

Abstract
Integrin-mediated adhesion promotes cell survival in vitro, whereas integrin antagonists induce apoptosis of adherent cells in vivo. Here, we demonstrate that cells adherent within a three-dimensional extracellular matrix undergo apoptosis due to expression of unligated integrins, the β subunit cytoplasmic domain, or its membrane proximal sequence KLLITIHDRKEF. Integrin-mediated death requires initiator, but not stress, caspase activity and is distinct from anoikis, which is caused by the loss of adhesion per se. Surprisingly, unligated integrin or β integrin tails recruit caspase-8 to the membrane, where it becomes activated in a death receptor–independent manner. Integrin ligation disrupts this integrin–caspase containing complex and increases survival, revealing an unexpected role for integrins in the regulation of apoptosis and tissue remodeling.
Keywords: cell adhesion; apoptosis; caspase; integrin; ligands
Introduction
Integrins are cell adhesion receptors that mediate cell survival, proliferation, and migration in response to cues from the surrounding extracellular matrix (ECM)* (Hynes, 1992; Giancotti and Ruoslahti, 1999). These extracellular cues are transmitted into the cells through the direct or indirect interactions of integrins with a diverse array of cytosolic mediators, including protein and lipid kinases (Auer and Jacobson, 1995; Hauck et al., 2001), adaptor proteins (Wary et al., 1996; Cho and Klemke, 2000), and other modulatory proteins (Du et al., 1995; Scatena et al., 1998). Integrin expression is regulated in response to cellular activation by a variety of agents, including growth factors and cytokines (Eliceiri et al., 1998), and is crucial to tissue remodeling events, such as wound healing, inflammation, reproductive cycling, and tumor establishment (Murray and Lessey, 1999; Tarone et al., 2000).
Unlike cytokine receptors, however, integrins function selectively as “solid state” receptors (Ingber, 1992). Whereas a substrate-immobilized ligand can serve as an integrin agonist, the same ligand in solution functions as an integrin antagonist. This characteristic of the integrin may relate to an altered capacity of the integrins to cluster (Broday, 2000), recruit kinases such as focal adhesion kinase (Hildebrand et al., 1993), tether adaptor proteins and cytoskeletal elements (Wary et al., 1996), and/or provide mechanical tension (Hocking et al., 2000). Irrespective of the mechanism, it is clear that both of these distinct integrin states provide information to the cell about the nature of its environment.
For example, de novo expression of integrin αvβ3 occurs on endothelial cells in response to angiogenic growth factors (Brooks et al., 1994a), allowing increased endothelial cell interaction with the deposited provisional ECM. Soluble antagonists of this receptor initiate endothelial cell apoptosis, suppressing angiogenesis in various animal models (Brooks et al., 1994b; Storgard et al., 1999). Notably, cell death does not result from the loss of adhesion, per se, as observed during anoikis (Frisch and Francis, 1994). Instead, apoptosis results from the selective blockade of a single integrin, in this case αvβ3, on adherent, tissue-associated cells. Similar observations have been made involving other integrins. Notably, the overexpression of unligated integrin α5β1 has been associated with apoptosis and reduced tumor cell growth in vitro and in vivo (Giancotti and Ruoslahti, 1990; Varner et al., 1995; Kim et al., 2000; Plath et al., 2000). These observations suggest that the expression of specific integrin complexes, in the absence of a suitable ligand, may promote death. Physiologically, this would provide a mechanism whereby cells that find themselves within an inappropriate ECM would be actively cued to undergo apoptosis.
To understand how integrins, in the ligated or unligated state, influence cell survival in a physiological environment, cells with a defined integrin profile were evaluated in the context of a homogenous three-dimensional ECM. The evidence provided demonstrates that simple expression of unligated integrin is sufficient to initiate cell death among adherent cells. This “integrin-mediated death” (IMD) was similarly induced by the cytoplasmic domain of β1 or β3 integrins, resulting in the recruitment of caspase-8 to the cell membrane and its subsequent activation. These results reveal an unexpected role for integrins as proactive mediators of cell death, and document a novel mechanism for the induction of apoptosis during tissue remodeling and homeostasis.
Results
The expression of unligated integrins leads to cell death in ECM-associated cells
It is evident that cell adhesion occurs in vivo among cells that are otherwise ECM adherent. To evaluate the effect of integrin ligation state upon the survival of adherent cells, we examined the αvβ3-expressing carcinoma, T24E, in a three-dimensional collagen ECM (Ilan et al., 1998). Collagen provides a ligand for specific β1 integrins, e.g., α1β1, α2β1, and α3β1, (Takada et al., 1988) but not for αvβ3 (Filardo et al., 1995), expressed on these cells. Within 48–72 h of collagen culture, T24E cells became apoptotic, exhibiting extensive blebbing, annexin-V staining, and nuclear collapse. In contrast, T24E cells sorted for the lack of integrin αvβ3 (T24E-L; Fig. 1 A) exhibited increased survival in this matrix (approximately twofold greater survival at 72 h; Fig. 1 B). To assess whether the αvβ3 expression level was responsible for these events, T24E-L cells were genetically reconstituted with αvβ3 (T24E-Lβ3). This resulted in accelerated cell death (50% death in 24 h vs. 72 h in the T24E-L cells lacking αvβ3; Fig. 1 B, left), suggesting that αvβ3 potentiates the death of these cells within this ECM. Importantly, the expression of αvβ3 was not generally deleterious to the cells. All of the T24 lines proliferated at a similar rate and exhibited comparable basal apoptotic rates (unpublished data). The expression of integrin αvβ3 was not generally harmful, since it conferred a survival advantage when growth factors were withdrawn (Fig. 1 B, right), resulting in a significant decrease in cell death. Thus, the expression of αvβ3 is associated with cell death only in the absence of an appropriate ligand.
Figure 1.
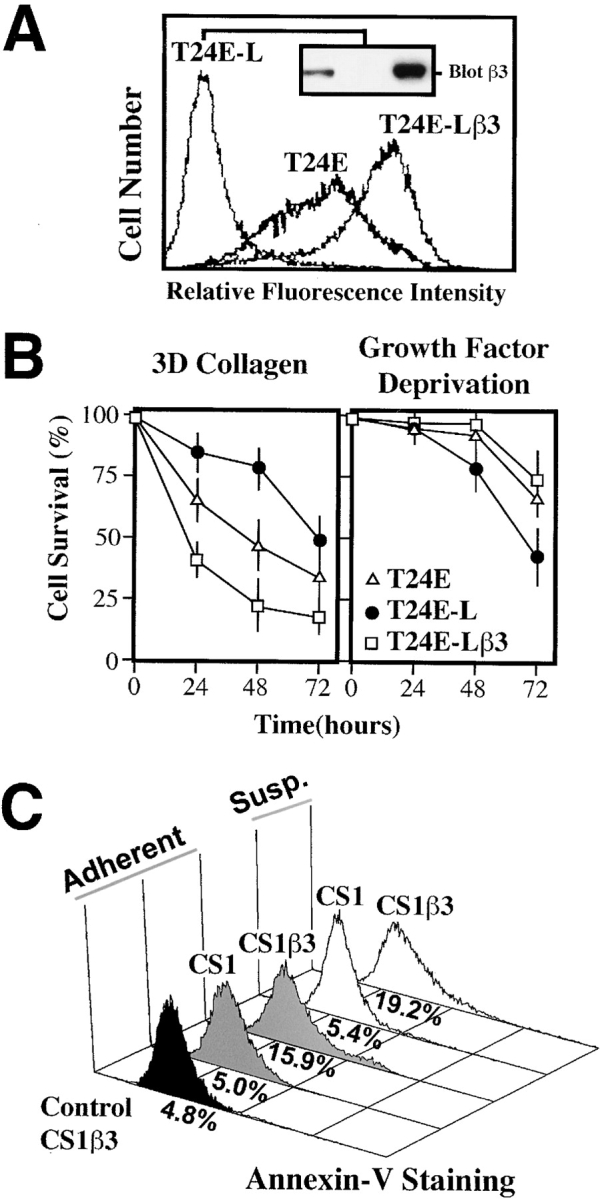
Expression of integrin αvβ3 increases death among cells attached to an ECM that does not ligate αvβ3. (A) T24E-L cells were derived from T24E cells by serial sorting to select a β3-lacking population. The T24E-Lβ3 cells, derived from T24E-L, were genetically reconstituted for αvβ3 expression. The relative αvβ3 integrin expression in these lines is shown by flow cytometry using monoclonal antibody LM609 (histograms), and by immunoblotting for the β3 integrin subunit with monoclonal antibody AP3 (inset). (B) Survival of these cell variants during culture in a collagen gel in the presence of serum (left) was determined. Death was scored by apoptotic morphology (condensation, satellite array; Cho and Klemke, 2000). Cell survival shown represents the mean ± SE of triplicate wells from a representative experiment. The viability of T24E, T24E-L, and T24E-Lβ3 cells were also assessed following serum deprivation (right). Viability was determined by immunofluorescent, microscopic assessment of PI exclusion at progressive time points. Data shown are from a representative experiment, and represent the mean ± SE viability of triplicate wells. (C) The stable expression of integrin αvβ3 in the CS1β3 cell line has been previously described (Filardo et al., 1995). CS1 parental cells (lacking αvβ3) or CS1β3 cells were grown as adherent to tissue culture (control CS1β3, black histogram), collagen cultured (gray histograms), or held in suspension (white histograms). After 20 h in complete media, cells were assessed for apoptosis by annexin-V staining and FACS® analysis. A representative experiment, of two, is shown.
To extend these observations, we next examined the anchorage-independent CS1 melanoma cell line, which survives and proliferates in suspension and, as such, exhibits no requirement for integrin-mediated signaling to maintain viability. However, CS1β3 cells, which have reconstituted expression of integrin αvβ3 (Filardo et al., 1995), underwent apoptosis when deprived of an αvβ3 ligand by culture on collagen gels or in suspension culture (Fig. 1 C), whereas parental CS1 cells did not. In this case, simple expression of unligated αvβ3 was proapoptotic, thus overcoming the anchorage-independent phenotype of these cells. These results reveal that the expression of an integrin, in this case αvβ3, can induce apoptosis in the absence of an appropriate ligand.
