

Abstract
Mutations in P0 (MPZ), the major myelin protein of the peripheral nervous system, cause the inherited demyelinating neuropathy Charcot-Marie-Tooth disease type 1B. P0 is a member of the immunoglobulin superfamily and functions as a homophilic adhesion molecule. We now show that point mutations in the cytoplasmic domain that modify a PKC target motif (RSTK) or an adjacent serine residue abolish P0 adhesion function and can cause peripheral neuropathy in humans. Consistent with these data, PKCα along with the PKC binding protein RACK1 are immunoprecipitated with wild-type P0, and inhibition of PKC activity abolishes P0-mediated adhesion. Point mutations in the RSTK target site that abolish adhesion do not alter the association of PKC with P0; however, deletion of a 14 amino acid region, which includes the RSTK motif, does abolish the association. Thus, the interaction of PKCα with the cytoplasmic domain of P0 is independent of specific target residues but is dependent on a nearby sequence. We conclude that PKC-mediated phosphorylation of specific residues within the cytoplasmic domain of P0 is necessary for P0-mediated adhesion, and alteration of this process can cause demyelinating neuropathy in humans.
Keywords: P0; adhesion; myelination; PKC; RACK1
Introduction
Myelin is a multilamellar structure that surrounds axons in both the central nervous system and the peripheral nervous system (PNS),* facilitating nerve conduction. P0, a transmembrane protein of the Ig superfamily, is the major myelin structural protein in the PNS, expressed exclusively by myelinating Schwann cells and necessary for normal myelin structure and function. In mice, the absence of P0 (Giese et al., 1992; Martini et al., 1995) or overexpression of P0 (Wrabetz et al., 2000) results in hypomyelination and peripheral neuropathy. In addition, mutations in the human P0 gene cause the demyelinating peripheral neuropathy Charcot-Marie-Tooth disease 1B, the more severe Dejerine-Sottas syndrome, and congenital hypomyelination, all associated with muscle weakness, atrophy, and sensory loss (for review see Nelis et al., 1999; Shy et al., 2001).
P0 functions as a homophilic adhesion molecule in vitro (Filbin et al., 1990; Filbin and Tennekoon, 1993), and this activity may mediate compaction of adjacent leaflets in peripheral nerve myelin. Analysis of the crystal structure of the P0 extracellular domain indicates that P0 molecules have the potential to interact in cis to form homotetramers, and these interact in trans to mediate homophilic adhesion (Shapiro et al., 1996). Consistent with this model, mutation of several of the amino acid residues critical for these putative cis and trans interactions can cause the inherited demyelinating peripheral neuropathy, CMT1B (Warner et al., 1996; Nelis et al., 1999).
The cytoplasmic domain of P0 is also important for its function, and mutations in this region result in loss of P0-mediated homophilic adhesion in vitro (Wong and Filbin, 1994). Coexpression of both wild-type and cytoplasmically truncated P0 causes loss of wild-type function, presumably due to a dominant negative effect of the truncated molecule (Wong and Filbin, 1996). Mutations within the P0 cytoplasmic domain are also found in patients with inherited demyelinating neuropathies, some of which have very severe clinical phenotypes (Nelis et al., 1999; Shy et al., 2001). These data demonstrate a critical role for the cytoplasmic domain of P0 in mediating homophilic adhesion and in the molecular pathogenesis of inherited demyelinating neuropathies.
We now demonstrate that deletion of a 14 amino acid sequence in the cytoplasmic domain of P0 abolishes adhesive function. Point mutations in this region that alter a PKC substrate motif (RSTK) and mutation of an adjacent serine residue (S204) are critical to P0 adhesive function. Moreover, PKCα and the receptor for activated C kinase (RACK1) are coimmunoprecipitated from cells expressing wild-type P0 but not P0 bearing a deletion of 14 amino acid sequence. In addition, inhibition of PKC activity results in loss of P0-mediated adhesion. We have also identified a mutation of the initial arginine of the PKC substrate motif to a serine residue (R198S) in a patient with CMT1B, and we find that cells expressing P0 bearing this mutation fail to form homophilic adhesions. These data indicate that PKC-mediated phosphorylation is an important component of the regulation of P0-mediated adhesion and demonstrate that alteration of this process can cause demyelinating neuropathy in humans. This is the first glimpse of the molecular machinery that regulates the function of P0 in myelination.
Results
A subterminal 13 amino acid sequence in the cytoplasmic domain of P0 is essential for adhesion
To define the regions of the cytoplasmic domain of P0 that are essential for homophilic adhesion, we stably transfected mouse L cells and HeLa cells with cDNAs encoding wild-type P0 or P0 deletion mutants lacking the COOH-terminal 13 (Δ13), 28 (Δ28), 43 (Δ43), or 59 amino acids (Δ59). Expression of the transfected P0 constructs was determined by reverse transcription (RT)-PCR using specific primers for each construct and by Western blotting. RT-PCR reveals that all constructs were expressed in relatively equal amounts in both cell types (Fig. 1 B shows L cells). To ensure that protein was expressed and inserted in the plasma membrane, we labeled live cells with a membrane impermeable biotinylation reagent followed by immunoprecipitation with anti-P0 antibody. The immunoprecipitated material was fractionated by SDS-PAGE, transferred to polyvinylene difluoride (PVDF) membranes, and probed with HRP-streptavidin. Each of the P0 constructs is expressed at the cell surface (Fig. 1 C shows L cells).
Figure 1.

