

Abstract
We have examined the mechanism and functional significance of hemidesmosome disassembly during normal epithelial cell migration and squamous carcinoma invasion. Our findings indicate that a fraction of EGF receptor (EGF-R) combines with the hemidesmosomal integrin α6β4 in both normal and neoplastic keratinocytes. Activation of the EGF-R causes tyrosine phosphorylation of the β4 cytoplasmic domain and disruption of hemidesmosomes. The Src family kinase inhibitors PP1 and PP2 prevent tyrosine phosphorylation of β4 and disassembly of hemidesmosomes without interfering with the activation of EGF-R. Coimmunoprecipitation experiments indicate that Fyn and, to a lesser extent, Yes combine with α6β4. By contrast, Src and Lck do not associate with α6β4 to a significant extent. A dominant negative form of Fyn, but not Src, prevents tyrosine phosphorylation of β4 and disassembly of hemidesmosomes. These observations suggest that the EGF-R causes disassembly of hemidesmosomes by activating Fyn, which in turn phosphorylates the β4 cytoplasmic domain. Neoplastic cells expressing dominant negative Fyn display increased hemidesmosomes and migrate poorly in vitro in response to EGF. Furthermore, dominant negative Fyn decreases the ability of squamous carcinoma cells to invade through Matrigel in vitro and to form lung metastases following intravenous injection in nude mice. These results suggest that disruption of hemidesmosomes mediated by Fyn is a prerequisite for normal keratinocyte migration and squamous carcinoma invasion.
Keywords: α6β4; fyn; EGF-R; hemidesmosomes; carcinoma invasion
Introduction
Normal cell migration and tumor invasion are both driven by changes in actin dynamics and integrin function. In essence, cells first extend actin-rich protrusions, named filopodia and lamellipodia, toward the direction of movement. Integrin-dependent adhesions, such as focal complexes and adhesion plaques, are then nucleated at the leading edge to derive the traction necessary for movement. Finally, cells pull forward by contracting the actin cytoskeleton and releasing integrin attachments at the rear end (Horwitz and Parsons, 1999). Whereas β1 and αv integrins are connected to the actin cytoskeleton and their role in cell migration and invasion is well established, the function of the α6β4 integrin and associated keratin cytoskeleton in these processes is poorly understood.
The α6β4 integrin is a laminin 5 receptor expressed in epithelial, Schwann, endothelial, and double-negative T cells (Giancotti, 1996; Borradori and Sonnenberg, 1999). In the basal cells of stratified and transitional epithelia, α6β4 is concentrated at hemidesmosomes, adhesive junctions connected to the keratin cytoskeleton (Carter et al., 1990; Sonnenberg et al., 1991). In addition to α6β4, hemidesmosomes contain the transmembrane element bullous pemphigoid antigen (BPAG)*-2, which is thought to interact with an unknown basement membrane component. Inside the cell, α6β4 and BPAG-2 interact as a functional unit with two plakins, plectin/HD-1 and BPAG-1, that form the inner plaque of hemidesmosomes and link to the keratin cytoskeleton (Rezniczek et al., 1998; Schaapveld et al., 1998; Geerts et al., 1999; Hopkinson and Jones, 2000). Although genetic analyses suggest that these proteins are essential to build the core structure of hemidesmosomes (Guo et al., 1995; McGrath et al., 1995; Dowling et al., 1996; Smith et al., 1996; van der Neut et al., 1996; Andra et al., 1997; Ryan et al., 1999), they are not sufficient to account for the dynamic regulation of these junctions. In particular, it is known that the hemidesmosomes are disassembled during keratinocyte migration, presumably in response to activation of the EGF receptor (EGF-R) (Gipson et al., 1993; Mainiero et al., 1996). In addition, squamous carcinoma cells often lack hemidesmosomes in vivo (Schenk, 1979). Because hemidesmosomes mediate stable adhesion, their disruption may be a prerequisite for both normal migration and cancer invasion. The mechanisms and regulatory components mediating the disassembly of hemidesmosomes are poorly understood.
The α6β4 integrin is characterized by the uniquely large cytoplasmic domain of its β4 subunit, which appears to interact directly with both BPAG-2 and plectin/HD-1, and which is necessary for the assembly of hemidesmosomes (Murgia et al., 1998; Schaapveld et al., 1998). Recent studies have revealed that α6β4 has also a signaling function. The integrin is associated with a tyrosine kinase and becomes phosphorylated on several tyrosine residues upon binding to laminin 5 or activation of the EGF-R (Mainiero et al., 1995; Mainiero et al., 1996). Tyrosine phosphorylation of β4 promotes recruitment of the signaling adaptor protein Shc. Upon tyrosine phosphorylation, Shc binds to the Grb2/mSOS complex and activates Ras and, hence, both the Raf–extracellular signal-regulated kinase (ERK) and phosphatidyl inositol-3 kinase (PI-3K)-Rac-JNK signaling cascades (Mainiero et al., 1997). Analysis of mice carrying a targeted deletion of the β4 cytoplasmic domain has indicated that this portion of the integrin is essential for both assembly of hemidesmosomes and activation of growth promoting signaling pathways (Murgia et al., 1998). Although signaling and assembly of hemidesmosomes by α6β4 are temporally distinguishable events, the relationship between the structural and signaling function of α6β4 is currently unclear.
We have shown previously that treatment of normal keratinocytes with EGF induces tyrosine phosphorylation of the cytoplasmic domain of β4 and disruption of hemidesmosomes (Mainiero et al., 1996). Others have suggested that protein kinase C may play a role in this process (Rabinovitz et al., 1999). Because the EGF-R and the α6β4 integrin are often overexpressed in highly invasive squamous carcinomas (Kimmel and Carey, 1986; Yamamoto et al., 1986; Tennenbaum et al., 1996), we have examined the effect of EGF-R activation on α6β4 function in squamous carcinoma cells. The results reported here indicate that a fraction of EGF-R combines with α6β4 and, through the tyrosine kinase Fyn, induces phosphorylation of the β4 tail and disruption of hemidesmosomes. Inhibition of Fyn increases the assembly or stability of hemidesmosomes and suppresses migration and invasion by squamous carcinoma cells. These findings suggest that the disassembly of hemidesmosomes mediated by Fyn is a prerequisite for normal cell migration and tumor invasion.
Results
The EGF-R induces phosphorylation of β4 through activation of a Src family kinase
To examine the mechanism by which the EGF-R induces tyrosine phosphorylation of β4, HaCaT keratinocytes, MSK-QLL-1, and A431 squamous carcinoma cells were subjected to immunoprecipitation and immunoblotting analysis. The HaCaT keratinocytes are immortalized but retain the ability to undergo differentiation in vitro (Boukamp et al., 1988); the MSK-QLL-1 squamous carcinoma cells express normal levels of EGF-R (unpublished data), and the A431 squamous carcinoma cells overexpress the EGF-R and secrete high amounts of TGF-α (Lin et al., 1984; Reiss et al., 1991).
As shown in Fig. 1, treatment with EGF caused tyrosine phosphorylation of β4 in HaCaT and MSK-QLL-1 cells, and it increased the extent to which β4 was already phosphorylated in A431 cells. The basal phosphorylation of β4 observed in A431 cells may be a consequence of the constitutive activation of the EGF-R in these cells (Van de Vijver et al., 1991). We noticed that a tyrosine phosphorylated protein with an apparent molecular mass similar to that of EGF-R coimmunoprecipitated with α6β4 from both MSK–QLL-1 and A431 cells (Fig. 1). Prolonged exposure of the blot revealed a similar band also in β4 immunoprecipitates from HaCaT keratinocytes. Immunoblotting with specific antibodies identified this protein as the EGF-R and provided evidence that a small but significant fraction of this receptor is associated with α6β4 in all three cell lines (Fig. 1). Densitometric analysis indicated that ∼1% of total EGF-R was coimmunoprecipitated with α6β4 from HaCaT keratinocytes and ∼2% from the squamous carcinoma cell lines. Control experiments indicated that neither normal mouse IgGs nor the anti-MHC monoclonal antibody W6.32 coprecipitated the EGF-R. Although α6β4 combines with the EGF-R family member ErbB-2 in breast carcinoma cells which overexpress both molecules (Falcioni et al., 1997), and to a much smaller extent in HaCaT cells (Hintermann et al., 2001), we were unable to detect such an association under our experimental conditions (see Materials and methods) (Fig. 1). These results suggest that a fraction of EGF-R associates specifically with α6β4 and induces phosphorylation of the β4 cytoplasmic domain.
Figure. 1.

A fraction of EGF-R is associated with α6β4 and induces tyrosine phosphorylation of the β4 cytoplasmic domain. The indicated cells were serum starved and then treated with EGF (25 ng/ml, 4 min) or left untreated. After solubilization in TX-DOC lysis buffer, the samples were immunoprecipitated with normal mouse IgGs (C) or the anti-β4 monoclonal antibody 3E1 (β4) followed by immunoblotting with the rabbit anti–β4-exo antiserum, the monoclonal antibody to P-Tyr RC-20, and rabbit antibodies to the COOH-terminus of the EGF-R or ErbB-2.