Endothelial cells survive when cultured in fibrin, an ECM that ligates integrin αvβ3 and at least two β1 integrins, yet undergo apoptosis when cultured in collagen (Fig. 2 A) (Filardo et al., 1995). Similarly, endothelial cells undergo apoptosis when cultured on the surface of collagen gels, but not on fibrin gels (Fig. 2 A, open bars). The induction of death was not simply due to an inability to attach and spread on collagen, since endothelial spreading on collagen and fibrin occurred to the same degree and with similar kinetics (Fig. 2 B). However, endothelial cells cultured on collagen were prone to apoptosis, leading to laemelopod retraction, blebbing, nuclear condensation, and eventual detachment.
Figure 2.

Decreasing the expression of endogenous integrin αvβ3 reduces endothelial cell apoptosis during collagen culture. (A) Endothelial cells undergo apoptosis when cultured in a three-dimensional collagen matrix in the presence of complete media. HUVECs were labeled with Cell Tracker green (2 μM) and cultured in a three-dimensional collagen gel, fibrin gel, or in suspension (filled bars) or, alternatively, on the surface of collagen or fibrin gels (open bars) for 24 h. ECM-associated cells were scored for apoptosis by the morphological analysis of digital images. A representative experiment is shown, each bar is the mean survival, ± SE, of six low power fields. (B) Cell spreading was assessed in HUVECs attached to the surface of collagen (Col) or fibrin (Fb) gels (right) after capture of digital images of Cell Tracker–labeled cells (left). Cells were segmented and area calculated using IP Lab software. Data shown for each point is the mean ± SE cell area calculated at each time point from 10 low power fields. (C) The effect of treatment with a human-specific β3 integrin antisense gene, delivered by adenovirus (AdASβ3), or that of a control, nonsense adenovirus (AdNS), upon the expression of native αvβ3 in HUVECs was determined using LM609 for FACS® analysis and AP3 for immunoblotting (inset) as described in Fig. 1 A. (D) AdASβ3-treated (or AdNS-treated) HUVECs were assessed for the percentage of surviving cells after a 24 h culture in a three-dimensional collagen gel (left) or fibrin gels (right) as described above. The results shown are the mean ± SE of two fibrin or four collagen experiments. (E) β3 antisense–treated HUVECs exhibited normal levels of ECM attachment. AdASβ3-treated HUVECs (filled bars) were assessed for adhesion to wells coated with ECM components vitronectin (VN), fibronectin (FN), laminin (LN), and collagen (COL). Adhesion was quantitated by staining of attached cells with crystal violet, followed by washing, reextraction, and quantitation of the bound dye (Filardo et al., 1995), and expressed relative to untreated HUVECs. AdNS-treated HUVECs (open bars) are shown as controls. The mean ± SE of two independent experiments is shown. (F) HUVEC survival was examined in collagen (left) or fibrin (right) gels, as described above, in the presence of 30 μg/ml monoclonal antibody P4C10 (anti-β1 integrin), monoclonal antibody LM609 (anti-αvβ3 integrin), both antibodies, or control monoclonal antibody 7G7B6 (anti-CD25). Survival shown is significantly decreased at 24 h (P < 0.05) in both ECMs in the presence of P4C10 or both antibodies. One of two similar experiments is shown.
To establish whether the presence of endogenous αvβ3 on these cells contributed to this apoptotic event, an antisense strategy was used to suppress endogenous αvβ3 integrin expression (Dallabrida et al., 2000). Decreasing the level of αvβ3 on these cells (40–60%; Fig. 2 C) resulted in a significant increase (twofold) in survival in collagen (Fig. 2 D, left), without significantly influencing survival in fibrin (Fig. 2 D, right) or cell adhesion to fibronectin, laminin, vitronectin, and, importantly, collagen (Fig. 2 E). As a second means to assess the role of integrins in cell survival, monoclonal antibodies directed against integrins αvβ3 or β1 were used as antagonists of integrin function. In this case, interfering with β1 integrin function accelerated apoptosis in collagen gels (Fig. 2 F, left), whereas blockade of either β1 or αvβ3 decreased survival in fibrin gels (Fig. 2 F, right). This result is consistent with previous observations that both αvβ3 and α5β1 bind fibrin (Yee et al., 2001). Together, these results indicate that a reduction of endogenous, unligated integrin expression can suppress apoptosis in ECM-attached cells, whereas an increase in integrin expression actively promotes apoptosis. The capacity to promote survival when ligated but to induce apoptosis when unligated is a hallmark of dependence receptors, such as nerve growth factor receptor, p75NGFR, and the netrin receptor, DCC (Bredesen et al., 1998; Mehlen et al., 1998). Our findings suggest that integrins may similarly be considered dependence receptors.
The cytoplasmic domain of the integrin β subunit is proapoptotic
Integrin-mediated signaling depends largely on the cytoplasmic domains of the α and β subunits. To determine whether the integrin cytoplasmic domain was sufficient to induce death, chimeric proteins composed of the extracellular domain of CD25 (IL2Ra, Tac) and the cytoplasmic domains of either integrin α5, β1, or β3 (LaFlamme et al., 1992) were expressed in COS7 cells. The expression of either Tac-β1 or Tac-β3 constructs resulted in increased death, while expression of the α5 chimera did not (Fig. 3 A), despite similar expression levels (Fig. 3 B). Tac-β3 expression produced a dose-dependent death among attached cells (Fig. 3 B). Importantly, the expression of these integrins was similar to, or less than, that of native integrins. Death occurred via apoptosis, as indicated by annexin-V reactivity (Fig. 3 C, top) and by processing of the caspase substrate, poly (ADP-ribose) polymerase (PARP), to the characteristic 85-kD apoptotic fragment (Fig. 3 C, bottom). IMD was induced efficiently by Tac-β3 or Tac-β1, yet only weakly by Tac-β5 (Fig. 3 D).
Figure 3.
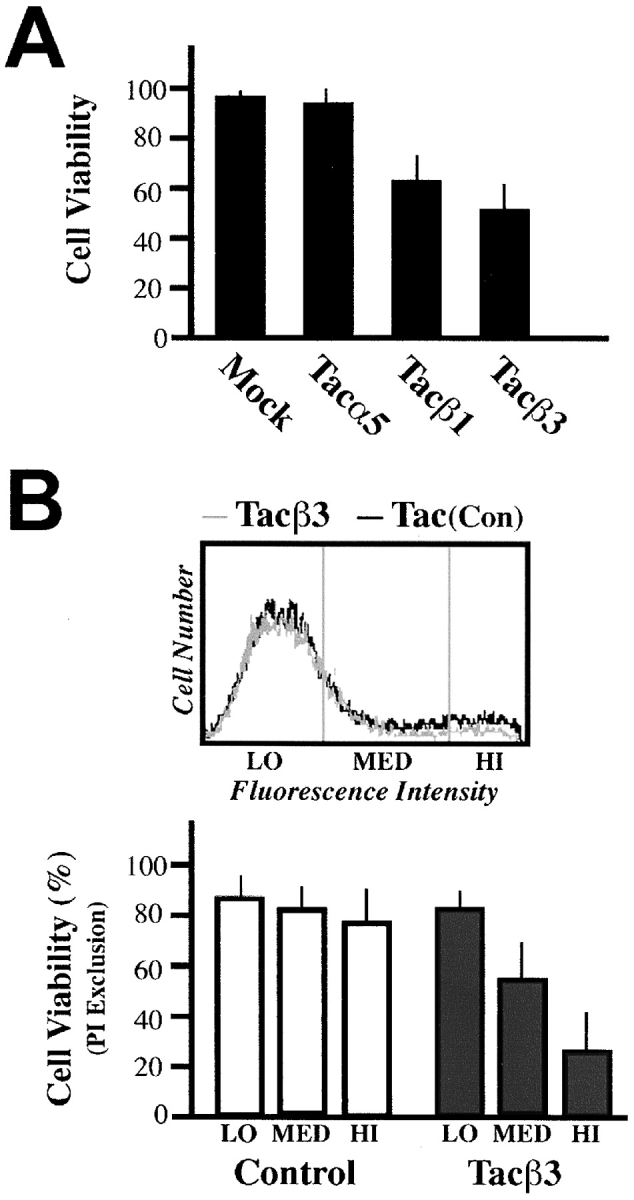
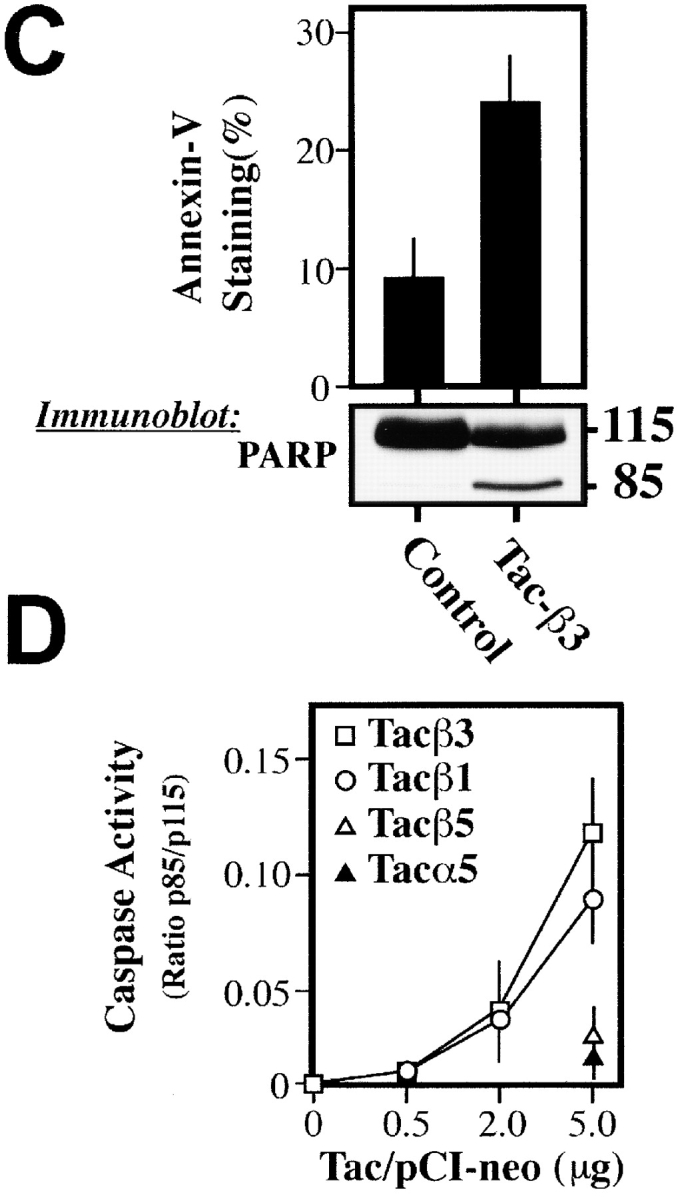
The integrin cytoplasmic domain is sufficient to induce apoptosis. (A) Chimeric constructs composed of the cytoplasmic domains of integrins α5, β1, or β3 and the extracellular and transmembrane regions of CD25 (Tac) were expressed in COS7 cells, and viability of cells positive for Tac expression (FITC-7G7B6 positive) was determined via PI exclusion 36 h later. (B) Analysis of the effect of increasing expression of Tac-β3 or control Tac-α5 on cell viability was performed. As shown, ∼25–35% of all cells expressed elevated Tac (top, MED and HI populations). To quantitate death, transfected cells were separated based on mean fluorescence intensity (LO, MED, and HI; top). The viability of these populations was determined by PI exclusion (bottom). Approximately 30–50% of the total Tac-β3–expressing cells (MED and HI) die during the course of the assay, or ∼8–16% of the total COS7 cell population. (C) Tac-β3–expressing COS7 cells exhibited classic apoptotic markers. 18 h after transfection, nonadherent cells were removed and discarded; only those remaining attached were assessed for the onset of apoptosis by staining with annexin-V–FITC (top). Each bar represents the mean percentage of annexin-positive cells (± SE) from three independent experiments. The cleavage of the executioner caspase substrate PARP was assessed by Western blotting of total cell lysates 36 h after transfection. Caspase-cleaved PARP was detected as the 85-kD fragment that is characteristic of apoptosis (bottom). The relative intensity of the cleaved fragment was quantitated as a ratio relative to the uncleaved PARP, including PARP from nonexpressing cells, to give a relative indication of the activation of executioner caspases. The derived PARP ratios shown are: lipofectamine control, 0.004; Tac-β3, 0.17. (D) Caspase activity, as measured by the relative cleavage of PARP, was determined as a dose-dependent quantity of pCI-NeoTac transfection. COS7 cells were transfected with cDNA-encoding integrin tail constructs. Approximately 25–40% of cells expressed Tac, and comparable expression was observed between constructs at each dose transfected. The effect of expressing Tac-β1, Tac-β3, and the control, Tac-α5, as well as a chimera expressing the cytoplasmic domain of β5 (Tac-β5) was determined as the mean ± SE of caspase activity (measured as the intensity of p85/p115 from three independent experiments, as described above).