Expression of full-length P0 and P0 deletion mutants in transfected L cells. (A) Diagram of P0 cDNA showing the relative positions of the primers used to clone full-length and mutant P0. The same 5′ primer (P0WT5) was used in all cases. The 3′ primers were designed to allow the cDNAs to be inserted in frame with the flag sequence in the pCMV-Tag4 vector. SP, signal sequence; EC, extracellular domain; TM, transmembrane domain; IC, intracellular domain. (B) P0 mRNA is expressed in L cells transfected with the P0 constructs as determined by RT-PCR using the specific primers shown in A. Primers specific to the enzyme HPRT were used to compare expression levels between cell lines. Numbers to the right of the figure represent the migration of molecular markers. (C) Full-length and mutant P0s are expressed at the cell surface in transfected L cells. Cell membrane proteins were labeled with biotin, and P0 was precipitated with an anti-P0 antibody. The immunoprecipitates were fractionated by SDS-PAGE, transferred to PVDF membranes, and probed with HRP-streptavidin. WT, full-length P0; Δ13, Δ28, Δ43, and Δ59, P0 constructs lacking the terminal 13, 28, 43, or 59 amino acids, respectively; Vect, L cells transfected with empty vector. Numbers to the left of the figure represent the migration of molecular markers.
Cells expressing each of the deletion mutants were assayed for their ability to form homophilic adhesions as measured by binding of single cells expressing P0 constructs to a confluent layer of cells expressing wild-type P0. Both L cells and HeLa cells expressing Δ13 were comparable to the same cell types expressing wild-type P0 (Fig. 2, A and B). However, P0-mediated adhesion was reduced dramatically in all cell lines with deletions of 28 amino acids or greater (Fig. 2, A and B). This suggests that the sequence between amino acids 193 and 206 is essential for P0-mediated cell–cell adhesion. This region contains three serine residues that are phosphorylated in mouse sciatic nerve: S197, S199, and S204 (Hilmi et al., 1995). These residues are conserved in the human P0 sequence. Furthermore, serine 199 is part of a PKC recognition motif (198-RSTK-201) (Kishimoto et al., 1985; Woodgett et al., 1986), suggesting that phosphorylation mediated by PKC is critical to P0 adhesive function.
Figure 2.
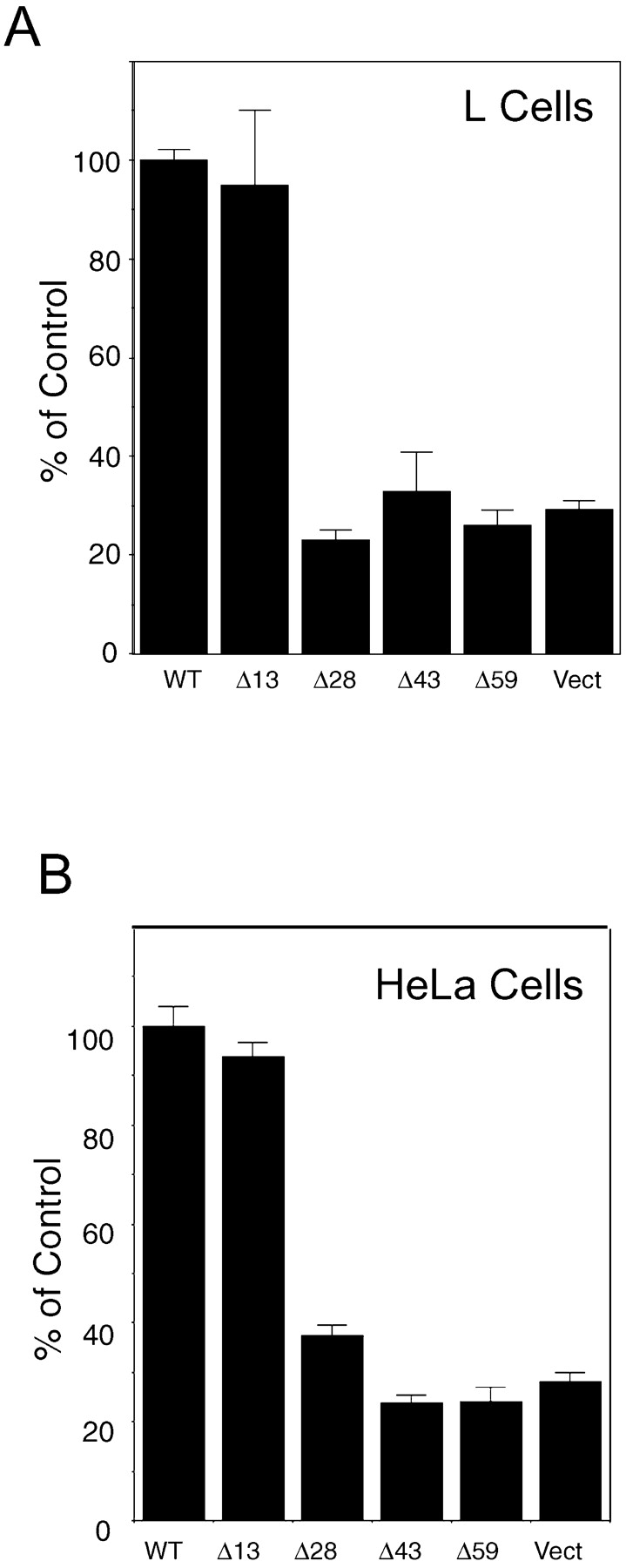
Identification of a region in the cytoplasmic domain of P0 that is essential for P0-mediated adhesion. (A) L cells stably expressing full-length or truncated P0 were labeled with a vital fluorescent dye and assayed for adhesion to monolayers of L cells expressing full-length P0. Adherent cells were determined by measuring the amount of fluorescence associated with the cell layers. Adhesion of L cells bearing full-length P0 was considered 100% and used to calculate adhesion of the other lines. Each cell type was assayed in triplicate. (B) HeLa cells stably expressing full-length or truncated P0 were assayed for adhesion as described in A. WT, full-length P0; Δ13, Δ28, Δ43, Δ59, P0 bearing deletions of the terminal 13, 28, 43, and 59 amino acids, respectively; Vect, L cells transfected with empty vector. Bars represent the standard deviation from the mean (p < 0.0001).