Fig. 1 also shows that exposure to EGF induced limited tyrosine phosphorylation of the fraction of EGF-R associated with α6β4 in HaCaT keratinocytes. By contrast, the fraction of EGF-R associated with α6β4 in MSK-QLL-1 and A431 cells was constitutively phosphorylated on tyrosine, perhaps because both cell lines secrete EGF-R ligands. Addition of EGF induced a coordinate increase in the tyrosine phosphorylation of β4 and associated EGF-R in both MSK-QLL-1 and A431 cells (Fig. 1). These observations suggest that overexpression of the EGF-R causes enhanced phosphorylation of β4 in squamous carcinoma cells. Immunofluorescence experiments indicated that HaCaT and MSK-QLL-1 cells form some hemidesmosome-like adhesions upon plating on a laminin 5–enriched matrix in the absence of EGF. By contrast, A431 cells failed to assemble these adhesive junctions under these conditions (unpublished data). Thus, increased EGF-R–mediated phosphorylation of β4 correlates with inhibition of hemidesmosome formation.
Because the EGF-R does not phosphorylate efficiently fusion proteins derived from the cytoplasmic domain of β4 in vitro (Mainiero et al., 1997), we asked if the EGF-R promotes phosphorylation of β4 by activating an intermediary kinase. It is known that the EGF-R can interact with and activate Src family kinases (Biscardi et al., 1999). In addition, our previous results had suggested that α6β4 is associated with a Src family kinase (Mainiero et al., 1995). Thus, we examined the role of Src kinases in EGF-R–induced phosphorylation of β4. As shown in Fig. 2 A, the two Src kinase inhibitors, PP1 and PP2, which display a higher affinity for Fyn than Src in vitro (Hanke et al., 1996), suppressed tyrosine phosphorylation of β4 in HaCaT keratinocytes treated with EGF. The effect of the two compounds was dose-dependent and specific, as both did not interfere with the activation and autophosphorylation of EGF-R. These results suggest that EGF-R–induced phosphorylation of β4 is mediated by a Src family kinase.
Figure 2.
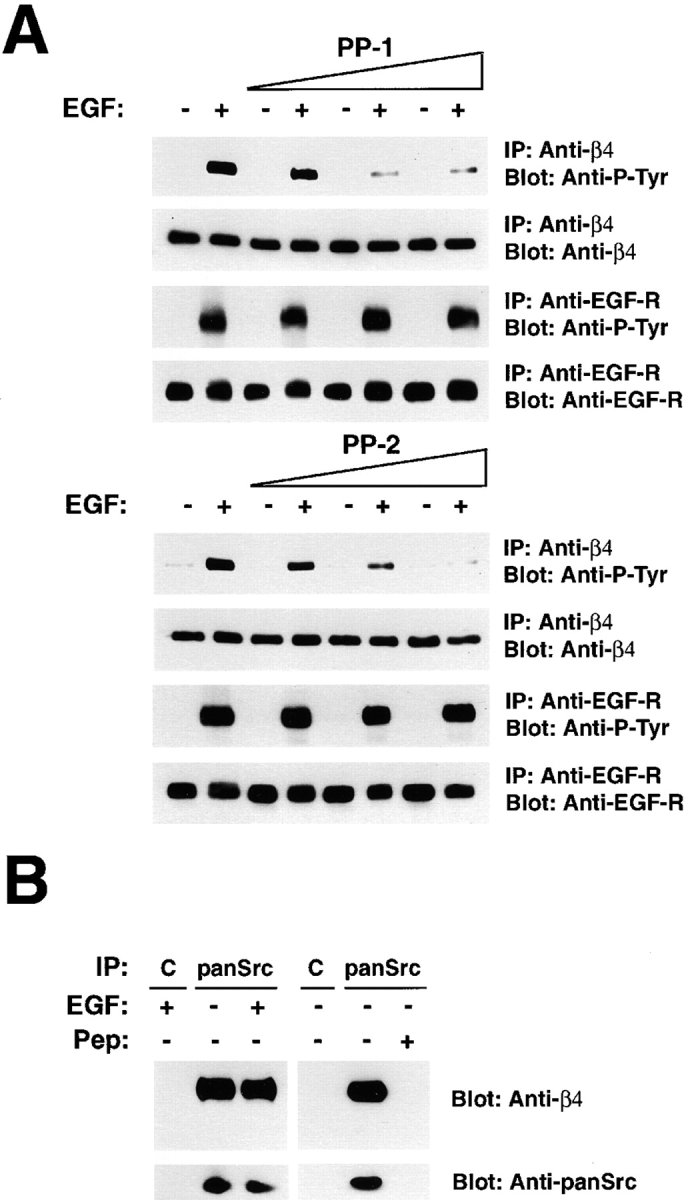
A Src family kinase is associated with α6β4 and phosphorylates β4 in response to activation of the EGF-R. (A) After serum starvation, HaCaT cells were incubated for 15 min with the Src family kinase inhibitor PP-1 or -2 (1, 2, and 4 μM), then treated with 25 ng/ml EGF for 4 min in presence of the inhibitors. The samples were immunoprecipitated with the anti-β4 monoclonal antibody 3E1 and probed by immunoblotting with the monoclonal antibody to P-Tyr RC-20 or the rabbit anti–β4-cyto serum. As a control, the samples were immunoprecipitated with the anti-EGFR monoclonal antibody 528 and probed by immunoblotting with the monoclonal antibody to P-Tyr RC-20 or the rabbit antiserum to the COOH terminus of the EGF-R. (B) Lysates from serum-starved HaCaT cells treated with EGF or left untreated (25 ng/ml, 4 min) were immunoprecipitated with normal rabbit IgGs (C) or the affinity-purified rabbit antibodies SRC2, which recognize Src, Fyn, and Yes (panSrc). The samples were probed by immunoblotting with rabbit anti–β4-cyto or anti-panSrc antibodies. The coimmunoprecipitation of α6β4 and Src kinases was blocked in presence of 20 μg of the peptide used as antigen to generate the anti-panSrc antibodies (pep).
The α6β4 integrin combines with Fyn
To directly test if α6β4 associates with a Src family kinase in vivo, we performed coimmunoprecipitation experiments. Immunoprecipitation with a high-affinity antipeptide antibody reacting with the three Src family kinases, Src, Fyn, and Yes (anti-panSrc), followed by immunoblotting with antibodies to β4, revealed that α6β4 is associated with Src family kinases in HaCaT keratinocytes (Fig. 2 B). Treatment with EGF did not modify the stoichiometry of the complex. The coimmunoprecipitation of α6β4 and Src kinases was blocked by excess of the antigenic peptide (Fig. 2 B). These results suggest that the α6β4 integrin is constitutively associated with one or more Src family kinases in HaCaT keratinocytes.
To identify the Src kinase(s) associated with α6β4, we performed immunoprecipitation and immunoblotting experiments in several epithelial cell lines. We varied extraction conditions and used antibodies reacting specifically with Src, Fyn, or Yes, either in the immunoprecipitation or the immunoblotting step of the coprecipitation assay. Despite several attempts, we were unable to detect a specific association of Src, Fyn, or Yes with α6β4 in cells expressing endogenous levels of Src kinases. We suspect that this negative result is due to the low stoichiometry or relative instability of the association of α6β4 with Src kinases and the low affinity of currently available antibodies reacting specifically with each Src kinase.
To overcome these problems, we decided to use an overexpression approach. 293-T HEK cells were transfected with vectors directing the expression of α6β4 and each one of several cytoplasmic tyrosine kinases. Preliminary experiments of immunoprecipitation followed by kinase assay and V8 peptide mapping indicated that α6β4 coprecipitated with Fyn, but to a much lesser or no extent with Src, Abl, focal adhesion kinase (FAK), or JAK1. No association of α6β1 with Fyn was detected by using the same assay (unpublished data), suggesting that α6β4 interacts with Fyn through the β4 subunit.
To further examine the association of α6β4 with Src kinases, 293-T cells were transfected with vectors directing the expression of α6β4 and wild-type or mutant versions of the Src family kinases known to be expressed in keratinocytes, namely Fyn, Src, and Yes (Fig. 3 A). To verify that each kinase was expressed at an equivalent level, we probed total lysates by immunoblotting with anti-Fyn and anti-panSrc (Fig. 3 B). Prior experiments had shown that this antibody displays similarly high affinity for recombinant Src and Fyn and reacts less efficiently with recombinant Yes (Fig. S1). The transfectants were subjected to immunoprecipitation and immunoblotting analysis. The results indicated that Fyn and, to a somewhat more limited extent Yes, associate with α6β4. By contrast, Src combined with α6β4 much less efficiently (Fig. 3 B).
Figure 3.

