

Abstract
At the time of fusion, membranes are packed with fusogenic proteins. Do adjacent individual proteins interact with each other in the plane of the membrane? Or does each of these proteins serve as an independent fusion machine? Here we report that the low pH–triggered transition between the initial and final conformations of a prototype fusogenic protein, influenza hemagglutinin (HA), involves a preserved interaction between individual HAs. Although the HAs of subtypes H3 and H2 show notably different degrees of activation, for both, the percentage of low pH–activated HA increased with higher surface density of HA, indicating positive cooperativity. We propose that a concerted activation of HAs, together with the resultant synchronized release of their conformational energy, is an example of a general strategy of coordination in biological design, crucial for the functioning of multiprotein fusion machines.
Keywords: viral fusion; influenza hemagglutinin; cooperativity; hemagglutinin interaction; inactivation
Introduction
Membrane remodeling in numerous cell biological processes is mediated by specialized fusion proteins. Most of the existing models of protein-mediated fusion suggest that the fusion site is surrounded by multiple proteins or protein machines providing conformational energy to drive the rearrangement of two lipid bilayers into one. Little is known about the mechanism by which fusion proteins coordinate their activity and also about the interactions, at present hypothetical, between fusion proteins in the plane of the membrane. These interactions must be transient and/or weak to allow the dissociation of protein clusters enclosing an expanding fusion pore. The very existence of these interactions still awaits confirmation, even for the well-characterized fusion reaction mediated by influenza hemagglutinin (HA)*(Wiley and Skehel, 1987; Skehel and Wiley, 2000).
HA is a homotrimeric envelope glycoprotein with individual monomers synthesized as a single polypeptide chain (referred to as HA0). Each monomer is cleaved by a trypsin-like protease into two disulfide-linked subunits, HA1 and HA2. Upon acidification of the endosome, this HA1–HA2 form undergoes major changes to acquire a fusion-competent conformation. In the initial HA conformation, the conserved, hydrophobic NH2-terminal peptide of HA2 (the fusion peptide) is hidden within the center of the trimeric stem (Wiley and Skehel, 1987). Low pH–dependent activation from this initial conformation to a fusion-competent one involves extrusion of the fusion peptide, i.e., its insertion into the target or viral membrane (for review see Gaudin et al., 1995), and extension of the triple-stranded, α-helical coiled-coil of HA2 together with 180° inversion of its viral, membrane-proximal part (Carr and Kim, 1993; Bullough et al., 1994; Wharton et al., 1995; Kim et al., 1998). This conformational change of HA also tilts the molecule from the normal orientation toward the membrane (Tatulian et al., 1995), relocates the HA1 subunit from its initial place at the top of HA molecule, and makes the S–S bond between HA1 and HA2 accessible to reducing agents such as dithiothreitol (DTT). This acidic form of HA is further susceptible to proteolysis by thermolysin and proteinase K (White and Wilson, 1987; Wiley and Skehel, 1987; Kemble et al., 1992).
Conformational changes in HA after low pH application take place in the absence of a target membrane and in a truncated fragment of HA, i.e., its solubilized ectodomain (Wiley and Skehel, 1987). Low pH pretreatment of an HA-expressing membrane (HA-membrane) in the absence of a target membrane causes HA inactivation, detected as a decrease in the fusion rate after the application of an additional pH pulse, in the presence of a target membrane (Puri et al., 1990). Studies of the specific HA activation and inactivation mechanisms have revealed a notable difference between HA molecules of two widely studied HA subtypes: H3 (e.g., X31 and Udorn strains) and H2 (e.g., the A/Japan/305/57 strain). Whereas X31 HA completely inactivates after brief acidification in the absence of a target membrane, Japan HA retains most of its fusogenic activity (Puri et al., 1990; Korte et al., 1999). This experiment has been interpreted as showing that X31 HA has a much faster kinetics of inactivation than Japan HA. This putative differential inactivation for the H3 and H2 subtypes has been used as a basis for revealing the pathways of protein refolding and membrane fusion (Puri et al., 1990; Ramalho-Santos et al., 1993; Korte et al., 1997; Korte et al., 1999).