Since it has been reported that expression of P0 in HeLa cells results in upregulation of cadherin expression (Doyle et al., 1995), we were careful to perform all P0 adhesion assays in the absence of Ca2+, which is necessary for cadherin-mediated adhesion. Additionally, we examined both HeLa and L cells bearing each of the P0 deletion constructs for cadherin expression using a pan cadherin antibody to blot equivalent amounts of cell lysate. In agreement with the previous observations, cadherin and the cadherin-associated protein β-catenin are upregulated in HeLa cells stably transfected with pCMV Tag4 containing the full-length wild-type P0 but not in L cells similarly transfected (unpublished data). Furthermore, among HeLa cells upregulation requires the same region of the molecule that is essential for adhesion function, amino acids 193–206 (unpublished data). The two cell lines do differ in their basal level of cadherin expression: HeLa cells express a small amount of cadherin, whereas L cells do not express detectable levels of cadherin. Thus, upregulation of cadherin expression in the presence of P0 is not universal and may depend on a basal level of cadherin expression and P0 adhesive function or signaling mediated by the cytoplasmic domain.
A PKC target motif and PKC activity are essential for P0-mediated adhesion
To investigate the importance of the RSTK motif and the adjacent serine, we assayed L cells transfected with P0 containing mutations in the RSTK motif (S199A and T200A) and in serine 204 (S204A) for their ability to form homophilic P0 adhesions. In addition, we assayed cells bearing a mutation at serine 197 (S197A), a site of low level in vivo phosphorylation (Hilmi et al., 1995), aspartic acid 195 (D195A), and tyrosine 191 (Y191A) as controls. Stable cell lines were created and tested for P0 expression by RT-PCR and immunoprecipitation of cell surface labeled P0 as above. All constructs were expressed at the cell surface (unpublished data). Cells expressing P0 mutated at serine sites 199 or 204 or threonine 200 failed to form P0-mediated adhesions (Fig. 3 A), indicating that both the PKC motif and serine residue 204 are functionally important. In contrast to these mutations, substitution of serine 197 or aspartic acid 195 have little or no effect (Fig. 3 A). Furthermore, replacement of tyrosine 191, a site shown recently to be phosphorylated in vivo (Xu et al., 2000a) with alanine, also has little or no effect on P0-mediated adhesion (Fig. 3 A).
Figure 3.
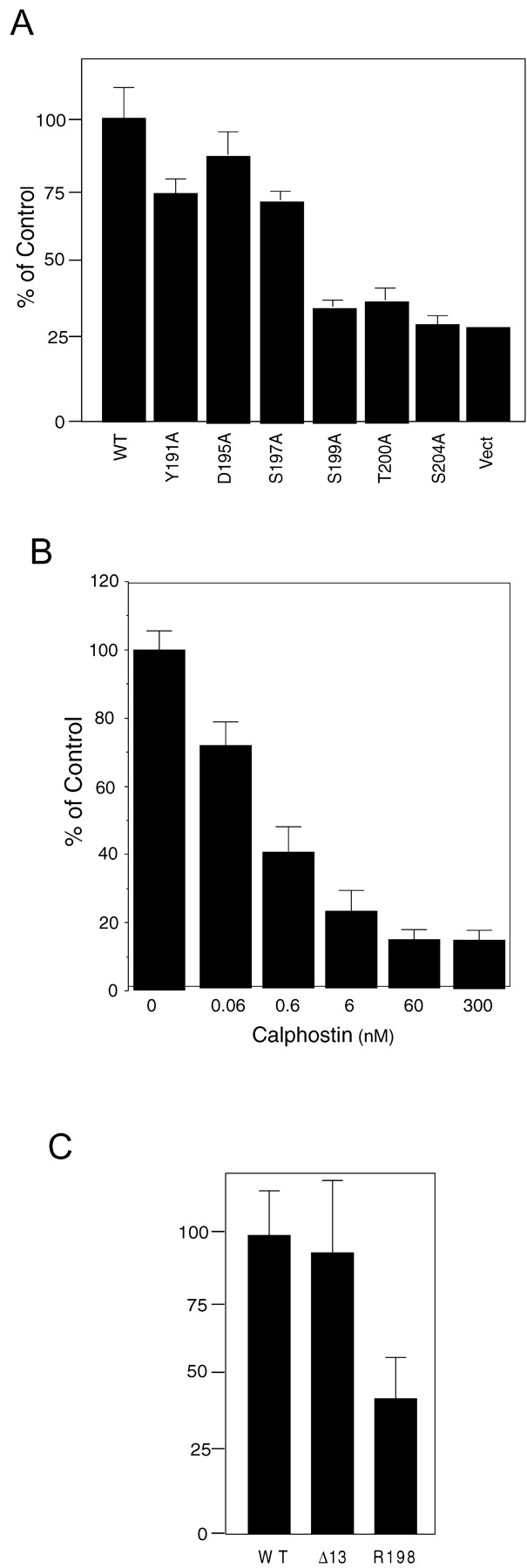
A PKC target motif and the adjacent serine 204 is essential for P0-mediated cell adhesion and myelination. (A) Full-length P0 constructs bearing point mutations at the indicated amino acids were prepared by PCR and transfected into L cells. Stable clones of cells expressing each P0 mutant were selected and assayed for P0-mediated adhesion as described in the legend to Fig. 2. Adhesion is expressed as percent of control, with the control being adhesion of L cells expressing full-length P0. Each cell type was assayed in triplicate. WT, cell expressing full-length P0. Mutant cell lines are indicated by the specific point mutation. Vect, cell transfected with empty vector. (B) Inhibition of PKC activity prevents P0-mediated adhesion. L cells expressing full-length P0 were assayed for adhesion in the presence of increasing concentrations of calphostin. Adhesion in the absence of the inhibitor was considered 100%. Each cell type was assayed in triplicate. (C) A point mutation in the PKC target motif results in human disease and loss of P0-mediated adhesion in transfected L cells. Stable clones of L cells expressing the R198S P0 mutant were assayed for P0-mediated adhesion as above. Adhesion is represented as percent of control. Bars represent the standard deviation from the mean (p < 0.0001).
