

Abstract
Proteins destined for the secretory pathway must first fold and assemble in the lumen of endoplasmic reticulum (ER). The pathway maintains a quality control mechanism to assure that aberrantly processed proteins are not delivered to their sites of function. As part of this mechanism, misfolded proteins are returned to the cytosol via the ER protein translocation pore where they are ubiquitinated and degraded by the 26S proteasome. Previously, little was known regarding the recognition and targeting of proteins before degradation. By tracking the fate of several mutant proteins subject to quality control, we demonstrate the existence of two distinct sorting mechanisms. In the ER, substrates are either sorted for retention in the ER or are transported to the Golgi apparatus via COPII–coated vesicles. Proteins transported to the Golgi are retrieved to the ER via the retrograde transport system. Ultimately, both retained and retrieved proteins converge at a common machinery at the ER for degradation. Furthermore, we report the identification of a gene playing a novel role specific to the retrieval pathway. The gene, BST1, is required for the transport of misfolded proteins to the Golgi, although dispensable for the transport of many normal cargo proteins.
Keywords: endoplasmic reticulum; vesicular transport; protein degradation; misfolded proteins; glycosylation
Introduction
Proteins destined for the secretory pathway first pass through the membranes of the endoplasmic reticulum (ER)*. To enter the lumen, they traverse a proteinaceous pore termed the “translocon” (Johnson and van Waes, 1999). Nascent soluble proteins are released into the lumen, whereas membrane proteins are integrated into the ER membrane. Since these proteins are translocated in an unfolded state, assembly into their native conformations occurs as a subsequent step in the ER. For this, the organelle maintains an inventory of raw materials, enzymes, and chaperones needed for proper protein folding and modification. Due to the localized nature of these functions, a mechanism termed “ER quality control” prevents transport of newly synthesized polypeptides to their sites of function until they reach their native conformation (Ellgaard et al., 1999).
The quality control mechanism also plays important roles when proteins fail to fold. Misfolded proteins are directed to a degradative pathway termed ER-associated protein degradation (ERAD) (Sommer and Wolf, 1997; Brodsky and McCracken, 1999). In this pathway, degradation does not occur in the lumen of the ER. Instead, proteins are transported back to the cytosol via the same translocon complex used for import (Wiertz et al., 1996; Pilon et al., 1997; Plemper et al., 1997; Zhou and Schekman, 1999). The process, termed retrotranslocation or dislocation, is usually coupled to ubiquitination, a requisite covalent modification of the substrate for degradation (Biederer et al., 1997). Ubiquitination takes place on the cytosolic surface of the ER, since the E2 and E3 enzymes Ubc7p and Hrd1p/Der3p, respectively, are localized there and may be positioned adjacent to the translocon (Hiller et al., 1996; Bordallo et al., 1998; Bays et al., 2001). Once marked, these proteins are rapidly degraded by the cytosolic 26S proteasome (Hiller et al., 1996).
Although much is known about the fate of ERAD substrates near the point of degradation, much less is understood regarding how they are recognized, retained, and targeted to the translocation/ubiquitination machinery. One model emerged that nascent polypeptides remain partially in the translocon after import. The polypeptide can only be released upon folding, whereas misfolded proteins are retrotranslocated via the same pore. The hypothesis was appealing, since it provided for a simple mechanism for retention and degradation. The model was brought into question when a well-established yeast soluble ERAD substrate, a mutant version of carboxypeptidase Y called CPY*, was shown to be translocated completely across the membrane (Plemper et al., 1999). However, the observation did not rule out the possibility that the nascent polypeptide remains associated with the translocon after translocation.
In mammalian cells, a mutant version of the well-characterized vesicular stomatitis virus G (VSV-G) protein, ts045, was observed to be localized to the ER of cells shifted to 39.5°C, a temperature that causes it to misfold (Kreis and Lodish, 1986). An elegant study using VSV-G ts045 fused to the green fluorescent protein provided direct evidence of an ER retention mechanism. Using photobleaching experiments in live cells, the integral membrane protein was shown to move freely in the plane of the membrane but did not leave the ER (Nehls et al., 2000). In cells overexpressing VSV-G ts045 through prolonged incubation at the restrictive temperature, a fraction of the protein escapes the ER and gets transported to the Golgi and retrieved (Hammond and Helenius, 1994). Although these earlier experiments were performed under more extreme conditions, they left open the possibility of a recycling mechanism for misfolded proteins. In yeast, the mechanism is less clear, but the efficient degradation of mutant versions of Ste6p and Yor1p integral membrane proteins in absence of ER-to-Golgi transport seems to support the mammalian view (Loayza et al., 1998; Katzmann et al., 1999).
A common quality control mechanism for both misfolded soluble and membrane proteins presents a spatial problem, since these two classes may occupy distinct regions of the ER (that is, luminal versus membrane). Therefore, it is plausible that different recognition and targeting mechanisms exist to direct the proteins into the degradation pathway. In this view, the ubiquitin/proteasome pathway used by both misfolded soluble and membrane proteins can be thought of either as an endpoint for an ER retention mechanism or a point of convergence for distinct mechanisms.
In this study, we examined the fate of several quality control substrates subject to ERAD-specific degradation in the budding yeast Saccharomyces cerevisiae. We demonstrate the coexistence of retention and retrieval mechanisms that define distinct classes of quality control substrates. For both pathways, a sorting step occurs in the ER whereby substrates of the retrieval pathway are packaged into COPII transport vesicles, whereas those to be retained are excluded. Furthermore, by using a genetic approach we've isolated mutants dissecting the two pathways. We report a mutant allele of the gene BST1 called per17–1 that prevents the ER-to-Golgi transport of misfolded proteins while preserving the transport of most normal proteins. In per17–1 cells, quality control is disrupted at an early step of the retrieval pathway as observed by the accumulation and stabilization of misfolded proteins in subcompartments associated with the ER.
Results
KHN is a misfolded protein retrieved from the Golgi apparatus for ERAD
Viral membrane proteins are excellent models to study protein folding and ER quality control (Gething et al., 1986; Machamer et al., 1990; Hammond and Helenius, 1994). To better understand quality control mechanisms, we sought to combine their advantages with the facile genetics of the budding yeast S. cerevisiae. We selected the simian virus 5 hemagglutinin neuraminidase (HN), since its folding state can be monitored using established methods (Ng et al., 1989). To express HN, we replaced the HN signal/anchor domain with the cleavable signal sequence from the yeast Kar2 protein and placed the fusion construct downstream of the moderate yeast PRC1 (CPY) promoter. This was done to bypass the poor utilization of the endogenous signal/anchor domain in yeast (unpublished data). The resulting protein, designated KHN, is similar to a soluble version of HN characterized previously in mammalian cells (Parks and Lamb, 1990).
We monitored the expression of KHN by metabolic pulse–chase analysis and made an unexpected observation. As shown in Fig. 1 A, KHN is lost rapidly after a 30-min chase and is nearly undetectable by 60 min. Since proteins from both cells and medium were combined for immunoprecipitation, secretion of KHN was ruled out to account for the loss. Alternatively, as a foreign protein KHN may fail to properly fold and be subject to quality control mechanisms leading to its degradation. Consistent with this notion, KHN fails to form disulfide-linked dimers and is not reactive to conformation-dependent anti-HN monoclonal antibodies (unpublished data). In a strain deleted of CUE1, a gene required for ubiquitination of proteins destined for ERAD (Biederer et al., 1997), KHN appeared to be stabilized during the same time course (Fig. 1 A, middle). We confirmed KHN as a bona fide ERAD substrate, since it is stabilized by multiple ERAD-specific mutants (see below). Interestingly, stabilization of KHN enhanced an unexpected characteristic for an ERAD substrate, that is, a time-dependent decrease in gel mobility (Fig. 1 A, p1 and p2). We next explored the nature of the altered forms.
Figure 1.

KHN is a rapidly degraded protein that is transported to the Golgi apparatus. (A) Wild-type and Δcue1 cells expressing KHN were metabolically pulse-labeled at 30°C with [35S]methionine/cysteine for 10 min followed by a cold chase for times indicated. KHN was immunoprecipitated from detergent lysates using anti-HN polyclonal antiserum and resolved by electrophoresis on a 10% SDS polyacrylamide gel. Where indicated, N-linked carbohydrates were removed by incubation of immunoprecipitated proteins with 500 U endoglycosidase H (Endo H) for 3 h. The positions of proteins immunoprecipitated nonspecifically are indicated by asterisks. (B) Wild-type, Δpmt1, and Δpmt2 cells expressing KHN were analyzed as described for A. (C) Wild-type, sec12–4, and sec18–1 cells expressing KHN were grown to log phase at 22°C and shifted to 37°C. After 30 min, the cells were pulse-labeled and chased for the times indicated. KHN was immunoprecipitated and analyzed as described for A. The positions of the KHN p1 and p2 forms are indicated (A), and arrows mark the position of the p1 form (B and C).