Do HA refolding and membrane fusion develop at the level of the individual trimer? Available crystallographic studies of initial and final HA conformations (for review see Skehel and Wiley, 2000) did not reveal any specific protein domains that might be involved in trimer–trimer interactions. On the other hand, the notion that viral fusion is mediated by a concerted action of multiple fusion proteins is supported by numerous functional studies (Ellens et al., 1990; Gutman et al., 1993; Blumenthal et al., 1996; Danieli et al., 1996; Gaudin et al., 1996; Plonsky and Zimmerberg, 1996; Chernomordik et al., 1998; Markovic et al., 1998; Leikina and Chernomordik, 2000; but see Gunther-Ausborn et al., 2000). Thus, the key question of whether HA trimers interact with each other during conformational rearrangement and fusion has remained open.
Here, we report that triggering the conformational change in an individual HA trimer is affected by the proximity of other HAs. We modified the surface density of Japan and X31 HA and assayed the transition of HA from its initial to its low pH conformation both as the development of HA susceptibility to S–S reduction and as the digestion of the exposed fusion peptide by thermolysin. Conformational change in HA was also detected functionally as inactivation of HA by low pH pretreatment in the absence of a target membrane. As expected, Japan HA-membranes retained fusogenic activity after longer low pH incubations than did X31 HA-membranes. Our results suggest that this difference reflects slow activation, rather than inactivation as formerly thought (Puri et al., 1990; Gutman et al., 1993; Korte et al., 1999). More importantly, we show that in both slow- and fast-activating strains, the percentage of activated HA increases with an increase in HA density, indicating that HA activation involves positive inter-trimer cooperativity. We propose that this concerted activation of adjacent proteins, which allows synchronized release of their conformational energy, is the mechanism by which multiple fusion proteins coordinate their activity at the fusion site.
Results
General approach
Upon acidification, HA molecules leave their initial, metastable conformation and undergo a transition toward their lowest energy state. Loss of the initial HA conformation was assayed after reneutralization. Hereafter, all HA conformations different from the initial one either in sensitivity to proteases and DTT or in ability to mediate fusion at low pH will be referred to as the low pH–activated conformations. We reasoned that if the conformational change of an HA trimer is somehow affected by its interactions with adjacent trimers, then the efficiency of HA activation will depend on the density of HA. To evaluate whether the observed dependencies are specific for the HA of a particular strain of influenza or instead reflect the general properties of HA activation, we first studied activation for two divergent influenza subtypes. Then, we focused on activation as a function of HA density, using a number of approaches in a number of systems.
Activation of Japan HA is much slower than that of X31 HA
In our fusion inactivation assay, cells expressing Japan or X31 HA were first treated with a low pH pulse in the absence of a target membrane (the activating pulse), and then were reneutralized and incubated with red blood cells (RBCs) for 15 min. To trigger fusion, the second low pH pulse (the triggering pulse) was necessary, as RBCs bound to HA-expressing cells (HA-cells) treated solely with the activating pulse gave no fluorescent dye redistribution. An irreversible conformational change of HA molecules at low pH leads to a complete loss of their fusogenic activity (Skehel and Wiley, 2000). Therefore, a higher degree of HA activation during the activating pulse resulted in a more profound HA inactivation, and thus a lower extent of fusion after the triggering pulse. An activating pulse at pH 4.9 for 10 min activated, and then inactivated, most of the available X31 HA molecules, giving no fusion after a 2-min fusion-triggering pulse of pH 4.9 (Fig. 1 A, closed symbols). In contrast, for Japan HA, the same activating pulse (pH 4.9, 10 min) had no effect on fusion observed after a 2-min triggering pulse of pH 4.9. The lack of fusion inhibition for Japan HA can be explained either by a low level of HA activation during the activating pulse or, as suggested by Puri et al. (1990), a very slow inactivation of Japan HA. If the former were true, one would expect to detect Japan HA inactivation if the pH of the fusion-triggering pulse were shifted from 4.9 to a suboptimal 5.3. Due to an excess of fusion-competent HA molecules, fusion is very robust at pH 4.9, and thus the extent of fusion is not sensitive to small changes in the number of HA molecules capable of mediating fusion. In contrast, the number of activated HA molecules available for fusion at pH 5.3 is significantly lower, and as a result, the system is much more sensitive to variation in pH. (Fusion at pH 5.3 is also more sensitive to membrane lipid composition [Chernomordik et al., 1997] and temperature [Melikyan et al., 1997a].) Thus, fusion induced by a pH 5.3 pulse is expected to be more sensitive to a small loss of fusion-competent HA due to prior inactivation, than is fusion induced by a pH 4.9 pulse. Indeed, increasing the pH of the fusion-triggering pulse to 5.3 (Fig. 1 A, open symbols) made inactivation easily detectable. The decline in fusion at prolonged low pH pretreatment proceeded similarly for X31 and Japan HA, indicating that although low pH activates X31 HA more efficiently than it does Japan HA, the subsequent inactivation rates are not notably dissimilar.
