

Abstract
Keratin 8 and 18 (K8/18) are the major components of intermediate filament (IF) proteins of simple or single-layered epithelia. Recent data show that normal and malignant epithelial cells deficient in K8/18 are nearly 100 times more sensitive to tumor necrosis factor (TNF)–induced cell death. We have now identified human TNF receptor type 1 (TNFR1)–associated death domain protein (TRADD) to be the K18-interacting protein. Among IF proteins tested in two-hybrid systems, TRADD specifically bound K18 and K14, type I (acidic) keratins. The COOH-terminal region of TRADD interacted with the coil Ia of the rod domain of K18. Endogenous TRADD coimmunoprecipitated with K18, and colocalized with K8/18 filaments in human mammary epithelial cells. Overexpression of the NH2 terminus (amino acids 1–270) of K18 containing the TRADD-binding domain as well as overexpression of K8/18 in SW13 cells, which are devoid of keratins, rendered the cells more resistant to killing by TNF. We also showed that overexpressed NH2 termini of K18 and K8/18 were associated with endogenous TRADD in SW13 cells, resulting in the inhibition of caspase-8 activation. These results indicate that K18 may sequester TRADD to attenuate interactions between TRADD and activated TNFR1 and moderate TNF-induced apoptosis in simple epithelial cells.
Keywords: apoptosis; keratin 8; keratin 18; TNF; TRADD
Introduction
Intermediate filaments (IFs)* are major components of the cytoskeleton and nuclear envelope in most types of eukaryotic cells (Franke, 1987; Fuchs and Weber, 1994; Fuchs and Cleveland, 1998). Although structural components of other major cytoskeletal proteins, e.g., actin and tubulin, are highly conserved in different cell types, constituent proteins of IFs show intriguing molecular diversities and are expressed in tissue-specific programs. The keratin subfamily, which is preferentially expressed in epithelial cells, has over 20 members (keratin 1–20) that form obligate noncovalent heteropolymers of at least one type I (keratin 9–20) and one type II keratin (keratin 1–8) (Moll et al., 1982). Keratin 8 and 18 (K8/18) are the major components of the IFs of simple or single-layered epithelia found in the gastrointestinal tract, liver, exocrine pancreas, and mammary gland, from which many carcinomas arise (Oshima et al., 1996). Gene targeting techniques have been used to elucidate the function of K8/18. K8 knockout mice in one strain died around day 12 from undetermined tissue damage (Baribault et al., 1993), whereas in a different strain, they survived to adulthood but colorectal hyperplasia and inflammation were present (Baribault et al., 1994). K18 null mice were fertile, whereas old K18 null mice developed a distinct liver pathology with abnormal hepatocytes containing K8-positive aggregates that resembled Mallory bodies seen in human livers with alcoholic hepatitis (Magin et al., 1998). Together with a report describing a mutation in the K18 gene in a patient with cryptogenic cirrhosis (Ku et al., 1997), these findings suggest that K18 mutations may possibly cause or result in a predisposition to liver disease (Omary and Ku, 1997).
A recent report from Oshima's group (Caulin et al., 2000) addressed a new, fascinating function of K8/18, and explained the phenotypes of K8 or K18 knockout mice. They stated that normal and malignant epithelial cells deficient in K8 and K18 are ∼100 times more sensitive to tumor necrosis factor (TNF)–induced cell death. K8 and K18 both bind to the cytoplasmic domain of TNF receptor type 2 (TNFR2) and moderate the effects of TNF. This diminution may be a fundamental function of K8/18 seen in liver regeneration, inflammatory bowel disease, hepatotoxin sensitivity, and the persistent expression of these keratins in many carcinomas (Caulin et al., 2000).
To further investigate the functions of K8/18, we screened a yeast two-hybrid library, using K8 or K18 as a bait in order to search for unidentified K8/18–associated proteins. Among the positive clones, we found a clone encoding the COOH-terminal portion of human TNFR1-associated death domain protein (TRADD), as a K18-interacting protein. We report here evidence for the in vivo association of TRADD with K18, and demonstrate that overexpression of the NH2 terminus (amino acids [aa] 1–270) of K18 containing the TRADD binding domain as well as overexpression of K8/18 in SW13 cells, which are devoid of keratins, rendered the cells more resistant to killing by TNF. We propose that sequestration of TRADD by K18 may be one molecular mechanism by which epithelial cells expressing keratins can be resistant to TNF-induced apoptosis.
Results
Identification of TRADD as a K18-interacting protein
To identify proteins interacting with K8/18, we screened a human liver cDNA library, using the yeast two-hybrid procedure with full-length K8 or K18 as a bait. In the library screening for K18, we earlier found that human Mrj, a DnaJ/heat shock protein 40 (hsp40) family protein, directly interacted with K18 as a cochaperone to regulate K8/18 filament organization (Izawa et al., 2000). We have also identified a clone that encodes the COOH-terminal region of TRADD as a K18-interacting protein. TRADD is a 34-kD protein that interacts specifically with TNFR1 (Hsu et al., 1995). In the initial step of TNFR1 signaling, TNF binds to the extracellular domain of TNFR1 and induces receptor trimerization (Banner et al., 1993). Next, the death domain of TNFR1 recruits the adaptor protein TRADD (Hsu et al., 1995). TRADD, in turn, binds Fas-associated death domain (FADD), TNFR-associated factor 2, and RIP, and activates the downstream signaling pathway leading to apoptosis (Hsu et al., 1996; Yeh et al., 1997), JNK/SAPK activation (Hsu et al., 1996; Liu et al., 1996; Yeh et al., 1997), and nuclear factor-κB (NF-κB) activation (Hsu et al., 1996; Kelliher et al., 1998). To determine the role of K8/18 in TNF signaling, we further analyzed the TRADD–K18 interaction. The human TRADD cDNA encodes a 312-aa protein, and the clone derived from the liver library contained residues 245 to 312 of TRADD, hereafter termed TRADD-C (Fig. 1 A). The TRADD-C fragment is the COOH-terminal part of the death domain of TRADD mediating the interaction between TRADD and the death domain of TNFR1, FADD, and RIP. To confirm that full-length TRADD (TRADD-F) interacts with K18 and to determine if TRADD can bind to other IF proteins, we examined the interaction of TRADD-F and TRADD-C with K5, K8, K14, K18, and type III IF proteins in the two-hybrid system (Fig. 1 B). TRADD-C strongly interacted with K14 and K18, type I (acidic) keratins, but did not interact with K5, K8, vimentin, glial fibrillary acidic protein (GFAP), or desmin. In the same fashion, TRADD-F specifically interacted with K14 and K18. We next analyzed the region of K18 that contained the TRADD interaction site. For this, the binding between a series of truncations of K18 and TRADD-F or TRADD-C was determined using the two-hybrid system (Fig. 1 C). The coil Ia region of the rod domain of K18 specifically interacted with both TRADD-F and TRADD-C.
Figure 1.

Identification of TRADD as a K18-interacting protein. (A) Domain organization of human TRADD protein. Death domain (DD) is indicated. Position of the original clone encoding the COOH-terminal region of TRADD (TRADD-C) is also indicated. Numbers refer to amino acid position. (B) Interactions of TRADD-C or full-length TRADD (TRADD-F) with K18 or other IFs in two- hybrid system. Y190 cells cotransformed with various pGBD-C1-IFs and pGAD-C1-TRADD-F or pGAD-C1-TRADD-C were selected in minus tryptophan (Trp)-leucine (Leu) media and subjected to β-galactosidase filter assay. The numbers of plus signs represent the relative rates at which the transformed yeast colonies turned blue after incubation at 30°C on filters, +++, <3 h; ++, 3–8 h; +, >8 h. Minus sign represents colonies remaining white at 24 h. (C) Identification of the coil Ia region of K18 that is sufficient for binding to TRADD. Y190 cells cotransformed with various pGBD-C1-K18 deletion mutants and pGAD-C1-TRADD-F or pGAD-C1-TRADD-C were selected in minus Trp-Leu media and subjected to β-galactosidase filter assay. Plus and minus signs are the same as in B. Numbers refer to amino acid position.