A membrane-proximal sequence in the integrin tail is sufficient to induce apoptosis in attached cells
As an initial means to address the regions of the β integrin tail required for IMD, successive truncations were made in the cytoplasmic domain of Tac-β3 (Fig. 4 A). Cells expressing each of the truncated constructs were analyzed for cell death by propidium iodide (PI) exclusion (Fig. 4 B), and for the activation of executioner caspases as indicated by PARP cleavage (Fig. 4 C). A minimal sequence required to induce apoptosis was identified within the membrane-proximal sequence of β3 718–727, KLLITIHDRKEF (Fig. 4, B and C). Interestingly, this region is insufficient to mediate signaling through previously characterized survival pathways, including via Src, Shc, or focal adhesion kinase (Wary et al., 1996; Tahiliani et al., 1997; Hauck et al., 2001). Thus, it is unlikely that IMD proceeds by the disruption of known survival signaling pathways.
Figure 4.

IMD requires a membrane localized region of the integrin cytoplasmic domain. (A) A series of mutant integrin chimeras with truncations of the native integrin β3 cytoplasmic domain (residues 715–762) and the extracellular domain of Tac were constructed in pCI-Neo and expressed in COS7 cells. (B) Expression of integrin tail mutant constructs leads to cell death. COS7 cells expressing the integrin constructs were assessed for viability based on PI exclusion by flow cytometry. Within each experiment, the relative capacity of each construct to induce death was normalized to the death induced by the full length tail (which ranged from 20 to 40% of expressing cells in these studies). (C) Executioner caspase activity is induced by integrin truncation mutants. For each mutant, the relative caspase activation (assessed by the p85/p115 ratio) was assessed and normalized to that induced by intact Tac-β3. The results shown are the mean ± SD of the normalized PARP ratios for three experiments. (D) Mutation of the NPXY motif within the tail of integrin β3 blocks αvβ3-directed cell migration, but does not inhibit IMD. T24-EL cells reconstituted to express wild type β3 (T24-ELβ3) or a point mutant in the NPXY motif (T24-ELN744A; shown in bold, Fig. 4 A) at similar levels were assessed for their migration on vitronectin (left) or for survival within a collagen ECM (right). Migration was measured microscopically as the average wound closure plotted, as the mean ± SE of six sites on a vitronectin-coated plate after 24 h. Collagen survival was assessed microscopically by direct cell counting to assess exclusion of PI (five fields per gel, three gels). The mean ± SE of three experiments are shown. (E) Membrane-anchored forms of the integrin cytosolic domain induce apoptosis. COS7 cells were transfected with three different β3 cytoplasmic domain constructs; Tac-β3, as well as a second membrane-anchored, GFP-β3, and an unanchored, His-β3, variant of the β3 cytoplasmic domain. Detergent lysates of these cells were subjected to immunoblotting to assess PARP cleavage (top) and to confirm expression of the individual constructs (GFP, top; Tac/His, bottom). One of two similar experiments is shown. (F) Cell spreading was assessed via analysis of digital images of cells expressing GFP-β3 or GFP-TM (control, GFP with a signal sequence and the transmembrane domain from CD25) as described above (Fig. 2 B) 12, 24, and 48 h after transfection. (G) Ligation of β3 constructs suppresses IMD. COS7 cells expressing submaximal levels of integrin tails were allowed to attach to substrates coated with anti-Tac monoclonal antibody (Ligation +) or maintained as a normal, adherent tissue culture (Ligation −). Adherent cells were lysed and assessed for the relative expression of death-inducing construct, i.e., integrin, present, as well as to determine executioner caspase activation (through quantitation of p85/p115 ratios) via immunoblot analysis. Bars represent the mean ± SE of three experiments.
A number of regions of the integrin tail have been implicated in the induction of signaling by integrins. In particular, the NPXY site, β3 744–747, has been identified as a critical motif involved in signaling downstream of integrin β3 (Filardo et al., 1995; Liu et al., 2000). In agreement, a mutant β3 integrin constructed with a loss of function mutation in this region (N744A) expressed in T24E-L cells (T24E-L744) resulted in complete loss of αvβ3-mediated migration (Fig. 4 D, left). However, this mutant still induced the IMD of cells within a three-dimensional collagen matrix (Fig. 4 D, right). Together, the results indicate that divergent mechanisms are required for IMD and “classical” integrin signaling. Membrane localization of the β3 tail appeared to be required to initiate IMD, since expression of the integrin tail without a transmembrane domain (His-β3) failed to cause death (Fig. 4 E), whereas expression of a second membrane-anchored integrin construct (green fluorescent protein [GFP]-β3) led to PARP cleavage (Fig. 4 E) and apoptosis. This finding provides further evidence that integrin expression does not cause death in a simple “dominant negative” manner.
The expression of GFP-β3 also allowed integrin-transfected cells to be monitored to assess whether cell spreading was compromised. In agreement with the previous endothelial cell spreading studies (Fig. 2 B), no difference in cell spreading was observed between GFP-β3 and control GFP-expressing cells until after apoptosis was evident (Fig. 4 F). Thus, the onset of apoptosis can initiate cell retraction and eventual detachment (Harrington et al., 2001).
Integrin-mediated substrate attachment, accomplished by adhesion to ECM proteins (Ilic et al., 1998; Scatena et al., 1998) or by integrin binding to substrate-immobilized antibodies (Stromblad et al., 1996), suppresses apoptosis. In contrast, antagonized (Brassard et al., 1999) and/or nonligated integrins (Stromblad et al., 1996; and this manuscript) promote apoptosis among otherwise adherent cells. To evaluate whether the integrin chimeras would promote death when substrate-immobilized, Tac-β3–expressing COS7 cells were replated on surfaces coated with an anti-Tac monoclonal antibody. This attachment and cell spreading enriched the number of integrin chimera–expressing cells (Fig. 4 G, top), yet these cells demonstrated a dramatic reduction in apoptosis (60% decrease in PARP cleavage) relative to the adherent control population (Fig. 4 G, bottom). In other experiments, we also tested whether cells treated with soluble anti-Tac or anti-Tac on microbeads could prevent IMD, but neither impacted this event (unpublished data). Our result suggests that only substrate-localized integrin “clustering” was capable of suppressing the death-promoting activity of the β3 tail.