Stepwise increases in molecular weight are commonly observed during the maturation of many yeast secretory pathway proteins. The increase is due to elaboration of carbohydrates attached initially in the ER (Herscovics and Orlean, 1993). The delay reflects the time needed to transport nascent polypeptides to the Golgi apparatus where the modifying enzymes reside (Gemmill and Trimble, 1999; Strahl-Bolsinger et al., 1999). With this in mind, the observed modification raised the intriguing possibility that KHN is transported to the Golgi and retrieved to the ER for degradation. We addressed this possibility by first determining whether the shifts are actually due to carbohydrate modification. Endoglycosidase H digestion was used to remove N-linked carbohydrates from KHN. If the gel mobility shifts were due solely to modification of N-linked sugars, all forms of KHN after endoglycosidase H treatment would migrate equally. As shown in Fig. 1 A (right), removal of N-linked sugars did not eliminate the mobility differences. We next tested for O-linked carbohydrates by using mutants specifically defective at the first step of O-mannosylation. O-mannosylation begins in the ER with the transfer of a single mannose residue from Man-P-dolichol to the polypeptide. Enzymes of the protein mannosyltransferase (PMT) family catalyze this reaction. Strains deleted of individual PMT genes exhibit substrate-specific defects in glycosylation, reflecting the nonredundant nature of these genes (Gentzsch and Tanner, 1996). We expressed KHN in strains singly deleted of each PMT family member (PMT1–PMT6). As shown in Fig. 1 B, strains deleted of PMT1 and PMT2 prevented KHN mobility shifts such that p1 remained the predominant form that is degraded eventually. These data show that KHN O-glycosylation is dependent on PMT1 and PMT2 whose products were shown previously to work together as a complex (Gentzsch et al., 1995). KHN processing was unaffected in strains singly deleted of PMT3-PMT6 (unpublished data).
Proteins O-mannosylated in the ER are usually modified through lengthening of the carbohydrates in the Golgi (Lussier et al., 1997). To test whether the KHN gel mobility shift is due to post-ER processing, we expressed KHN in the well-characterized ER-to-Golgi transport mutants sec12–4 and sec18–1 (Eakle et al., 1988; Nakano et al., 1988; Barlowe and Schekman, 1993). When transport is blocked in these strains, KHN remains in the p1 form over an extended time course (Fig. 1 C). This is consistent with formation of the p2 form in the Golgi apparatus where the modifying enzymes reside. From these data, we designate the ER form as p1 and the Golgi form as p2. Interestingly, the turnover of KHN appears to be impaired in these mutants, suggesting that transport out of the ER may be a required step for degradation. Unfortunately, nonspecific immunoprecipitation of proteins overlapping the p1 and p2 forms made the kinetics of KHN turnover difficult to measure. Thus, the extent of the stabilization was inconclusive from these experiments.
To accurately measure the kinetics of KHN turnover, a modified version was constructed bearing a COOH-terminal triple HA epitope tag (KHNt). When using the anti-HA monoclonal antibody, immunoprecipitations of KHNt were free of background, and the yields were otherwise indistinguishable from experiments using the anti-HN polyclonal antisera (Fig. 2 A; unpublished data). KHNt is modified and degraded similarly to KHN except that the rate of turnover seems to be reduced slightly (Fig. 2 A, top, compared with Fig. 1 A, left). Although preliminary results suggested that KHN might be a substrate of the ERAD pathway, its transport to the Golgi raised the possibility that a fraction might continue forward and degrade in the vacuole (the yeast equivalent of lysosomes). This was ruled out when KHNt was degraded similarly to wild type in a mutant deficient in functional vacuolar proteases (Fig. 2, A and B, Δpep4). To establish firmly that KHN is a substrate of ERAD, we measured the stability of KHNt in several mutants defective specifically in the pathway. As shown in Fig. 2, when KHNt is expressed in strains deleted of the CUE1 (role in ubiquitination by anchoring Ubc7p to the ER membrane), DER1 (encodes an ER membrane protein required for ERAD), or HRD1/DER3 (encodes an ER-localized E3 ubiquitin ligase) genes, its degradation is impaired to the extent similar to other established ERAD substrates (Hampton et al., 1996; Knop et al., 1996; Biederer et al., 1997; Bordallo et al., 1998). Western blot analysis shows the steady-state accumulation of higher molecular weight (p2) forms of KHNt in each of the mutants, confirming that it is these species that are preferentially degraded in wild-type cells (Fig. 2 C).
Figure 2.


KHN t is a substrate for degradation by the ERAD pathway. (A) Wild-type and mutant strains expressing KHNt were pulse-labeled for 10 min with [35S]methionine/cysteine and followed by a cold chase as indicated. Immunoprecipitation of KHNt was performed using anti-HA monoclonal antibody (HA.11; BabCo) and normalized by total TCA precipitable counts. Proteins were analyzed by SDS-PAGE and visualized by autoradiography. (B) The experiments described for A were quantified by PhosphorImager analysis using the same gels that generated the autoradiograms shown in A. (C) Relative steady-state levels of KHNt in wild-type and ERAD mutants were analyzed by immunoblotting. Equal amounts of cell lysate (0.2 OD600 equivalents of cells) were loaded in each lane, separated by electrophoresis, transferred to nitrocellulose, and probed using HA.11 monoclonal antibody. Proteins were visualized using chemiluminescence (Pierce Chemical Co.). (D) Immunolocalization of KHNt in wild-type and ERAD mutant cells were performed using fixed and permeabilized cells on glass slides. KHNt and BiP were detected using α-HA monoclonal antibody and α-Kar2p polyclonal antiserum, respectively. After binding of fluorescent secondary antibodies, KHNt was visualized in the red channel (a, b, and c), and BiP was visualized in the green channel (d, e, and f). In each channel, images were captured using identical exposure times. Bar, 2 μm.
Misfolded proteins accumulate in the ER of cells defective for ERAD functions (Knop et al., 1996; Loayza et al., 1998). Since KHNt is transported to the Golgi before degradation, we wondered where it accumulates when ERAD is disrupted. By performing indirect immunofluorescence, we found that KHNt also accumulates in the ER of ERAD mutant cells as shown by its colocalization with the ER marker BiP (Fig. 2 D). These data show that KHNt behaves similarly to some established ERAD substrates and point to the possibility of a retrieval pathway for its degradation.
Two distinct mechanisms for the quality control of proteins destined for ERAD
The expression of KHN in ER-to-Golgi mutants led to an unexpected observation—transport may be an obligatory step for its degradation. This was surprising since other ERAD substrates, including mutant Ste6p and Yor1p, were observed to degrade normally under the same conditions Loayza et al., 1998; Katzmann et al., 1999). The apparent contradiction could be resolved if different mechanisms exist to target aberrant proteins for degradation: a static (nonrecycling) ER retention mechanism for proteins like Ste6p and Yor1p (both integral membrane proteins) and a transport and retrieval mechanism for others like KHN. To test this possibility, we applied complementary in vivo and in vitro approaches to assess the fate of substrates before degradation.
First, we examined the effect of preventing ER-to-Golgi transport. It was reported that the ERAD substrate Ste6–166p is degraded in a sec18 mutant, suggesting that ERAD functions normally even if transport is blocked (Loayza et al., 1998). We confirmed the observation in both sec12 and sec18 cells by finding the stability of Ste6–166p is identical to wild type (Fig. 3 A). As a control, we showed that an ERAD defective mutant stabilizes Ste6–166p under the same conditions (Fig. 3 A). We also analyzed Sec61–2p, another membrane protein subject to ERAD (Sommer and Jentsch, 1993; Biederer et al., 1996). Since Sec61p itself plays a role in ERAD, Sec61–2p was expressed ectopically and distinguished from wild type with an HA epitope tag. As with Ste6–166p, Sec61–2p is degraded normally under the restrictive conditions in each strain (Fig. 3 B). By contrast, the degradation of KHNt was strongly impaired (Fig. 3 C). Since core ERAD functions are normal in these strains, the defect is likely a consequence of perturbing the KHNt trafficking pattern that precedes degradation. We wondered whether this requirement is unique to KHNt or reflects a more general feature of ER quality control. For this, we examined an HA epitope–tagged version of another well-characterized soluble substrate, CPY* (Finger et al., 1993). Although it is well established that CPY*HA uses the core ERAD machinery, it was unclear whether it is retained or undergoes a retrieval cycle. As shown in Fig. 3 D, CPY*HA is stabilized strongly in both sec12 and sec18 mutants, suggesting that it too is dependent on the vesicular transport pathway. However, we were somewhat surprised with the effect of these mutants, since it was reported previously that CPY* is degraded in a sec18 mutant (Finger et al., 1993). There, the degradation was most pronounced after a long chase period of 3 h. We also observed some degradation in the transport mutants so we might expect a substantial fraction of the substrate to be degraded if we applied a similarly extended chase.
Figure 3.

ER-to-Golgi transport is required for degradation of soluble but not membrane-bound ERAD substrates. (A–D) Wild-type and ER transport mutant strains sec12–4 and sec18–1 expressing HA-tagged ERAD substrates were grown to log phase at 22°C and shifted to the restrictive temperature of 37°C for 30 min. Time courses were performed and analyzed as described in the legend to Fig. 2. The data is plotted to compare rates of degradation for each substrate in various strain backgrounds. A Δcue1 strain was included as a positive control for Ste6–166p and Sec61–2p.