To further examine the importance of the PKC function in P0-mediated adhesion, we tested the effect of the PKC inhibitor, calphostin C (Tamaoki and Nakano, 1990; Svetlov and Nigam, 1993). Calphostin C inhibits P0-mediated adhesion in a dose-dependent manner with maximum inhibition at 60 nM (Fig. 3 B). At this concentration (60 nM), calphostin has no detectable effect on cell viability as measured by Sytox green (Molecular Probes).
Mutation within the RSTK domain (R198S) causes the demyelinating peripheral neuropathy CMT1B and loss of P0 homophilic adhesion
We have identified an individual with CMT1B who is heterozygous for a P0 mutation producing a substitution of arginine for serine at amino acid 198 (R198S) within the PKC binding motif. The patient had normal growth, development, and motor function until 5 yr ago, in his late 30s, when he noted the onset of lower extremity clumsiness; however, these symptoms have not progressed notably since that time. On neurological examination, he had minimal symmetrical foot dorsiflexor weakness, a mild decrease in both large and small fiber sensory modalities (vibration and pin prick) in his feet, and diminished Achilles and patellar deep tendon reflexes. His gait appeared normal, but he was unable to walk on his heels, confirming the mild weakness of the foot dorsiflexors. In addition, he had bilateral pes cavus, a common finding in patients with inherited neuropathy, probably caused by weakness of the intrinsic muscles of the feet. On electrophysiological testing, the nerve conduction velocities of peroneal and tibial motor nerves were slowed bilaterally to ∼25 m/s, whereas his right sural sensory NCV was slowed to 28 m/s. The left sural potential was unobtainable. These neurological signs and symptoms and electrophysiological findings are consistent with the presence of a mild demyelinating sensory and motor peripheral neuropathy.
To determine if this mutation affected P0 homophilic adhesion, we transfected L cells with P0 bearing this mutation. Expression was assayed as above and was found approximately equivalent to other P0 constructs in L cells (unpublished data). These cells showed a much reduced ability to form P0 homophilic adhesions (Fig. 3 C). Thus, elimination of PKC target residues or alteration of the PKC target motif not only abolishes P0 adhesive function in vitro but also causes demyelinating disease in humans.
PKCα and the PKC binding protein RACK1 are associated with P0 in situ
The functional requirement for the PKC target motif in P0 and its importance to human disease led us to determine which of the many isoforms of PKC is present in sciatic nerve. We find that two conventional PKCs, α and γ, are detected, as well as the novel isoforms δ, ε, and η (Table I).
Table I. PKC expression in mouse sciatic nerve.
| Isoform | Expression | Isoform | Expression |
|---|---|---|---|
| Conventional | Novel | ||
| α | + | δ | + |
| β | − | ɛ | + |
| γ | + | η | + |
| Atypical | |||
| ι | − | ||
| λ | − |
All of these isoforms are also detected in P0-transfected L cells (Fig. 4, none). However, P0 immunoprecipitates from L cells lysed in neutral detergent reveal that only PKCα is present in association with P0 (Fig. 4). Furthermore, PKCα is present and associated with P0 in cultured Schwann cells (Fig. 4, SC).
Figure 4.

PKCα coimmunoprecipitates specifically with P0 from Schwann cells and L cells expressing full-length P0. Confluent cultures of Schwann cells (SC), L cells expressing full-length P0 (P0wt), and L cells transfected with empty vector (Vect) were lysed in nonionic detergent. 1 ml of each cleared cell lysate was immunoprecipitated with anti-P0 antibody (P0), and equal aliquots of resuspended precipitates were fractionated by SDS-PAGE, transferred to PVDF, and probed with antibody to the PKC isoform indicated. 20 μl of each cell lysate, before immunoprecipitation, was also fractionated by SDS-PAGE, transferred to PVDF, and blotted with the indicated antibody (lanes labeled none). Numbers to the left indicate the migration of prestained standards. Bands due to PKC and Ig heavy chain are indicated at the right.
We further probed P0 immunoprecipitates derived from each of our deletion mutants for the association of PKC and the PKC binding protein RACK1. RACKs bind activated PKC at a site distinct from the substrate-binding site (Mochly-Rosen et al., 1991) and are thought to target the enzyme to specific cellular sites independent of PKC substrate recognition (Sim and Scott, 1999; for reviewed see Jaken and Parker, 2000). As can be seen in Fig. 5, deletion of the same 14 amino acid sequence containing the RSTK motif that is essential for P0-mediated adhesion eliminates the association of PKCα and RACK1 with P0 (Fig. 5, compare Δ13 with Δ28 and Δ59). However, point mutations altering the RSTK substrate recognition motif or the adjacent serine 204, mutations that do eliminate P0-mediated adhesion, do not alter PKC or RACK1 binding. Thus, substrate recognition by PKC and binding, presumably mediated by RACK1, are two independent events occurring through distinct sites within a 14 amino acid domain of P0.
Figure 5.

A 14 amino acid sequence in the cytoplasmic domain of P0 is essential for its association with PKCα and the PKC binding protein RACK1. L cells expressing wild-type P0 (WT) or P0 with point mutations or progressive truncations of its cytoplasmic domain were lysed in neutral detergent and immunoprecipitated with anti-P0 antibody (P0) or preimmune serum (Co). The precipitates were fractionated by SDS-PAGE and transferred to PVDF. The membranes were cut at the 55 kD marker and the top half probed with anti-PKCα antibody, whereas the bottom half was probed with anti-RACK1 monoclonal antibody. Cell line indicates L cells transfected with empty vector (Vec), wild-type P0 (WT), P0 lacking the 13 COOH-terminal amino acids (Δ13), P0 lacking the 28 COOH-terminal amino acids (Δ28), P0 lacking the 59 COOH-terminal amino acids (Δ59), P0 bearing a S197A mutation (S197), P0 bearing a S199A mutation (S199), or P0 bearing a S204A mutation (S204).