Direct association of TRADD with K8/18 in vitro
Next we analyzed the in vitro direct association of TRADD, using a cosedimentation assay (Fig. 2). As the separation of K8, vimentin or desmin, and recombinant TRADD protein (produced as a glutathione S-transferase [GST] fusion; GST–TRADD) in the Coomassie brilliant blue–stained gel after SDS-PAGE was not clear (Fig. 2 A), the samples were also detected with an anti-GST antibody. After immunoblotting, the proportions of supernatant and precipitated GST–TRADD were revealed (Fig. 2 B). Incubation in filament assembly buffer induced a rapid polymerization of K8 and K18 (Fig. 2 A, lane 1). As a control, recombinant GST was not sedimented in the presence of K8/18 (Fig. 2 A and B, lane 3). In the absence of K8/18, GST–TRADD was not sedimented (Fig. 2 A and B, lane 4), but in the presence of K8/18, a substantial portion of GST–TRADD was precipitated (Fig. 2 A and B, lane 5). Because TRADD interacted with K18, but not with K8 in the two-hybrid analysis, the TRADD sedimenting with K8/18 was thought to be associated with the K8/18 polymer via direct binding to K18. In addition, TRADD did not sediment with vimentin (Fig. 2 A and B, lane 6) or desmin (Fig. 2 A and B, lane 7), findings consistent with results obtained using the two-hybrid method.
Figure 2.

Cosedimentation of TRADD with K8/18. K8/18 filaments were assembled in the absence (lane 1) or presence of GST (lane 3), or GST–TRADD (lane 5). As a control, only GST (lane 2) or GST–TRADD protein (lane 4) was incubated in the assembly buffer. Vimentin (lane 6) or desmin filaments (lane 7) were assembled in the presence of GST–TRADD. The supernatant (S) and pellet (P) fractions were analyzed by SDS-PAGE, and stained with Coomassie brilliant blue (A), or detected with anti-GST antibody by immunoblotting (B). Substantial portions of GST–TRADD were sedimented with K8/18. GST–TRADD was not sedimented with either vimentin or desmin under these conditions. The arrowhead and arrow indicate positions of GST and GST–TRADD, respectively. Molecular size markers are shown on the left.
Overexpressed myc-tagged TRADD colocalizes with K8/18 filaments
In attempts to characterize the K18–TRADD association in vivo, we first determined the subcellular localization of overexpressed myc-tagged TRADD in various epithelial cells. We transfected HeLa cells, T24 cells, MDCK cells, and immortalized human mammary epithelial cells (HMEC) with a myc-tagged vector encoding the full-length TRADD. 16 h later, the cells were fixed and doubly stained with anti-myc polyclonal antibody and anti-K18 monoclonal antibody (Fig. 3 A). In various cells, overexpressed myc–TRADD exhibited a filamentous pattern, and these myc–TRADD filaments markedly colocalized with K8/18 filaments (Fig. 3 A). Overexpressed myc–TRADD did not colocalize with either actin filaments or microtubules (unpublished data). The overexpression of the death domain of TRADD also resulted in a cytosolic filamentous distribution (unpublished data). Because it is reported that FADD and caspase-8/FLICE (Perez and White, 1998; Siegel et al., 1998), containing death effector domains, and caspase-2 (Colussi et al., 1998) and Bcl10 (Guiet and Vito, 2000), containing a caspase recruitment domain, form cytoplasmic filamentous structures when overexpressed, we examined whether overexpressed FADD, caspase-8, or caspase-2 colocalized with K8/18. To do this, we overexpressed myc-tagged, full-length TRADD, FADD, caspase-8, or caspase-2 in HeLa cells in the presence of 20 μM zVAD-fmk, a caspase inhibitor that inhibits apoptosis, and the cells were doubly stained for myc and K18 (Fig. 3 B). In the presence of zVAD-fmk, overexpressed myc–TRADD colocalized with K8/18 filaments, as described above. Overexpressed myc–FADD formed filamentous structures, but these filaments did not colocalize with K8/18 filaments; a result consistent with a report that the filamentous structures formed by FADD and caspase-8 did not colocalize with known cytoskeletal elements including IFs (Siegel et al., 1998). As it was reported that death effector domain mutants of caspase-8 could form filamentous structures but full-length caspase-8 showed a diffuse distribution (Perez and White, 1998; Siegel et al., 1998), overexpressed myc–full-length caspase-8 showed a diffuse distribution, and did not show colocalization with K8/18 filaments. Overexpressed myc–caspase-2 showed both cytoplasmic and nuclear localization as previously reported (Colussi et al., 1998), and did not colocalize with K8/18 filaments. These results suggest that among the apoptosis-related proteins overexpressed in this study, TRADD is specifically associated with K8/18 filaments.
Figure 3.


Overexpressed, myc-tagged TRADD colocalizes with K8/18 filaments. (A) Subcellular distribution of overexpressed myc-tagged TRADD in various epithelial cells. pRK5–myc-TRADD vectors were transfected into HeLa cells, T24 cells, MDCK cells, and immortalized HMEC. The transfected cells were fixed 16 h later and doubly stained with anti-myc antibody and anti-K18 antibody. The merged images are also shown. (B) Subcellular distribution of overexpressed myc-tagged TRADD, FADD, caspase-2, and caspase-8 in HeLa cells. These myc-tagged proteins were overexpressed in HeLa cells in the presence of 20 μM zVAD-fmk. The transfected cells were doubly stained with anti-myc antibody and anti-K18 antibody. The merged images are also shown. Bars, 10 μm.
Endogenous TRADD is associated with K18 in vivo
To explore the interaction between native TRADD and K18, polyclonal antibodies were raised against TRADD, using the purified histidine (His)6-tagged NH2-terminal region of TRADD (TRADD-N) as the immunogen. Affinity-purified rabbit anti-TRADD antibody specifically recognized a polypeptide with a relative molecular mass of 34 kD in lysates obtained from immortalized HMEC (Fig. 4 A), as well as in those from HeLa, T24, and MDCK cells (unpublished data). Preincubation of the antibody with the immunogen selectively inhibited the immunoreactivity (Fig. 4 A). Since HMEC, which are immortalized by retroviral expression of the catalytic component of human telomerase, maintain normal natures of HMEC (Kiyono et al., 1998) and express the highest level of TRADD among the cells we tested, we mainly used HMEC in the following experiments. Using the rabbit anti-TRADD antibody, we precipitated complexes containing TRADD proteins from 1% NP-40 soluble fraction of HMEC lysates (Fig. 4 B). TRADD was immunoprecipitated with anti-TRADD antibody, but not with control rabbit IgG. K18 was coimmunoprecipitated with TRADD, using the anti-TRADD antibody. These results indicate that TRADD is associated with K18, in vivo. To determine the intracellular distribution of TRADD and to check if TRADD colocalizes with K8/18 filament networks, HMEC were fixed with 50% methanol/50% acetone and double stained with anti-TRADD polyclonal antibody and anti-K18 monoclonal antibody. TRADD was present in the cytoplasm and showed some filamentous patterns, and there was a significant overlap between filamentous TRADD and K18 (Fig. 4 C, a–c). When we determined if TRADD colocalized with other major cytoskeletal components, e.g., actin filaments and microtubules, double staining showed no apparent codistribution of TRADD with actin filaments or with microtubules (unpublished data). To further confirm the colocalization of TRADD with K18, we transfected HMEC with a K8 deletion mutant to disrupt K8/18 filaments, and then observed the distribution of TRADD (Fig. 4 C, d–f). Cells expressing the myc-tagged K8 deletion mutant (aa 1–239 of K8) showed disorganization of K8/18 filaments (unpublished data) and the disappearance of the filamentous distribution of TRADD. These results confirm the colocalization of TRADD with K8/18 filaments, and suggest that K8/18 filament organization affects the subcellular distribution of TRADD.
Figure 4.