Figure 1.
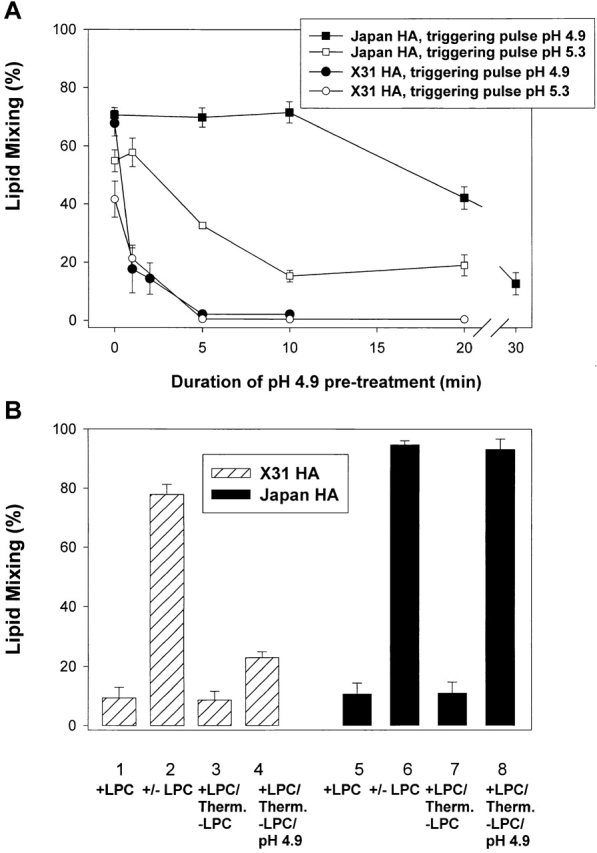
X31 and Japan HA differ in activation rather than inactivation rates. (A) The time course of Japan (squares) and X31 HA (circles) activation/inactivation at low pH in the absence of target membrane. HA-cells were treated with a pH 4.9 activating pulse for 1 to 30 min. Then, after RBC binding, cells were treated with a fusion-triggering pulse of pH 4.9 for 2 min (closed symbols) or pH 5.3 for 5 min (open symbols). A higher degree of HA activation during the activating pulse resulted in a lower extent of lipid mixing after the triggering pulse. (B) X31 (bars 1–4) or Japan HA-cells (bars 5–8) with bound RBCs were incubated for 5 min in pH 4.9 medium containing LPC. Although no lipid mixing was observed in the presence of LPC (bars 1 and 5), its removal completely restored fusion (bars 2 and 6). In the experiments represented in bars 3, 4, 7, and 8, cells at the LPC-arrested fusion stage were treated with thermolysin to cleave low pH–activated HA. Lipid mixing was assayed either directly after LPC removal (bars 3 and 7) or after application of an additional 5-min pulse of pH 4.9 (bars 4 and 8). Points are means ± SE, n > 3.
Further evidence that Japan and X31 HA differ in their efficiency of low pH activation came from functional experiments in which the first low pH pulse was applied in the presence of a target membrane. HA-cells with bound RBCs were exposed to low pH in the presence of lysophosphatidylcholine (LPC), which reversibly blocks fusion (Fig. 1 B). Then, cells were treated with thermolysin to cleave activated HA molecules. Removal of LPC at this stage did not result in fusion, confirming that most of the low pH–activated HA molecules in the contact zone were cleaved by the enzyme. The remaining fusogenic activity of HA was evaluated after a second low pH pulse. The majority of X31 HA molecules, but not Japan HA molecules, were restructured after the activating pulse and cleaved by thermolysin, as deduced from the decrease in fusion (Fig. 1 B, bar 4 vs. 2 and bar 8 vs. 6). Therefore, a single low pH pulse activates only a small portion of the available Japan HA, whereas most of the X31 HA is being activated. Such an activation–thermolysin cleavage cycle in Japan HA can be repeated at least twice without a measurable decrease in the extent of fusion. Similar results were obtained when fusion was blocked by lowering the temperature to 4°C instead of by LPC application. In brief, functional experiments indicated that the rate of activation of X31 HA significantly exceeds that of Japan HA.