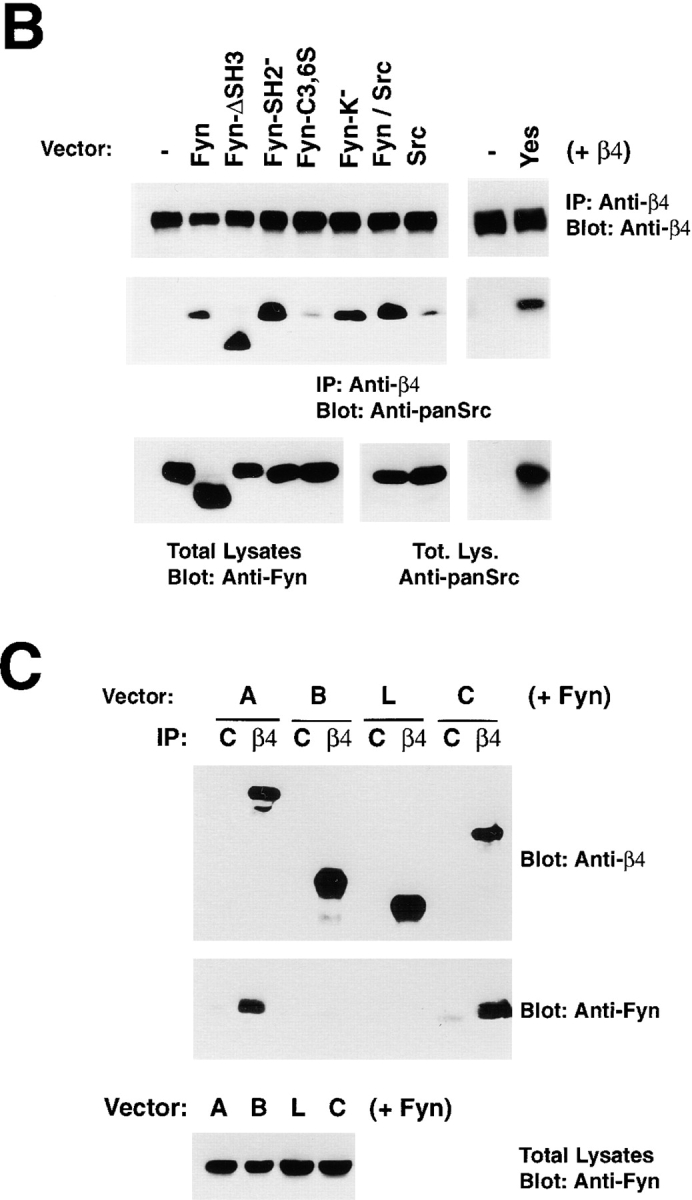
Mutational analysis of the association of Fyn with α6β4. (A) Structure of the Src family kinases (top) and wild-type and mutant β4 subunits (bottom) used to define the association of Fyn with α6β4. *, point mutations. β4 constructs: black boxes, Fn type III modules; grey box, connecting segment. (B) 293 T cells were transiently transfected with plasmids encoding wild-type α6 and β4 in combination with the indicated wild-type and mutant Src family kinases. After extraction in RIPA buffer, the lysates were immunoprecipitated with the monoclonal antibody 3E1 covalently linked to Sepharose beads. The top part of the blot was probed with the rabbit antiserum β4-exo and the bottom with affinity purified anti-panSrc antibodies (SRC2). Total lysates were probed by immunoblotting with affinity-purified anti-Fyn (FYN3) antibodies to verify the level of expression of wild-type and mutant Fyn proteins (with the exception of the Fyn–Src chimera) and anti-panSrc (SRC2) antibodies to verify the level of expression of Src, the chimera Fyn–Src, and Yes; IgG H, immunoglobulin heavy chain. (C) 293 T cells were transiently transfected with plasmids encoding wild-type Fyn and α6 in combination with the indicated wild-type and mutant β4 subunits. After extraction in RIPA buffer, the lysates were immunoprecipitated with the monoclonal antibody 3E1 covalently linked to Sepharose beads. The top part of the blot was probed with the rabbit antiserum β4-exo and the bottom with affinity-purified antibodies to Fyn (Fyn3). Total lysates were probed by immunoblotting with the same antibodies to verify equal expression of Fyn in the four samples.
To examine the structural requirements of the interaction of Fyn with α6β4, we introduced various mutant forms of Fyn, in combination with wild-type α6β4 (Fig. 3 A), into 293 T cells. Immunoprecipitation and immunoblotting analysis revealed that the association of Fyn with α6β4 was not impaired by deletion of the SH3 domain (Fyn-ΔSH3), inactivation of the SH2 domain (Fyn-SH2), or impairment of the kinase function (Fyn-K−) (Fig. 3 B). By contrast, mutation of the two cysteine residues required for palmitoylation (Fyn-C3, 6S) reduced the association of Fyn with α6β4 considerably. Thus, we expressed a chimeric molecule comprising the NH2-terminus of Fyn fused to Src (Fyn1-13/Src14-533). The Fyn–Src chimera associated with α6β4 much more efficiently than Src (Fig. 3 B). These results suggest that the association of Fyn with α6β4 is not mediated by the SH3 domain and does not require the phosphotyrosine binding function of the SH2 domain or the kinase activity of the molecule. However, it requires the NH2-terminal segments of Fyn, including Cys 3 and 6, which are palmitoylated. However, palmitoylation is not the only structural determinant required for association with α6β4, because Lck is palmitoylated but does not combine with α6β4. It is possible that palmitoylation promotes localization of Fyn at the plasma membrane in close proximity to α6β4 and thereby facilitates a protein–protein interaction between the two molecules.
To examine the sequences of β4 required for the association with Fyn, 293 T cells were transfected with various deletion mutants of β4 (Fig. 3 A) in combination with wild-type Fyn. Immunoprecipitation and immunoblotting analysis indicated that a recombinant β4 subunit lacking almost the entire COOH-terminal domain, which includes all Fn-III repeats (mutant C), associated with Fyn as efficiently as wild-type β4 (molecule A). By contrast, recombinant forms of β4 lacking the entire (mutant L), or almost the entire, cytoplasmic domain of β4 (mutant B) did not combine with Fyn (Fig. 3 C). These observations indicate that the membrane proximal portion of the cytoplasmic domain of β4, comprising amino acid residues 854–1183, is required for association with Fyn.
Taken together, these results indicate that Fyn interacts specifically with α6β4, and that this interaction requires the membrane-proximal NH2-terminal segment of Fyn and the corresponding portion of the cytoplasmic domain of β4.
Fyn mediates EGF-R–induced phosphorylation of β4 and disruption of hemidesmosomes
To examine the role of Fyn in regulation of hemidesmosomes, we used the rat bladder carcinoma 804G cells, which form relatively well structured hemidesmosome-like adhesions in vitro (Riddelle et al., 1991). 804G clones stably expressing a dominant negative (kinase dead) form of Fyn or a corresponding mutant version of Src were generated. Immunoblotting with anti-Fyn indicated that the clones F1, 2, and 3 express levels of kinase-dead Fyn significantly higher than those of endogenous wild-type Fyn in control clone C1 (Fig. 4 A). Similar results were obtained with the clones S1 and 2, which express dominant negative Src (Fig. 4 A). Endogenous Fynand Src in clone C1 were detected only upon overexposure of the blot. Densitometric analysis of films exposed for different times indicated that kinase-dead Src and Fyn were similarly overexpressed as compared to the levels of corresponding wild-type endogenous proteins (unpublished data). In addition, immunoblotting with anti-panSrc, which reacts with Src slightly better than wth Fyn (Fig. S1), indicated that kinase-dead Src was expressed at levels somewhat higher than kinase-dead Fyn (Fig. 4 A).
Figure 4.

Dominant negative Fyn, but not Src, suppresses EGF-mediated tyrosine phosphorylation of β4. (A) Total lysates of 804G clones stably transfected with empty vector (C1) or dominant negative Fyn (F1, 2, and 3) were probed by immunoblotting with affinity purified anti-Fyn (FYN3) or anti-panSrc antibodies (SRC2). Total lysates of 804G clones stably transfected with empty vector (C1) or dominant negative Src (S1 and 2) were probed by immunoblotting with affinity purified anti-Src (N-16) or anti-panSrc antibodies (SRC2). (B) After serum starvation, the indicated 804G clones were treated with 50 ng/ml EGF for 5 min or left untreated. The lysates were immunoprecipitated with the rabbit antiserum β4 cyto and probed by immunoblotting with the anti–P-Tyr monoclonal antibody RC-20 or the rabbit antiserum β4 cyto.
To examine the role of Fyn in EGF-R–induced phosphorylation of β4, clones expressing dominant negative Fyn or Src were treated with EGF and subjected to immunoprecipitation with anti-β4, followed by immunoblotting with anti–P-Tyr. As shown in Fig. 4 B, expression of dominant negative Fyn suppressed EGF-induced tyrosine phosphorylation of β4, whereas dominant negative Src did not inhibit this process. These experiments suggest that Fyn mediates phosphorylation of β4 upon activation of the EGF-R. Src is unable to function in this pathway presumably because it is unable to associate efficiently with α6β4.
We next evaluated the effect of dominant negative Fyn and Src on hemidesmosomes. At the immunofluorescent microscope, hemidesmosomes appear as punctuate structures, which tend to coalesce around circular areas of unknown composition. The resulting staining is commonly referred to as “swiss cheese”–like. As shown in Fig. 5, staining with antibodies to BPAG-2 revealed that 804G clones expressing dominant negative Fyn display a significantly higher number of hemidesmosomes than control cells. The clones expressing dominant negative Src did not have an increased number of hemidesmosomes; however, these junctions tended to be somewhat more clustered than in control cells. Similar results were obtained after staining with antibodies to β4 (unpublished data). Treatment with EGF caused an almost complete disruption of hemidesmosomes in control cells and clones expressing dominant negative Src. By contrast, the hemidesmosomes of clones expressing dominant negative Fyn were resistant to the effect of EGF to a significant extent (Fig. 5). Parental 804G cells and HaCaT keratinocytes treated with PP2 displayed a similar stabilization of hemidesmosomes (unpublished data). These results imply that Fyn prevents the assembly and/or causes disassembly of hemidesmosomes and this effect requires the catalytic activity of the kinase.