IMD requires initiator caspases
To begin to address the molecular mechanism responsible for IMD, checkpoint-specific inhibitors of caspase activation were expressed in COS7 cells undergoing IMD. Expression of dominant negative caspase-9 or bclxl, which inhibit the stress-mediated caspase cascade (Finucane et al., 1999; Soengas et al., 1999; Wolf and Green, 1999), did not prevent IMD, but did block stress-induced apoptosis of these cells (Fig. 5 A). In contrast, crmA, a serpin that inhibits activation by death receptor–activated caspases (Zhou et al., 1997), protected cells from IMD (Fig. 5, A and B). Surprisingly, a negative-acting variant of the death receptor adaptor, FADD (Fas-associating protein with death domain), which inhibits Fas-initiated apoptosis mediated by death receptors (Chinnaiyan et al., 1996), was not protective (Fig. 5, A and B), despite the fact that it effectively blocked the Fas-mediated death of these cells (Fig. 5 A). Apoptosis could also be prevented by treatment of integrin tail–expressing cells with peptide inhibitors of initiator and executioner caspases (Fig. 5 C) (Thornberry et al., 1997), but was unaffected by a stress caspase–selective inhibitor (zLEHDfmk).
Figure 5.

IMD depends on initiator caspase activity. (A) Executioner caspase activity is blocked by crmA but not by inhibitors of stress or Fas-mediated apoptosis. Coexpression of crmA, DN-FADD, Bclxl, or catalytically inactive caspase-9, DN Casp 9 (4 μg) with the proapoptotic Tac-β3 construct (2 μg) was performed to determine whether these checkpoint-specific apoptosis inhibitors could suppress IMD. Cells were assessed after 36 h using the activation of executioner caspase activity (PARP cleavage immunoblot assay) as an indicator. Data are expressed as a ratio of PARP cleavage found in (Tac-β3 + inhibitor) to that observed in (Tac-β3 + control vector) transfected cells, and represents the normalized results of three to four independent experiments. To confirm that constructs with no activity in IMD were functional, apoptosis was also induced via Fas overexpression (2 μg; hatched bars), or Bad expression (1 μg; gray bars). (B) Cell viability is maintained by crmA. Cells were allowed to express constructs (as per A, above), but in this case apoptosis was quantified by FACS®. Cells harvested 36 h after transfection were labeled with Alexa dye–conjugated anti-CD25 (7G7B6-Alexa488) to label Tac-expressing cells. Dead cells were identified by uptake of PI (500 ng/ml). Results are expressed as the percentage of dead, Tac-expressing cells in each group, and represent the mean ± SE of two experiments. (C) Peptides that inhibit stress-induced apoptosis are not active in IMD. COS7 cells, induced to die via expression of Tac-β3, were treated with 40 μM of caspase-inhibitory peptides (zIETDfmk, zDEVDfmk, or zLEHDfmk) as a single dose 12 h after transfection, and then monitored for executioner caspase activation, i.e., PARP cleavage, 24 h later. Data are expressed relative to cells treated with diluent (DMSO) alone and represent the mean ± SD of two experiments. (D) IMD induced by endogenous integrins is blocked by crmA. COS7 cells coexpressing Ds-RED fluorescent protein, a transfection marker, and checkpoint- specific inhibitors of apoptosis, as listed above, were assessed for survival after a 48 h culture on collagen gels in complete growth medium via morphologic scoring, as described (Fig. 2). Data shown is the mean survival ± SE. A representative experiment of two is shown.
To confirm that the apoptotic pathways initiated by the integrin chimeras were the same as those induced by endogenous integrins, COS7 cells expressing these checkpoint specific inhibitors were examined for their survival on collagen gels. Similar to our previous findings (Fig. 5, A and B), crmA expression suppressed IMD, whereas DN–FADD, DN–caspase-9, and bclxl were unable to block this form of apoptosis (Fig. 5 D). Together, these findings suggest that initiation of IMD does not require the components of the stress caspase cascade, but does depend upon the activation of initiator caspases in a manner independent of death receptors.
Unligated integrins activate caspase-8–like activity
To explore the mechanism(s) by which unligated integrins might activate caspase and induce IMD, we next examined the subcellular localization of activated caspases during IMD. Integrin-initiated caspase activity was visualized by labeling with FAM-VADfmk, which covalently and fluorescently labels activated caspases. Visual assessment of cells undergoing IMD revealed a punctate distribution of caspase-associated fluorescence that colocalized with unligated β3 integrin tail on the cell surface (Fig. 6 A). In contrast, Tac-α5, which does not induce IMD, appeared diffusely on the cell surface and did not colocalize with caspase activity (Fig. 6 A). Notably, caspase activity was also found to be reduced in or absent from Tac-β3–expressing cells that were attached to substrate-immobilized Tac antibody (Fig. 6 A, left). In these cells, Tac-β3 expression was found to be redistributed and predominantly at the cell–substrate interface, as determined by confocal z-section analysis. The series of collected, integrin-containing z-sections were subjected to digital image analysis by blinded observers to assess possible colocalization between the integrin and active caspase (Fig. 6 B). Only under conditions that induced IMD (unligated Tac-β3), was significant colocalization observed between caspase and integrin (coefficient of colocalization = 0.49 ± 0.07; P < 0.01).
Figure 6.


Caspase-8 is recruited to the membrane and becomes activated during IMD. (A) Integrin tails colocalize with caspase activity during IMD. The cellular location of death-inducing integrin tails and active caspases was assessed by immunofluorescence using a 1024 Bio-Rad confocal microscope. Cells were induced to undergo apoptosis by integrin cytoplasmic domain expression, or were rescued by ligation (as described in Fig. 4 G). Before fixation in PBS/4% paraformaldehyde, cells were labeled with FAM-VAD in serum free media for 60 min to label active caspases (fluorescein, green channel). Cells were then probed for Tac expression (monoclonal antibody 7G7B6, red channel). Signals for each image were gathered serially as independent scans using the 488 nm (middle row) and 566 nm (top row) laser lines. Colocalization within the z-sections is shown as a yellow/orange signal (bottom row). (B) Confocal images of COS7 cells expressing the Tac–integrin chimeras described in A were acquired by blinded observers. The coefficient of colocalization between the 488 and 566 nm signal within the integrin-containing z-sections was determined (Bio-Rad Lasersharp Software). The mean ± SE of 10 random fields for each group are plotted. (C) Caspase-8 and -3 are labeled during IMD. COS7 cells induced to die by the expression of integrin tail were incubated with biotin-VAD to label active caspase. Active caspases were then isolated with avidin-Sepharose and resolved via SDS-PAGE on 8–16% nonreducing gradient gels. Immunoblotting was performed with polyclonal rabbit antisera to caspases-3 (lane 2) or -6 (lane 4) (control and not detected, respectively) or monoclonal antisera to caspase-8 (lane 3). (D) Caspase-8–like activity is elevated during IMD. Lysates from COS7 cells expressing Tac constructs with either the integrin α5 cytoplasmic domain (gray circles), the integrin β3 cytoplasmic domain (open circles), or mock transfectants (open squares) were assessed for their relative ability to cleave the caspase-8 substrate, IETD-pNA. The induced IETDase activity in the Tac-β3 expressing cells was suppressed by substrate ligation of the chimera (as described in Fig. 2 G), as shown (black circle). (E) The subcellular distribution and predominant forms of caspase-8 change in cells undergoing IMD. Cell fractionation of COS7 cells expressing GFP-β3 (induces IMD) or GFP-β3Δ717 (inactive truncation) was performed 36 h after transfection with the individual constructs (4 μg). The relative distribution and forms of caspase-8 were assessed by immunoblot analysis of the cytosolic (Csol), membrane (Mem), and cytoskeletal/nuclear (Cskn) fractions. Relative molecular mass (kD) of the detected species is indicated. Bar, 10 μm.
To identify the caspases activated during this process, COS7 cells undergoing IMD were labeled with a biotinylated caspase inhibitor (biotin-VADfmk), lysed, and analyzed for the presence of biotinylated products. Biotin-VAD labeling revealed several discernible caspase forms, including zymogen and activated forms of caspase-3 and caspase-8 (Fig. 6 C). Characterization of the caspase activity present in cells undergoing IMD confirmed a caspase-8–like (“IETDase”) activity that was suppressed by substrate ligation of the integrin chimera (Fig. 6 D). Caspase activity was not elevated in cells expressing the control α5 integrin chimera (Fig. 6 D). However, IETDase activity is not absolutely specific to caspase-8 and could be mediated by caspase-3. Therefore, subsequent cell fractionation studies (Chen et al., 2000) were performed to confirm that caspase-8 was processed to its active form in these cells. Interestingly, the processed form of caspase-8 became highly enriched within the membrane and cytoskeletal/nuclear fractions in cells expressing the β3 integrin tail, but not in cells expressing a truncation mutant that fails to induce apoptosis, GFP-β3Δ717 (Fig. 6 E).
Recruitment of caspase-8 by integrin β subunit tails
The external clustering of death receptors, such as Fas, leads to formation of a death-inducing signaling complex (DISC), which recruits and activates caspase-8 (Scaffidi et al., 1999). To determine if integrins induced apoptosis in a manner analogous to death receptors, COS7 cells were held in suspension and treated with anti-αvβ3 integrin–antagonist antibodies. The cells were lysed and integrin complexes were then immunoprecipitated from the resulting lysates and subjected to analysis for DISC components. Although both the zymogen and active forms of caspase-8 were observed in integrin immunoprecipitates (Fig. 7 A), FADD was not detected (unpublished data). Clustering of a control transmembrane protein, LDL-like receptor protein (LRP), which is expressed at levels similar to integrin αvβ3 on COS7 cells, did not recruit either form of caspase-8 (Fig. 7 A).
Figure 7.