The data suggest two classes of ERAD substrates, one uses the vesicular trafficking machinery for quality control and the other depends on static ER retention. This distinction predicts that sorting takes place in the ER to segregate misfolded proteins to be transported from those to be retained. The differences in degradation rates in the sec12 and sec18 mutants provided only suggestive evidence and did not rule out the possibility of indirect effects. To test the hypothesis directly, we performed in vitro assays that reproduce COPII-coated vesicle budding and cargo selection from ER membranes (Barlowe et al., 1994). For these experiments, microsomes were prepared from wild-type strains expressing KHNt, CPY*HA, and Ste6–166p. COPII budded vesicles from these microsomes were isolated, and the level of individual proteins packaged into vesicles were monitored by immunoblots (Fig. 4). The efficiency of incorporation for each protein was calculated as a percentage of the total by densitometry. For KHNt and CPY*HA, we found both proteins packaged into COPII vesicles at 1–2%, whereas the negative control Sec61p was not packaged. Although the amount of misfolded proteins packaged in COPII vesicles is less relative to other secretory proteins, it is consistent with the slower transport of KHNt compared with other cargo proteins (see Fig. 6 B). For the analysis of KHNt and CPY*HA by this method, membranes were treated with trypsin to ensure detection of protease-protected luminal species. These data provide independent confirmation that a subset of proteins destined for ERAD are first exported from the ER using the standard membrane trafficking machinery.
Figure 4.

Soluble ERAD substrates are contained in COPII vesicles. Recon stituted COPII budding reactions were performed on ER membranes isolated from wild-type strains expressing KHNt (A), CPY*HA (B), and Ste6–166p (C). Lanes labeled T represent one tenth of the total membranes used in a budding reaction, minus (−) lanes indicate the amount of vesicles formed in the absence of the purified COPII components, and plus (+) lanes indicate vesicles produced when COPII proteins are added. Total membranes and budded vesicles were collected by centrifugation, resolved on a polyacrylamide gel, and immunoblotted for indicated proteins. The amount of glyco-pro-α-factor (gpαf) was detected using fluorography.
Figure 6.


per17–1 is a mutant specific to the retrieval pathway, which blocks the transport of misfolded proteins but not properly folded proteins. (A) The turnover of KHNt, CPY*HA, Ste6–166p, and Sec61–2p in wild-type and per17–1 cells were measured by metabolic pulse–chase analysis as described in the legend to Fig. 2. Experiments were performed at 30°C except for strains expressing Sec61–2p. Strains expressing Sec61–2p were grown to log phase at 30°C, shifted to 37°C for 30 min, and continued for the pulse–chase. (B) Autoradiograms generated from gels of the KHNt time course shown in part A are shown at the top. The positions of the p1 (ER) and p2 (Golgi-modified) forms are indicated. Endogenous CPY and Gas1p were immunoprecipitated in parallel from aliquots of lysates prepared from the KHNt time course. The proteins were separated by gel electrophoresis and visualized by autoradiography (P1, ER proCPY; P2, Golgi proCPY; mCPY, mature CPY; ER Gas1p, ER form of Gas1p; mGas1, mature Golgi-modified Gas1p). (C) Wild-type and per17–1 cells were pulse labeled for 10 min and chased for times indicated. CPS and ALP were immunoprecipitated and analyzed by gel electrophoresis followed by autoradiography. The pro (proCPS and proALP) and mature (mCPS and mALP) forms of each protein are indicated.
Next, we examined Ste6–166p. There already exists evidence that Ste6–166p is targeted for degradation using an ER retention mechanism (Loayza et al., 1998). However, the nature of the retention was unclear. When we applied the ER vesicle budding assay to Ste6–166p, we found it remained exclusively in ER membranes even as other integral membrane proteins were incorporated efficiently into COPII-coated vesicles (Fig. 4 C). These data show that the plasma membrane protein Ste6p, when misfolded, is retained in the ER by exclusion from transport vesicles. Together, these results reveal a novel facet of ER quality control. As part of its surveillance mechanism, the cell sorts misfolded proteins for ER retention or transport.
After transport to the Golgi, degradation of KHNt and CPY*HA by ERAD would require retrograde trafficking to the ER. The reverse flow of membranes and proteins from the Golgi is driven by the formation of coated vesicles of the COPI class. To examine whether the trafficking of misfolded proteins use COPI-coated vesicles, we expressed KHNt and CPY*HA in the γ-COP mutant sec21–1 and measured their turnover. At the permissive temperature of 30°C, where forward transport is unaffected and retrograde transport is little affected (Letourneur et al., 1994), we observed a small but reproducible delay in KHNt and CPY*HA degradation (unpublished data). However, at the semipermissive temperature of 33°C, which partially disrupts retrograde transport with only a minor delay in forward transport (Letourneur et al., 1994), degradation of both proteins is inhibited (Fig. 5, A and B). We confirmed the proficiency of forward transport by analyzing endogenous CPY (unpublished data) and the formation of the KHNt p2 Golgi form (Fig. 5 A). We also ruled out indirect effects on ERAD function, since Ste6–166p degradation is normal in sec21–1 cells (Fig. 5 C). Taken together, these data demonstrate that misfolded proteins are sorted for ER retention or transport and retrieval from the Golgi. Ultimately, both pathways converge in the ER for degradation by the ERAD pathway.
Figure 5.

Degradation of KHN t and CPY* HA but not Ste6–166p requires Golgi-to-ER transport. Pulse–chase analysis was performed on wild-type and sec21–1 strains expressing (A) KHNt, (B) CPY*HA, and (C) Ste6–166p as described in the legend to Fig. 2 except that strains were grown to log phase at 22°C and pulse-labeled immediately after a shift to 33°C. Incubation at 33°C was continued for the cold chase (times as indicated). Gels were visualized by autoradiography (left) and quantified by PhosphorImager analysis (right). In C, the gel images were from PhosphorImager scans.
A gene required for ER quality control early in the retrieval pathway
Recent studies have demonstrated that some cargo proteins leaving the ER are actively sorted into transport vesicles (Muniz et al., 2001). Although the molecular mechanisms of these sorting events are not well understood, specific genes have been implicated for the transport of just a subset of proteins (Belden and Barlowe, 1996; Muniz et al., 2000). Since KHNt and CPY*HA may represent a new class of cargo proteins, we wondered whether dedicated factors function to sort and package misfolded proteins into transport vesicles. To address this question, we employed a genetic approach. If such factors exist, we reasoned that their loss of function would cause the retention and stabilization of misfolded proteins normally transported out of the ER. We reported previously a genetic screen based on synthetic lethality with the unfolded protein response pathway as a powerful means of identifying genes associated with ER quality control (Ng et al., 2000). As the original screen was far from exhausted, we expanded the scope with the intent of dissecting the ER retention and recycling mechanisms of quality control. Although the details of the genetic screen will be presented elsewhere (unpublished data), we report here the discovery of a gene needed for the anterograde transport of misfolded proteins in the retrieval pathway.
Starting with a pool of 152 recessive protein processing in the ER ( per) mutants, those exhibiting general processing defects of normal proteins including glycosylation and transport were excluded (Ng et al., 2000). Of the remaining 107, we analyzed ERAD activity by measuring the stability of CPY*HA and Sec61–2p as described in the legend to Fig. 3. Although still in progress, of the mutants showing defects in only CPY*HA degradation we began analyzing the stability and processing of KHNt. For one mutant, per17–1, both KHNt and CPY*HA are defective for degradation (Fig. 6 A). However, unlike other ERAD mutants (Fig. 2), KHNt remains in the ER p1 form in per17–1 cells consistent with a transport block to the Golgi (Fig. 6 B, top). Gas1p (Fig. 6 B, bottom) and chitinase (unpublished data) carbohydrate processing in per17–1 cells is normal and serves to control for functional O-mannosylation and modification in per17–1 cells (Nuoffer et al., 1991; Gentzsch and Tanner, 1996). This shows that the prevalence of the KHNt p1 form reflects a transport defect rather than an indirect effect on glycosylation. Interestingly, transport of folded cargo proteins showed differential effects. CPY transport was similar to wild type, whereas Gas1p was slower than normal (Fig. 6 B). Since Gas1p is anchored in the membrane, we examined two additional integral membrane cargo proteins, carboxypeptidase S (CPS) and alkaline phosphatase (ALP) (Cowles et al., 1997; Spormann et al., 1992). As shown in Fig. 6 C, both proteins are transported indistinguishably to wild type, confirming that the per17–1 mutation does not cause general defects in ER-to-Golgi transport.
The data suggest that the per17–1 mutation inhibits degradation by failing to promote the transport of misfolded proteins destined for the retrieval pathway. To reinforce this view, we analyzed the fate of ERAD substrates that are sorted for ER retention. If PER17 plays such a distinct role in ER quality control, we expect the retention pathway to be functional and these substrates to turn over normally in per17–1 cells. As shown in Fig. 6 A (bottom), Ste6–166p and Sec61–2p are degraded with wild-type kinetics in per17–1 cells. These data show that the per17–1 allele is specific to the recycling pathway and validates our genetic strategy. Although these data are similar to those obtained using the sec12 and sec18 mutants, they extend the evidence that transport is an important step for degradation, since the per17–1 transport block affects misfolded soluble proteins while leaving the transport of several normal cargo proteins intact.