This increased efficiency of X31 HA activation was confirmed by cell surface enzyme-linked immunosorbent assay (CELISA), showing an increase in antifusion peptide antibody binding to acidified X31 with no measurable increase for Japan HA. CELISA-derived binding ratios of low to neutral pH HA after a 10-min application of pH 4.9 at 37°C were 2.01 ± 0.07 for X31 and 1.03 ± 0.16 for Japan, n = 3. Neither functional assays nor CELISA allowed a quantitative evaluation of the percentage of HA molecules activated under different conditions. To measure this percentage, we monitored HA activation by means of DTT-induced HA1–HA2 S–S bond reduction and HA1 release (Graves et al., 1983). The percentage of activation was detected by Western blotting under nonreducing conditions (shown in Fig. 2 A for viral particles) either as a loss in the intensity of the HA1–HA2 band or as the ratio of HA2 to the sum of HA2 and HA1–HA2 band intensities. (Both calculation methods gave statistically indistinguishable results.) A 10-min application of pH 4.9 resulted in almost complete disappearance of the HA1–HA2 band for X31 HA, compared with a minor loss of Japan HA1–HA2 (73–85% of X31 vs. 10–20% of Japan for HA-cells). More efficient activation of X31 HA than Japan HA was also found for a membrane-free preparation of bromelain-cleaved HA ectodomain (i.e., 76% vs. 31%) and for viral particles (80% vs. 40%; data in Fig. 2 A). Thus, our biochemical experiments with membrane-anchored HA and soluble HA ectodomain confirmed a lower efficiency of activation for Japan HA. The alternative possibility that low pH forms of X31 HA inactivate faster and, in addition, are more sensitive than Japan HA to both DTT and thermolysin, although feasible, seems unlikely.
Figure 2.

Activation of X31 and Japan HA assessed by Western blotting as exposure of HA1–HA2 S–S bond. (A) Japan or X31 HA viral particles (the two left and two right panels, respectively) were acidified (pH 4.9, 10 min, 22°C) or not and reduced with DTT. Minor loss of the HA1–HA2 band for Japan is in apparent contrast to X31 HA1–HA2 disappearance. (B) Time course of activation for Japan and X31 HA expressed in cells. Cells were incubated at 22°C in pH 4.9 medium for a given time interval in the presence of protease inhibitors.
X31 HA and Japan HA have a similar pH dependence for activation (unpublished data; see also Korte et al., 1999). However, the rate of activation was notably different for these two strains (Fig. 2 B). For instance, a 10-min application of pH 4.9 to X31 HA-cells transformed 84% of the HA into a DTT-susceptible form. Longer exposure of X31 HA to pH 4.9, up to 1 h, did not notably increase the level of activation. In contrast, 1 h of acidification of Japan HA-cells yielded only 22% activated HA. Very slow and inefficient Japan HA activation was confirmed in viral particles, where the percentage of activated HA slowly increased with the time of incubation at pH 4.9 (51.2%, 61%, and 82% for 10 min, 6 h, and 24 h, respectively). Leveling off the activation at pH 4.9 after the first 30 min, which was observed for Japan HA-cells and not observed for Japan virus, can reflect a deterioration of the cells caused by long, low pH treatments, visible as obvious changes in cell morphology.
In brief, low pH–triggered activation of Japan HA is notably slower than that of X31 HA. This difference was observed for the proteins expressed in the stable cell lines (HAb2 and HA300a), in viral particles, and in solubilized HA ectodomain. In addition, the higher efficiency of Japan HA activation in influenza virus (vs. that observed in a stable cell line expressing HA) was consistent with the hypothesis that the rate of HA activation increased at higher HA surface density, which is characteristic for viral particles.