Figure 5.

Dominant negative Fyn promotes assembly and/or inhibits disassembly of hemidesmosomes. The indicated 804G clones were plated on glass coverslips for 24 h and then treated for 12 h with 50 ng/ml EGF in serum-free medium or left untreated. After a mild extraction with Triton X-100, the cells were fixed and stained with affinity purified antibodies to BPAG-2 to visualize hemidesmosomal structures. Results similar to those shown here were obtained with all three clones expressing dominant negative Fyn and the two clones expressing dominant negative Src.
Src kinases are required for migration and invasion
It is reasonable to assume that a cell must disassemble stable adhesions with the extracellular matrix, such as hemidesmosomes, in order to migrate and invade (Giancotti and Mainiero, 1994). Thus, we examined if inhibition of Fyn suppressed cell migration. As shown in Fig. 6, 804G clones expressing dominant negative Fyn migrated across an artificial in vitro wound much less efficiently than control clones. The migratory ability of clones expressing dominant negative Src was also reduced, but to a lesser extent than that of clones expressing dominant negative Fyn. This effect of dominant negative Src may result from stabilization of focal adhesions (Fincham and Frame, 1998). Immunofluorescent staining with antibodies to BPAG-2 revealed disassembly of hemidesmosomes at the rear end of control cells entering the wound area. Similar results were obtained with cells expressing dominant negative Src. By contrast, these posterior hemidesmosomes were not disassembled in cells expressing dominant negative Fyn (Fig. 6). These observations suggest that dominant negative Fyn inhibits cell migration by enhancing the stability of hemidesmosomes.
Figure 6.

Dominant negative Fyn inhibits disassembly of hemidesmosomes and suppresses cell migration. The indicated 804G clones were grown until confluent and then serum starved. After wounding, the monolayers were treated with EGF (50 ng/ml) for 5 or 10 h or left untreated for 5 h, and then either stained with Crystal violet or subjected to immunofluorescent staining with affinity purified anti–BPAG-2 antibodies. Cell migration results were quantitated as described in the Materials and methods section and plotted graphically. The graph shows the mean and SD of values from a representative experiment performed in triplicate. Results similar to those shown here were obtained with all three clones expressing dominant negative Fyn and the two clones expressing dominant negative Src. Bar, 250 μm.
We next used chemical inhibitors to examine the role of Src kinases in cell invasion through the reconstituted basement membrane Matrigel. As shown in Fig. 7, the Src kinase inhibitors Herbimycin and PP2 inhibited Matrigel invasion by A431 and MSK-QLL-1 squamous carcinoma cells. The effect of these two inhibitors was comparable or superior to that of the PI-3K inhibitor LY294002, which previous studies have shown to profoundly affect tumor invasion in vitro (Keely et al., 1997). In contrast, an inihibitor of the p38 MAP kinase did not exert a significant effect on Matrigel invasion by these cells.
Figure 7.

Src-family kinases are required for squamous carcinoma invasion through Matrigel. A431 and MSK-QLL-1 cells were plated on Matrigel together with the Src family kinase inhibitors Herbimycin A (100 ng/ml) and PP-2 (1, 5, and 10 μM), the PI-3K inhibitor LY294002 (25 μM), or the p38 kinase inhibitor SB203580 (10 μM). Serum-free medium containing EGF (25 ng/ml) was added to the lower chamber. After 24 h, cells that had invaded through Matrigel and remained attached to the lower side of the filter were stained with Crystal violet and counted under a microscope. The graphs show the mean and SD of values from a representative experiment performed in triplicate.
Inhibition of Fyn suppresses experimental metastasis formation
The 804G cells did not produce lung metastases upon intravenous injection in nude mice (unpublished data). To test the role of Fyn in squamous carcinoma metastasis, we generated A431 clones expressing dominant negative Fyn. These clones grew in culture at the same rate of control cells and did not undergo apoptosis when maintained in suspension for 24 h (unpublished data). In addition, they continued to proliferate in suspension at the same rate of control cells, as shown by the presence of similar number of mitotic figures after 24 h of suspension culture (unpublished data). These results suggest that dominant negative Fyn does not inhibit growth or survival of A431 cells.
Next, we sought to verify that dominant negative Fyn prevented tyrosine phosphorylation of β4 and stabilized hemidesmosomes also in A431 cells. As shown in Fig. 8 A, EGF treatment caused significant tyrosine phosphorylation of β4 in the control clone, but not in those expressing dominant negative Fyn. In addition, whereas A431 clones expressing dominant negative Fyn were able to form numerous hemidesmosome-like adhesions upon plating on a laminin 5 matrix, control cells were much less able to do so (Fig. 8 B). Finally, clones expressing dominant negative Fyn migrated into an artificial wound and invaded through Matrigel less efficiently than control clones (Fig. 8 C). These results indicate that dominant negative Fyn stabilizes hemidesmosomes and suppresses migration and Matrigel invasion by A431 squamous carcinoma cells.
Figure 8.
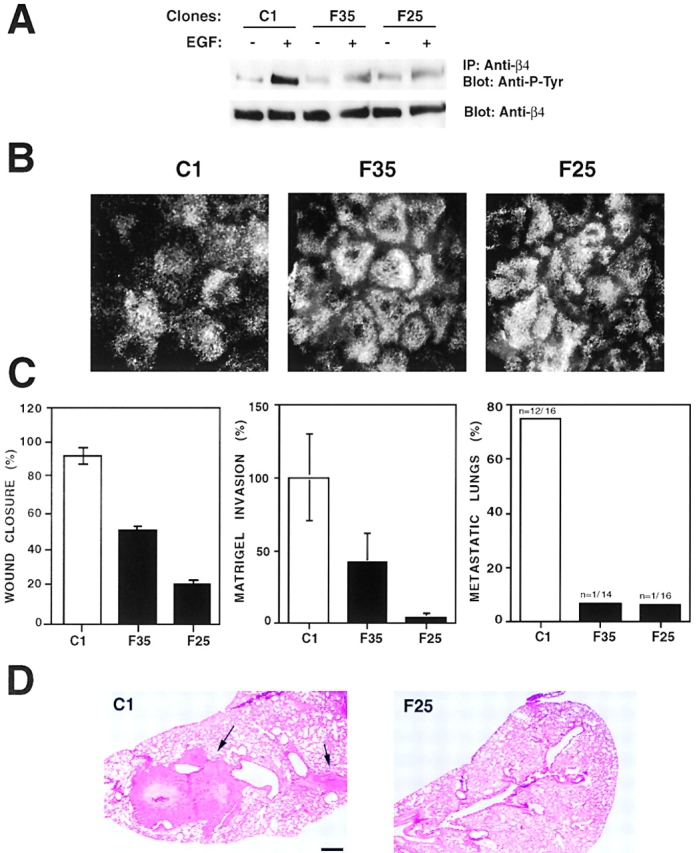
Dominant negative Fyn restores hemidesmosomes and inhibits both Matrigel invasion and experimental metastasis in A431 cells. (A) Two A431 clones expressing dominant negative Fyn (F35 and F25) and a control clone (C1) were starved and then treated with EGF (25 ng/ml) for 10 min or left untreated. RIPA lysates were immunoprecipitated with the anti-β4 monoclonal antibody 3E1 and probed by immunoblotting with the anti–P-Tyr monoclonal antibody RC-20 or the rabbit antiserum β4 cyto. The indicated A431 clones were plated on laminin 5 matrix–coated glass coverslips for 48 h. Cells were mildly permeabilized with Triton X-100, fixed, and stained with the anti-β4 monoclonal antibody 3E1. (C) The indicated A431 clones were tested for their ability to migrate in an in vitro wound assay, to invade through Matrigel in response to EGF (25 ng/ml) as described in the legends to Figs. 6 and 7, and to form lung metastases upon injection in the tail vein of nude mice. The number of metastatic over total number of lungs examined is shown at the top of each bar. (D) Hematoxylin and eosin staining of lung sections from mice injected with C1 or F25 cells. The arrows point to a large and a smaller lung metastasis in a mouse injected with C1 cells; no metastases are visible in the lung section from a mouse injected with F25 cells. Bar, 500 μm.
Intravenous injection of control A431 cells in nude mice resulted in the appearance of lung metastases within 5 wk. Histologically, these metastases appeared as large, often bilateral nodules occupying a significant area of lung parenchyma (Fig. 8 D). They were often necrotic in the center. Their identity as lung metastasis of A431 cells was confirmed by immunostaining with antibodies to β4 and keratins 5 and 14 (unpublished data). We evaluated the number of lung metastases formed by A431 clones expressing dominant negative Fyn and control clones by histological analysis of serial sections spanning the entire lungs of each injected mouse. The results indicated that A431 cells expressing dominant negative Fyn have a considerably reduced ability to colonize the lung upon intravenous injection in nude mice (Fig. 8 C and D). This observation implies that inhibition of Fyn impairs squamous carcinoma invasion in vivo.