To better understand the nature of the per17–1 transport block, we performed indirect immunofluorescence to localize KHNt and CPY*HA stabilized in per17–1 cells. As shown in Fig. 7, both KHNt and CPY*HA are concentrated within punctate structures throughout the cell. This differs from transport competent ERAD mutants, since they accumulate these substrates diffusely throughout the ER (Knop et al., 1996; Fig. 2 D). Interestingly, the punctate distribution is reminiscent of the pattern observed for cargo proteins blocked for transport in sec12 mutant cells (Nishikawa et al., 1994). In those cells, the ER chaperone BiP colocalized with cargo proteins at discrete sites within the ER. Since the misfolded proteins are similarly blocked for transport, we also examined the distribution of BiP in the per17–1 cells. As shown in Fig. 7, BiP was found in the same punctate structures as KHNt and CPY*HA (Fig. 7 A, b and e). Although BiP is widely used as a marker for ER morphology, we wondered whether the pattern reflected subdomains of the ER as the case in sec12 cells or a general reorganization of ER membranes. To address this, we chose an alternative ER marker, the signal recognition particle receptor β subunit (SRβ). SRβ is an integral membrane protein that is distributed throughout the ER (Ogg et al., 1998). As shown in Fig. 7 B, SRβ staining in per17–1 cells is similar to wild type, indicating that there are no gross changes in ER morphology (Fig. 7 B, e). This is in good agreement with ultrastructural analysis performed with the same strains (unpublished data). In double-label experiments, the punctate structures are always coincident with the ER as defined by SRβ (Fig. 7 B). These data show that misfolded proteins accumulate with BiP at distinct ER sites in per17–1 cells. Whether BiP forms a complex with the misfolded proteins at these sites remains to be shown.
Figure 7.


Immunolocalization of misfolded proteins in per17–1 cells. (A) per17–1 cells expressing KHNt (a–c) and CPY*HA (d–f) and Δder1 cells expressing CPY*HA (g–i) were fixed and permeabilized from logarithmic cultures. The cells were stained with α-HA and α-Kar2p antibodies followed by Alexa Fluor 546 goat α-mouse (a, d, and g) and Alexa Fluor 488 goat α-rabbit (b, e, and h) secondary antibodies. Staining with DAPI (c, f, and i) indicates the positions of nuclei. Arrows mark specific points of colocalization. (B) Wild-type and per17–1 cells expressing HA epitope–tagged SRβ were processed and bound to primary antibodies as in A. Alexa Fluor 546 goat α-rabbit and Alexa Fluor 488 goat α-mouse were used such that BiP was visualized in the red channel (a and d), whereas SRβ was visualized in the green channel (b and e). Bars, 2 μM.
We next determined the identity of the PER17 gene. A yeast genomic library based on the centromeric YCp50 vector was transformed into the per17–1 mutant. By restoration of the sectoring phenotype, a complementing clone was obtained (Ng et al., 2000). Through deletion mapping, a single ORF encoding the BST1 (bypass of sec thirteen) was identified as the PER17 gene. We were intrigued, since BST1 encodes an ER integral membrane protein first cloned through genetic interaction with SEC13, a component of the COPII vesicle coat (Elrod-Erickson and Kaiser, 1996). Thus, BST1 is believed to play a role in ER-to-Golgi transport. However, its precise role was unclear, since a BST1 gene deletion did not seem to affect the transport of two prototypic cargo proteins, CPY and invertase. Our data suggest a novel function for BST1 in ER quality control. Since per17–1 and Δbst1 cells prevent the transport of misfolded but not most properly folded proteins, the data suggest a role in cargo protein sorting (Fig. 6 B; unpublished data). Interestingly, the exhaustive genetic screen that previously identified BST1 also identified EMP24 and ERV25 (Elrod-Erickson and Kaiser, 1996). EMP24 and ERV25 are members of the p24 family of integral membrane proteins believed to be involved in cargo protein sorting (Kaiser, 2000). However, they appear to function differently than BST1, since the transport and degradation of both KHNt and CPY*HA occur normally, if not slightly accelerated, in strains deleted of those genes (unpublished data).
Discussion
A cellular surveillance system that monitors the folding state of nascent proteins in the ER was first observed nearly a quarter century ago. Those pioneering studies showed that viral membrane proteins, when misfolded, were not transported to the plasma membrane but retained at the site of synthesis (Gething et al., 1986; Kreis and Lodish, 1986). Subsequently, the phenomenon was appropriately termed “ER quality control” and led to the realization that several human diseases, including cystic fibrosis, owed their molecular basis to the retention and degradation of mutant proteins (Carrell and Gooptu, 1998; Kim and Arvan, 1998; Kopito and Ron, 2000). More recently, important strides have improved our understanding of ER quality control. Most notably, the degradation step, or ERAD, is now known to involve the retrotranslocation of substrates to the cytosol through the ER translocon pore (Wiertz et al., 1996; Pilon et al., 1997; Plemper et al., 1997; Zhou and Schekman, 1999). During or after retrotranslocation, substrates are ubiquitinated and degraded by the 26S proteasome (Ward et al., 1995; Hiller et al., 1996). Despite these advances, the events upstream to ERAD remained unclear.
In this paper, we report the collaboration of two distinct mechanisms to assure the quality control of protein biosynthesis in the yeast secretory pathway. By combining biochemical and genetic approaches, we reconfirm the retention mechanism while uncovering another that uses established ER-to-Golgi vesicle transport and retrieval pathways (Fig. 8). The dual mechanistic nature of ER quality control may have evaded characterization, since both pathways converge at the degradation step and a block there results in the steady-state accumulation of substrates in the ER. However, several studies have hinted that vesicle trafficking mechanisms might have a role in quality control. It was observed that CPY* can acquire α1,6-mannose in a strain deleted of the DER1 gene. α1,6-mannose is a carbohydrate modification that occurs only in the Golgi apparatus, suggesting a transport mechanism. However, since only a small fraction was affected and the modification was not detected in wild-type cells, it was concluded that the majority of CPY* is retained, but some can leave in DER1-deleted cells (Knop et al., 1996). More recently, during the preparation of this article a paper available electronically reported that ERAD substrates show retarded degradation rates in ER-to-Golgi transport mutants (Caldwell et al., 2001). Although in agreement with our findings, no direct evidence was presented that misfolded proteins are transported as part of their degradative pathway. Here, we provide direct evidence of ER-to-Golgi transport of misfolded proteins in vivo and in vitro and a requirement for retrograde transport.
Figure 8.
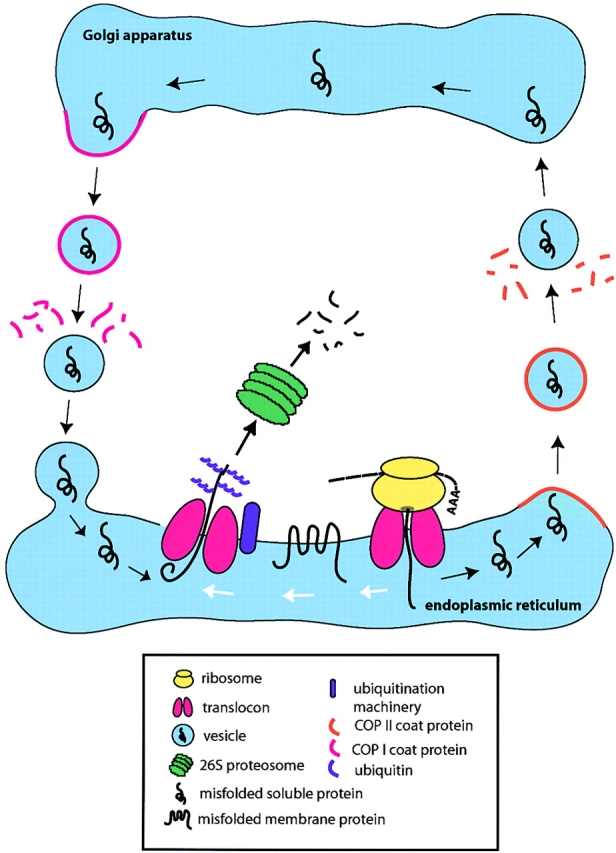
Proposed model of ER quality control in budding yeast. After translocation, proteins that misfold are sorted for the retention pathway (white arrows) or the retrieval pathway (black arrows). In the retrieval pathway, proteins are packaged into COPII vesicles, transported to the Golgi apparatus, and retrieved via the retrograde transport pathway. In the ER, substrates of both pathways converge for ERAD. The proteins cross the ER membrane via the translocon complex, marked by ubiquitination and degraded by the cytosolic 26S proteasome.