Discussion
In this study, we present data correlating specific amino acid residues in the cytoplasmic domain of P0 with adhesion function and ultimately with human disease. By creating a series of deletions in the cytoplasmic domain, we find that the region between amino acids 193 and 206 is essential for P0-mediated adhesion. This region contains a PKC substrate motif (RSTK) that is one of two major sites of in situ phosphorylation on serine residues (serine 199) (Hilmi et al., 1995). Using site-directed mutagenesis, we demonstrate that this motif is important for adhesive function. A second serine residue at position 204, closely juxtaposed to the RSTK site, is also critical for adhesion function, and this residue is also phosphorylated in the sciatic nerve (Hilmi et al., 1995). Another serine (197) that is phosphorylated in situ (Hilmi et al., 1995) does not appear to be necessary for adhesion function, nor is a tyrosine residue in the cytoplasmic domain, Y191, which is also phosphorylated in sciatic nerve (Iyer et al., 1996).
Of the PKC isoforms present in sciatic nerve and our L cell model, PKCα is specifically immunoprecipitated with P0, indicating an in situ association between the two molecules. This interaction may be mediated by RACK1, the receptor for activated C kinase, since we find that under our experimental conditions the association of these two proteins is absolutely correlated. Furthermore, the association of both proteins with P0 is dependent on a 14 amino acid sequence in the cytoplasmic domain, amino acids 193–206. This is the same region containing the PKC substrate motif; however, the substrate site and the binding site are distinct, since point mutations in the substrate site that eliminate adhesion function do not eliminate binding of PKC or RACK1. RACK1 is one member of a group of proteins that bind PKC at a site distinct from the substrate-binding site (Mochly-Rosen et al., 1991; Sim and Scott, 1999; for review see Jaken and Parker, 2000). We suggest that RACK1 binds activated PKC and interacts with P0 either directly or through an additional adaptor to bring activated PKCα into substrate proximity.
PKC has been implicated previously in phosphorylation of P0 in vitro (Suzuki et al., 1990; Rowe-Rendleman and Eichberg, 1994) and tumor promoters that activate PKC accentuate phosphorylation in situ (Agrawal and Agrawal, 1989). Most pertinent to our identification of PKCα as the critical isoform, abolition of phorbol 12,13 dibutyrate–induced upregulation of PKC in sciatic nerve is accompanied by a reduction in PKCα (Rowe-Rendleman and Eichberg, 1994). However, these prior studies were not able to correlate the PKC-dependent phosphorylation of P0 with a specific function. Our data indicate for the first time that PKC-mediated phosphorylation on serine residues is important for P0 adhesion function: P0-mediated adhesion is abolished by mutations of a PKC target motif, PKCα and the adaptor RACK1 are immunoprecipitated with wild-type P0, and P0-mediated adhesion is decreased dramatically by calphostin C, a kinase inhibitor that preferentially blocks PKC activity (IC50 for PKC = 50 nM).
There are two possible mechanisms by which phosphorylation of the cytoplasmic domain of P0 may regulate adhesion and/or myelination. One is a direct mechanism in which phosphorylation of P0 alters its conformation, allowing the cis or trans interactions required for adhesion (Shapiro et al., 1996). The second is an indirect mechanism in which phosphorylation of P0 allows or facilitates the binding of effector or adaptor molecules essential for adhesion. In either case, dynamic changes in the phosphorylation state of P0 would have significant effects on P0-mediated adhesion and/or myelination. Further experiments will clearly be required to distinguish between these two possibilities.
Our deletion and mutational analysis are consistent with some of the mutations known to give rise to human neuropathies. There are no known human mutations in the COOH-terminal 13 amino acids that give rise to pathology (Fig. 6); the single amino acid change identified in this region (R215L) appears to have no physiological ramifications (Nelis et al., 1999). Accordingly, we find that deletion of this entire region has no effect on adhesion. Five human mutations causing neuropathy produce a shift in the reading frame, and two result in introduction of a stop codon, all altering or deleting the PKC target site and/or serine 204. One of the frame shifts leaves the RSTK motif intact but deletes serine 204. This mutation gives rise to a severe form of CMT1, emphasizing the importance of this residue. In addition, we have identified a patient with CMT1B in which an arginine residue has been changed to a serine within this PKC target motif (RSTK→SSTK), potentially eliminating the ability of PKC to phosphorylate residues critical to adhesive function (Fig. 6). Indeed, mimicking this mutation in our in vitro assay abolishes P0 homophilic adhesion. Thus, these data implicate PKC-mediated phosphorylation of the cytoplasmic domain of P0 in both the regulation of homophilic adhesion and myelination. Consistent with this possibility, mutations in a dual specificity phosphatase, myotubularin-related protein-2, cause an autosomal recessive form of demyelinating CMT, called CMT4B (Bolino et al., 2000). Although it is not yet known whether myotubularin-related protein-2 cleaves phosphate groups on P0, these results clearly demonstrate that a phosphorylation/dephosphorylation cascade plays a role in the regulation of normal PNS myelination.
Figure 6.

Diagram showing human mutations found in the cytoplasmic domain of P0 and the corresponding syndrome. The site and type of mutation are indicated. (1) Y152 to Stop; (2) nucleotide deletion; (3) 17 nucleotide insertion; (4) Q186 to Stop; (5) nucleotide insertion; (6) nucleotide insertion; (7) R198 to S; (8) four nucleotide deletions including S204. The amino acid sequences deleted to create the several P0 truncated mutants used in Fig. 1 are represented as alternating blocks of red and blue. The amino acids that prove to be essential for P0-mediated adhesion are shown in black, and the point mutations that have no effect are shown in green.