In vivo association of K18 with TRADD. (A) Detection of TRADD in HMEC. Lysates from HMEC were stained with Coomassie brilliant blue (CBB), detected with anti-TRADD antibody by immunoblotting (α-TRADD), or immunostained with anti-TRADD antibody preabsorbed by an immunogen (Absorbed). Anti-TRADD antibody specifically recognized bands of ∼34 kD. Molecular size markers are shown on the left. (B) Interactions of TRADD with K18 in vivo. Immunoprecipitates were prepared from lysates of HMEC, using control rabbit IgG and anti-TRADD polyclonal antibody. Each precipitate, separated into two parts, was subjected to Western blotting with anti-TRADD monoclonal or anti-K18 monoclonal antibody. (C) (a–c) Colocalization of TRADD and K8/18 filaments in HMEC. HMEC were double stained with anti-TRADD antibody (a) and anti-K18 antibody (b). The merged image of a and b is shown (c). (d–f) Effects of the expression of the myc-K8 deletion mutant on the subcellular distribution of TRADD in HMEC. pRK5–myc-K8 deletion mutant vectors were transfected into HMEC. The cells were fixed 16 h later and doubly stained with anti-TRADD antibody (d) and anti-myc antibody (e). The merged image of d and e is shown (f). A transfected cell (arrow) and an untransfected cell (arrowhead) are shown. Bar, 20 μm.
A high dose of TNF treatment leads to release of TRADD from K18
Since TRADD is a TNFR1 adaptor protein recruited to the TNFR1 in a TNF-dependent manner, we next determined if TNF stimulation would affect K18–TRADD association. To observe effects of TNF treatment on the subcellular distribution of TRADD, HMEC were untreated or incubated for 20 min with increasing concentrations of TNF, and then doubly stained for TRADD and K18 (Fig. 5 A). At the physiological concentration of TNF (3 ng/ml), there were no apparent changes in the colocalization of TRADD and K18. At 30 ng/ml of TNF, the filamentous pattern of TRADD distribution began to diminish (unpublished data). At the high dose of TNF (100 ng/ml), TRADD dissociated from K8/18 filaments, and dispersed diffusely (Fig. 5 A). We next checked the binding of TRADD with K18 after TNF treatment by immunoprecipitation (Fig. 5 B). Lysates prepared from HMEC treated for 20 min with various doses of TNF, or left untreated, were immunoprecipitated with anti-TRADD antibody, and the presence of K18 or TNFR1 in the TRADD immune complex was examined. Coprecipitating K18 was readily detected in lysates from untreated (control) cells and physiological dose TNF (3 ng/ml)–treated cells but was barely visible in the 100 ng/ml TNF-treated samples. Immunoprecipitates formed with control rabbit IgG contained a much smaller amount of K18 than those immunoprecipitated with anti-TRADD antibody (Fig. 4 B). The negligible amount of K18 immunoprecipitated by control rabbit IgG did not change with various doses of TNF treatment (unpublished data). In contrast, coprecipitating TNFR1 was barely visible in the case of 3 ng/ml of TNF but was readily visible at 100 ng/ml of TNF. Thus, K18 and TRADD are preassociated in unstimulated conditions. Furthermore, the dissociation of TRADD from K18 and the recruitment of TRADD to activated TNFR1 became obvious in the presence of high concentrations of TNF in HMEC. We then examined the induction of apoptosis after treatment of TNF in HMEC. HMEC were treated for 24 h with increasing concentrations of TNF in the presence of 1 μg/ml cycloheximide (CHX), to inhibit the production of protective proteins, and cell viability was evaluated by Trypan blue staining. Most HMEC were viable after incubation with physiological concentrations (1–3 ng/ml) of TNF, but viable cells markedly decreased in cases of high concentrations (100–300 ng/ml) of TNF (Fig. 5 C). Thus, high concentrations of TNF are necessary for the full induction of apoptosis in HMEC, a notion consistent with the observation that normal and malignant epithelial cells containing K8 and K18 are ∼100 times more insensitive to TNF-induced death compared with the cells deficient in K8 and K18 (Caulin et al., 2000). Taken together, these data show that high concentrations of TNF are necessary for the release of TRADD from K18, the binding of TRADD with activated TNFR1, and full induction of apoptosis. Because TRADD is an indispensable adaptor molecule for TNFR1 signal transmission to downstream cascades, K18 may have an inhibitory effect on TNF-induced apoptosis by competing with activated TNFR1 for binding to TRADD. In TNFR1-induced cell death signaling, FADD couples the TNFR1–TRADD complexes to activation of caspase-8 by recruiting procaspase-8 (also called a FLICE or MACH) to the receptor complex (Boldin et al., 1996; Muzio et al., 1996). The recruitment of procaspase-8 appears to result in its oligomerization, activation of its autoproteolytic activity, and ultimately the generation of enzymatically active caspase-8 (Yeh et al., 1998; Zhang et al., 1998), which in turn cleaves other caspases, thereby initiating apoptosis. Since TNFR1 did not induce apoptosis in FADD-deficient embryonic fibroblasts (Yeh et al., 1998), the TRADD–FADD–caspase-8 complex formation and the activation of caspase-8 are thought to be early and central events in TNFR1-induced apoptosis. The complex formation was first described as the death-inducing signaling complex (DISC) formation in CD95/APO-1/Fas-induced apoptosis (Kischkel et al., 1995; Medema et al., 1997). To further characterize the time course of the TNFR1-induced apoptosis in HMEC, we prepared an affinity-purified rabbit antibody (anti-h8D401 antibody) that recognizes the active form but not the proform of human caspase-8. The anti-h8D401 antibody showed the same specificity as the anti-m8D387 antibody that was raised against a cleavage site of mouse caspase-8 (Kouroku et al., 2000a,b), and did not react with the active form of other caspases (unpublished data). Using the anti-h8D401 antibody, we compared the time courses of the subcellular distribution of TRADD and the appearance of the active form of caspase-8 (Fig. 5 D). 30 min after the treatment of HMEC with 100 ng/ml TNF and 1 μg/ml CHX, TRADD dispersed diffusely as described above, and this diffuse distribution did not change during the observed period (Fig. 5 D, a). The active form of caspase-8 began to be detected in ∼5% of the cells 3 h after treatment (Fig. 5 D, b). Some of the activated caspase-8–positive cells showed the abnormal appearance of a nucleus. These results demonstrated that the activation of caspase-8 occurred after the dissociation of TRADD from K18 in HMEC, which is consistent with our model that K18 may have an inhibitory effect on TNF-induced apoptosis by competing with activated TNFR1 for binding to TRADD.
Figure 5.
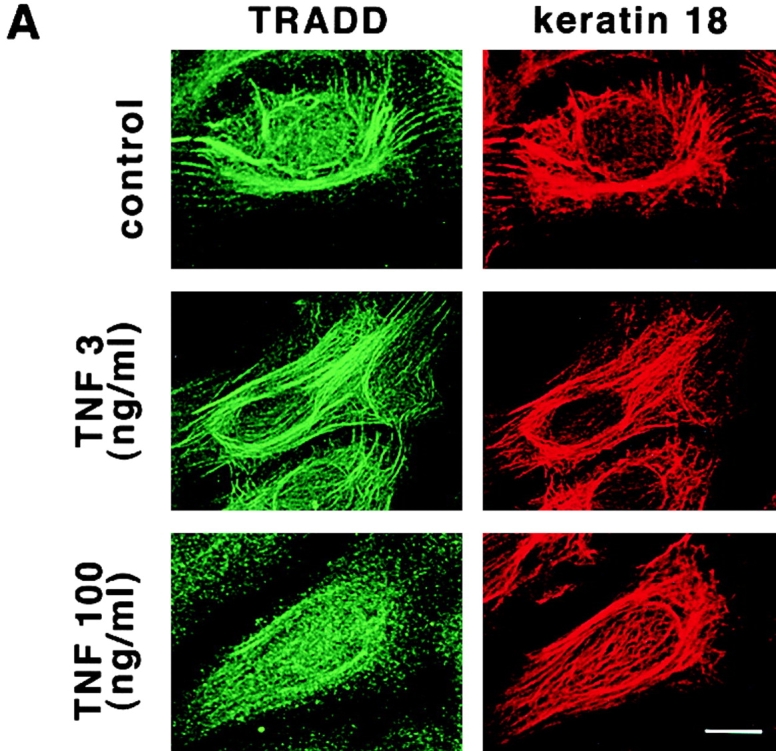


Dissociation of TRADD from K18 after TNF treatment. (A) Subcellular distribution of TRADD after TNF treatment. HMEC were untreated or incubated for 20 min with increasing concentrations of TNF, and then costained for TRADD and K18. (B) TRADD is released from K18 and recruited to TNFR1 after TNF stimulation. Lysates prepared from HME cells treated for 20 min with the indicated doses of TNF were immunoprecipitated with anti-TRADD antibody. Coprecipitating K18 or TNFR1 were detected with anti-K18, or anti-TNFR1 antibody, respectively. (C) Effects of TNF treatment on cell viability. HMEC were treated with various doses of TNF for 12 h in the presence of 1 μg/ml of CHX. After the treatment, cell viability was determined by Trypan blue staining. Experiments were repeated at least three times per condition. Standard error is represented by bars. (D) Time course of the subcellular distribution of TRADD and the appearance of the active form of caspase-8 after TNF treatment in HMEC. HMEC were treated with 100 ng/ml TNF in the presence of 1 μg/ml CHX for 0, 0.5, 1, 3, and 6 h, and stained with anti-TRADD antibody (a) or doubly stained with anti-h8D401 antibody, which recognizes the active form of caspase-8 and propidium iodide (PI) (b). The active form of caspase-8 began to be detected 3 h after the treatment (arrow). Some of activated caspase-8–positive cells showed the abnormal appearance of nuclei (arrowhead). Bars, 10 μm.