Key to our approach was the characterization of KHN as a novel ERAD substrate. Unlike other misfolded proteins commonly studied, KHN allows the use of O-linked sugar modifications to monitor its transport (Fig. 1). The native HN protein is not O-glycosylated in mammalian cells so it seems likely that the modifications are due to promiscuous O-mannosylation that can occur when proteins misfold in yeast (Harty et al., 2001). The processing of these carbohydrates shows that most, if not all, of the protein uses a retrieval mechanism before ERAD. Furthermore, we found that disruption of either forward or retrograde transport compromised KHN degradation. The transport requirement is not peculiar, since the well-characterized substrate CPY* is affected similarly under all circumstances. Since substrates subject to retention are degraded normally in these mutants, the data strongly suggest that transport and retrieval are obligatory steps for efficient KHN and CPY* degradation.
An in vitro vesicle budding assay using purified components provided direct evidence that KHN and CPY* are packaged into COPII-coated vesicles, whereas Ste6–166p is excluded. These experiments were important, since the assay was established previously to reflect early events in ER-to-Golgi transport. Although the data serve to confirm and extend the in vivo experiments, they also reveal a novel ER sorting mechanism for misfolded proteins at or just before the formation of COPII vesicles. The retrieval pathway largely uses the standard vesicle transport machinery, but we do not know whether misfolded proteins occupy the same vesicles as folded cargo proteins. Recently, it was shown that different classes of folded cargo proteins occupy distinct vesicle populations (Shimoni et al., 2000; Muniz et al., 2001). Thus, it seems possible that misfolded proteins are sorted into specialized vesicles for transport to the Golgi.
The retention and retrieval mechanisms appear to be highly selective. Of the substrates examined, each used only one pathway. This raises the important question of what specific structural features determine selection of substrates into their respective pathways. Although we examined only four substrates for this study, we propose for most substrates membrane proteins are retained, whereas soluble proteins are retrieved. Although the appeal is obvious from a mechanistic point of view, there already exists a possible exception. A mutant form of pro-α-factor, a soluble protein, was degraded in vitro using purified ER microsomal membranes (McCracken and Brodsky, 1996). The lack of Golgi membranes in the assay suggested that a vesicle transport pathway is not required. Although these studies suggest retention, mutant pro-α-factor may represent a distinct mechanism altogether, since it does not require ubiquitination for degradation in vitro and differs from CPY* in its requirements for chaperones (Werner et al., 1996; Nishikawa et al., 2001). Therefore, alternative strategies to those described here are probably needed to clear all forms of aberrant proteins from the ER.
Recently, it became clear that the unfolded protein response (UPR) pathway plays an important role in regulating ERAD (Casagrande et al., 2000; Friedlander et al., 2000; Ng et al., 2000; Travers et al., 2000). It was gratifying to learn that an ER signaling pathway sensitive to the accumulation of misfolded proteins plays a direct role in ridding them. By whole genome microarray analysis, it was also found that many genes of the secretory pathway are UPR targets including those involved in retrograde transport from the Golgi (Travers et al., 2000). The significance of these target genes in UPR function was unclear previously. A retrieval mechanism for ERAD substrates provides a logical physiological basis for their regulation. However, it remains to be tested whether induction of these specific genes by the UPR is required for the correct processing of ERAD substrates during periods of ER stress.
A retrieval mechanism adds layers of complexity to our current view of ER quality control and raises numerous new questions. First, misfolded proteins are sorted in the ER for transport or retention. How is that achieved? For misfolded membrane proteins, there is evidence of retention factors. Mutants of the plasma membrane ATPase Pma1p are retained and degraded in the ER. Chang and coworkers reported a genetic approach that uncovered a protein disulfide isomerase-related protein, Eps1p, needed to prevent the transport of mutant Pma1p (Wang and Chang, 1999). In light of our results, their data suggest retention to be an active process using specific factors to restrict the partitioning of proteins into transport vesicles. What role Eps1p plays in this process is currently unknown. Similarly, our genetic analysis of the retrieval pathway suggests an active mechanism for recognition and transport. In the absence of BST1 function, KHN and CPY* are retained and stabilized in the ER, whereas the retention pathway substrate Ste6–166p is degraded normally (Fig. 6). Since BST1 is not essential for the transport of most folded cargo proteins examined, the data suggest a possible role in cargo protein sorting. In support of this hypothesis, a genetic link was found between BST1 and SEC13, a gene encoding a component of COPII vesicle coats (Elrod-Erickson and Kaiser, 1996). Experiments are in progress to directly test the assertion. In addition to per17 alleles, other mutants with similar phenotypes were isolated from our genetic screen, suggesting the collaboration of other factors at this early step (unpublished data).
Although the precise function of BST1 is unclear currently, we have performed additional experiments that reinforce its vital role in ER quality control. The unfolded protein response activates in response to the accumulation of misfolded proteins in the ER (Kozutsumi et al., 1988). We observed that loss of BST1 function strongly induces the UPR in the absence of ectopically expressed proteins (unpublished data). We interpret the response to reflect the accumulation of endogenous misfolded proteins that normally depend on BST1 for degradation. Furthermore, in contrast to other ERAD mutants examined in this study the viability of BST1 mutants is hypersensitive to the overexpression of misfolded proteins (unpublished data). This effect suggests a role at a physiologically important point in ER quality control. Taken together, BST1 represents a new class of genes for ER quality control. It acts in the retrieval pathway far upstream the point of convergence for both pathways.
Although both pathways converge at the translocon for retrotranslocation and degradation, the events just preceding this last step are unclear. Since it is essentially a protein translocation mechanism, established paradigms may serve to guide future studies and have been discussed eloquently in a recent essay (Johnson and Haigh, 2000). In typical translocation pathways across membranes, nascent polypeptides contain signal sequences recognized by specific targeting factors. For retrotranslocation of misfolded proteins, it is unlikely that signal sequence motifs will be found, since they would also exist on newly synthesized proteins. In mammalian cells, the ER lectins calnexin and calreticulin bind to carbohydrates of incompletely folded proteins and thus may serve as potential targeting factors (Hauri et al., 2000). Indeed, specific N-linked carbohydrate moieties are necessary for the degradation of CPY* in yeast (Jakob et al., 1998). In experiments comparing the carbohydrate requirements of KHN and CPY*, inhibition of O-mannosylation reduces the rate of degradation for KHN but has no effect on CPY* (unpublished data). It is conceivable that Golgi modification of KHN O-mannosylated residues signals its degradation upon return to the ER. We are currently exploring this possibility. Despite these examples, there are numerous substrates that are not glycosylated but nevertheless degraded efficiently by ERAD (Sommer and Jentsch, 1993; Biederer et al., 1996; McCracken and Brodsky, 1996; Loayza et al., 1998). Thus, carbohydrates may play differential roles depending on the specific substrate but cannot serve as general signals. Alternatively, molecular chaperones are known to bind exposed hydrophobic domains on the surfaces of misfolded proteins. With this, they are qualified to be general targeting factors that bring substrates to the translocon. The ER chaperone BiP could serve such a role, since it is required for ERAD and can interact directly with Sec63p, a component of the translocon (Brodsky and Schekman, 1993; Plemper et al., 1997; Brodsky et al., 1999). Since BiP binds to nascent polypeptides, the cell must be able to distinguish proteins in the process of folding from those that cannot fold. The retrieval pathway may provide the means to partition misfolded proteins destined for ERAD from those in the process of folding. Our data are consistent with an active sorting mechanism to transport misfolded proteins out of the ER. Although just one of many possible scenarios, misfolded proteins may be returned to the regions of the ER specialized for ERAD. Although this notion is highly speculative, in sec12 and per17–1 mutants misfolded proteins are retained in the ER. However, the proteins are degraded very poorly, and we find that the proteins are in distinct regions of the ER (Fig. 7; unpublished data). Since retention pathway substrates are degraded efficiently in these mutants, it seems possible that the defect is attributable to their aberrant localization.
The delineation of two separate mechanisms for ER quality control raises far more questions than are answered. The distinction is still important, since it impacts the experimental designs of future studies in ER quality control. Already, we have applied the principles to our genetic approaches to identify mutants and genes specific to the retention and retrieval mechanisms and after their point of convergence (unpublished data). By monitoring the processing of KHN, we have also begun to dissect the mechanisms that underlie the retrieval pathway.
Materials and methods
Plasmids used in this study
Plasmids were constructed using standard cloning protocols (Sambrook et al., 1989). For pDN431 and pDN436, HA epitope–tagged CPY* expression vectors were described previously (Ng et al., 2000). For pSM1083 and pSM1346, HA epitope–tagged Ste6–166p expression vectors were gifts from S. Michaelis (Johns Hopkins University, Baltimore, MD) (Loayza et al., 1998).