Discussion
It is likely that changes in integrin expression facilitate tumor invasion because of their combined effects on cell adhesion and signaling (Giancotti and Mainiero, 1994; Varner and Cheresh, 1996; Boudreau and Bissell, 1998). The α6β4 integrin is expressed at higher levels in squamous carcinomas of the skin, larynx, cervix, and lung than in corresponding normal tissues (Kimmel and Carey, 1986; Costantini et al., 1990). In addition, whereas normal thyroid epithelium does not express α6β4, the integrin appears to be present at significant levels on the surface of a large proportion of thyroid carcinomas (Serini et al., 1996). Several studies suggest that α6β4 facilitates tumor invasion (Wolf et al., 1990; Carico et al., 1993; Tani et al., 1996; Shaw et al., 1997). If this is true, a mechanism must exist to inactivate the ability of α6β4 to mediate stable adhesion to the basement membrane. Our results indicate that a fraction of EGF-R combines with α6β4 and, through the integrin-associated Src family kinase Fyn, induces tyrosine phosphorylation of the β4 cytoplasmic domain and disruption of hemidesmosomes. Dominant negative Fyn, but not Src, stabilizes hemidesmosomes and prevents their disassembly in response to EGF. Accordingly, carcinoma cells expressing dominant negative Fyn migrate and invade in vitro much less than control cells and are unable to form lung metastases upon injection in the tail vein of nude mice. These findings identify a mechanism of hemidesmosome disassembly important for both normal cell migration and carcinoma invasion.
Previous studies have indicated that cell migration and invasion require assembly of focal adhesions and stress fibers at the leading edge of the cell and their disassembly at the trailing edge (Horwitz and Parsons, 1999). These processes are jointly regulated by β1 and αv integrins and by growth factor receptors through the focal adhesion kinase FAK (Sieg et al., 2000) and the adaptor protein Shc (Collins et al., 1999; Gu et al., 1999). Upon activation, FAK combines with Src, which phosphorylates a number of focal adhesion components, including p130CAS and paxillin (Giancotti and Ruoslahti, 1999). Genetic evidence implies that Src signaling stimulates cell migration by promoting spreading of the leading edge and turnover of focal adhesions at the trailing edge (Kaplan et al., 1995; Fincham and Frame, 1998; Klinghoffer et al., 1999). Shc is activated by Fyn and possibly other palmitoylated Src family kinases (Wary et al., 1998) and plays a role in cell migration through activation of ERK and disassembly of focal adhesions (Collins et al., 1999; Gu et al., 1999). In addition to, or instead of, focal adhesions, many epithelial cells form hemidesmosomes. Our results argue that their disassembly in response to activation of the EGF-R and phosphorylation of the β4 tail is required for epithelial cell migration and invasion.
What is the mechanism by which EGF causes phosphorylation of the β4 cytoplasmic domain? Our results indicate that a fraction of EGF-R associates with α6β4 and induces phosphorylation of the β4 cytoplasmic domain through the Src family kinase Fyn (Fig. 9). Dominant negative studies suggest that Src can not replace Fyn in this pathway. This is most likely because Fyn combines with α6β4, but Src does so much less efficiently. By contrast, Yes combines relatively efficiently with α6β4. The NH2-terminal domains of both Fyn and Yes are known to be palmitoylated and we have observed that the association of Fyn with α6β4 requires this lipid modification. It is likely that the association of Yes with α6β4 similarly requires palmitoylation of the kinase.
Figure 9.

Relationship between α6β4 signaling and assembly of hemidesmosomes. Hypothetical model of the underlying pathways. We have observed that the palmitoylated fraction of α6β4 is localized in lipid rafts and is preferentially associated with Fyn and Yes (unpublished data). We hypothesize that this fraction of α6β4 mediates recruitment of Shc and activation of Ras and PI-3K. The EGF-R induces α6β4 signaling by activating Fyn and Yes and causing phosphorylation of the β4 cytoplasmic domain. It is likely that the signaling fraction of α6β4 exists in dynamic equilibrium with the cytoskeletal fraction involved in assembly of hemidesmosomes. We hypothesize that assembly of hemidesmosomes requires dephosphorylation of the β4 tail (Dans et al., 2001).
Previous studies have indicated that palmitoylation targets Fyn and other signaling molecules to defined membrane microdomains enriched in cholesterol and glycosphingolipids (membrane rafts) (van't Hof and Resh, 1997; Brown and London, 1998). Increasing evidence suggests that targeting of signaling molecules to membrane rafts promotes compartmentalization of signaling complexes, thus ensuring signaling specificity. A prime example is provided by the T cell receptor. Upon T cell receptor activation, its ζ chains accumulate in membrane rafts, where they interact with Fyn and other signaling molecules (Baldari et al., 2000). Palmitoylation of Fyn is required to localize the kinase to rafts, where it phosphorylates the ζ chain immunoreceptor tyrosine-based activation motifs (ITAMs). The SH2 domain of Fyn then combines with the phosphorylated ITAMs stabilizing the interaction with the ζ chain (van't Hof and Resh, 1999). Interestingly, we have recently observed that a significant fraction of α6β4 is palmitoylated and localized to membrane rafts (unpublished data). It is possible that palmitoylation of α6β4 mediates localization to membrane rafts thus facilitating interaction with Fyn (Fig. 9).
Our mutagenesis results indicate that the association of α6β4 with Fyn requires the membrane proximal segment of the cytoplasmic domain of β4, but not the SH2 domain, the SH3 domain, or the enzymatic function of Fyn, suggesting that palmitoylation of α6β4 and Fyn may be required but not sufficient to mediate the interaction between the two molecules. It is likely that the membrane proximal portion of the cytoplasmic domain of β4 and the NH2-terminus of Fyn establish a protein–protein interaction. However, future studies will be required to identify more precisely the protein sequences involved in this interaction. In addition, although our data are most consistent with the hypothesis that Fyn interacts with and phosphorylates β4 directly, this remains to be established formally.
The cytoplasmic domain of β4 has both a signaling and a cytoskeletal function. What is the relationship between these two functions? Ligation of α6β4 causes a rapid phosphorylation of the β4 tail, followed by recruitment of Shc and activation of ERK (Mainiero et al., 1995, 1997). In addition, there is evidence that α6β4 activates PI-3K, either through Ras or IRS-1 and 2, rapidly (Mainiero et al., 1997; Shaw et al., 1997; Shaw, 2001). Finally, our results suggest that the EGF-R can activate α6β4 signaling, by acting on Fyn, with a similarly rapid kinetics. By contrast, assembly of hemidesmosomes is a much slower process, at least in cultured cells (Riddelle et al., 1991). In addition, because exposure to EGF induces both phosphorylation of β4 and disruption of hemidesmosomes, it is likely that α6β4-dependent signaling and nucleation of hemidesmosomes are mutually exclusive processes. It is possible that phosphorylation of β4 by Fyn prevents incorporation of α6β4 in hemidesmosomes (Fig. 9).
Previous studies have indicated that mutation of the four tyrosine residues in β4 necessary for binding to Shc prevents disassembly of hemidesmosomes in cells exposed to the tyrosine phosphatase inhibitor pervanadate (Dans et al., 2001). However, this mutation does not interfere with hemidesmosome disruption in response to EGF in both 804G cells (Dans et al., 2001), and in keratinocytes (unpublished data). It is possible that EGF prevents assembly of hemidesmosomes by promoting phosphorylation of additional tyrosine residues, as suggested by the fact that mutation of the four tyrosines necessary for binding to Shc reduces but does not suppress tyrosine phosphorylation of β4 in response to EGF (unpublished data). In addition, it is known that the EGF-R activates PKC through PLC-γ (Chen et al., 1996), and there is evidence suggesting that inhibition of PKC prevents EGF-mediated disruption of hemidesmosomes (Rabinovitz et al., 1999). Thus, it is also possible that PKC contributes to disruption of hemidesmosomes by phosphorylating the β4 tail on serine residues (Rabinovitz et al., 1999) or by contributing to the activation of Fyn (Shanmugam et al., 1998). Finally, although we did not detect tyrosine phosphorylation of BPAG-1 or -2, or plectin/HD-1 in cells treated with EGF (unpublished data), activation of the EGF-R could affect the function of other hemidesmosomal components or the stability of other interactions within hemidesmosomes.
Introduction of α6β4 in breast carcinoma cells that have lost its expression promotes activation of the PI-3K–Rac pathway and invasion through Matrigel (Shaw et al., 1997). Because it is well established that PI-3K–Rac signaling is important for tumor invasion (Keely et al., 1997), it is possible that, in addition to promoting disruption of hemidesmosomes and thus releasing the cell's brakes, α6β4-associated Fyn promotes migration and invasion by activating the PI-3K–Rac pathway or signaling to ERK (Fig. 9).
In squamous carcinoma cells, the fraction of EGF-R associated with α6β4 appears to be activated even in the absence of exogenous EGF. Hence, these cells display enhanced tyrosine phosphorylation of β4 and reduced ability to assemble hemidesmosomes. These observations imply that the signaling pathway that prevents assembly of hemidesmosomes is constitutively activated in squamous carcinoma cells. Dominant negative Fyn suppresses the ability of these cells to migrate and invade both in vitro and in vivo suggesting that disruption of hemidesmosomes is required for carcinoma invasion. Because in our experimental metastasis assay the cells were injected directly into the blood stream, Fyn is probably required in one or more steps of the extravasation process, perhaps traversal of the endothelial basement membrane and/or underlying interstitial matrix. To our knowledge, our study provides the first evidence that inhibition of a Src family kinase suppresses a complex phenomenon such as metastasis. It is plausible that strategies aimed at inhibiting the kinase activity of Fyn may be useful to control squamous carcinoma progression.