Effects of various apoptotic stimuli on the intracellular distribution of TRADD
To search for the role of K18–TRADD interaction in cellular responses to various apoptotic stimuli, we treated HMEC with 1 μM aclarubicin (ACR), a topoisomerase inhibitor, 10 μg/ml neocarzinostatin (NCS), a chemical that directly causes DNA breaks, 1 μM taxol, a microtubule stabilizer, and UV irradiation (100 J/m2). 20 min after treatment with these stimuli, we examined the distribution of TRADD using immunostaining techniques (Fig. 6 A). In contrast to TNF treatment (Fig. 5 A), these stimuli did not alter subcellular distribution of TRADD. We also checked TRADD–K18 binding after these treatments using immunoprecipitation methods (Fig. 6 B). TRADD remained associated with K18 and was not recruited to TNFR1. Immunoprecipitates formed with control rabbit IgG contained a much smaller amount of K18 than those formed with anti-TRADD antibody (Fig. 4 B). The negligible amount of K18 immunoprecipitated by control rabbit IgG did not change after these treatments (unpublished data). 24 h after treatment, we examined the induction of apoptosis in HMEC by counting the number of cells showing apoptotic, condensed nuclei. We found that all these treatments induced marked apoptosis as shown in Fig. 6 C. These results indicate that among various apoptotic stimuli, the release of TRADD from K18 occurs specifically after TNF treatment.
Figure 6.


Other apoptotic stimuli do not affect TRADD–K18 interaction. (A) Effects of various apoptotic stimuli on the subcellular distribution of TRADD. HMEC were incubated for 20 min with 1 μM of ACR, 10 μg/ml of NCS, or 1 μM of taxol, and then costained for TRADD and K18. HMEC were also subjected to immunostaining 20 min after exposure to 100 J/m2 UVC. (B) Various apoptotic stimuli, except TNF, did not alter association of TRADD with K18. Lysates prepared from HMEC treated with various stimuli, as described in A, were immunoprecipitated with anti-TRADD antibody. Coprecipitating K18 or TNFR1 were detected with ant-K18 or anti-TNFR1 antibody, respectively. (C) Induction of apoptosis after the treatment with various apoptotic stimuli. To check for the induction of apoptosis, HMEC were fixed 24 h after treatment, and counted for the presence of condensed chromatin and fragmented nuclei (DAPI stain). Bar, 10 μm.
Overexpressing the NH2 terminus (aa 1–270) of K18 inhibits TNF-induced apoptosis
To further show the involvement of K18 in TNF-induced cell death in HMEC, we examined the effects of overexpressing either K8/18 or the NH2 terminus of K18 (aa 1–270), which contains the TRADD binding site, on TNF-induced apoptosis. We transiently transfected Epstein-Barr virus–based vectors into cells expressing the trans-acting Epstein-Barr virus–associated nuclear antigen (EBNA)-1 gene to obtain extremely high transformation efficiency as well as high expression level of transferred genes (Yasui et al., 1998). We first obtained EBNA-expressing SW13 cells, which lack endogenous keratin and vimentin. These EBNA-expressing SW13 cells were then transiently transfected with pDR2 vectors expressing myc-tagged K8 and K18, myc-tagged NH2 terminus of K18 (aa 1–270), or vimentin, using lipofection. 24 h after the transfection, the cells were treated with 1 ng/ml TNF in the presence of 1 μg/ml CHX or incubated only with 1 μg/ml CHX. After another 24 h, the cells were stained with anti-myc antibody or antivimentin antibody to detect the transfected cells, and with DAPI to observe nuclei. To determine the percentage of viable cells, the number of transfected cells showing no apoptotic nuclei (condensed chromatin and fragmented nuclei) was determined, and cell viability was expressed as a ratio of the percentage of viable cells in the presence of TNF to those in the absence of TNF. As shown in Fig. 7 A, cells expressing the NH2 terminus of K18 as well as those expressing K8/18 were more resistant to TNF-induced cytotoxicity than the cells transfected with control vectors or vimentin expression vectors. We also checked the expression of TNFR1 and TRADD in EBNA-expressing SW13 cells by immunoblotting. The expression of endogenous TNFR1 and TRADD was detected in EBNA-expressing SW13 cells (Fig. 7 B) at a level almost comparable to that found in parent SW13 cells and HMEC (unpublished data). To confirm that the resistance to TNF-induced apoptosis was caused by the association of TRADD with overexpressed K8/18 or the NH2 terminus of K18, we observed the subcellular localization of endogenous TRADD and overexpressed K8/18 or NH2 terminus of K18 in the SW13 cells. In untransfected SW13 cells, which are devoid of keratins, native TRADD was diffusely present in the cytoplasm (Fig. 7 C, a). In K8/18–transfected SW13 cells, TRADD was recruited to overexpressed K8/18 filaments (Fig. 7 C, d–f). In cells overexpressing the NH2 terminus of K18, TRADD markedly colocalized with the overexpressed NH2 terminus (Fig. 7 C, g–i). These results indicate that overexpressed K8/18 or K18 NH2 terminus could sequester endogenous TRADD, leading to the inhibition of TNF-induced apoptosis in SW13 cells. We then determined if overexpressing K8/18 or the NH2 terminus of K18 could affect the activation of caspase-8, an early and central event of TNFR1-induced cell death signaling as described earlier (Fig. 7 D). Using anti-h8D401 antibody, which recognizes the active form but not the proform of human caspase-8, the activation of caspase-8 began to be detected in ∼6% of SW13 cells 4 h after the treatment with 1 ng/ml TNF in the presence of 1 μg/ml CHX. 24 h after the transfection, the cells were treated with 1 ng/ml TNF in the presence of 1 μg/ml CHX for 4 h, and then fixed and doubly stained with anti-myc antibody or antivimentin antibody, to detect the transfected cells, and with anti-h8D401 antibody, to detect the activation of caspase-8. As shown in Fig. 7 D, cells overexpressing the NH2 terminus of K18 as well as those overexpressing K8/18 showed less positive staining for the active form of caspase-8 than the cells transfected with control vectors or vimentin expression vectors. After the treatment with 1 μg/ml CHX only, <0.5% of both SW13 cells and cells transfected with these vectors showed positive staining for activated caspase-8 (unpublished data). These results indicate that K18 could affect TNF-induced death signaling in a step upstream of caspase-8 activation, which strongly supports our model that K18 sequesters TRADD and inhibits the recruitment of TRADD to activated TNFR1, leading to the inhibition of DISC formation (TRADD–FADD–caspase-8) and caspase-8 activation. Next, we asked if the overexpression of the NH2 terminus of K18 or K8/18 only protect cells from TNF-induced apoptosis. To this end, 24 h after the transfection, in the same fashion as described above, the cells were left untreated or were treated with 0.3 μM ACR, 1 μg/ml NCS, 1 μM taxol, or UV irradiation (30 J/m2). After another 24 h, the cells were fixed and the percentages of viable cells were recorded (Fig. 7 E). We observed no effects from the overexpression of K8/18, the NH2 terminus of K18, or vimentin on the survival rates of cells after treatment with these various apoptotic stimuli. These results demonstrate that K8/18 protects the cell specifically from TNF cytotoxicity, as reported by Oshima's group (Caulin et al., 2000). Furthermore, these results support our model that K18 may exert antiapoptotic functions via association with TRADD.