Construction of the HA epitope–tagged Sec61–2p expression vector pDN1002
The promoter and coding sequences of sec61–2 were cloned from strain RSY533 (MATα, sec61–2, leu2, ade2, ura3, pep4–3) by amplification of genomic DNA using Vent polymerase (New England Biolabs, Inc.) performed according to manufacturer's protocol. Using the primers N182 (5′-CGAATCCGTCGTTCGTCACC-3′) and N183 (5′-TTCCCATGGAATCAGAAAATCCTGG-3′), the amplified 2,016-bp fragment was digested with HindIII and NcoI, and the 1,931-bp fragment was purified. The purified fragment was ligated into pDN333 digested with the same enzymes. pDN333 was generated by inserting the HA-tagged insert from pDN280 (Ng et al., 1996) into pRS315 (Sikorski and Hieter, 1989). An NcoI site from N183 places the Sec61–2p coding sequence in frame with vector sequences encoding a single HA tag followed by ACT1 terminator sequences.
Construction of KHN expression vectors pSM31, pSM56, pSM70, and pSM72
The KHN fusion gene was constructed by ligating the sequences encoding the first 45 amino acids of Kar2p (signal sequence and signal peptidase cleavage site) to the COOH-terminal 528 amino acids of the SV5 HN gene. Both fragments were amplified by PCR using Vent polymerase and inserted into pDN251 to generate pSM31. pDN251 is identical to the yeast expression vector pDN201 (Ng et al., 1996) except it contains the moderate PRC1 promoter in place of the TDH3 promoter. pSM70 is identical to pSM31 except for the addition of a triple HA epitope tag inserted in-frame to the COOH terminus of KHN. Sequences encoding the triple HA epitope tag were excised from pCS124 (a gift from C. Shamu, Harvard University, Cambridge, MA). pSM56 and pSM72 are similar to pSM31 and pSM70, respectively, except that the KHN gene sequences were subcloned into pRS315.
pES69 was constructed by inserting a NotI/KpnI fragment containing the gene for HA epitope–tagged SRβ from pSO459 (Ogg et al., 1998) into pRS426 (Sikorski and Hieter, 1989).
Strains and antibodies
Yeast strains used in this study are described in Table I. Anti-HA monoclonal antibody (HA.11) was purchased from BabCo. Anti-Kar2p antibody was provided by Peter Walter (University of California, San Francisco, CA). Anti-CPY antiserum was provided by Reid Gilmore (University of Massachusetts, Worcester, MA). Anti-Gas1p was a gift from Howard Riezman (University of Basel, Basel, Switzerland). Anti-ALP and anti-CPS antisera were gifts from Chris Burd and Scott Emr (University of California, San Diego, CA). Anti-HN antiserum was described previously (Ng et al., 1990). Secondary antibodies labeled with Alexa Fluor 488 or 546 were purchased from Molecular Probes, Inc.
Table I. Strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| W303a | MAT a, leu2-3,112, his3-11, trp1-1, ura3-1, can1-100, ade2-1 | P. Waltera |
| SMY248 | MAT a, W303, pSM70 | This study |
| SMY249 | MAT a, cue1::TRP1, pSM70, W303 background | This study |
| SMY 250 | MAT a, der1::KANMX, pSM70, W303 background | This study |
| SMY251 | MAT a, der3::KANMX, pSM70, W303 background | This study |
| SMY252 | MAT a, per17-1, pSM70, W303 background | This study |
| SMY254 | MATα, pep4::HIS3, pSM70, W303 background | This study |
| SMY258 | MAT a, sec12-4, pSM70, W303 background | This study |
| SMY259 | MAT a, sec18-1, pSM70, W303 background | This study |
| RSY255 | MATα, leu2-3,112, ura3-52 | R. Schekmanb |
| RSY277 | MATα, sec21-1, ura3-52 | R. Schekman |
| SMY359 | MATα, RSY255, pSM70 | This study |
| SMY360 | MATα, RSY255, pDN431 | This study |
| SMY361 | MATα, RSY277, pSM70 | This study |
| SMY362 | MATα, RSY277, pDN431 | This study |
| SMY390 | MAT a, RSY255, pSM1083 | This study |
| SMY391 | MAT a, RSY277, pSM1083 | This study |
| SMY301 | MAT a, W303, pSM31 | This study |
| SMY302 | MAT a, sec12-4, pSM31, W303 background | This study |
| SMY303 | MAT a, sec18-1, pSM31, W303 background | This study |
| SMY312 | MAT a, cue1::TRP1, pSM31, W303 background | This study |
| SY114 | MATα, leu2-3, ura3-52, his3-Δ200, lys2-801, trp1-Δ90, suc2-Δ9 | S. Sandersc |
| SY655 | MATα, pmt1::URA3, SY114 background | S. Sanders |
| SY656 | MATα, pmt2::LEU2, SY114 background | S. Sanders |
| SY657 | MATα, pmt3::HIS3, SY114 background | S. Sanders |
| SY415 | MATα, pmt4::TRP1, SY114 background | S. Sanders |
| SY658 | MATα, pmt5::URA3, SY114 background | S.Sanders |
| SY659 | MATα, pmt6::URA3, SY114 background | S. Sanders |
| SMY319 | MATα, pmt1::URA3, pSM56, W303 background | This study |
| SMY347 | MATα, pmt2::LEU2, pSM56, W303 background | This study |
| SMY313 | MATα, SY656, pSM31 | This study |
| SMY320 | MAT a, SMY237, pSM56, W303 background | This study |
| SMY326 | MAT a, SMY239, pSM31, W303 background | This study |
| SMY348 | MAT a, W303, pSM1346, W303 background | This study |
| SMY349 | MAT a, per17-1, SM1346, W303 background | This study |
| SMY225 | MAT a, W303, pSM1083, W303 background | This study |
| SMY226 | MAT a, cue1::TRP1, pSM1083, W303 background | This study |
| SMY227 | MAT a, sec12-4, pSM1083, W303 background | This study |
| SMY228 | MAT a, sec18-1, pSM1083, W303 background | This study |
| WKY4 | MAT a, sec12-4, pDN431, W303 background | This study |
| WKY20 | MAT a, per17-1, pDN436, W303 background | This study |
| WKY21 | MAT a, per17-1, pDN1002, W303 background | This study |
| WKY23 | MAT a, sec12-4, pDN1002, W303 background | This study |
| WKY25 | MAT a, sec18-1, pDN1002, W303 background | This study |
| WKY108 | MAT a, per17-1, pSM1346, W303 background | This study |
| WKY110 | MAT a, W303, pSM1346, W303 background | This study |
| WKY135 | MAT a, sec18-1, pDN436, W303 background | This study |
| SMY340 | MAT a, W303, pSM70, pSM72 | This study |
| SMY342 | MAT a, per17-1, pSM70, pSM72, W303 background | This study |
| SMY383 | MAT a, der1::KANMX, pSM70, pSM72, W303 background | This study |
| SMY384 | MAT a, der3:KANMX, pSM70, pSM72, W303 background | This study |
| WKY213 | MAT a, W303, pDN436 | This study |
| SMY385 | MAT a, W303, pES69 | This study |
| SMY387 | MAT a, per17-1, pES69, W303 background | This study |
University of California, San Francisco, CA.
University of California, Berkeley, CA.
Massachusetts Institute of Technology, Cambridge, MA.
Cell labeling and immunoprecipitation
Typically, 2 A600 OD U of log phase cells were pelleted and resuspended in 1.0 ml of synthetic complete medium lacking methionine and cysteine. After 30 min of incubation at the appropriate temperature, cells were labeled with 480 μCi of Tran35S-label (ICN Biomedicals). A chase was initiated by adding cold methionine/cysteine to a final concentration of 2 mM. The chase was initiated 30 s before the end of the pulse to exhaust intracellular pools of unincorporated label. Labeling/chase was terminated by the addition of trichloroacetic acid to 10%. Preparation of cell lysates, immunoprecipitation procedures, gel electrophoresis, and quantification of immunoprecipitated proteins were performed as described previously (Ng et al., 2000).
In vitro budding assays
Vesicle budding from the ER was reproduced in vitro by incubation of microsomes (Wuestehube and Schekman, 1992) with purified COPII proteins (Sar1p, Sec23p complex, and Sec13p complex) as described (Barlowe et al., 1994). Microsomes were prepared from cells expressing misfolded KHNt, CPY*HA, and Ste6–166p (SMY248, WKY114, and SMY225). To measure incorporation of proteins into COPII vesicles, a 15-μl aliquot of the total budding reaction and 150 μl of a supernatant fluid containing budded vesicles were centrifuged at 100,000 g in a TLA100.3 rotor (Beckman Coulter) to collect membranes. The resulting membrane pellets were solubilized in 30 μl of SDS-PAGE sample buffer, and 10–15 μl were resolved on 12.5% polyacrylamide gels. For measurement of KHNt and CPY* contained in COPII vesicles, membranes were treated with trypsin (100 μg/ml) for 10 min on ice followed by trypsin inhibitor (100 μg/ml) to ensure detection of a protease-protected species. The percentages of individual proteins (KHNt, CPY*, Ste6–166p, Bos1p, Erv25p, and Sec61p) packaged into vesicles from a total reaction were determined by densitometric scanning of immunoblots. Protease protected [35S]glyco–pro-α-factor packaged into budded vesicles was measured by precipitation with concanavalin A–Sepharose after posttranslational translocation of [35S]-prepro-α-F into microsomes (Wuestehube and Schekman, 1992). [35S]glyco–pro-α-factor was also visualized by PhosphorImager analysis (Molecular Dynamics) after transfer to nitrocellulose membranes and exposure to a phosphor screen.