Given the central role that P0 plays in myelination, it is likely that other components interact with the cytoplasmic domain to further regulate the process of myelination. Thus, P0 mutations that may have profound effects on the process of myelination independent of its adhesive function will render incomplete any correlation between adhesion itself and severity of disease. Recent work from our group emphasizes this point, since the expression of P0 has dramatic effects on the levels of expression of other myelin proteins important for myelination (Xu et al., 2000b). Additionally, the possibility that tyrosine 191 plays a role independent of adhesive function is suggested by the fact that mutation has no effect on adhesion, yet the array of polypeptides precipitated with anti-P0 antibody is altered by its phosphorylation (Xu et al., 2000a). We expect that a full accounting of the molecular machinery associated with the cytoplasmic domain of P0 will reveal many more effectors and adaptors as is the case for the cytoplasmic domains of other Ig superfamily adhesion molecules (for review see Crossin and Krushel, 2000) and for cadherin (Vleminckx and Kemler, 1999) and integrin (Aplin et al., 1998) adhesion molecules.
Materials and methods
Antibodies
Polyclonal anti-P0 was prepared using a synthetic peptide specific to a 12 amino acid fragment (TWRYQPEGGRDA) from the extracellular domain of rat P0 as immunogen (Bio-Synthesis Inc.). The antiserum was affinity purified on immobilized peptide and recognizes an ∼27 KD P0 molecule in the rat sciatic nerve (unpublished data). Antipan cadherin antibody is from Sigma-Aldrich. Antibodies to PKC isoforms and RACK1 were from Transduction Laboratories. HRP-conjugated anti–mouse or –rabbit IgG were from Cappel Laboratories (ICN Biomedicals). The secondary antibodies conjugated to magnetic beads used in immunoprecipitations were obtained from PerSeptive Biosystems.
Cloning the wild-type P0 and creating a series of mutated P0 constructs
The coding sequences of full-length wild-type P0 (P0WT) and P0 COOH-terminal deletion mutants were cloned into the pCMV-Tag4 expression vector (Stratagene) using a PCR-based technique. These various mutants contain deletions at the COOH terminus of the P0 cytoplasmic domain lacking the last 13 (Δ13), 28 (Δ28), 43 (Δ43), or 59 (Δ59) amino acids. All constructs were created using the same forward primer (P0WT5, ATAGGATCCCACCATGGCTCCTGGGGCTC), containing a BamHI restriction site and a Kozak consensus sequence. The reverse 3′ primers were designed to contain overlapping DNA sequence according to the position of the deletion followed by a stop codon and a XhoI restriction site (Fig. 1 A): P0WT3, ATACTCGAGTTTCTTATCCTTGCGAGAC; Δ13flag, ATACTCGAGGTCGTCCTCGCCAC; Δ28flag, ATACTCGAGATACAGCACTGGCGTCT; Δ43flag, ATACTCGAGAGACTTGTGAAATTTCCCCT; and Δ59flag, ATACTCGAGGGCAGCCTGCCTGCGCAG.
All constructs were cloned into pCMV-Tag4 and tested by restriction enzyme cleavage and sequenced to ensure that the insertions were in frame and that no mutations were introduced during PCR.
Site-directed mutagenesis
Recombinant PCR was used to introduce point mutations in the P0 cytoplasmic domain substituting the natural amino acids with alanine residues. The oligonucleotide primers for point mutations were as follows. The underlined bases indicate the changes from the naturally occurring nucleotides: forward Y191A, 5′-AGACCCCAGTGCTGGCTGCCAT-3′; reverse Y191A, 5′-TGGTCCAGCATGGCAGCCAGCACT-3′; forward D195A, 5′-TGCTGGCCCACAGCCGAA-3′; reverse D195A, 5′-TGCTTCGGCTGTGGGCCAGCA-3′; forward S197A, 5′-CTGGACCACGCTCGAAGCACCAAA-3′; reverse S197A, 5′-AGCTTTGGTGCTTCGAGCGTGGT-3′; forward S199A, 5′-CTGGACCACAGCCGAGCTACCAAA-3′; reverse S199A, 5′-AGCTTTGGTAGCTCGGCTGTGGT-3′; forward T200A, 5′-CACAGCCGAAGCGCCAAAGCT-3′; reverse T200A, 5′-TGGCAGCTTTGGCGCTTCGGCT-3′; forward S204A, 5′-AAGCACCAAAGCTGCCGCTGAGAA-3′; and reverse S204A, 5′-ATTTCTTCTCAGCGGCAGCTT-3′.
All mutant constructs were confirmed by sequencing. The constructs were named Y191A, D195A, S197A, S199A, T200A, S204A, and R198, respectively.
Transfection of L and HeLa cells
All of the constructs and pCMV-Tag4 without inserts (Vect) were transfected into mouse L cells. WTP0, Δ13, Δ28, Δ43, Δ59, and Vect were also transfected into HeLa cells. Stable cell lines were selected for 2 wk in the presence of 1 mg/ml and maintained in the presence of 200 μg/ml of G418 (GIBCO BRL). Single clones were isolated and expanded. Clones of cells expressing high levels of transfected proteins as determined by immunoblotting with anti-P0 serum were chosen for adhesion assays.
RT-PCR
Expression of transfected constructs was assayed by RT-PCR. In brief, total RNA was isolated from cultured cells using a QIAGEN kit and reverse transcribed with Superscript II and oligo dT primers (GIBCO BRL). The resulting cDNA was used to amplify the transfected construct using the same primers that were used for cloning. For RT-PCR, the hypoxanthine phosphoribosyl transferase (HPRT) cDNA was used as an internal control. The two primers for HPRT amplification were: forward primer, 5′-CACAGGACTAGAACACCTGC-3′, and reverse primer, 5′-GCTGGTGAAAAGGACCTCT-3′.