Figure 7.



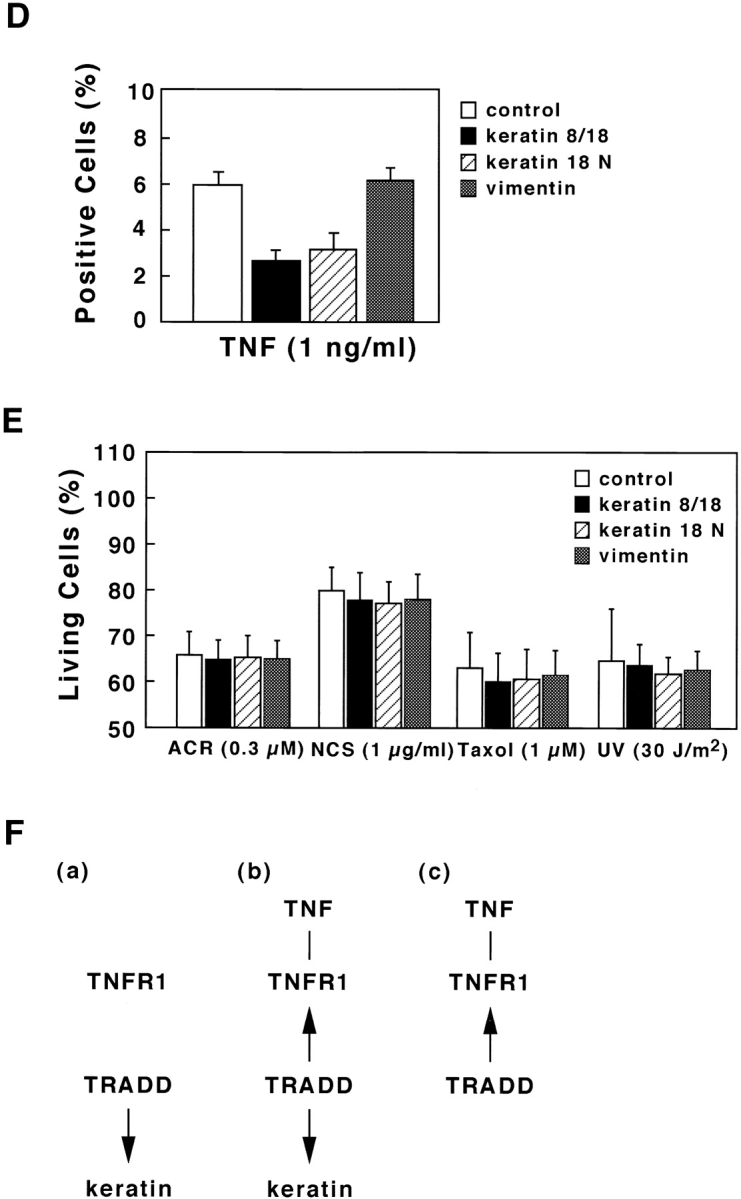
Overexpression of the NH 2 terminus of K18 inhibits TNF-induced apoptosis. (A) SW13 cells expressing K8/18 and the NH2 terminus (aa 1–270) of K18 exhibit decreased sensitivity to TNF-induced apoptosis compared with cells transfected with control vectors or vimentin expression vectors. The EBNA-expressing SW13 cells were transiently transfected with pDR2 vectors expressing myc-tagged K8 and K18 (K8/18), myc-tagged NH2 terminus (residues 1–270) of K18 (K18 N), or vimentin, using lipofectamine. 24 h after the transfection, the cells were treated with 1 ng/ml TNF in the presence of 1 μg/ml of CHX or only incubated with 1 μg/ml of CHX. After another 24 h, the cells were stained with anti-myc antibody or antivimentin antibody, to detect the transfected cells, and with DAPI, to see nuclei. To obtain the percentage of living cells, the number of transfected cells showing no apoptotic nuclei (condensed chromatin and fragmented nuclei) was determined, and cell viability was expressed as a ratio of percentage of viable cells in the presence of TNF to those in the absence of TNF. Experiments were repeated at least three times per condition. Standard error is represented by bars. (B) Expression of TNFR1 and TRADD in EBNA-expressing SW13 cells. Lysates prepared from EBNA-expressing SW13 cells were subjected to immunoblotting with anti-TNFR1 antibody (lane 1) or anti-TRADD antibody (lane 2). The arrowhead and arrow indicate positions of TNFR1 and TRADD, respectively. Molecular size markers are shown on the left. (C) Subcellular distribution of endogenous TRADD and overexpressed myc-tagged K8/18 or NH2 terminus of K18 (K18 N) in SW13 cells. The untransfected, EBNA-expressing SW13 cells were doubly stained with anti-TRADD antibody (a) and anti-myc antibody (b). The myc-tagged K8/18–transfected cells were doubly stained with anti-TRADD antibody (d) and anti-myc antibody (e). The cells expressing myc-tagged K18 N were doubly stained with anti-TRADD antibody (g) and anti-myc antibody (h). The merged images of a and b (c), d and e (f), and g and h (i) are shown. (D) Overexpression of K8/18 or the NH2 terminus of K18 (K18 N) inhibits the activation of caspase-8 in SW13 cells. The EBNA-expressing SW13 cells were transiently transfected as in A. 24 h after the transfection, the cells were treated with 1 ng/ml TNF in the presence of 1 μg/ml CHX for 4 h, and then doubly stained with anti-myc antibody or a ntivimentin antibody, to detect the transfected cells, and with anti-h8D401 antibody, which recognizes the active form of caspase-8. The percentages of transfected cells showing positive staining for the active form of caspase-8 were determined. Experiments were repeated at least three times per condition. Standard error is represented by bars. (E) K18 has cell-protective effects specifically on TNF cytotoxicity. 24 h after the transfection, in the same fashion as described in A, the cells were left untreated or treated with 0.3 μM of ACR, 1 μg/ml of NCS, 1 μM of taxol, or 30 J/m2 of UVC irradiation. After another 24 h, the cells were fixed and the percentages of living cells were determined as in A. (F) Models for K18-dependent moderation of TNF–TNFR1 signaling in epithelial cells. TRADD is associated with K18, but not with TNFR1 in unstimulated conditions (a). Upon TNF ligation, the binding of TRADD to K18 may serve as a negative regulatory mechanism, governing the rate of TRADD association with activated TNFR1 (b). Lack or dysfunction of K18 may free TRADD to facilitate the association of TRADD with activated TNFR1 (c). Bar, 10 μm.
Discussion
The recent study by Oshima's group (Caulin et al., 2000) provided evidence for K8/18's key role in moderating the signaling of TNF. They found that decreasing levels of K8 and K18 in cultured epithelial cells increases cellular sensitivity to killing by TNF due to binding of both K8 and K18 with the cytoplasmic domain of TNFR2. They also demonstrated that K8 and K18 diminish the TNF-dependent activation of Jun NH2-terminal kinase (JNK) and the NF-κB transcriptional factor. Using an in vivo model, these same authors found that mice without K8 or K18 are more sensitive to TNF-mediated apoptotic liver damage (Caulin et al., 2000). Here we uncovered an alternative molecular mechanism by which epithelial cells can be resistant to TNF-induced cytotoxicity. Our data obtained in vitro and in vivo provide support for the notion of a direct association of K18 with TRADD, an indispensable adaptor molecule for TNFR1 signaling. Thus, K18 may sequester TRADD to attenuate the interaction of TRADD with activated TNFR1, leading to diminution of TNF-induced apoptosis.