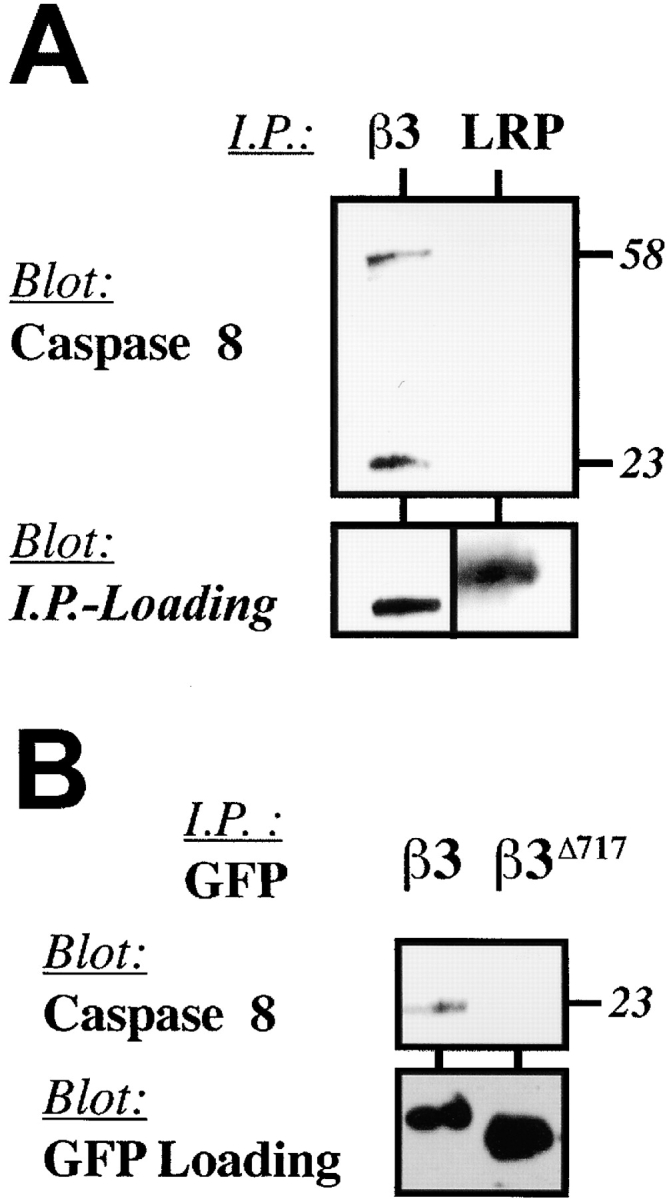

Caspase-8 associates with integrins during IMD. (A) Caspase-8 associates with native integrin αvβ3. Integrin αvβ3 and LRP complexes are expressed at similar levels on COS7 cells (unpublished data) and were subjected to clustering and immunoprecipitation followed by immunoblot analysis for caspase-8 to determine if caspase-8 was recruited to an integrin DISC-like complex (Scaffidi et al., 1999). (B) Integrin cytosolic domains are sufficient to recruit caspase-8. Expressed GFP-β3 (proapoptotic) or GFP-β3Δ717 (inactive) constructs were subjected to immunoprecipitation with GFP-Sepharose followed by immunoblot analysis for the presence of associated caspase-8 (top) or the GFP chimera (bottom). (C) Ligation of integrin disrupts caspase association. Integrin-expressing COS7 cells (Tac-β1, unligated Tac-β3, Tac-α5) and Tac-β3–expressing cells replated onto anti-Tac substrate (ligated Tac-β3) (as described in Fig. 4 F) were lysed and subjected to immunoprecipitation with anti-Tac monoclonal antibody. The precipitates were resolved via SDS-PAGE and analyzed for the presence of associated caspase-8 (top) or Tac (bottom) via immunoblotting. (D) An αvβ3 integrin ligand rescues apoptosis and interrupts caspase-8 recruitment to integrins in a three-dimensional ECM. HUVECs were cultured in three-dimensional fibrin or collagen gels, or maintained in suspension (apoptosis positive control) for 16 h. Endothelial cells were then recovered from the gels, lysed, and subjected to immunoprecipitation using LM609 (anti-αvβ3). The precipitates were resolved and analyzed for the presence of caspase-8 by immunoblotting, as described above.
Since the cytoplasmic domain of the integrin was sufficient to induce apoptosis (Fig. 3) as well as recruitment and activation of caspase-8 at the cell membrane (Fig. 6, A and E), we next determined whether the transmembrane domain and cytoplasmic tail of β3 integrin was sufficient to mediate association with caspase-8. Immunoprecipitation of GFP-β3 expressed in COS7 cells revealed associated caspase-8 (Fig. 7 B), which was not observed in immunoprecipitates from cells expressing the inactive GFP-β3Δ717 mutant. Moreover, association of the integrin tail with caspase-8 was functionally important, as no association was observed with nonapoptotic integrin α5, and integrin ligation events that blocked apoptosis also disrupted the association of caspase-8 with integrin β3 (Fig. 7 C). Finally, integrin and caspase-8–containing complexes did not include caspases-1, -3, -6, -7, and -10 as detected by immunoblot analysis (unpublished data).
To determine whether endogenous integrins induced IMD by recruitment of caspase-8, native αvβ3 complexes were isolated from endothelial cells undergoing apoptosis in collagen gels and assessed for the presence of caspase-8. Endothelial cells cultured in collagen or in suspension (positive apoptosis control) were enriched for the presence of caspase-8 (Fig. 7 D), whereas complexes isolated from endothelial cells cultured in a fibrin gel displayed a dramatic decrease in both caspase-8 (Fig. 7 D) and apoptosis (Fig. 2 A). Thus, cells expressing high levels of endogenous αvβ3, in an inappropriate ECM, can be induced to die by the death receptor–independent, and presumably, FADD-independent, recruitment and activation of caspase-8. Conversely, this process is prevented in the presence of an integrin-appropriate ECM.
Discussion
Integrins are known to promote the survival of various cell types in culture, and have been shown to be sufficient at mediating the “anchorage dependent” component of cell growth (Schwartz, 1997). Recently, integrin-mediated adhesion has also been demonstrated to promote cell survival in response to stress-associated apoptotic triggers such as starvation, growth factor withdrawal, and chemical mediators (Ilic et al., 1998; Moro et al., 1998; Bonfoco et al., 2000). In these cases, integrin signaling proceeds via focal adhesion kinase to the nucleus, allowing sustained cell survival under conditions that might otherwise induce death, i.e., survival signaling (Ilic et al., 1998). In contrast, data is provided here that demonstrates that integrins themselves, when unligated, can trigger apoptosis of adherent cells in the absence of any other death-inducing stimuli. This form of apoptosis, termed IMD, is biologically and biochemically distinct from anoikis, which is defined as death due to cell detachment from substrate (Frisch and Francis, 1994).
IMD
IMD depends upon the ability of the integrin β subunit to recruit caspase-8 to the cell surface in a FADD-independent manner. These findings may explain our previous observations in vivo, where angiogenesis was suppressed in tissues treated with αvβ3 integrin antagonists due to an induction of apoptosis of angiogenic endothelial cells (Brooks et al., 1994b; Stromblad et al., 1996; Storgard et al., 1999). In recent studies, we observed that inhibitors of caspase-8, but not caspase-9, restored neovascularization in αvβ3 antagonist–treated angiogenic tissues (unpublished data). Thus, cells that interact with an inappropriate ECM, or those exposed to integrin antagonists may succumb to IMD. Interestingly, while β1 and β3 integrins can promote this process, β5 apparently does not (Fig. 3 D). Therefore, distinct integrins may have different capacities to induce apoptosis due to regulatory regions present elsewhere in the integrin heterodimer.
IMD was observed in several cell lines, including COS7, CS1, HEK, HTB, and HUVEC. However, it is noteworthy that some cells examined, including CHO-K1 and HeLa, were resistant to IMD (unpublished observations). Resistance to IMD may occur, in vivo or in vitro, for several reasons. Clearly, cell type–specific or clonal variations result in cultured cells having varied capacities to secrete or otherwise remodel their immediate ECM. Furthermore, not all cells express appropriate caspases, such as caspase-8, or may express inhibitors such as c-flip (Aoudjit and Vuori, 2001). In these cases, integrin antagonism may induce apoptosis by other means. For example, integrin antagonism also leads to the activation of the tumor suppressor p53 (Stromblad et al., 1996). Since p53 complexes with caspase-8 (Ding et al., 2000), it may have a direct role in IMD. However, transcriptional activation of p53 also elicits stress-mediated death via activation of caspase-9 (Soengas et al., 1999). Accordingly, neurons that express little or no caspase-8 eventually undergo apoptosis in response to integrin antagonism via the stress–caspase-9 pathway (Bonfoco et al., 2000).
In addition, a variety of other proteins that can play key regulatory roles in the induction of, or resistance to, apoptosis have been identified. For example, tumor cells that are anchorage independent may be resistant to IMD. However, the expression of integrin αvβ3 on CS1β3 cells was able to induce IMD in an otherwise anchorage-independent cell (Fig. 1 D). These findings suggest that specific integrin antagonists may be useful as antitumor agents, as suggested by others (Ruoslahti, 1997; Curley et al., 1999).
Physiological roles for IMD
IMD would be predicted to occur during tissue remodeling associated with development, wound repair, or inflammation to ensure that only appropriate cells remain viable within a given tissue microenvironment. For example, the ECM is dramatically remodeled during the involution/epithelial cell regression phase following weaning (Sympson et al., 1994). During this apoptotic process, epithelial cells undergo caspase activation in a β1 integrin–dependent manner (Boudreau et al., 1996). Antagonism of β1 also results in chondrocyte apoptosis during ECM remodeling associated with differentiation (Hirsch et al., 1997). In agreement with these studies, we show that the cytoplasmic domain is sufficient to induce IMD. However, it is not clear why apoptosis is more often associated with specific β1 integrins. In particular, the overexpression of integrin α5β1 has been documented to suppress tumorigenicity (Giancotti and Ruoslahti, 1990; Varner et al., 1995) and/or angiogenesis (Kim et al., 2000), possibly through the induction of apoptosis (Plath et al., 2000). The effect is reversed by fibronectin, a ligand for α5β1 (Varner et al., 1995). In contrast, proliferative tumors with an elevated expression of α5β1 in vivo, which would be IMD resistant, might therefore be associated with a poorer prognostic outcome (Adachi et al., 2000). Thus, IMD may be an important regulatory event under many physiological circumstances in vivo.