Indirect immunofluorescence microscopy
Cells were grown in synthetic complete medium to an OD600 of 0.5–0.9. Formaldehyde (EM grade; Polysciences, Inc.) was added directly to the medium to 3.7% at 30°C for 1 h. After fixation, cells were collected by centrifugation and washed with 5 ml 0.1 M potassium phosphate buffer (pH 7.5). Cells were incubated 30 min at 30°C in spheroplasting buffer (1.0 mg/ml zymolyase 20T [ICN Biomedicals], 0.1 M potassium phosphate, pH 7.5, 0.1% 2-mercaptoethanol) to digest the cell wall. Digestion was terminated by washing cells once in PBS. 30 μl of cell suspension was applied to each well of a poly-l-lysine–coated slide for 1 min and washed three times with PBS. Slides were immersed in acetone for 5 min at –20°C and allowed to air dry. Subsequent steps were performed at room temperature. 30 μl of PBS block (3% BSA in PBS) were added to each well and incubated for 30 min. Primary antibodies α-HA or α-Kar2p were applied and used at 1:1,000 or 1:5,000 dilutions for in PBS block, respectively, for 1 h. Wells were washed three to five times with PBS block. 30 μl secondary antibodies (Alexa Fluor 488 goat α-mouse or α-rabbit and Alexa Fluor 546 goat α-mouse or α-rabbit; Molecular Probes, Inc.) were added to wells and incubated for 45 min in the dark. Wells were washed five to seven times with PBS block and two times with PBS. Each well is sealed with 5 μl mounting medium (PBS, 90% glycerol, 0.025 μg/ml DAPI) and a glass coverslip. Samples were viewed on a ZEISS Axioplan epifluorescence microscope. Images were collected using a Spot 2 cooled digital camera (Diagnostic Instruments) and archived using Adobe Photoshop® 4.0. In experiments using KHNt, two copies of the gene were introduced into each strain to enhance detection. Low expression levels at single copy were likely due to suboptimal codon usage of this mammalian viral gene by yeast cells. By increasing gene dosage, the expression level was similar to CPY*HA at single copy and had no effect on its processing as an ERAD substrate (unpublished data).
Acknowledgments
We thank Jeff Brodsky and members of the Ng lab for discussion of and comments on the article. We thank Bob Lamb and Reay Paterson for suggesting the use of SV5 HN as a folding substrate. We thank Chris Burd, Scott Emr, Reid Gilmore, Susan Michaelis, Steve Ogg, Howard Riezman, Sylvia Sanders, Randy Schekman, and Peter Walter for gifts of antibodies, plasmids, and strains. We thank Christie Blackman for assistance in rendering artwork. We acknowledge Joel Graham, Geoff Knudson, and Zhenjian Zhang for contributions to this project during their rotations.
This research was supported by grants from the National Institutes of Health to D.T.W. Ng (GM059171) and C. Barlowe (GM052549).
Footnotes
Abbreviations used in this paper: ALP, alkaline phosphatase; CPS, carboxypeptidase S; CPY, carboxypeptidase Y; ER, endoplasmic reticulum; ERAD, ER-associated protein degradation; HN, hemagglutinin neuraminidase; PMT, protein mannosyltransferase; SRβ, signal recognition particle receptor β subunit; UPR, unfolded protein response; VSV-G, vesicular stomatitis virus G.
References
- Barlowe, C., and R. Schekman. 1993. SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature. 365:347–349. [DOI] [PubMed] [Google Scholar]
- Barlowe, C., L. Orci, T. Yeung, M. Hosobuchi, S. Hamamoto, N. Salama, M.F. Rexach, M. Ravazzola, M. Amherdt, and R. Schekman. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 77:895–907. [DOI] [PubMed] [Google Scholar]
- Bays, N.W., R.G. Gardner, L.P. Seelig, C.A. Joazeiro, and R.Y. Hampton. 2001. Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 3:24–29. [DOI] [PubMed] [Google Scholar]
- Belden, W.J., and C. Barlowe. 1996. Erv25p, a component of COPII-coated vesicles, forms a complex with Emp24p that is required for efficient endoplasmic reticulum to Golgi transport. J. Biol. Chem. 271:26939–26946. [DOI] [PubMed] [Google Scholar]
- Biederer, T., C. Volkwein, and T. Sommer. 1996. Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin-proteasome pathway. EMBO J. 15:2069–2076. [PMC free article] [PubMed] [Google Scholar]
- Biederer, T., C. Volkwein, and T. Sommer. 1997. Role of Cue1p in ubiquitination and degradation at the ER surface. Science. 278:1806–1809. [DOI] [PubMed] [Google Scholar]
- Bordallo, J., R.K. Plemper, A. Finger, and D.H. Wolf. 1998. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell. 9:209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, J.L., and R. Schekman. 1993. A Sec63p-BiP complex from yeast is required for protein translocation in a reconstituted proteoliposome. J. Cell Biol. 123:1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, J.L., and A.A. McCracken. 1999. ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 10:507–513. [DOI] [PubMed] [Google Scholar]
- Brodsky, J.L., E.D. Werner, M.E. Dubas, J.L. Goeckeler, K.B. Kruse, and A.A. McCracken. 1999. The requirement for molecular chaperones during endoplasmic reticulum-associated protein degradation demonstrates that protein export and import are mechanistically distinct. J. Biol. Chem. 274:3453–3460. [DOI] [PubMed] [Google Scholar]
- Caldwell, S.R., K.J. Hill, and A.A. Cooper. 2001. Degradation of ER quality control substrates require transport between the ER and Golgi. J. Biol. Chem. In press. 276:23296–23303. [DOI] [PubMed] [Google Scholar]
- Carrell, R.W., and B. Gooptu. 1998. Conformational changes and disease—serpins, prions and Alzheimer's. Curr. Opin. Struct. Biol. 8:799–809. [DOI] [PubMed] [Google Scholar]
- Casagrande, R., P. Stern, M. Diehn, C. Shamu, M. Osario, M. Zuniga, P.O. Brown, and H. Ploegh. 2000. Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Mol. Cell. 5:729–735. [DOI] [PubMed] [Google Scholar]
- Cowles, C.R., W.B. Snyder, C.G. Burd, and S.D. Emr. 1997. Novel Golgi to vacuole delivery pathway in yeast: identification of a sorting determinant and required transport component. EMBO J. 16:2769–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eakle, K.A., M. Bernstein, and S.D. Emr. 1988. Characterization of a component of the yeast secretion machinery: identification of the SEC18 gene product. Mol. Cell. Biol. 8:4098–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard, L., M. Molinari, and A. Helenius. 1999. Setting the standards: quality control in the secretory pathway. Science. 286:1882–1888. [DOI] [PubMed] [Google Scholar]
- Elrod-Erickson, M.J., and C.A. Kaiser. 1996. Genes that control the fidelity of endoplasmic reticulum to Golgi transport identified as suppressors of vesicle budding mutations. Mol. Biol. Cell. 7:1043–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger, A., M. Knop, and D.H. Wolf. 1993. Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur. J. Biochem. 218:565–574. [DOI] [PubMed] [Google Scholar]
- Friedlander, R., E. Jarosch, J. Urban, C. Volkwein, and T. Sommer. 2000. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2:379–384. [DOI] [PubMed] [Google Scholar]
- Gemmill, T.R., and R.B. Trimble. 1999. Overview of N- and O-linked oligosaccharide structures found in various yeast species. Biochim. Biophys. Acta. 1426:227–237. [DOI] [PubMed] [Google Scholar]
- Gentzsch, M., and W. Tanner. 1996. The PMT gene family: protein O-glycosylation in Saccharomyces cerevisiae is vital. EMBO J. 15:5752–5759. [PMC free article] [PubMed] [Google Scholar]
- Gentzsch, M., T. Immervoll, and W. Tanner. 1995. Protein O-glycosylation in Saccharomyces cerevisiae: the protein O-mannosyltransferases Pmt1p and Pmt2p function as heterodimer. FEBS Lett. 377:128–130. [DOI] [PubMed] [Google Scholar]
- Gething, M.J., K. McCammon, and J. Sambrook. 1986. Expression of wild-type and mutant forms of influenza hemagglutinin: the role of folding in intracellular transport. Cell. 46:939–950. [DOI] [PubMed] [Google Scholar]
- Hammond, C., and A. Helenius. 1994. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 126:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton, R.Y., R.G. Gardner, and J. Rine. 1996. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell. 7:2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty, C., S. Strahl, and K. Romisch. 2001. O-mannosylation protects mutant alpha-factor precursor from endoplasmic reticulum-associated degradation. Mol. Biol. Cell. 12:1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauri, H., C. Appenzeller, F. Kuhn, and O. Nufer. 2000. Lectins and traffic in the secretory pathway. FEBS Lett. 476:32–37. [DOI] [PubMed] [Google Scholar]
- Herscovics, A., and P. Orlean. 1993. Glycoprotein biosynthesis in yeast. FASEB J. 7:540–550. [DOI] [PubMed] [Google Scholar]
- Hiller, M.M., A. Finger, M. Schweiger, and D.H. Wolf. 1996. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 273:1725–1728. [DOI] [PubMed] [Google Scholar]