In HMEC, in unstimulated conditions, TRADD is associated with K18, but not with TNFR1 (Fig. 5 and Fig. 7 F, a). Treatment with high concentrations of TNF stimuli resulted in dissociation of TRADD from K18, the recruitment of TRADD to TNFR1, and the full induction of apoptosis, whereas treatment with the physiological concentration of TNF did not lead to a release of TRADD from K18 or marked induction of apoptosis (Fig. 5). These observations suggest that upon TNF ligation, the binding of TRADD to K18 may serve as a negative regulatory mechanism, governing the rate of TRADD association with activated TNFR1 (Fig. 7 F, b). Lack or dysfunction of K18 may free TRADD to facilitate the association of TRADD with activated TNFR1 (Fig. 7 F, c). As shown in Fig. 7 A, B, and E, we also found that overexpression of K8/18 and the NH2-terminus (aa 1–270) of K18, which contains the TRADD binding domain, could specifically inhibit TNF-induced apoptosis. We further showed that overexpressed K8/18 or NH2-terminal fragment of K18 could sequester endogenous TRADD in SW13 cells (Fig. 7 C), and that K18 could affect TNF-induced cell death signaling in a step upstream of caspase-8 activation (Fig. 7 D). These results suggest a molecular mechanism where K18 sequesters TRADD and affects the recruitment of TRADD to activated TNFR1, leading to the inhibition of the formation of DISC (TRADD–FADD–caspase-8) and the activation of caspase-8. Our data, described above, clearly show that K18 could attenuate TNF-induced cell death through association with TRADD in at least some kinds of simple epithelial cells. Since effects of TNF are complex and vary in cells of different lineage and differentiation stage, it is necessary to investigate roles of K18–TRADD interactions in simple epithelial cells of various lineage and differentiation stages in future research.
The sequestering of TRADD by K18 seems reminiscent of the sequestering of Smads by microtubules (Dong et al., 2000). In the TGFβ pathway, microtubules serve as a cytoplasmic sequestering network for Smads in unstimulated cells. TGFβ triggers dissociation of Smads from microtubules, and phosphorylation and nuclear localization of Smad2 and 3 with consequent activation of transcription inside the nucleus (Dong et al., 2000). In addition, it was reported that MIP-T3 sequesters TNFR-associated factor 3 to microtubules (Ling and Goeddel, 2000). In this context, it was discovered that depolymerization of microtubules activates NF-κB, findings that suggest a role for NF-κB in sensing changes in the state of the cytoskeleton and converting them to changes in gene activity (Rosette and Karin, 1995). Taken together, these observations suggest that the sequestering of adaptor molecules for receptors by cytoskeletal proteins may represent a general mechanism by which cytoskeletal proteins affect cellular signaling and thereby transmit morphology-related information.
In a two-hybrid system, TRADD specifically bound K18 and K14, type I (acidic) keratins, but did not bind type II keratins, including K5 and K8, or type III IFs, including vimentin, GFAP, and desmin (Fig. 1 B). K14 is preferentially expressed in the basal layer of epidermis, and in the same fashion as K18, it is possible that epidermal K14 may function as an inhibitor of TNF–TNFR1 signaling through an association with TRADD. Accumulating evidence, including data on mice that are null for the molecules involved in NF-κB signaling, suggests that NF-κB plays a particularly central role in epidermal biology (Kaufman and Fuchs, 2000). By linking cell cycle withdrawal and protection from apoptosis, NF-κB can coordinate and monitor both homeostasis within the epidermis as well as transition of the epidermal cell from basal to suprabasal layers (Kaufman and Fuchs, 2000). Our present data shed new light on a role of keratins in the regulation of NF-κB signaling in the skin.
By way of summary, we found that K18 directly interacts with TRADD, and we postulate that K18 may suppress TNF–TNFR1-induced apoptosis by associating with TRADD. These observations pave the way for ongoing research on the dissection of cross talk between keratin IF and intracellular signaling as well as for a better understanding of the biology of cancer cells transformed from simple epithelial cells.
Materials and methods
Cell culture and reagents
HeLa cells, human bladder cell carcinoma T24 cells, and MDCK cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and penicillin in an air-5% CO2 atmosphere with constant humidity. HMEC were immortalized by retroviral gene transfer of a cDNA encoding the catalytic component of human telomerase (pLXSN-fhTERT), which was provided by Dr. D.A. Galloway (Fred Hutchinson Cancer Research Center, Seattle, WA), and grown as described (Kiyono et al., 1998). The human adenocarcinoma cell line SW13, which is devoid of both keratins and vimentin, was grown as described (Sarria et al., 1990). TNFα, CHX, and taxol were obtained from Sigma-Aldrich. zVAD-fmk was from Promega. ACR and NCS were gifts from R. Ishida (Aichi Cancer Center Research Institute).
Yeast two-hybrid screening
cDNA encoding the full-length K8 or K18, provided by H. Eto (Massachusetts General Hospital, Boston, MA) was cloned into the yeast GAL4 DNA–binding domain vector pYTH9α as described (Izawa et al., 2000). The resulting plasmids, pYTH9α-K8 and -K18 were used in the two-hybrid screen of a human liver cDNA library fused to pACT2 vector (CLONTECH Laboratories, Inc.), following the Matchmaker Two-Hybrid System Protocol (CLONTECH Laboratories, Inc.). Positive clones were screened for their potential to grow on selective medium containing 25 mM 3-aminotriazole and for the expression of β-galactosidase. Subsequent two-hybrid interaction analyses were performed by cotransformation of plasmids, provided by P. James (The University of Wisconsin, Madison, WI), containing the GAL4 DNA–binding (pGBD-C1) and –activation (pGAD-C1) domains into Saccharomyces cerevisiae strain Y190. K5 and K14 cDNAs, gifts from E. Fuchs (The University of Chicago, Chicago, IL), were cloned into pGBD-C1 vectors. K8, K18, vimentin, GFAP, and desmin cDNAs were also cloned into pGBD-C1 plasmids. A series of truncations of K18 were constructed into the pGBD-C1 plasmid by means of enzyme digestion and use of PCR techniques.
Cloning of full-length TRADD and DNA constructs
The full-length cDNA of human TRADD was amplified using a human liver cDNA library (CLONTECH Laboratories, Inc.), PyroBest polymerase (Takara), and a set of primers designed according to the human TRADD cDNA sequence (Hsu et al., 1995). Various TRADD fragments, including TRADD-N (aa 1–174), and TRADD-C (aa 245–312), were also produced by PCR. Full-length TRADD and TRADD-C were cloned into yeast pGAD-C1 vectors. Sequences of these constructs were verified by DNA sequencing.
Purification of recombinant proteins
Recombinant TRADD-N protein was expressed as six consecutive His6-tagged proteins, using pQE30 vectors (QIAGEN). Expression and purification of the His6–TRADD-N protein were performed according to the manufacturer's protocol (QIAGEN). Full-length TRADD and TRADD-N were also expressed as GST fusion proteins in Escherichia coli and purified, essentially as described (Smith and Johnson, 1988).
Filament assembly and cosedimentation assay
IF assembly in vitro was performed as described (Inagaki et al., 1987; Yano et al., 1991). Briefly, purified K8 and K18 were dissolved at a 1:1 (mol/mol) ratio in 10 mM Tris/HCl (pH 8.8), containing 50 mM 2-mercaptoethanol, 2 mM EGTA, 1 mM PMSF and 7 M urea. The mixture in 7 M urea solution was dialyzed against a buffer of 10 mM Tris/HCl (pH 8.8), containing 50 mM 2-mercaptoethanol, 2 mM EGTA, and 1 mM PMSF, for 24 h at 4°C. These heterotypic complexes of K8 and K18 (0.5 mg/ml) were incubated, with or without GST or GST–TRADD in a buffer of 20 mM imidazole (pH 7.0), containing 1 mM MgCl2 at 25°C for 1 h. Purified vimentin or desmin was assembled in the presence or absence of GST–TRADD in a buffer of 25 mM Tris/HCl (pH 7.5), containing 50 mM NaCl, at 25°C for 1 h. The reassembled complexes were subjected to centrifugation at 15 krpm for 30 min. The supernatants and precipitates were then analyzed using SDS-PAGE.
Generation of anti-TRADD antibody and immunoblotting
Anti-TRADD antibody was produced in rabbits injected with recombinant His6–TRADD-N protein, and affinity purified with recombinant GST–TRADD-N fusion protein. Lysates prepared from HMEC were separated by SDS-PAGE, and transferred onto a polyvinylidene difluoride membrane (Atto). The blots were incubated with anti-TRADD antibody and a horseradish peroxidase–conjugated second antibody, and immunoreactive bands were visualized using chemiluminescence detection reagents (Renaissance; NEN Life Science Products).