HA activation increases with the increase in HA surface density
Both slow- and fast-activating strains of HA were next used to study the role of trimer–trimer interaction in HA activation. If low pH–dependent activation develops at the level of individual trimers, then increasing the number of HA trimers at the cell surface should not change the percentage of activated molecules (i.e., the ratio of activated HA to total HA). In contrast, if HA activation involves positive cooperativity, the efficiency of activation should increase with HA density.
We increased surface density of Japan HA by growing HAb2 cells in the presence of different concentrations of sodium butyrate (NaBut). Flow cytometry indicated that the surface density of HA at all NaBut concentrations varies broadly between cells. However, this assay and two other assays, trypsinization and surface biotinylation, confirmed that preincubation with NaBut shifts the distributions to higher HA densities (Fig. 3 A). The extent of HA expression promotion at 5 mM NaBut was in excellent agreement with that reported in Danieli et al. (1996) (i.e., a 4.9-fold increase in HA from 0 to 5 mM NaBut). The reason for the quantitative discrepancy between our three assays for 9 mM NaBut is unclear to us. Importantly, none of the conclusions in the further discussion depend upon the exact differences in the surface densities.
Figure 3.
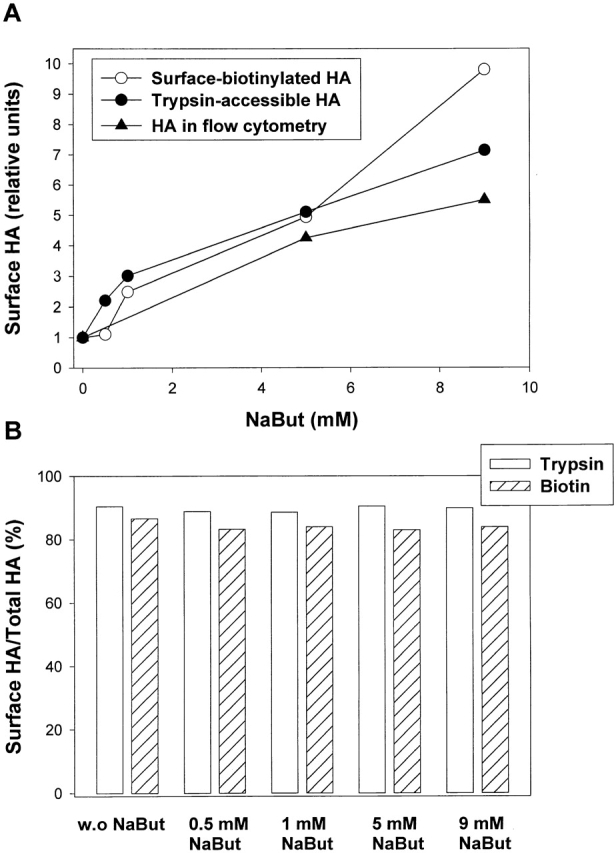
NaBut-induced increase in the surface density of Japan HA. (A) Increase in the surface HA expression after preincubation with NaBut was detected by biotinylation (open circles) and trypsinization (closed circles) of surface HA and using flow cytometry (closed triangles). (B) The percentage of surface HA among total cellular HA for cells treated with 0–9 mM NaBut was determined by cell-surface biotinylation and trypsinization. Note that the majority of HA is expressed at the cell surface.
The NaBut-induced changes in the surface HA expression were proportional to those in the total cellular HA expression, as evidenced by the constant percentage of total HA (83–90%) that was accessible for trypsin cleavage and surface biotinylation (Fig. 3 B). Thus, NaBut-induced changes in the total HA expression provide reliable measure for the changes in the surface HA.
Increasing surface density of HA notably accelerated HA activation (assayed by DTT susceptibility; Fig. 4 A) supporting the hypothesis of HA interaction during activation. Furthermore, for a low pH pulse of a given duration (i.e., 10 min), a higher percentage of HA molecules become activated at progressively higher levels of HA expression (Fig. 4 B). Note that HA density in HA-cells at high NaBut concentrations (e.g., 12.6 × 103 HA/μm2 at 5 mM NaBut; Danieli et al., 1996) approached that in viral particles (15–30 × 103 HA/μm2). In cells with the highest level of Japan HA expression, the level of activation (38%) approached the level observed in viral particles (40%) (Fig. 4 B).