Role of caspase-8 in IMD
Our findings indicate that caspases function downstream of unligated integrins in the induction of IMD. Thus, inhibition of caspase-8 via peptide or protein (crmA) antagonists results in reduced IMD, whereas increased expression of unligated integrins increases caspase activation and apoptosis. Evidence is provided that caspase-8 can directly or indirectly interact with integrin β subunit cytoplasmic domain, and this interaction contributes to the activation of the caspase. Unligated integrins tend to cluster on the cell surface in a manner dependent upon integrin expression level, however, these integrins are not associated with focal adhesion kinase or tethered by the cytoskeleton (unpublished data). These untethered clusters of integrin may serve to recruit sufficient numbers of caspase-8 zymogens to facilitate proximity-induced activation of the initiator caspase cascade (induced proximity model; Salvesen and Dixit, 1999) in a death receptor–independent manner. Although integrins may be less efficient than death receptors in this respect, it is noteworthy that other methods of “clustering” caspase-8 serve to activate it, including mutation, to add multimerization domains (MacCorkle et al., 1998; Memon et al., 1998; Muzio et al., 1998; Fan et al., 1999), and overexpression strategies (Stennicke and Salvesen, 1999). Supporting this notion, integrin cytoplasmic domain constructs that provide no localization signal, e.g., His-β3, also fail to induce IMD, whereas, increasing the number of unligated integrins (Figs. 1 B and 3 B) or blocking multiple integrins (αvβ3 and β1 integrins; Fig. 2 F), augmented the incidence of IMD. Thus, the increased apoptotic rate seen with combined anti-αvβ3/anti-β1 treatment in collagen gels (Fig. 2 F) may be due to the increased sum quantity of unligated integrins (in this case, both β3 and β1).
Integrin- or caspase-deficient mice frequently show developmental defects and/or lethality (Fassler et al., 1996; Zheng and Flavell, 2000). Although αvβ3 is proapoptotic when unligated (this work), or antagonized (Brooks et al., 1994b; Brassard et al., 1999; Storgard et al., 1999), it is, nonetheless, nonessential for the viability of humans (Coller et al., 1991) or mice (Hynes and Hodivala-Dilke, 1999). Why then, should αvβ3 continue to be expressed on invasive tissues in the broad range of species observed? Integrin αvβ3 may function during invasive processes as a biosensor, providing positive feedback to the cell during “productive” interactions, i.e., in a permissive ECM (Ilic et al., 1998), while inducing death by triggering caspase recruitment/activation in those cells that enter an inappropriate microenvironment. Disrupting expression of integrin αvβ3 would thus only remove one of several possible triggers for apoptosis. This capacity to selectively suppress or promote apoptosis distinguishes integrins from the death receptor and stress apoptotic pathways, and suggests that integrins may be grouped within a third emerging category of apoptotic triggers, the dependence receptors (Bredesen et al., 1998).
It is still not known whether accessory molecules may be involved in the regulation of this interaction. However, not all cells were subject to IMD, and not all integrins are proapototic. Thus, it is likely that extended investigations will reveal both cell and integrin-specific factors that govern the induction of this cascade. Nevertheless, this novel apoptotic trigger, which is regulated by integrin repertoire and local ECM composition, provides a new means to account for cell death and survival associated with tissue remodeling and homeostasis.
Materials and methods
Cell lines and three-dimensional cell culture
T24-E carcinoma cells (formerly designated ECV-304) (catalog number 1998; American Type Culture Collection) were serially sorted to isolate stable, β3 integrin–lacking (T24-EL) populations. β3 integrin expression was reconstituted by transfection of pcDNA-Neoβ3 or pcDNA-Neoβ3(N744A) followed by neomycin selection (500 μg/ml) with FACS® sorting to eliminate neomycin-resistant but nonexpressing cells from the population as we have previously done for the CS1 cell line (Filardo et al., 1995). Integrin function was also assessed via transwell migration (Cho and Klemke, 2000), wound assay (Hauck et al., 2001), or adhesion assays (Filardo et al., 1995), as described. T24E, T24EL, T24-ELβ3, COS7, or HUVE cells were microcultured in complete medium either within a three-dimensional collagen (Vitrogen; Cohesion Technologies) (Cho and Klemke, 2000) or thrombin-elicited fibrin gel (American Diagnostica) (500 cells/10 μl), or on the surface of these immobilized gels after washing/equilibrating with complete medium for 30 min. Cells were subsequently cultured in complete media (M199, 20% FBS, endothelial cell growth supplement for HUVECs, RPMI 1640, 5% FCS for CS1 lines, DME, 5% FBS for all other cell lines, all supplemented with gentamycin and glutamine, minimal amino acids and pyruvate; Sigma-Aldrich). Three methods were used to confirm that death occurred via apoptosis in the gels. Dead or dying cells were identified using nuclear dyes (PI) to visualize nuclear collapse. Morphological scoring (satellite/condensation) assays (Cho and Klemke, 2000) were performed using Cell Tracker-Green–labeled cells (Molecular Probes). Cell Tracker labeling was proceeded by a 10-min incubation with 2 μM Cell Tracker in serum-free medium before assay. Annexin-V (CLONTECH Laboratories, Inc.) reactivity was used as an early marker of apoptosis and identified dying cells before spreading was lost. These methods yielded comparable results. Biochemical analysis of PARP cleavage was performed in scaled-up gels (250,000 cells/5 ml × 10, 100 mm plates); cells were extracted from collagen by brief treatment with PBS/0.25% bacterial collagenase (Sigma-Aldrich) or from fibrin by brief plasmin (Sigma-Aldrich) treatment at 37°C before washing twice in PBS and immunoprecipitation studies, described below.
Constructs and gene delivery
Deficient adenoviral antisense constructs containing a CMV-driven ORF complementary to the first 508 bp of β3 or nonsense (empty read through) constructs were generated by recombination of pAdCI/pJM17 plasmids in U293 (E1 complementary) cells. HUVECs were infected at a multiplicity of infection of ∼100 pfu/cell, yielding 100% infection as assessed by infection with adenoviruses encoding an Ad-β gal reporter gene. The pCI-NeoTac-β3, pCI-NeoTac-β5, and pCI-NeoTac-β1 constructs were created by ligation of integrin cytoplasmic domains to the extracellular and transmembrane domains of Tac/CD25, as originally reported (LaFlamme et al., 1992). The truncated chimeras were created by introduction of inframe stop codons in the integrin coding sequence, as described (Filardo et al., 1995). The His-β3 construct was constructed by in frame expression of the β3 cytoplasmic domain after the His tag in pcDNA3.1/V5/His A vector (Invitrogen). The GFP-β3 chimeras were constructed essentially as described for β1 (Smilenov et al., 1999) but included the signal sequence and transmembrane/cytoplasmic domains of integrin β3, rather than β1, expressed in the pEGFP-C3 vector (CLONTECH Laboratories, Inc.). The bclxl, bad, and crmA constructs in pCMV5 were the gifts of S. Huang (The Scripps Research Institute, La Jolla, CA), independent DN-FADD constructs that lack the DED domains were obtained from H. Duan (University of Michigan, Ann Arbor, MI) and E. Li (The Scripps Research Institute, La Jolla, CA). DN–caspase-9 and Fas were provided by G. Salvesen (The Burnham Institute, La Jolla, CA). For the induction of death by chimeras, subconfluent (∼30% confluent) cell cultures were transfected via lipofectamine or lipofectamine plus, as per the manufacturer's instructions (Invitrogen), and harvested at the time points described. For PARP cleavage studies, cells were lysed on the plate in complete RIPA. For PI studies, cells were briefly trypsinized and collected. Cells were labeled with Alexa-488–conjugated anti-CD25 (7G7B6), and PI was added to assess loss of viability. To avoid misinterpretation of DNA toxicity in coexpression experiments, only 2 μg of pCI-NeoTac-β3 was cotransfected with 4 μg of the specific apoptotic inhibitor, as described in the cotransfection/rescue studies. Expression of these constructs was confirmed by FACS® and/or immunoblotting, as described in the legends.
Assessment of apoptotic indicators
For FACS® analysis of apoptosis, only cells remaining adherent on tissue culture plastic after washing with PBS were trypsinized briefly, quenched in complete media, and again washed in PBS, before annexin-V staining and assessment on a FACScan® flow cytometer (Becton Dickinson). Tac-expressing cells (Alexa 594, FL3) were identified as apoptotic by annexin staining (FITC, FL1). For PARP cleavage studies, cells were lysed in RIPA (Filardo et al., 1995), cell debris was pelleted at 16000 g, and lysates were analyzed by immunoblotting (2 μg/ml PARP; catalog number 66391A; PharMingen), 5 μg/ml Tac mAb 7G7B6 (American Type Culture Collection), rabbit antisera to GFP (Santa Cruz Biotechnology, Inc.) via species-specific secondary HRP-conjugated antiserum (Jackson ImmunoResearch Laboratories) and Lumigen PS-3 improved chemiluminescent substrate (Lumigen). The 115- and 85-kD caspase-processed forms were quantitated (IP Lab Spectrum Software) within each experiment. Data was expressed as a ratio of p85/p115 for the lane (construct expressing and nonexpressing cells, inclusive). Caspase activity isolated from total cell populations was also assessed using LEHD-pNA (unpublished data) and IETD-pNA colorimetric assays according to the manufacturer's protocol (Chemicon).
Cell spreading
Digital images of cells were captured serially during spreading using a Princeton Instruments Micromax CCD camera (Roper Scientific, Inc.). Cell spreading was quantified by segmentation of GFP or GFP-β3–expressing cells (COS7) or Cell Tracker-Green–labeled cells (HUVEC) with automated area calculations using IP Lab Spectrum Software (Scanalytics). The average cell spreading was determined from the cell area on 10 random fields for each time point.