Materials and methods
Cell culture and DNA constructs
The immortalized human keratinocytes HaCaT (Boukamp et al., 1988), epidermoid squamous carcinoma cells A431 (from American Type Culture Collection) and MSK-QLL-1 (Xu et al., 1994), human embryonic kidney cells 293-T, provided by D. Levy (New York University School of Medicine, New York, NY), rat bladder carcinoma cells 804G (Izumi et al., 1981), and mouse mammary carcinoma cells RAC-11P/SD (Sonnenberg et al., 1993) were cultured in DME supplemented with 10% fetal bovine serum (Sigma-Aldrich). 293-T cells were transiently transfected by using the calcium coprecipitation method and lysed 24 h later. A431 and 804G cells were transfected with Lipofectamine (GIBCO BRL) according to the manufacturer's instructions. Transfected cells were selected in Hygromycin B (Calbiochem).
All eucaryotic expression vectors were based on the cytomegalovirus promoter. Vectors encoding human α6, β4, β4 deletion mutants (B, C, L), Src, Fyn, Fyn ΔSH3, kinase-dead Fyn or Src, and the palmitoylation-defective Fyn mutant (FynC3,6S) have been described previously (Spinardi et al., 1993; Alland et al., 1994; Mainiero et al., 1997; Wary et al., 1998). The vector encoding Yes was provided by M. Sudol (Mount Sinai Medical School, New York, NY). The Fyn-SH2 point mutant (FynR176E) was generated by two-step PCR using the oligonucleotides 5′-ACCTTGCGTACGAGAGGA, 5'-TCACTCTCTTCGATAAGAAAG, 5′-TTTCTT-ATCGAAGAGAGTGAA, and 5′GATTCTCGAGGGATTTCCCAG; the PCR product was cloned into the SplI/XhoI sites of pRK5-c-Fyn. The chimera Fyn–Src (Fyn1-13/Src14-533) was generated by PCR with the oligonucleotides 5′-ATCGGATCC-GGAAATTTTGGAATTTAG and 5′-ATGCCGGCGTTTTGTTGCTTCTTTAT; the PCR product was cloned into the HindIII/NaeI site of pRK5-c-Src.
Antibodies and chemical compounds
The monoclonal antibody 3E1, reacting with the extracellular portion of human β4, and the rabbit polyclonal antiserum to the COOH-terminal peptide of β4, have been described previously (Giancotti et al., 1992). The rabbit polyclonal antiserum to the COOH-terminal of the EGF receptor was obtained from J. Schlessinger (New York University School of Medicine, New York, NY). The affinity- purified rabbit polyclonal antibodies to Fyn (FYN3), Src (N-16), Src kinases (anti-pan Src SRC2), and ErbB-2 (C-18), and the mouse monoclonal antibody to the EGF-R (528) were from Santa Cruz Biotechnology, Inc. The anti-phosphotyrosine monoclonal antibody RC20 was from BD Transduction Laboratories. The rabbit antiserum β4-exo, reacting with the extracellular portion of β4, and affinity-purified rabbit antibodies to BPAG2 have been described previously (Murgia et al., 1998). The Src kinase inhibitors PP1, PP2 (Calbiochem), and Herbimycin A (GIBCO BRL), the PI-3K inhibitor LY 294002 (Biomol), and the p38 kinase inhibitor SB203580 (Stratagene) were dissolved in DMSO and further diluted in culture medium.
Biochemical methods
Prior to treatment with EGF (25 ng/ml, 4 min) (Intergen), HaCaT and MSK-QLL-1 cells were serum starved overnight; A431 cells were starved for 40 h. The cells were then lysed in TX-DOC lysis buffer (50 mM Hepes, 150 mM NaCl, 4 mM EDTA, 1 mM EGTA, 1% Triton X-100, and 0.5% sodium deoxycolate) containing 1 mM sodium orthovanadate, 20 mM sodium pyrophosphate, 100 mM NaF, 0.02% aprotinin, 20 μg/ml leupeptin, and 4 μg/ml pepstatin A (all from Sigma-Aldrich). 804G cells were stimulated with 50 ng/ml EGF for 4 min and lysed in modified RIPA buffer (50 mM Hepes, 150 mM NaCl, 4 mM EDTA, 1 mM EGTA, 10% glycerol, 1% Triton X-100, 1% sodium deoxycolate, and 0.1% sodium dodecylsulfate) containing protease and phosphatase inhibitors as above. 293-T cells were lysed in modified RIPA buffer. The Src family kinase inhibitors PP1 and PP2 were added to the culture medium at the indicated concentrations, and cells were incubated for 15 min at 37°C before the addition of EGF. Immunoprecipitation and immunoblotting were performed as described previously (Mainiero et al., 1995). To avoid aspecific binding of the EGF-R to the beads during immunoprecipitation experiments, cell lysates were precleared with protein A Agarose (Upstate Biotechnology) and normal Mouse IgGs (Sigma-Aldrich) covalently linked to CNBr-activated Sepharose™ 4B (Amersham Pharmacia Biotech) for 1.5 h at 4°C. After centrifugation, the supernatants were immunoprecipitated using the 3E1 monoclonal antibody covalently linked to CNBr-activated beads. The immobilized immunocomplexes were washed extensively in lysis buffer before SDS-PAGE. To cross-link the antibodies to the Sepharose beads, the antibodies were dialyzed in Spectra/Por MWCO:6–8,000 dialysis bags (Spectrum Medical Industries) against coupling buffer (carbonate-bicarbonate buffer, pH 9.2, 0.5 M NaCl) overnight at 4°C. CNBr-activated Sepharose beads (400 mg/5 mg of antibody) were incubated in 1 mM HCl for 15 min at room temperature. The beads were then washed with 1 mM HCl and then once in coupling buffer before incubation with the antibody. The suspension was incubated overnight at 4°C on a rotator, then spun down, and the pellet was washed two times in 0.1 M Tris HC, pH 8, containing 0.5 M NaCl, and stored in TBS at 4°C. The efficiency of cross-linking was checked by measuring the concentration of the dialyzed antibody before and after incubation with the activated beads. This method allows to minimize the amount of beads used (about 10 μl/sample) and to use high quantities of antibody (25–30 μg/sample).
Immunofluorescence
804G clones were plated on glass coverslips for 24 h and then treated with EGF (50 ng/ml) in serum-free medium. The cells were permeabilized in 0.2% Triton X-100 in PBS for 4 min on ice and fixed with cold methanol for 10 min. A431 clones were plated on glass coverslips coated with the laminin 5 matrix deposited by RAC-11P/SD cells (Sonnenberg et al., 1993) for 48 h before permeabilization and fixation. The cells were incubated for 1 h with the primary antibody, washed with 0.1% Triton X-100 in PBS, and then stained with FITC-conjugated goat anti–mouse or goat anti–rabbit IgGs (Jackson ImmunoResearch Laboratories) for 45 min. After washing, the coverslips were mounted in Citifluor mounting medium (Ted Pella).
In vitro wound assay and invasion assay
After overnight starvation, wounds were made on confluent cell monolayers with a plastic tip. 804G cells were stimulated with EGF (50 ng/ml) for 5 or 10 h, and A431 cells were treated with EGF (25 ng/ml) for 24 h, before staining in 0.25% crystal violet. To quantify cell migration, three randomly chosen regions of a wound (1 mm long) were photographed at a magnification of 40X; a mean wound width was measured every 20 μm, and an average percent wound closure was calculated. Three independent wounds were examined per sample and a mean percent wound closure was calculated.
For in vitro invasion assays, 50 μl of Matrigel (Collaborative Biomedical Products) (1 mg/ml in serum-free DME) was allowed to gelify on the upper side of the filter (8 μm pore size) in a Transwell chamber (Corning Costar Corp.) at 37°C for 2 h. Cells were then plated on the Matrigel (80,000 cells per chamber in 100 μl of serum-free DME), let attach for 2 h, and the lower chamber was filled with serum-free medium containing EGF (25 ng/ml). After 24 h, the Matrigel and the cells that did not invade were removed from the upper side of the filter with a cotton swab, and the cells attached to the lower side of the filter were stained with crystal violet and counted. Each experiment was performed in triplicate. For invasion assays in presence of chemical inhibitors, cells were seeded on Matrigel and, when attached, the indicated compound was added to the medium. The inhibitor was present in the upper chamber for the entire duration of the assay; at the end of the assay, cell viability in the upper chamber was assessed by Trypan blue.
In vivo metastasis formation experiment
3–6-wk-old athymic nude mice (Nu/nu Harlan Sprague Dawley) were injected with 2 × 106 cells suspended in 200 μl of PBS. After 5 wk, the mice were sacrificed and their lungs removed, fixed in formalin, and paraffin-embedded. 8-μm sections were stained with Gill's hematoxylin and eosin, and examined for the presence of lung metastases.