Expression of transfected cDNAs
To analyze protein expression of the transfected cDNAs and determine plasma membrane localization, cell surface molecules were labeled by biotinylation of intact cells followed by immunoprecipitation. Confluent cell layers in 100-mm dishes were rinsed free of serum and labeled with 0.1 mg/ml freshly prepared NHS-LC-biotin (Pierce Chemical Co.) in cold PBS for 30 min. After washing and quenching, cell lysates were prepared using 1 ml of lysis buffer (1.5% NP-40, 0.15 M NaCl, 10 mM Tris, pH 7.2, 5 mM EDTA, 1 mM PMSF, 100 μg/ml protease inhibitor cocktail [Sigma-Aldrich], and 100 μg/ml DNase) per 100-mm plate, cleared by centrifugation at 14,000 g for 5 min, and the supernatant immunoprecipitated with anti-P0 antibody. The immunoprecipitated proteins were separated by SDS-PAGE, transferred to PVDF membrane, and biotinylated proteins were detected with HRP-streptavidin.
Immunoprecipitation and Western blotting
To analyze the expression of PKC isoforms in sciatic nerve, equal weights of mouse sciatic nerve were solubilized in 4% SDS, fractionated by SDS-PAGE, and transferred to PVDF membranes. The membranes were probed with the indicated anti-PKC isoform antibody followed by HRP-conjugated anti–mouse IgG and visualized using the extended duration HRP substrate (Pierce Chemical Co.). For L cells transfected with wild-type P0, cultures were grown to confluence on 100-mm plates, washed, and incubated free of serum for 2 h before lysis as indicated above. An aliquot of the lysate was fractionated by SDS-PAGE and immunoblotted with antibodies to the PKC isoforms detected in sciatic nerve. The remaining lysate was preincubated with preimmune rabbit serum followed by goat anti–rabbit magnetic beads. The beads were discarded, and the cleared supernatant was incubated with anti-P0 antibody. The immunoprecipitated P0 and associated molecules were then fractionated by SDS-PAGE, transferred to PVDF, and probed with anti-PKC and anti-RACK1 antibodies. The immunoblots were processed as described above.
Adhesion assays
Cell layers were washed free of serum, harvested using 0.1% trypsin (GIBCO BRL) in PBS buffer, and resuspended in serum-free medium (DME) containing 10 mM EDTA at 5 × 106 cells/ml. 2 μl/ml of fluorogenic dye calcein acetoxymethyl ester (calcein AM) (Molecular Probes) were added to the cell suspensions and incubated at 37°C for 30 min. After two washes, 5 × 105 single calcein-labeled cells were added to prepared microplate wells containing confluent monolayers of cells expressing wild-type P0 or BSA-coated wells as controls. After 1 h incubation at 37°C, the nonadherent calcein-labeled cells were removed by carefully washing several times with DME containing 10 mM EDTA until the BSA-coated wells had no cells left. The number of cells adhering to the monolayer was measured with a molecular device microplate reader equipped with a fluorescein filter set at 494 nm.
To determine the importance of the PKC motif in the P0 sequence, calphostin C (Calbiochem) was used to specifically inhibit PKC. Wild-type P0-transfected cells were preincubated with increasing concentrations of calphostin C for 1 h and then assayed for adhesion as described above.
Acknowledgments
We wish to express our appreciation to Athena Diagnostics (Worcester, MA) for referring the new CMT1B patient to us, and to Beth Hjertos for help with transfection and culture of cells expressing mutant forms of P0.
This research was supported by a grant from the National Multiple Sclerosis Society to J. Kamholz and M. Shy.
W. Xu's present address is Dept. of Neurology, University of Michigan, Ann Arbor, MI 48105.
Footnotes
Abbreviations used in this paper: HPRT, hypoxanthine phosphoribosyl transferase; PNS, peripheral nervous system; PVDF, polyvinylene difluoride; RACK, receptor for activated C kinase; RT, reverse transcription.