Preparation of antiserum against cleavage site of caspase-8
Recently, we prepared antiserum against a cleavage site of mouse caspase-8 (TLEVD387, anti-m8D387) (Kouroku et al., 2000a). Antiserum against a cleavage site of human caspase-8 at D401 was prepared basically as previously described (Kouroku et al., 1998). In brief, CYLEMD401, a peptide corresponding to a COOH-terminal processing site (D401) of human caspase-8 and cysteine, was synthesized (Sawady Technology Co., LTD.). Antiserum against YLEMD (anti-h8D401) was generated by injecting CYLEMD conjugated to keyhole limpet hemocyanin (KLH) into rabbits. Anti-h8D401 antibody was purified by YLEMD-peptide affinity column chromatography.
Mammalian expression vectors and transfection
A cDNA–encoding full-length TRADD was cloned into the mammalian expression vector pRK5-myc for expression of myc epitope–tagged proteins. cDNAs encoding full-length mouse FADD, mouse caspase-2 (Nedd2), and human caspase-8 were provided by H. Ichijo (Tokyo Medical and Dental University, Tokyo, Japan), M. Noda (Kyoto University, Kyoto, Japan), and S. Yonehara (Kyoto University, Kyoto, Japan), respectively, and were cloned into pCMV-Tag3 vectors (Stratagene) for expression of myc epitope–tagged proteins. A cDNA encoding K8 deletion mutant (aa 1–239) was also cloned into the pRK5-myc vector. Cells were seeded onto 13-mm glass coverslips in six-well dishes the day before lipofection with 1 μg of each plasmid, using LipofectAMINE PLUS or 2000 (GIBCO BRL) according to the manufacturer's protocols. 16 h after lipofection, the cells were fixed for immunofluorescence studies. SW13 cells were cotransfected with pCMVEBNA and pSV2neo plasmids (CLONTECH Laboratories, Inc.) using LipofectAMINE PLUS as described (Yasui et al., 1998). Cells expressing pCMVEBNA were cloned and maintained in the presence of G418 sulfate (GIBCO BRL). The EBNA-expressing SW13 cells were transiently transfected with myc-tagged K8 and K18, myc-tagged NH2-terminus of K18 (aa 1–270), or vimentin cDNAs in pDR2 plasmids, using LipofectAMINE PLUS, and then were treated with various cytotoxic stimuli 24 h after the transfection.
Immunoprecipitation
Cells were lysed on ice for 20 min in lysis buffer consisting of 1% NP-40, 20 mM Tris/HCl (pH 7.5), 50 mM NaCl, 1 mM EDTA, 10 μM PMSF, and 10 μg/ml leupeptin. Lysates, clarified by centrifugation at 15 krpm for 30 min, were then incubated with anti-TRADD antibody or control rabbit IgG, and immunocomplexes were immobilized on protein A agarose beads (Sigma-Aldrich). Immunoprecipitates were analyzed by immunoblotting with anti-TRADD monoclonal antibody (Transduction Laboratory), anti-K18 monoclonal antibody (CY-90; Sigma-Aldrich), or anti-TNFR1 monoclonal antibody (Santa Cruz Biotechnology, Inc.).
Immunofluorescence
Cells grown on 13-mm coverslips were fixed by incubation for 10 min in 50% methanol/50% acetone (vol/vol) at -20°C. For double staining for TRADD and K18, cells were first incubated with anti-TRADD polyclonal antibody followed by Alexa 488-labeled anti–rabbit antibody (Molecular Probes). Next, the cells were incubated with anti-K18 antibody (CY-90; Sigma-Aldrich) followed by FluoroLink Cy3–linked anti–mouse antibody (Amersham Pharmacia Biotech). To visualize myc tags, cells were reacted with anti-myc monoclonal antibody (9E10) or anti-myc polyclonal antibody (Medical and Biological Laboratories Co., Ltd.), followed by FluoroLink Cy3–linked anti–mouse antibody or Alexa 488–labeled anti–rabbit antibody, respectively. The coverslips were examined on an Olympus LSM-GB200 microscope.
UV irradiation and apoptosis assay
Cells were irradiated with UVC for 77 s (HMEC) or 23 s (EBNA-expressing SW13 cells) after removal of medium and one wash with PBS, using a UVC (254 nm) germicidal lamp, 1.3 J/m2/s. The viability of cells obtained under the different conditions was evaluated by Trypan blue staining. Nuclear morphologic changes were evaluated by DAPI staining, and the number of cells with no apoptotic nuclei (condensed chromatin and fragmented nuclei) was determined and the percentages of living cells were recorded.
Acknowledgments
We thank E. Fuchs for providing pET8c-K5 and -K14 plasmids, H. Eto for providing pGEX-2TH-K8 and -K18 plasmids, D.A. Galloway for providing a retroviral vector harboring a cDNA encoding the catalytic component of human telomerase (pLXSN-fhTERT), P. James for providing pGBD-C1 and pGAD-C1 plasmids, M. Noda for providing mouse Nedd2 cDNA, S. Yonehara for suggestions and providing human caspase-8 cDNA, H. Ichijo for helpful discussion and providing mouse FADD cDNA, R. Ishida for providing ACR and NCS, K. Ishizaki from Aichi Cancer Center Research Institute for advice on UV irradiation, and K. Nagata and Y. Yasui in our laboratory for helpful discussions and technical assistance. We are grateful to M. Ohara for the critique of the manuscript.
This research was supported in part by Grants-in-Aid for Scientific Research and Cancer Research from the Ministry of Education, Science, Technology, Sports, and Culture of Japan; by the Japan Society of the Promotion of Science Research for the Future; by a grant-in-aid for the Second Term Comprehensive 10-Year Strategy for Cancer Control from the Ministry of Health and Welfare, Japan; and by a Research Grant of the Princess Takamatsu Cancer Research Foundation.
H. Inada, I. Izawa, and M. Nishizawa contributed equally to this work.
Footnotes
Abbreviations used in this paper: aa, amino acid(s); ACR, aclarubicin; CHX, cycloheximide; DISC, death-inducing signaling complex; EBNA, Epstein-Barr virus–associated nuclear antigen; FADD, Fas-associated death domain; GFAP, glial fibrillary acidic protein; GST, glutathione S-transferase; His, histidine; HMEC, human mammary epithelial cells; IF, intermediate filament; K8/18, keratin 8 and 18; NCS, neocarzinostatin; NF-κB, nuclear factor-κB; TNF, tumor necrosis factor; TNFR, TNF receptor; TRADD, TNFR1-associated death domain protein.