Online supplemental material
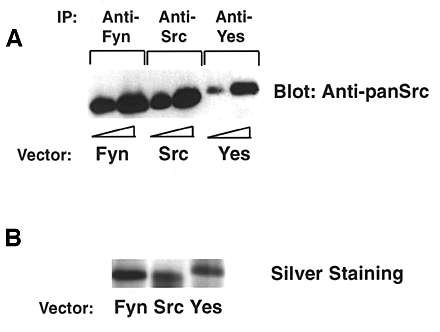
Fig. S1 is available at http://www.jcb.org/cgi/content/full/jcb.200105017/DC1 and shows reactivity of the anti-pan Src antibodies with recombinant Fyn, Src, and Yes.
Supplemental Material
Acknowledgments
We thank D. Levy, M. Resh, and P. Sacks, M. Sudol, and J. Schlessinger for reagents (Memorial Sloan-Kettering Cancer Center, New York, NY), K. Manova of the Molecular Cytology Facility for her help in the analysis of lung metastasis, and members of the Giancotti laboratory for comments on the manuscript.
This study was supported by National Institutes of Health grants R01 CA58976 (to F.G. Giancotti) and P30 CA08748 (to Memorial Sloan-Kettering Cancer Center). A. Mariotti was recipient of a fellowship from the American-Italian Cancer Foundation. F.G. Giancotti was an established investigator of the American Heart Association.
The online version of this article contains supplemental material.
P.A. Kedeshian's present address is UCLA Medical Center, Division of Head and Neck Surgery, Los Angeles, CA 90502.
L. Gagnoux-Palacios's present address is INSERM U385, Nice, France.
Footnotes
Abbreviations used in this paper: BPAG, bullous pemphigoid antigen; ERK, extracellular signal-regulated kinase; FAK, focal adhesion kinase; ITAM, immunoreceptor tyrosine-based activation motif; PI-3K, phosphatidylinositol-3 kinase.
References
- Alland, L., S.M. Peseckis, R.E. Atherton, L. Berthiaume, and M.D. Resh. 1994. Dual myristylation and palmitylation of Src family member p59fyn affects subcellular localization. J. Biol. Chem. 269:16701–16705. [PubMed] [Google Scholar]
- Andra, K., H. Lassmann, R. Bittner, S. Shorny, R. Fassler, F. Propst, and G. Wiche. 1997. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 11:3143–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldari, C.T., J.L. Telford, and O. Acuto. 2000. Lymphocyte antigen receptor and coreceptor signaling. Embo J. 19:4857–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscardi, J.S., D.A. Tice, and S.J. Parsons. 1999. c-Src, receptor tyrosine kinases, and human cancer. Adv. Cancer Res. 76:61–119. [DOI] [PubMed] [Google Scholar]
- Borradori, L., and A. Sonnenberg. 1999. Structure and function of hemidesmosomes: more than simple adhesion complexes. J. Invest. Dermatol. 112:411–418. [DOI] [PubMed] [Google Scholar]
- Boudreau, N., and M.J. Bissell. 1998. Extracellular matrix signaling: integration of form and function in normal and malignant cells. Curr. Opin. Cell Biol. 10:640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukamp, P., R.T. Petrussevska, D. Breitkreutz, J. Hornung, A. Markham, and N.E. Fusenig. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, D.A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell. Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- Carico, E., D. French, B. Bucci, R. Falcioni, A. Vecchione, and R. Mariani-Costantini. 1993. Integrin beta 4 expression in the neoplastic progression of cervical epithelium. Gynecol. Oncol. 49:61–66. [DOI] [PubMed] [Google Scholar]
- Carter, W.G., P. Kaur, S.G. Gil, P.J. Gahr, and E.A. Wayner. 1990. Distinct functions for integrins α3 β1 in focal adhesions and α6 β4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: relation to hemidesmosomes. J. Cell Biol. 111:3141–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P., H. Xie, and A. Wells. 1996. Mitogenic signaling from the egf receptor is attenuated by a phospholipase C-gamma/protein kinase C feedback mechanism. Mol. Biol. Cell. 7:871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, L.R., W.A. Ricketts, L. Yeh, and D. Cheresh. 1999. Bifurcation of cell migratory and proliferative signaling by the adaptor protein Shc. J. Cell Biol. 147:1561–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini, R.M., R. Falcioni, P. Battista, G. Zupi, S.J. Kennel, A. Colasante, I. Venturo, C.G. Curio, and A. Sacchi. 1990. Integrin (alpha 6/beta 4) expression in human lung cancer as monitored by specific monoclonal antibodies. Cancer Res. 50:6107–6112. [PubMed] [Google Scholar]
- Dans, M., L. Gagnoux-Palacios, P. Blaikie, S. Klein, A. Mariotti, and F.G. Giancotti. 2001. Tyrosine phosphorylation of the beta 4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J. Biol. Chem. 276:1494–1502. [DOI] [PubMed] [Google Scholar]
- Dowling, J., Q.C. Yu, and E. Fuchs. 1996. β4 integrin is required for hemidesmosome formation, cell adhesion and cell survival. J. Cell Biol. 134:559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcioni, R., A. Antonini, P. Nistico, S. Di Stefano, M. Crescenzi, P.G. Natali, and A. Sacchi. 1997. Alpha6beta4 and alpha6beta1 integrins associate with ErbB2 in human carcinoma cell lines. Exp. Cell Res. 236:76–85. [DOI] [PubMed] [Google Scholar]
- Fincham, V.J., and M.C. Frame. 1998. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J. 17:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerts, D., L. Fontao, M.G. Nievers, R.Q. Schaapveld, P.E. Purkis, G.N. Wheeler, E.B. Lane, I.M. Leigh, and A. Sonnenberg. 1999. Binding of integrin α6β4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filamet binding. J. Cell Biol. 147:417–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giancotti, F.G. 1996. Signal transduction by the alpha 6 beta 4 integrin: charting the path between laminin binding and nuclear events. J. Cell Sci. 109:1165–1172. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G., and F. Mainiero. 1994. Integrin-mediated adhesion and signaling in tumorigenesis. Biochim. Biophys. Acta. 1198:47–64. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G., and E. Ruoslahti. 1999. Integrin signaling. Science. 285:1028–1032. [DOI] [PubMed] [Google Scholar]
- Giancotti, F.G., M.A. Stepp, S. Suzuki, E. Engvall, and E. Ruoslahti. 1992. Proteolytic processing of endogenous and recombinant β4 integrin subunit. J. Cell Biol. 118:951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson, I.K., S. Spurr-Michaud, A. Tisdale, J. Elwell, and M.A. Stepp. 1993. Redistribution of the hemidesmosome components alpha 6 beta 4 integrin and bullous pemphigoid antigens during epithelial wound healing. Exp. Cell Res. 207:86–98. [DOI] [PubMed] [Google Scholar]
- Gu, J., M. Tamura, R. Pankov, E.H. Danen, T. Takino, K. Matsumoto, and K.M. Yamada. 1999. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell Biol. 146:389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, L., L. Degenstein, J. Dowling, Q.C. Yu, R. Wollmann, B. Perman, and E. Fuchs. 1995. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell. 81:233–243. [DOI] [PubMed] [Google Scholar]
- Hanke, J.H., J.P. Gardner, R.L. Dow, P.S. Changelian, W.H. Brissette, E.J. Weringer, B.A. Pollok, and P.A. Connelly. 1996. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem. 271:695–701. [DOI] [PubMed] [Google Scholar]
- Hintermann, E., M. Bilban, A. Sharabi, and V. Quaranta. 2001. Inhibitory role of α6β4‴associated erbB-2 and phosphoinositide 3-kinase in keratinocyte haptotactic migration dependent on α3β1 integrin. J. Cell Biol. 153:465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkinson, S.B., and J.C. Jones. 2000. The N terminus of the transmembrane protein BP180 interacts with the N-terminal domain of BP230, thereby mediating keratin cytoskeleton anchorage to the cell surface at the site of the hemidesmosome. Mol. Biol. Cell. 11:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz, A.R., and J.T. Parsons. 1999. Cell migration—movin' on [comment]. Science. 286:1102–1103. [DOI] [PubMed] [Google Scholar]
- Izumi, K., Y. Hirao, L. Hopp, and R. Oyasu. 1981. In vitro induction of ornithine decarboxylase in urinary bladder carcinoma cells. Cancer Res. 41:405–409. [PubMed] [Google Scholar]
- Kaplan, K.B., J.R. Swedlow, D.O. Morgan, and H.E. Varmus. 1995. c-Src enhances the spreading of src-/- fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 9:1505–1517. [DOI] [PubMed] [Google Scholar]
- Keely, P.J., J.K. Westwick, I.P. Whitehead, C.J. Der, and L.V. Parise. 1997. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 390:632–636. [DOI] [PubMed] [Google Scholar]
- Kimmel, K.A., and T.E. Carey. 1986. Altered expression in squamous carcinoma cells of an orientation restricted epithelial antigen detected by monoclonal antibody A9. Cancer Res. 46:3614–3623. [PubMed] [Google Scholar]
- Klinghoffer, R.A., C. Sachsenmaier, J.A. Cooper, and P. Soriano. 1999. Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 18:2459–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C.R., W.S. Chen, W. Kruiger, L.S. Stolarsky, W. Weber, R.M. Evans, I.M. Verma, G.N. Gill, and M.G. Rosenfeld. 1984. Expression cloning of human EGF receptor complementary DNA: gene amplification and three related messenger RNA products in A431 cells. Science. 224:843–848. [DOI] [PubMed] [Google Scholar]
