051–1055. [DOI] [PubMed] [Google Scholar]
- Gaidarov, I., and J.H. Keen. 1999. Phosphoinositide–AP-2 interactions required for targeting to plasma membrane clathrin-coated pits. J. Cell Biol. 146:755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidarov, I., F. Santini, R.A. Warren, and J.H. Keen. 1999. Spatial control of coated-pit dynamics in living cells. Nat. Cell Biol. 1:1–7. [DOI] [PubMed] [Google Scholar]
- Goode, B.L., J.J. Wong, A.C. Butty, M. Peter, A.L. McCormack, J.R. Yates, D.G. Drubin, and G. Barnes. 1999. Coronin promotes the rapid assembly and cross-linking of actin filaments and may link the actin and microtubule cytoskeletons in yeast. J. Cell Biol. 144:83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman, O.B., J.G. Krupnick, V.V. Gurevich, J.L. Benovic, and J.H. Keen. 1997. Arrestin/clathrin interaction: localization of the arrestin binding locus to the clathrin terminal domain. J. Biol. Chem. 272:15017–15022. [DOI] [PubMed] [Google Scholar]
- Heuser, J.E. 1983. Procedure for freeze-drying molecules adsorbed to mica flakes. J. Mol. Biol. 169:155–195. [DOI] [PubMed] [Google Scholar]
- Heuser, J.E. 1989. a. Protocol for 3-D visualization of molecules on mica via the quick-freeze, deep-etch technique. J. Electron Microsc. Tech. 13:244–263. [DOI] [PubMed] [Google Scholar]
- Heuser, J.E. 1989. b. Effects of cytoplasmic acidification on clathrin lattice morphology. J. Cell Biol. 108:401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser, J.E. 2000. a. Membrane traffic in anaglyph stereo. Traffic. 1:35–37. [DOI] [PubMed] [Google Scholar]
- Heuser, J.E. 2000. b. The production of ‘cell cortices’ for light and electron microscopy: follow the middle road. Traffic. 1:545–552. [DOI] [PubMed] [Google Scholar]
- Heuser, J.E. 2000. c. How to convert a traditional electron microscopy laboratory to digital imaging. Traffic. 1:614–621. [DOI] [PubMed] [Google Scholar]
- Hisanaga S., H. Murofushi, K. Okuhara, R. Sato, Y. Masuda, H. Sakai, and N. Hirokawa. 1989. The molecular structure of adrenal medulla kinesin. Cell Motil. Cytoskeleton. 12:264–272. [DOI] [PubMed] [Google Scholar]
- Holtzman, D.A., S. Yang, and D.G. Drubin. 1993. Synthetic-lethal interactions identify two novel genes, SLA1 and SLA2, that control membrane cytoskeleton assembly in Saccharomyces cerevisiae. J. Cell Biol. 122:635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, T., S. Koshiba, T. Kigawa, A. Kikuchi, S. Yokoyama, and T. Takenawa. 2001. Role of the ENTH domain in phosphatidylinositol-4,5-bisphosphate binding and endocytosis. Science. 291:1047–1051. [DOI] [PubMed] [Google Scholar]
- Jarousse, N., and R.B. Kelly. 2001. Endocytic mechanism in synapses. Curr. Opin. Cell Biol. 13:461–469. [DOI] [PubMed] [Google Scholar]
- Kalchman, M.A., H.B. Koide, K. McCutcheon, R.K. Graham, K. Nichol, K. Nishiyama, P. Kazemi-Esfarjani, F.C. Lynn, C. Wellington, M. Metzler et al. 1997. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat. Genet. 16:44–53. [DOI] [PubMed] [Google Scholar]
- Kay, B.K., M. Yamabhai, B. Wendland, and S.D. Emr. 1999. Identification of a novel domain shared by putative components of the endocytic and cytoskeletal machinery. Protein Sci. 8:435–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen, J.H. 1990. Clathrin and associated assembly and disassembly proteins. Annu. Rev. Biochem. 59:415–438. [DOI] [PubMed] [Google Scholar]
- Keen, J.H. 1987. Clathrin assembly proteins: affinity purification and model for coat assembly. J. Cell Biol. 105:1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen, J.H., K.A. Beck, T. Kirchhausen, and T. Jarrett. 1991. Clathrin domains involved in recognition by assembly protein AP-2. J. Biol. Chem. 266:7950–7956. [PubMed] [Google Scholar]