References
- Agrawal, H.C., and D. Agrawal. 1989. Tumor promoters accentuate phosphorylation of P0: evidence for the presence of protein kinase C in purified PNS myelin. Neurochem. Res. 14:409–413. [DOI] [PubMed] [Google Scholar]
- Aplin, A.E., A. Howe, S.K. Alahari, and R.L. Juliano. 1998. Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharm. Rev. 5:197–263. [PubMed] [Google Scholar]
- Bolino, A., M. Muglia, F.L. Conforti, E. LeGuern, M.A. Salih, D.M. Georgiou, K. Christodoulou, I. Hausmanowa-Petrusewicz, P. Mandich, A. Schenone, et al. 2000. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat. Genet. 25:17–20. [DOI] [PubMed] [Google Scholar]
- Crossin, K.L., and L.A. Krushel. 2000. Cellular signaling by neural cell adhesion molecules of the immunoglobulin superfamily. Develop. Dyn. 218:260–279. [DOI] [PubMed] [Google Scholar]
- Doyle, J.P., J.G. Stempak, P. Cowin, D.R. Colman, and D. D'Urso. 1995. Protein zero, a nervous system adhesion molecule, triggers epithelial reversion in host carcinoma cells. J. Cell Biol. 131:465–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin, M.T., and G.I. Tennekoon. 1993. Homophilic adhesion of the myelin P0 protein requires glycosylation of both molecules in the homophilic pair. J. Cell Biol. 122:451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin, M.T., F.S. Walsh, B.D. Trapp, J.A. Pizzey, and G.I. Tennekoon. 1990. Role of P0 protein as a homophilic adhesion molecule. Nature. 344:871–872. [DOI] [PubMed] [Google Scholar]
- Giese, K.P., R. Martini, G. Lemke, P. Soriano, and M. Schachner. 1992. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. 17:565–576. [DOI] [PubMed] [Google Scholar]
- Hilmi, S., M. Fournier, H. Valeins, J.C. Gandar, and J. Bonnet. 1995. Myelin P0 glycoprotein: identification of the site phosphorylated in vitro and in vivo by endogenous protein kinases. J. Neurochem. 64:902–907. [DOI] [PubMed] [Google Scholar]
- Iyer, S., C.L. Rowe-Rendleman, R. Bianchi, and J. Eichberg. 1996. Tyrosine phosphorylation of myelin protein P0. J. Neurosci. Res. 46:531–539. [DOI] [PubMed] [Google Scholar]
- Jaken, S., and P.J. Parker. 2000. Protein kinase C binding partners. Bioessays. 22:245–254. [DOI] [PubMed] [Google Scholar]
- Kishimoto, A., K. Nishiyama, H. Nakanishi, Y. Uratsuji, H. Nomura, Y. Takeyama, and Y. Nishizuka. 1985. Studies on the phosphorylation of myelin basic protein by protein kinase C and adenosine 3′:5′-monophosphate-dependent protein kinase. J. Biol. Chem. 260:12492–12499. [PubMed] [Google Scholar]
- Martini, R., J. Zielasek, K.V. Toyka, K.P. Giese, and M. Schachner. 1995. Protein zero (P0)-deficient mice show myelin degeneration in peripheral nerves characteristic of inherited human neuropathies. Nat. Genet. 11:281–286. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen, D., H. Khaner, and J. Lopez. 1991. Identification of intracellular receptor proteins for activated protein kinase C. Proc. Natl. Acad. Sci. USA. 88:3997–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelis, E., H. Neva, and C. Van Broeckhoven. 1999. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Human Mutation. 13:11–28. [DOI] [PubMed] [Google Scholar]
- Rowe-Rendleman, C.L., and J. Eichberg. 1994. P0 phosphorylation in nerves from normal and diabetic rats: role of protein kinase C and turnover of phosphate groups. Neurochem. Res. 19:1023–1031. [DOI] [PubMed] [Google Scholar]
- Shapiro, L., J.P. Doyle, P. Hensley, D.R. Coleman, and W.A. Hendrickson. 1996. Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron. 17:435–449 [DOI] [PubMed] [Google Scholar]
- Sim, A.T.R., and J.D. Scott. 1999. Targeting of PKA, PKC and protein phosphatases to cellular microdomains. Cell Cal. 26:209–217. [DOI] [PubMed] [Google Scholar]
- Shy, M.E., J. Balsamo, J. Lilien, and J. Kamholz. 2001. A molecular basis for hereditary motor and sensory neuropathy disorders. Curr. Neurol. Neurosci. Rep. 1:77–88. [DOI] [PubMed] [Google Scholar]
- Suzuki, M., Y. Sakamoto, K. Kitamura, K. Fukunaga, H. Yamamoto, E. Miyamoto, and K. Uyemura. 1990. Phosphorylation of Po glycoprotein in peripheral nerve myelin. J. Neurochem. 55:1966–1971. [DOI] [PubMed] [Google Scholar]
- Svetlov, S., and S. Nigam. 1993. Calphostin C, a specific protein kinase C inhibitor, activates human neutrophils: effect on phospholipase A2 and aggregation. Biochim. Biophys. Acta. 1177:75–78. [DOI] [PubMed] [Google Scholar]
- Tamaoki, T., and H. Nakano. 1990. Potent and specific inhibitors of protein kinase C of microbial origin. Biotech. 8:732–735. [DOI] [PubMed] [Google Scholar]
- Vleminckx, K., and R. Kemler. 1999. Cadherins and tissue formation integrating adhesion and signaling. Bioessays. 21:211–220. [DOI] [PubMed] [Google Scholar]
- Warner, L.E., M.J. Hilz, S.H. Appel, J.M. Killian, E.H. Kolodry, G. Karpati, S. Carpenter, G.V. Watters, C. Wheeler, D. Witt, et al. 1996. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron. 17:451–460. [DOI] [PubMed] [Google Scholar]
- Wong, M.-H., and M.T. Filbin. 1994. The cytoplasmic domain of the myelin Po protein influences the adhesive interactions of its extracellular domain. J. Cell Biol. 126:1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, M.-H., and M.T. Filbin. 1996. Dominant negative effect on adhesion by myelin Po protein truncated in its cytoplasmic domain. J. Cell Biol. 134:1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett, J.R., K.L. Gould, and T. Hunter. 1986. Substrate specificity of protein kinase C. Use of synthetic peptides corresponding to physiological sites as probes for substrate recognition requirements. Eur. J. Biochem. 161:177–184. [DOI] [PubMed] [Google Scholar]
- Wrabetz, L., M.L. Feltri, A. Quattrini, D. Imperiale, D. Previtali, M. D'Antonio, X. Yin, B.D. Trapp, L. Zhou, S.Y. Chiu, et al. 2000. P(0) glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J. Cell Biol. 148:1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, M., R. Zhao, X. Sui, F. Xu, and Z.J. Zhao. 2000. a. Tyrosine phosphorylation of myelin P0 and its implication in signal transduction. Biochem. Biophys. Res Commun. 267:820–825. [DOI] [PubMed] [Google Scholar]
- Xu, W., D. Manichella, H. Jiang, J.M. Vallat, J. Lilien, P. Baron, G. Scarlato, J. Kamholz, and M.E. Shy. 2000. b. Absence of P0 leads to dysregulation of myelin gene expression and myelin morphogenesis. J. Neurosci. Res. 60:714–724. [DOI] [PubMed] [Google Scholar]