- Jakob, C.A., P. Burda, J. Roth, and M. Aebi. 1998. Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J. Cell Biol. 142:1223–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, A.E., and M.A. van Waes. 1999. The translocon: a dynamic gateway at the ER membrane. Annu. Rev. Cell Dev. Biol. 15:799–842. [DOI] [PubMed] [Google Scholar]
- Johnson, A.E., and N.G. Haigh. 2000. The ER translocon and retrotranslocation: is the shift into reverse manual or automatic. Cell. 102:709–712. [DOI] [PubMed] [Google Scholar]
- Kaiser, C. 2000. Thinking about p24 proteins and how transport vesicles select their cargo. Proc. Natl. Acad. Sci. USA. 97:3783–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmann, D.J., E.A. Epping, and W.S. Moye-Rowley. 1999. Mutational disruption of plasma membrane trafficking of Saccharomyces cerevisiae Yor1p, a homologue of mammalian multidrug resistance protein. Mol. Cell. Biol. 19:2998–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, P.S., and P. Arvan. 1998. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr. Rev. 19:173–202. [DOI] [PubMed] [Google Scholar]
- Knop, M., A. Finger, T. Braun, K. Hellmuth, and D.H. Wolf. 1996. Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J. 15:753–763. [PMC free article] [PubMed] [Google Scholar]
- Kopito, R.R., and D. Ron. 2000. Conformational disease. Nat. Cell Biol. 2:E207–E209. [DOI] [PubMed] [Google Scholar]
- Kozutsumi, Y., M. Segal, K. Normington, M.J. Gething, and J. Sambrook. 1988. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 332:462–464. [DOI] [PubMed] [Google Scholar]
- Kreis, T.E., and H.F. Lodish. 1986. Oligomerization is essential for transport of vesicular stomatitis viral glycoprotein to the cell surface. Cell. 46:929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneur, F., E.C. Gaynor, S. Hennecke, C. Demolliere, R. Duden, S.D. Emr, H. Riezman, and P. Cosson. 1994. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 79:1199–1207. [DOI] [PubMed] [Google Scholar]
- Loayza, D., A. Tam, W.K. Schmidt, and S. Michaelis. 1998. Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway in Saccharomyces cerevisiae. Mol. Biol. Cell. 9:2767–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lussier, M., A.M. Sdicu, F. Bussereau, M. Jacquet, and H. Bussey. 1997. The Ktr1p, Ktr3p, and Kre2p/Mnt1p mannosyltransferases participate in the elaboration of yeast O- and N-linked carbohydrate chains. J. Biol. Chem. 272:15527–15531. [DOI] [PubMed] [Google Scholar]
- Machamer, C.E., R.W. Doms, D.G. Bole, A. Helenius, and J.K. Rose. 1990. Heavy chain binding protein recognizes incompletely disulfide-bonded forms of vesicular stomatitis virus G protein. J. Biol. Chem. 265:6879–6883. [PubMed] [Google Scholar]
- McCracken, A.A., and J.L. Brodsky. 1996. Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 132:291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniz, M., C. Nuoffer, H.P. Hauri, and H. Riezman. 2000. The Emp24 complex recruits a specific cargo molecule into endoplasmic reticulum-derived vesicles. J. Cell Biol. 148:925–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniz, M., P. Morsomme, and H. Riezman. 2001. Protein sorting upon exit from the endoplasmic reticulum. Cell. 104:313–320. [DOI] [PubMed] [Google Scholar]
- Nakano, A., D. Brada, and R. Schekman. 1988. A membrane glycoprotein, Sec12p, required for protein transport from the endoplasmic reticulum to the Golgi apparatus in yeast. J. Cell Biol. 107:851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehls, S., E.L. Snapp, N.B. Cole, K.J. Zaal, A.K. Kenworthy, T.H. Roberts, J. Ellenberg, J.F. Presley, E. Siggia, and J. Lippincott-Schwartz. 2000. Dynamics and retention of misfolded proteins in native ER membranes. Nat. Cell Biol. 2:288–295. [DOI] [PubMed] [Google Scholar]
- Ng, D.T.W., J.D. Brown, and P. Walter. 1996. Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J. Cell Biol. 134:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, D.T.W., R.E. Randall, and R.A. Lamb. 1989. Intracellular maturation and transport of the SV5 type II glycoprotein hemagglutinin-neuraminidase: specific and transient association with GRP78-BiP in the endoplasmic reticulum and extensive internalization from the cell surface. J. Cell Biol. 109:3273–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, D.T.W., S.W. Hiebert, and R.A. Lamb. 1990. Different roles of individual N-linked oligosaccharide chains in folding, assembly, and transport of the simian virus 5 hemagglutinin-neuraminidase. Mol. Cell. Biol. 10:1989–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, D.T.W., E.D. Spear, and P. Walter. 2000. The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J. Cell Biol. 150:77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa, S., A. Hirata, and A. Nakano. 1994. Inhibition of endoplasmic reticulum (ER)-to-Golgi transport induces relocalization of binding protein (BiP) within the ER to form the BiP bodies. Mol. Biol. Cell. 5:1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa, S., S.W. Fewell, Y. Kato, J.L. Brodsky, and T. Endo. 2001. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 153:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuoffer, C., P. Jeno, A. Conzelmann, and H. Riezman. 1991. Determinants for glycophospholipid anchoring of the Saccharomyces cerevisiae GAS1 protein to the plasma membrane. Mol. Cell. Biol. 11:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg, S.C., W.P. Barz, and P. Walter. 1998. A functional GTPase domain, but not its transmembrane domain, is required for function of the SRP receptor beta-subunit. J. Cell Biol. 142:341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, G.D., and R.A. Lamb. 1990. Folding and oligomerization properties of a soluble and secreted form of the paramyxovirus hemagglutinin-neuraminidase glycoprotein. Virology. 178:498–508. [DOI] [PubMed] [Google Scholar]
- Pilon, M., R. Schekman, and K. Romisch. 1997. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 16:4540–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper, R.K., S. Bohmler, J. Bordallo, T. Sommer, and D.H. Wolf. 1997. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature. 388:891–895. [DOI] [PubMed] [Google Scholar]
- Plemper, R.K., P.M. Deak, R.T. Otto, and D.H. Wolf. 1999. Re-entering the translocon from the lumenal side of the endoplasmic reticulum. Studies on mutated carboxypeptidase yscY species. FEBS Lett. 443:241–245. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.M. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Plainview, NY.
- Shimoni, Y., T. Kurihara, M. Ravazzola, M. Amherdt, L. Orci, and R. Schekman. 2000. Lst1p and Sec24p cooperate in sorting of the plasma membrane ATPase into COPII vesicles in Saccharomyces cerevisiae. J. Cell Biol. 151:973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski, R.S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer, T., and S. Jentsch. 1993. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature. 365:176–179. [DOI] [PubMed] [Google Scholar]
- Sommer, T., and D.H. Wolf. 1997. Endoplasmic reticulum degradation: reverse protein flow of no return. FASEB J. 11:1227–1233. [DOI] [PubMed] [Google Scholar]
- Spormann, D.O., J. Heim, and D.H. Wolf. 1992. Biogenesis of the yeast vacuole (lysosome). The precursor forms of the soluble hydrolase carboxypeptidase yscS are associated with the vacuolar membrane. J. Biol. Chem. 267:8021–8029. [PubMed] [Google Scholar]
- Strahl-Bolsinger, S., M. Gentzsch, and W. Tanner. 1999. Protein O-mannosylation. Biochim Biophys Acta. 1426:297–307. [DOI] [PubMed] [Google Scholar]
- Travers, K.J., C.K. Patil, L. Wodicka, D.J. Lockhart, J.S. Weissman, and P. Walter. 2000. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 101:249–258. [DOI] [PubMed] [Google Scholar]
- Wang, Q., and A. Chang. 1999. Eps1, a novel PDI-related protein involved in ER quality control in yeast. EMBO J. 18:5972–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, C.L., S. Omura, and R.R. Kopito. 1995. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 83:121–127. [DOI] [PubMed] [Google Scholar]
- Werner, E.D., J.L. Brodsky, and A.A. McCracken. 1996. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA. 93:13797–13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiertz, E.J., D. Tortorella, M. Bogyo, J. Yu, W. Mothes, T.R. Jones, T.A. Rapoport, and H.L. Ploegh. 1996. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 384:432–438. [DOI] [PubMed] [Google Scholar]
- Wuestehube, L.J., and R.W. Schekman. 1992. Reconstitution of transport from endoplasmic reticulum to Golgi complex using endoplasmic reticulum-enriched membrane fraction from yeast. Methods Enzymol. 219:124–136. [DOI] [PubMed] [Google Scholar]
- Zhou, M., and R. Schekman. 1999. The engagement of Sec61p in the ER dislocation process. Mol. Cell. 4:925–934. [DOI] [PubMed] [Google Scholar]