- Kessels, M.M., A.E.Y. Engqvist-Goldstein, D.G. Drubin, and B. Qualmann. 2001. Mammalian Abp1, a signal-responsive F-actin-binding protein, links the actin cytoskeleton to endocytosis via the GTPase dynamin. J. Cell Biol. 153:351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen, T., and S.C. Harrison. 1981. Protein organization in clathrin trimers. Cell. 23:755–761. [DOI] [PubMed] [Google Scholar]
- Lamaze, C., L.M. Fujimoto, H.L. Yin, and S.L. Schmid. 1997. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J. Biol. Chem. 272:20332–20335. [DOI] [PubMed] [Google Scholar]
- Marsh, M., and H.T. McMahon. 1999. The structural era of endocytosis. Science. 285:215–220. [DOI] [PubMed] [Google Scholar]
- McCann, R.O., and S.W. Craig. 1997. The I/LWEQ module: a conserved sequence that signifies F-actin binding in functionally diverse proteins from yeast to mammals. Proc. Natl. Acad. Sci. USA. 94:5679–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann, R.O., and S.W. Craig. 1999. Functional genomic analysis reveals the utility of the I/LWEQ module as a predictor of protein:actin interaction. Biochem. Biophys. Res. Commun. 266:135–140. [DOI] [PubMed] [Google Scholar]
- Morgan, J.R., K. Prasad, W. Hao, G.J. Augustine, and E.M. Lafer. 2000. A conserved clathrin assembly motif essential for synaptic vesicle endocytosis. J. Neurosci. 20:8667–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, J.E., and J.H. Keen. 1992. Recognition sites for clathrin-associated proteins AP-2 and AP-3 on clathrin triskelia. J. Biol. Chem. 267:10850–10855. [PubMed] [Google Scholar]
- Qualmann, B., M.M. Kessels, and R.B. Kelly. 2000. Molecular links between endocytosis and the actin cytoskeleton. J. Cell Biol. 150:F111–F116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever, S., H. Damke, and S.L. Schmid. 2000. Dynamin:GTP controls the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J. Cell Biol. 150:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Haar, E., S.C. Harrison, and T. Kirchhausen. 2000. Peptide-in-groove interactions link target protein to the beta-propeller of clathrin. Proc. Natl. Acad. Sci. USA. 97:1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell, E., and L. Oestergaard. 1989. Identification of the clathrin assembly protein AP180 in crude calf brain extracts by two-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal. Biochem. 179:352–356. [DOI] [PubMed] [Google Scholar]
- Ungewickell, E., and H. Ungewickell. 1991. Bovine brain clathrin light chains impede heavy chain assembly in vitro. J. Biol. Chem. 266:12710–12714. [PubMed] [Google Scholar]
- Wanker, E.E., C. Rovira, E. Scherzinger, R. Hasenbank, S. Wälter, D. Tait, J. Colicelli, and H. Lehrach. 1997. HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum. Mol. Genet. 6:487–495. [DOI] [PubMed] [Google Scholar]
- Wendland, B., S.D. Emr, and H. Riezman. 1998. Protein traffic in the yeast endocytic and vacuolar protein sorting pathways. Curr. Opin. Cell Biol. 10:513–522. [DOI] [PubMed] [Google Scholar]
- Wesp, A., L. Hicke, J. Palecek, R. Lombardi, T. Aust, A.L. Munn, and H. Riezman. 1997. End4p/Sla2p interacts with actin-associated proteins for endocytosis in Saccharomyces cerevisiae. Mol. Biol. Cell. 8:2291–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, S., M.J. Cope, and D.G. Drubin. 1999. Sla2p is associated with the yeast cortical actin cytoskeleton via redundant localization signals. Mol. Biol. Cell. 10:2265–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ybe, J.A., B. Greene, S.-H. Liu, U. Pley, P. Parham, and F.M. Brodsky. 1998. Clathrin self-assembly is regulated by three light-chain residues controlling the formation of critical salt bridges. EMBO J. 17:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba, S., and J.H. Keen. 1983. Assembly polypeptides from coated vesicles mediate reassembly of unique clathrin coats. J. Cell Biol. 97:1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.