- Mainiero, F., C. Murgia, K.K. Wary, A.M. Curatola, A. Pepe, M. Blumemberg, J.K. Westwick, C.J. Der, and F.G. Giancotti. 1997. The coupling of alpha6beta4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO J. 16:2365–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainiero, F., A. Pepe, K.K. Wary, L. Spinardi, M. Mohammadi, J. Schlessinger, and F.G. Giancotti. 1995. Signal transduction by the alpha 6 beta 4 integrin: distinct beta 4 subunit sites mediate recruitment of Shc/Grb2 and association with the cytoskeleton of hemidesmosomes. EMBO J. 14:4470–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainiero, F., A. Pepe, M. Yeon, Y. Ren, and F.G. Giancotti. 1996. The intracellular functions of alpha6beta4 integrin are regulated by EGF. J. Cell Biol. 134:241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J.A., B. Gatalica, A.M. Christiano, K. Li, K. Owaribe, J.R. McMillan, R.A. Eady, and J. Uitto. 1995. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat. Genet. 11:83–86. [DOI] [PubMed] [Google Scholar]
- Murgia, C., P. Blaikie, N. Kim, M. Dans, H.T. Petrie, and F.G. Giancotti. 1998. Cell cycle and adhesion defects in mice carrying a targeted deletion of the integrin beta4 cytoplasmic domain. EMBO J. 17:3940–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitz, I., A. Toker, and A.M. Mercurio. 1999. Protein kinase C-dependent mobilization of the alpha6beta4 integrin from hemidesmosomes and its association with actin-rich cell protrusions drive the chemotactic migration of carcinoma cells. J. Cell Biol. 146:1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss, M., E.B. Stash, V.F. Vellucci, and Z.L. Zhou. 1991. Activation of the autocrine transforming growth factor alpha pathway in human squamous carcinoma cells. Cancer Res. 51:6254–6262. [PubMed] [Google Scholar]
- Rezniczek, G.A., J.A. de Pereda, S. Reipert, and G. Wiche. 1998. Linking integrin α6β4-based cell adhesion to the intermediate filament cytoskeleton: direct interaction between the β4 subunit and plectin at multiple sites. J. Cell Biol. 141:209–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddelle, K.S., K.J. Green, and J.C. Jones. 1991. Formation of hemidesmosomes in vitro by a transformed rat bladder cell line. J. Cell Biol. 112:159–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, M.C., K. Lee, Y. Miyashita, and W.G. Carter. 1999. Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J. Cell Biol. 145:1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaapveld, R.Q., L. Borradori, D. Geerts, M.R. van Leusden, I. Kuikman, M.G. Nievers, C.M. Niessen, R.D. Steenbergen, P.J. Snijders, and A. Sonnenberg. 1998. Hemidesmosome formation is initiated by the β4 integrin subunit, requires complex formation of β4 and HD1/plectin, and involves a direct interaction between β4 and the bullous pemphigoid antigen 180. J. Cell Biol. 142:271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk, P. 1979. The fate of hemidesmosomes in laryngeal carcinoma. Arch. Otorhinolaryngol. 222:187–198. [DOI] [PubMed] [Google Scholar]
- Serini, G., L. Trusolino, E. Saggiorato, O. Cremona, M. De Rossi, A. Angeli, F. Orlandi, and P.C. Marchisio. 1996. Changes in integrin and E-cadherin expression in neoplastic versus normal thyroid tissue. J. Natl. Cancer Inst. 88:442–449. [DOI] [PubMed] [Google Scholar]
- Shanmugam, M., N.L. Krett, C.A. Peters, E.T. Maizels, F.M. Murad, H. Kawakatsu, S.T. Rosen, and M. Hunzicker-Dunn. 1998. Association of PKC delta and active Src in PMA-treated MCF-7 human breast cancer cells. Oncogene. 16:1649–1654. [DOI] [PubMed] [Google Scholar]
- Shaw, L.M. 2001. Identification of insulin receptor substrate 1 (IRS -1) and IRS-2 as signaling intermediates in the α6β4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol. Cell Biol. 21:5082–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, L.M., I. Rabinovitz, H.H. Wang, A. Toker, and A.M. Mercurio. 1997. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 91:949–960. [DOI] [PubMed] [Google Scholar]
- Sieg, D.J., C.R. Hauck, D. Ilic, C.K. Klingbeil, E. Schaefer, C.H. Damsky, and D.D. Schlaepfer. 2000. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell. Biol. 2:249–256. [DOI] [PubMed] [Google Scholar]
- Smith, F.J., R.A. Eady, I.M. Leigh, J.R. McMillan, E.L. Rugg, D.P. Kelsell, S.P. Bryant, N.K. Spurr, J.F. Geddes, G. Kirtschig, G. Milana, A.G. de Bono, K. Owaribe, G. Wiche, L. Pulkkinen, J. Uitto, W.H. McLean, and E.B. Lane. 1996. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat. Genet. 13:450–457. [DOI] [PubMed] [Google Scholar]
- Sonnenberg, A., J. Calafat, H. Daams, L.M. van der Raaij-Helmer, R. Falcioni, S.J. Aplin, J. Baker, M. Loizidou, et al. 1991. Integrin α6/β4 complex is located in hemidesmosomes, suggesting a major role in epidermal cell-basement membrane adhesion. J. Cell Biol. 113:907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg, A., A.A. de Melker, A.M. Martinez de Velasco, H. Janssen, J. Calafat, and C.M. Niessen. 1993. Formation of hemidesmosomes in cells of a transformed murine mammary tumor cell line and mechanisms involved in adherence of these cells to laminin and kalinin. J. Cell Sci. 106:1083–1102. [DOI] [PubMed] [Google Scholar]
- Spinardi, L., Y.L. Ren, R. Sanders, and F.G. Giancotti. 1993. The beta 4 subunit cytoplasmic domain mediates the interaction of alpha 6 beta 4 integrin with the cytoskeleton of hemidesmosomes. Mol. Biol. Cell. 4:871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani, T., T. Karttunen, T. Kiviluoto, E. Kivilaakso, R.E. Burgeson, P. Sipponen, and I. Virtanen. 1996. Alpha 6 beta 4 integrin and newly deposited laminin-1 and laminin-5 form the adhesion mechanism of gastric carcinoma. Continuous expression of laminins but not that of collagen VII is preserved in invasive parts of the carcinomas: implications for acquisition of the invading phenotype. Am. J. Pathol. 149:781–793. [PMC free article] [PubMed] [Google Scholar]
- Tennenbaum, T., A.J. Belanger, V. Quaranta, and S.H. Yuspa. 1996. Differential regulation of integrins and extracellular matrix binding in epidermal differentiation and squamous tumor progression. J. Investig. Dermatol. Symp. Proc. 1:157–161. [PubMed] [Google Scholar]
- Van de Vijver, M.J., R. Kumar, and J. Mendelsohn. 1991. Ligand-induced activation of A431 cell epidermal growth factor receptors occurs primarily by an autocrine pathway that acts upon receptors on the surface rather than intracellularly. J. Biol. Chem. 266:7503–-7508. [PubMed] [Google Scholar]
- van der Neut, R., P. Krimpenfort, J. Calafat, C.M. Niessen, and A. Sonnenberg. 1996. Epithelial detachment due to absence of hemidesmosomes in integrin beta 4 null mice. Nat. Genet. 13:366–369. [DOI] [PubMed] [Google Scholar]
- van't Hof, W., and M.D. Resh. 1997. Rapid plasma membrane anchoring of newly synthesized p59fyn: selective requirement for NH2-terminal myristoylation and palmitoylation at cysteine-3. J. Cell Biol. 136:1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Hof, W., and M.D. Resh. 1999. Dual fatty acylation of p59(Fyn) is required for association with the T cell receptor zeta chain through phosphotyrosine-Src homology domain-2 interactions. J. Cell Biol. 145:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varner, J.A., and D.A. Cheresh. 1996. Integrins and cancer. Curr. Opin. Cell Biol. 8:724–730. [DOI] [PubMed] [Google Scholar]
- Wary, K.K., A. Mariotti, C. Zurzolo, and F.G. Giancotti. 1998. A requirement for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell. 94:625–634. [DOI] [PubMed] [Google Scholar]
- Wolf, G.T., T.E. Carey, S.P. Schmaltz, K.D. McClatchey, J. Poore, L. Glaser, D.J. Hayashida, and S. Hsu. 1990. Altered antigen expression predicts outcome in squamous cell carcinoma of the head and neck. J. Natl. Cancer Inst. 82:1566–1572. [DOI] [PubMed] [Google Scholar]
- Xu, L., B.J. Davidson, V.V. Murty, R.G. Li, P.G. Sacks, P. Garin-Chesa, S.P. Schantz, and R.S. Chaganti. 1994. TP53 gene mutations and CCND1 gene amplification in head and neck squamous cell carcinoma cell lines. Int. J. Cancer. 59:383–387. [DOI] [PubMed] [Google Scholar]
- Yamamoto, T., N. Kamata, H. Kawano, S. Shimizu, T. Kuroki, K. Toyoshima, K. Rikimaru, N. Nomura, R. Ishizaki, I. Pastan, et al. 1986. High incidence of amplification of the epidermal growth factor receptor gene in human squamous carcinoma cell lines. Cancer Res. 46:414–416. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}