Figure 4.
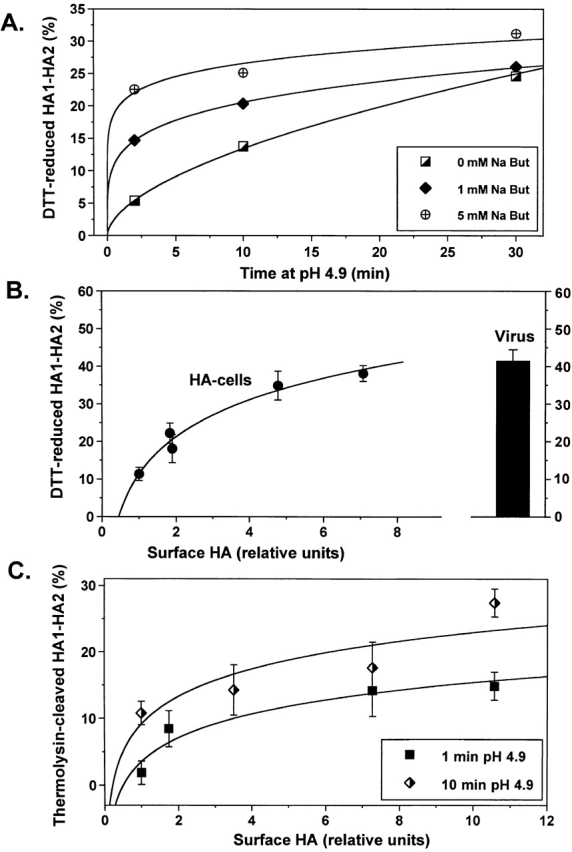
Cooperative activation of Japan HA. Japan HA activation was monitored by Western blotting as HA sensitivity to DTT (A and B) or thermolysin (C). (A) Time course of HA activation for different HA expression levels. HA expression was varied by preincubation of HA-cells with 0, 1, or 5 mM NaBut. The duration of the pH 4.9 pulse ranged from 1 to 30 min (B) Efficiency of Japan HA activation after a 10-min pulse of pH 4.9 as a function of the surface density of HA. Relative surface density of HA after cell preincubation with 0 to 9 mM NaBut was assayed by measuring the changes in total cellular HA and normalized by that in NaBut-untreated HAb2 cells. Points are means ± SE, n = 4. (C) The percentage of low pH–activated HA molecules that reached the early stage of fusion peptide exposure was assayed by means of thermolysin cleavage. HA expression in cells was altered by pretreatment with 0 to 9 mM NaBut. Cells were incubated at pH 4.9 for 1 or 10 min, reneutralized, and treated with thermolysin to cleave exposed fusion peptides. Cleavage of activated HA resulted in a decrease in the HA1–HA2 band, which was normalized by the pH 7.4 band taken as 100%.
Does the cooperative activation stage precede or follow the exposure of the fusion peptide, an early sign of activation (White and Wilson, 1987; White, 1996)? To address this question, we took advantage of the fact that exposed fusion peptides can be cleaved by thermolysin. Cells with different levels of Japan HA expression were treated with a 1- or 10-min low pH pulse immediately followed by thermolysin application (Fig. 4 C). Similar to our results with DTT, we found that the percentage of thermolysin-cleaved HA detected as a loss of HA1–HA2 band intensity increases with increasing HA expression, indicating that HA density affects an early stage of HA activation.
Was the dependence of activation upon HA density conserved among HA subtypes? Since NaBut did not increase HA density in HA300a-cells, we used different approaches to vary the expression levels. These experiments also test whether cooperativity is artifactually dependent upon any particular way of boosting surface density. All strains and approaches yielded the same essential result: positive cooperative activation. In one approach, we varied the concentration of trypsin used to cleave X31 HA0 into the activation- and fusion-competent HA1–HA2 form and found higher levels of activation with higher numbers of activatable HA molecules per cell (Fig. 5 A). Note that the interpretation of this particular experiment is complicated by the possibility that mild trypsinization may result in partial cleavage of HA, such that some monomers in the same trimer may be present in the HA0 and some in the HA1–HA2 form.
Figure 5.