Adhesion to antibody-coated substrate
Anti-Tac (5 μg/ml in PBS, pH 8.1) was allowed to coat nontissue culture dishes or coverslips overnight at 4°C. Surfaces were washed twice with PBS before cell adhesion. COS7 cells expressing Tac 12 h after transfection with 40 ng Tac-β3 pCI-Neo/cm2 were trypsinized, quenched, and allowed to seed the anti-Tac–coated plates in serum-free media. After 1 h at 37°C, unattached cells were removed by washing with serum-free media, and complete media was added. Cells that were lysed and analyzed for integrin expression and PARP cleavage (as described) were compared 24 h later to control populations still adherent on their original 100-mm dish.
Membrane isolation and immunoprecipitations
Cellular fractionation was performed as previously described (Chen et al., 2000). Pelleted membranes were resolubilized in Laemmli buffer for gel analysis, or in 1× protease inhibitor cocktail (1% Triton X-100, 25 mM Hepes, pH 7.4; Boehringer) for immunoprecipitation with anti-GFP agarose (Santa Cruz Biotechnology Inc.). Alternately, integrin–caspase complexes were immunoprecipitated by adding precipitating antisera (polyclonal anti-β3, polyclonal anti-GFP, or polyclonal anti-LRP) to form DISC-like structures, followed by lysis on ice in 1% triton, as described (Scaffidi et al., 1999). Caspase immunoblotting was performed with anticaspase-8 polyclonal (Chemicon; StressGen Biotechnologies) or monoclonal (Calbiochem; mAb3) antibodies. These antibodies recognize both the zymogen and active forms of the caspase. The potential presence of other caspases was excluded by immunoblotting for the presence of caspases-1, -3, -6, -7, -9, and -10 using specific rabbit polyclonal antisera (Chemicon; StressGen Biotechnologies) that recognize both zymogen and active forms of the caspase. Even though caspases-3, -8, and -9 were the predominant caspases in COS7 cells, all but caspase-1 and -10 were detected.
Caspase labeling and inhibition with peptides
The methyl ester–derived peptide inhibitors zIETDfmk, zDEVDfmk, and zLEHDfmk (Calbiochem) or zVADfmk (Bachem) were reconstituted in DMSO (50 mM) and diluted as appropriate. Apoptosis was assessed in subconfluent 33-mm dishes of COS7 cells expressing submaximal quantities of death-inducing constructs (1–2 μg of Tac-β3/pCI-NEO) using 40–80 μM peptides (or DMSO diluent) by visual scoring (not shown) and via the cleavage of the executioner caspase substrate PARP. Fluorescent FAM-VAD (InterGen) labeling of caspase in Tac-expressing cells was performed according to the manufacturer's specifications, followed by fixation (PBS/1% paraformaldehyde) and blocking (PBS/3%BSA) for 1 h at 37°C, and detection of Tac with Alexa-594–conjugated (Molecular Probes) mAb 7G7B6 (0.5 μg/ml) for 1 h. Cells were washed three times with PBS and examined with a Bio-Rad Laboratories 1024 confocal microscope. Biotinylation of active caspases with biotin-VADfmk was performed by first removing serum-containing media, washing the cells once, and incubating at a final concentration of 10 μM biotin-VAD in DME for the final hour. Biotinylated complexes were isolated with avidin-Sepharose, identified by probing with antibiotin horseradish peroxidase–conjugated monoclonal antibody (Sigma-Aldrich), and developed via CDP-star reagent (Applied Biosystems). Membranes were subsequently reprobed with caspase-specific antisera (-3, -6, -7, -8, -9) as described.
Acknowledgments
The authors wish to thank Drs. R. Klemke, G. Salvesen, S. Shattil, and H. Stennickle for helpful discussions.
This work was supported by grants CA45726, CA50286, and CA78045 (D.A. Cheresh) and AR02089 (C.M. Storgard) from the National Institutes of Health. X.S. Puente is a recipient of a Human Frontier Science Program fellowship. This is Scripps Research Institute manuscript number 12974-IMM.
Footnotes
Abbreviations used in this paper: DISC, death-inducing signaling complex; ECM, extracellular matrix; FADD, Fas-associating protein with death domain; GFP, green fluorescent protein; HUVEC, human umbilical vein endothelial cell; IMD, integrin-mediated death; LRP, LDL-like receptor protein; PI, propidium iodide, PARP, poly (ADP-ribose) polymerase.
References
- Adachi, M., T. Taki, M. Higashiyama, N. Kohno, H. Inufusa, and M. Miyake. 2000. Significance of integrin alpha5 gene expression as a prognostic factor in node-negative non-small cell lung cancer. Clin. Cancer Res. 6:96–101. [PubMed] [Google Scholar]
- Aoudjit, F., and K. Vuori. 2001. Matrix attachment regulates Fas-induced apoptosis in endothelial cells. A role for c-flip and implications for anoikis. J. Cell Biol. 152:633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer, K.L., and B.S. Jacobson. 1995. Beta 1 integrins signal lipid second messengers required during cell adhesion. Mol. Biol. Cell. 6:1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco, E., W. Chen, R. Paul, D.A. Cheresh, and N.R. Cooper. 2000. Beta1 integrin antagonism on adherent, differentiated human neuroblastoma cells triggers an apoptotic signaling pathway. Neuroscience. 101:1145–1152. [DOI] [PubMed] [Google Scholar]
- Boudreau, N., Z. Werb, and M.J. Bissell. 1996. Suppression of apoptosis by basement membrane requires three-dimensional tissue organization and withdrawal from the cell cycle. Proc. Natl. Acad. Sci. USA. 93:3509–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brassard, D.L., E. Maxwell, M. Malkowski, T.L. Nagabhushan, C.C. Kumar, and L. Armstrong. 1999. Integrin alpha(v)beta(3)-mediated activation of apoptosis. Exp. Cell Res. 251:33–45. [DOI] [PubMed] [Google Scholar]
- Bredesen, D.E., X. Ye, A. Tasinato, S. Sperandio, J.J. Wang, N. Assa-Munt, and S. Rabizadeh. 1998. p75NTR and the concept of cellular dependence: seeing how the other half die. Cell Death Differ. 5:365–371. [DOI] [PubMed] [Google Scholar]
- Broday, D.M. 2000. Diffusion of clusters of transmembrane proteins as a model of focal adhesion remodeling. Bull. Math. Biol. 62:891–924. [DOI] [PubMed] [Google Scholar]
- Brooks, P.C., R.A. Clark, and D.A. Cheresh. 1994. a. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 264:569–571. [DOI] [PubMed] [Google Scholar]
- Brooks, P.C., A.M. Montgomery, M. Rosenfeld, R.A. Reisfeld, T. Hu, G. Klier, and D.A. Cheresh. 1994. b. Integrin alpha v beta 3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 79:1157–1164. [DOI] [PubMed] [Google Scholar]
- Chen, F., B.M. Hersh, B. Conradt, Z. Zhou, D. Riemer, Y. Gruenbaum, and H.R. Horvitz. 2000. Translocation of C. elegans CED-4 to nuclear membranes during programmed cell death. Science. 287:1485–1489. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan, A.M., C.G. Tepper, M.F. Seldin, K. O'Rourke, F.C. Kischkel, S. Hellbardt, P.H. Krammer, M.E. Peter, and V.M. Dixit. 1996. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J. Biol. Chem. 271:4961–4965. [DOI] [PubMed] [Google Scholar]
- Cho, S.Y., and R.L. Klemke. 2000. Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J. Cell Biol. 149:223–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller, B.S., D.A. Cheresh, E. Asch, and U. Seligsohn. 1991. Platelet vitronectin receptor expression differentiates Iraqi-Jewish from Arab patients with Glanzmann thrombasthenia in Israel. Blood. 77:75–83. [PubMed] [Google Scholar]
- Curley, G.P., H. Blum, and M.J. Humphries. 1999. Integrin antagonists. Cell. Mol. Life Sci. 56:427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallabrida, S.M., M.A. De Sousa, and D.H. Farrell. 2000. Expression of antisense to integrin subunit beta 3 inhibits microvascular endothelial cell capillary tube formation in fibrin. J. Biol. Chem. 275:32281–32288. [DOI] [PubMed] [Google Scholar]
- Ding, H.F., Y.L. Lin, G. McGill, P. Juo, H. Zhu, J. Blenis, J. Yuan, and D.E. Fisher. 2000. Essential role for caspase-8 in transcription-independent apoptosis triggered by p53. J. Biol. Chem. 275:38905–38911. [DOI] [PubMed] [Google Scholar]
- Du, X., T.C. Saido, S. Tsubuki, F.E. Indig, M.J. Williams, and M.H. Ginsberg. 1995. Calpain cleavage of the cytoplasmic domain of the integrin beta 3 subunit. J. Biol. Chem. 270:26146–26151. [DOI] [PubMed] [Google Scholar]
- Eliceiri, B.P., R. Klemke, S. Stromblad, and D.A. Cheresh. 1998. Integrin αvβ3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 140:1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, L., K.W. Freeman, T. Khan, E. Pham, and D.M. Spencer. 1999. Improved artificial death switches based on caspases and FADD. Hum. Gene Ther. 10:2273–2285. [DOI] [PubMed] [Google Scholar]
- Fassler, R., E. Georges-Labouesse, and E. Hirsch. 1996. Genetic analyses of integrin function in mice. Curr. Opin. Cell Biol. 8:641–646. [DOI] [PubMed] [Google Scholar]
- Filardo, E.J., P.C. Brooks, S.L. Deming, C. Damsky, and D.A. Cheresh. 1995. Requirement of the NPXY motif in the integrin beta 3 subunit cytoplasmic tail for melanoma cell migration in vitro and in vivo. J. Cell Biol. 130:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane, D.M., E. Bossy-Wetzel, N.J. Waterhouse, T.G. Cotter, and D.R. Green. 1999. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J. Biol. Chem. 274:2225–2233. [DOI] [PubMed] [Google Scholar]