References
- Banner, D.W., A. D'Arcy, W. Janes, R. Gentz, H.-J. Schoenfeld, C. Broger, H. Loetscher, and W. Lesslauer. 1993. Crystal structure of the soluble human 55 kd TNF receptor-human TNFβ complex: implications for TNF receptor activation. Cell. 73:431–445. [DOI] [PubMed] [Google Scholar]
- Baribault, H., J. Price, K. Miyai, and R.G. Oshima. 1993. Mid-gestational lethality in mice lacking keratin 8. Genes Dev. 7:1191–1202. [DOI] [PubMed] [Google Scholar]
- Baribault, H., J. Penner, R.V. Iozzo, and M. Wilson-Heiner. 1994. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 8:2964–2973. [DOI] [PubMed] [Google Scholar]
- Boldin, M.P., T.M. Goncharov, Y.V. Goltsev, and D. Wallach. 1996. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 85:803–815. [DOI] [PubMed] [Google Scholar]
- Caulin, C., C.F. Ware, T.M. Magin, and R.G. Oshima. 2000. Keratin-dependent, epithelial resistance to tumor necrosis factor-induced apoptosis. J. Cell Biol. 149:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colussi, P.A., N.L. Harvey, and S. Kumar. 1998. Prodomain-dependent nuclear localization of the caspase-2 (Nedd2) precursor. A novel function for a caspase prodomain. J. Biol. Chem. 273:24535–24542. [DOI] [PubMed] [Google Scholar]
- Dong, C., Z. Li, R. Alvarez, Jr., X.-H. Feng, and P.J. Goldschmidt-Clermont. 2000. Microtubule binding to Smads may regulate TGFβ activity. Mol. Cell. 5:27–34. [DOI] [PubMed] [Google Scholar]
- Franke, W.W. 1987. Nuclear lamins and cytoplasmic intermediate filament proteins: a growing multigene family. Cell. 48:3–4. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and K. Weber. 1994. Intermediate filaments: structure, dynamics, function, and disease. Annu. Rev. Biochem. 63:345–382. [DOI] [PubMed] [Google Scholar]
- Fuchs, E., and D.W. Cleveland. 1998. A structural scaffolding of intermediate filaments in health and disease. Science. 279:514–519. [DOI] [PubMed] [Google Scholar]
- Guiet, C., and P. Vito. 2000. Caspase recruitment domain (CARD)–dependent cytoplasmic filaments mediate bcl10-induced NF-κB activation. J. Cell Biol. 148:1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, H., J. Xiong, and D.V. Goeddel. 1995. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell. 81:495–504. [DOI] [PubMed] [Google Scholar]
- Hsu, H., H.-B. Shu, M.-G. Pan, and D.V. Goeddel. 1996. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 84:299–308. [DOI] [PubMed] [Google Scholar]
- Inagaki, M., Y. Nishi, K. Nishizawa, M. Matsuyama, and C. Sato. 1987. Site-specific phosphorylation induces disassembly of vimentin filaments in vitro. Nature. 328:649–652. [DOI] [PubMed] [Google Scholar]
- Izawa, I., M. Nishizawa, K. Ohtakara, K. Ohtsuka, H. Inada, and M. Inagaki. 2000. Identification of Mrj, a DnaJ/Hsp40 family protein, as a keratin 8/18 filament regulatory protein. J. Biol. Chem. 275:34521–34527. [DOI] [PubMed] [Google Scholar]
- Kaufman, C.K., and E. Fuchs. 2000. It's got you covered: NF-κB in the epidermis. J. Cell Biol. 149:999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelliher, M.A., S. Grimm, Y. Ishida, F. Kuo, B.Z. Stanger, and P. Leder. 1998. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity. 8:297–303. [DOI] [PubMed] [Google Scholar]
- Kischkel, F.C., S. Hellbardt, I. Behrmann, M. Germer, M. Pawlita, P.H. Krammer, and M.E. Peter. 1995. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 14:5579–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyono, T., S.A. Foster, J.I. Koop, J.K. McDougall, D.A. Galloway, and A.J. Klingelhutz. 1998. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 396:84–88. [DOI] [PubMed] [Google Scholar]
- Kouroku, Y., K. Urase, E. Fujita, K. Isahara, Y. Ohsawa, Y. Uchiyama, M.Y. Momoi, and T. Momoi. 1998. Detection of activated caspase-3 by a cleavage site-directed antiserum during naturally occurring DRG neurons apoptosis. Biochem. Biophys. Res. Commun. 247:780–784. [DOI] [PubMed] [Google Scholar]
- Kouroku, Y., E. Fujita, A. Jimbo, T. Mukasa, T. Tsuru, M.Y. Momoi, and T. Momoi. 2000. a. Localization of active form of caspase-8 in mouse L929 cells induced by TNF treatment and polyglutamine aggregates. Biochem. Biophys. Res. Commun. 270:972–977. [DOI] [PubMed] [Google Scholar]
- Kouroku, Y., E. Fujita, K. Urase, T. Tsuru, R. Setsuie, T. Kikuchi, Y. Yagi, M.Y. Momoi, and T. Momoi. 2000. b. Caspases that are activated during generation of nuclear polyglutamine aggregates are necessary for DNA fragmentation but not sufficient for cell death. J. Neurosci. Res. 62:547–556. [DOI] [PubMed] [Google Scholar]
- Ku, N.-O., T.L. Wright, N.A. Terrault, R. Gish, and M.B. Omary. 1997. Mutation of human keratin 18 in association with cryptogenic cirrhosis. J. Clin. Invest. 99:19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, L., and D.V. Goeddel. 2000. MIP-T3, a novel protein linking tumor necrosis factor receptor-associated factor 3 to the microtubule network. J. Biol. Chem. 275:23852–23860. [DOI] [PubMed] [Google Scholar]
- Liu, Z.-G., H. Hsu, D.V. Goeddel, and M. Karin. 1996. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 87:565–576. [DOI] [PubMed] [Google Scholar]
- Magin, T.M., R. Schröder, S. Leitgeb, F. Wanninger, K. Zatloukal, C. Grund, and D.W. Melton. 1998. Lessons from keratin 18 knockout mice: formation of novel keratin filaments, secondary loss of keratin 7 and accumulation of liver-specific keratin 8–positive aggregates. J. Cell Biol. 140:1441–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema, J.P., C. Scaffidi, F.C. Kischkel, A. Shevchenko, M. Mann, P.H. Krammer, and M.E. Peter. 1997. FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 16:2794–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll, R., W.W. Franke, D.L. Schiller, B. Geiger, and R. Krepler. 1982. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 31:11–24. [DOI] [PubMed] [Google Scholar]
- Muzio, M., A.M. Chinnaiyan, F.C. Kischkel, K. O'Rourke, A. Shevchenko, J. Ni, C. Scaffidi, J.D. Bretz, M. Zhang, R. Gentz, et al. 1996. FLICE, a novel FADD-homologus ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell. 85:817–827. [DOI] [PubMed] [Google Scholar]
- Omary, M.B., and N.-O. Ku. 1997. Intermediate filament proteins of the liver: emerging disease association and functions. Hepatology. 25:1043–1048. [DOI] [PubMed] [Google Scholar]
- Oshima, R.G., H. Baribault, and C. Caulín. 1996. Oncogenic regulation and function of keratins 8 and 18. Cancer Metastasis Rev. 15:445–471. [DOI] [PubMed] [Google Scholar]
- Perez, D., and E. White. 1998. E1B 19K inhibits Fas-mediated apoptosis through FADD-dependent sequestration of FLICE. J. Cell Biol. 141:1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosette, C., and M. Karin. 1995. Cytoskeletal control of gene expression: depolymerization of microtubules activates NF-κB. J. Cell Biol. 128:1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarria, A.J., S.K. Nordeen, and R.M. Evans. 1990. Regulated expression of vimentin cDNA in cells in the presence and absence of a preexisting vimentin filament network. J. Cell Biol. 111:553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, R.M., D.A. Martin, L. Zheng, S.Y. Ng, J. Bertin, J. Cohen, and M.J. Lenardo. 1998. Death effector filaments: novel cytoplasmic structures that recruit caspases and trigger apoptosis. J. Cell Biol. 141:1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, D.B., and K.S. Johnson. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 67:31–40. [DOI] [PubMed] [Google Scholar]
- Yano, T., T. Tokui, Y. Nishi, K. Nishizawa, M. Shibata, K. Kikuchi, S. Tsuiki, T. Yamauchi, and M. Inagaki. 1991. Phosphorylation of keratin intermediate filaments by protein kinase C, by calmodulin-dependent protein kinase and by cAMP-dependent protein kinase. Eur. J. Biochem. 197:281–290. [DOI] [PubMed] [Google Scholar]
- Yasui, Y., M. Amano, K.-I. Nagata, N. Inagaki, H. Nakamura, H. Saya, K. Kaibuchi, and M. Inagaki. 1998. Roles of Rho-associated kinase in cytokinesis: mutations in Rho- associated kinase phosphorylation sites impair cytokinetic segregation of glial filaments. J. Cell Biol. 143:1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh, W.-C., A. Shahinian, D. Speiser, J. Kraunus, F. Billia, A. Wakeham, J.L. de la Pompa, D. Ferrick, B. Hum, N. Iscove, et al. 1997. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 7:715–725. [DOI] [PubMed] [Google Scholar]
- Yeh, W.-C., J.L. de la Pompa, M.E. McCurrach, H.-B. Shu, A.J. Elia, A. Shahinian, M. Ng, A. Wakeham, W. Khoo, K. Mitchell, et al. 1998. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 279:1954–1958. [DOI] [PubMed] [Google Scholar]
- Zhang, J., D. Cado, A. Chen, N.H. Karba, and A. Winoto. 1998. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 392:296–300. [DOI] [PubMed] [Google Scholar]