- Frisch, S.M., and H. Francis. 1994. Disruption of epithelial cell–matrix interactions induces apoptosis. J. Cell Biol. 124:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1990. Elevated levels of the alpha 5 beta 1 fibronectin receptor suppress the transformed phenotype of Chinese hamster ovary cells. Cell. 60:849–859. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Harrington, E.O., A. Smeglin, J. Newton, G. Ballard, and S. Rounds. 2001. Protein tyrosine phosphatase-dependent proteolysis of focal adhesion complexes in endothelial cell apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 280:L342–L353. [DOI] [PubMed] [Google Scholar]
- Hauck, C.R., T. Hunter, and D.D. Schlaepfer. 2001. The v-Src SH3 domain facilitates a cell adhesion-independent association with FAK. J. Biol. Chem. 276:17653–17662. [DOI] [PubMed] [Google Scholar]
- Hildebrand, J.D., M.D. Schaller, and J.T. Parsons. 1993. Identification of sequences required for the efficient localization of the focal adhesion kinase, pp125FAK, to cellular focal adhesions. J. Cell Biol. 123:993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, M.S., L.E. Lunsford, V. Trinkaus-Randall, and K.K. Svoboda. 1997. Chondrocyte survival and differentiation in situ are integrin mediated. Dev. Dyn. 210:249–263. [DOI] [PubMed] [Google Scholar]
- Hocking, D.C., J. Sottile, and K.J. Langenbach. 2000. Stimulation of integrin-mediated cell contractility by fibronectin polymerization. J. Biol. Chem. 275:10673–10682. [DOI] [PubMed] [Google Scholar]
- Hynes, R.O. 1992. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 69:11–25. [DOI] [PubMed] [Google Scholar]
- Hynes, R.O., and K.M. Hodivala-Dilke. 1999. Insights and questions arising from studies of a mouse model of Glanzmann thrombasthenia. Thromb. Haemost. 82:481–485. [PubMed] [Google Scholar]
- Ilan, N., S. Mahooti, and J.A Madri. 1998. Distinct signal transduction pathways are utilized during the tube formation and survival phases of in vitro angiogenesis. J. Cell Biol. 111:3621–3631. [DOI] [PubMed] [Google Scholar]
- Ilic, D., E.A. Almeida, D.D. Schlaepfer, P. Dazin, S. Aizawa, and C.H. Damsky. 1998. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 143:547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingber, D.E. 1992. Extracellular matrix as a solid-state regulator in angiogenesis: identification of new targets for anti-cancer therapy. Semin. Cancer Biol. 3:57–63. [PubMed] [Google Scholar]
- Kim, S., K. Bell, S.A. Mousa, and J.A. Varner. 2000. Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am. J. Pathol. 156:1345–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFlamme, S.E., S.K. Akiyama, and K.M. Yamada. 1992. Regulation of fibronectin receptor distribution. J. Cell Biol. 117:437–447 (published erratum appears in J. Cell Biol. 1992 Jul;118(2):491). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S., D.A. Calderwood, and M.H. Ginsberg. 2000. Integrin cytoplasmic domain-binding proteins. J. Cell Sci. 113:3563–3571. [DOI] [PubMed] [Google Scholar]
- MacCorkle, R.A., K.W. Freeman, and D.M. Spencer. 1998. Synthetic activation of caspases: artificial death switches. Proc. Natl. Acad. Sci. USA. 95:3655–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlen, P., S. Rabizadeh, S.J. Snipas, N. Assa-Munt, G.S. Salvesen, and D.E. Bredesen. 1998. The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature. 395:801–804. [DOI] [PubMed] [Google Scholar]
- Memon, S.A., J. Hou, M.B. Moreno, and C.M. Zacharchuk. 1998. Apoptosis induced by a chimeric Fas/FLICE receptor: lack of requirement for Fas- or FADD-binding proteins. J. Immunol. 160:2046–2049. [PubMed] [Google Scholar]
- Moro, L., M. Venturino, C. Bozzo, L. Silengo, F. Altruda, L. Beguinot, G. Tarone, and P. Defilippi. 1998. Integrins induce activation of EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 17:6622–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, M.J., and B.A. Lessey. 1999. Embryo implantation and tumor metastasis: common pathways of invasion and angiogenesis. Semin. Reprod. Endocrinol. 17:275–290. [DOI] [PubMed] [Google Scholar]
- Muzio, M., B.R. Stockwell, H.R. Stennicke, G.S. Salvesen, and V.M. Dixit. 1998. An induced proximity model for caspase-8 activation. J. Biol. Chem. 273:2926–2930. [DOI] [PubMed] [Google Scholar]
- Plath, T., K. Detjen, M. Welzel, Z. von Marschall, D. Murphy, M. Schirner, B. Wiedenmann, and S. Rosewicz. 2000. A novel function for the tumor suppressor p16(INK4a): induction of anoikis via upregulation of the alpha(5)beta(1) fibronectin receptor. J. Cell Biol. 150:1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti, E. 1997. Integrins as signaling molecules and targets for tumor therapy. Kidney Int. 51:1413–1417. [DOI] [PubMed] [Google Scholar]
- Salvesen, G.S., and V.M. Dixit. 1999. Caspase activation: the induced-proximity model. Proc. Natl. Acad. Sci. USA. 96:10964–10967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi, C., P.H. Krammer, and M.E. Peter. 1999. Isolation and analysis of components of CD95 (APO-1/Fas) death-inducing signaling complex. Methods. 17:287–291. [DOI] [PubMed] [Google Scholar]
- Scatena, M., M. Almeida, M.L. Chaisson, N. Fausto, R.F. Nicosia, and C.M. Giachelli. 1998. NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J. Cell Biol. 141:1083–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz, M.A. 1997. Integrins, oncogenes, and anchorage independence. J. Cell Biol. 139:575–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilenov, L.B., A. Mikhailov, R.J. Pelham, E.E. Marcantonio, and G.G. Gundersen. 1999. Focal adhesion motility revealed in stationary fibroblasts. Science. 286:1172–1174. [DOI] [PubMed] [Google Scholar]
- Soengas, M.S., R.M. Alarcon, H. Yoshida, A.J. Giaccia, R. Hakem, T.W. Mak, and S.W. Lowe. 1999. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 284:156–159. [DOI] [PubMed] [Google Scholar]
- Stennicke, H.R., and G.S. Salvesen. 1999. Caspases: preparation and characterization. Methods. 17:313–319. [DOI] [PubMed] [Google Scholar]
- Storgard, C.M., D.G. Stupack, A. Jonczyk, S.L. Goodman, R.I. Fox, and D.A. Cheresh. 1999. Decreased angiogenesis and arthritic disease in rabbits treated with an alphavbeta3 antagonist. J. Clin. Invest. 103:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromblad, S., J.C. Becker, M. Yebra, P.C. Brooks, and D.A. Cheresh. 1996. Suppression of p53 activity and p21WAF1/CIP1 expression by vascular cell integrin alphaVbeta3 during angiogenesis. J. Clin. Invest. 98:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sympson, C.J., R.S. Talhouk, C.M. Alexander, J.R. Chin, S.M. Clift, M.J. Bissell, and Z. Werb. 1994. Targeted expression of stromelysin-1 in mammary gland provides evidence for a role of proteinases in branching morphogenesis and the requirement for an intact basement membrane for tissue-specific gene expression. J. Cell Biol. 125:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani, P.D., L. Singh, K.L. Auer, and S.E. LaFlamme. 1997. The role of conserved amino acid motifs within the integrin beta3 cytoplasmic domain in triggering focal adhesion kinase phosphorylation. J. Biol. Chem. 272:7892–7898. [DOI] [PubMed] [Google Scholar]
- Takada, Y., E.A. Wayner, W.G. Carter, and M.E. Hemler. 1988. Extracellular matrix receptors, ECMRII and ECMRI, for collagen and fibronectin correspond to VLA-2 and VLA-3 in the VLA family of heterodimers. J. Cell. Biochem. 37:385–393. [DOI] [PubMed] [Google Scholar]
- Tarone, G., E. Hirsch, M. Brancaccio, M. De Acetis, L. Barberis, F. Balzac, F. Retta, C. Botta, F. Altruda, and L. Silengo. 2000. Integrin function and regulation in development. Int. J. Dev. Biol. 44:725–731. [PubMed] [Google Scholar]
- Thornberry, N.A., T.A. Rano, E.P. Peterson, D.M. Rasper, T. Timkey, M. Garcia-Calvo, V.M. Houtzager, P.A. Nordstrom, S. Roy, J.P. Vaillancourt, et al. 1997. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272:17907–17911. [DOI] [PubMed] [Google Scholar]
- Varner, J.A., D.A. Emerson, and R.L. Juliano. 1995. Integrin alpha 5 beta 1 expression negatively regulates cell growth: reversal by attachment to fibronectin. Mol. Biol. Cell. 6:725–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wary, K.K., F. Mainiero, S.J. Isakoff, E.E. Marcantonio, and F.G. Giancotti. 1996. The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell. 87:733–743. [DOI] [PubMed] [Google Scholar]
- Wolf, B.B., and D.R. Green. 1999. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J. Biol. Chem. 274:20049–20052. [DOI] [PubMed] [Google Scholar]
- Yee, K.O., Y. Ikari, and S.M. Schwartz. 2001. An update of the Grutzbalg hypothesis: the role of thrombosis and coagulation in atherosclerotic progression. Thromb. Haemost. 85:207–217. [PubMed] [Google Scholar]
- Zheng, T.S., and R.A. Flavell. 2000. Divinations and surprises: genetic analysis of caspase function in mice. Exp. Cell Res. 256:67–73. [DOI] [PubMed] [Google Scholar]
- Zhou, Q., S. Snipas, K. Orth, M. Muzio, V.M. Dixit, and G.S. Salvesen. 1997. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J. Biol. Chem. 272:7797–7800. [DOI] [PubMed] [Google Scholar]