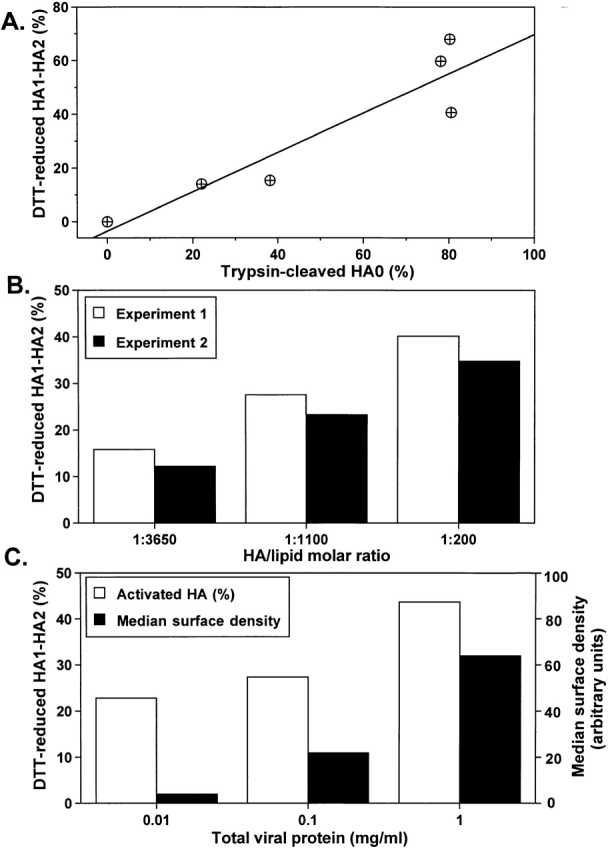
Cooperative activation of X31 and Udorn HA. In all experiments, activation was monitored by Western blotting as HA sensitivity to DTT. (A) The surface density of activation-competent X31 HA molecules in HA-cells was varied by altering trypsin concentration (1–10 μg/ml). The increase in the percentage of the HA1–HA2 form, out of total HA, gave the increase in percentage of activation. (B) X31 HA reconstituted in virosomes at a higher HA to lipid ratio, and thus at a higher surface density, activates to a higher level. The percentage of HA activated after a 10-min pulse of pH 4.9 is shown for two independent experiments (open and closed bars). (C) The surface density of Udorn HA was altered by varying the multiplicity of infection of CV1 cells with SV-40 recombinant virus carrying the HA gene. Increases in the median cell surface fluorescence assessed by FACS® analysis (closed bars) confirmed an increase in the average amount of HA per cell in the specific range of the recombinant virus concentrations (0.01–1 mg/ml total viral protein). Open bars represent the percentage of activated HA molecules after a 10-min pulse of pH 4.9.
In another approach, and to exclude the possibility that other membrane proteins assist in activation at different HA levels, we reconstituted X31 HA into virosomes at different HA to lipid ratios. Once again, the percentage of activated HA molecules increased with increasing HA density (Fig. 5 B). In control experiments, we verified that the change in HA to lipid ratio alters the HA surface density in virosomes rather than the ratio of virosomes to protein-free liposomes. Virosomes formed with different ratios of HA and lipid were characterized by ultracentrifugation in a sucrose density gradient. For each of the virosome preparations, HA and lipid peaked in the same fraction of the gradient, with sucrose densities of 1.12 and 1.05 g/cm3 for HA to lipid ratios of ∼1:200 (undiluted virosomes in which viral HA was reconstituted with no exogenous lipid added) and ∼1:3,650 (lipid-diluted virosomes). These results indicated that the decrease in activation efficiency for virosomes with lower HA to lipid ratios reflects the decrease in HA concentration. Quantitative analysis of these data is complicated by the possibility that some HA molecules can be clustered in the virosome membrane rather than be homogeneously distributed along the membrane according to the average surface density, and by the fact that some HA may have the wrong orientation.
In still another approach, we used Udorn HA, which is 96.7% homologous to X31 HA. In this case, HA density was altered by varying the multiplicity of CV1 cell infection with SV-40 recombinant virus carrying the Udorn HA gene. FACS® analysis confirmed that in a specific concentration range of recombinant SV-40 virus (0.01–1 mg/ml total viral protein), the number of HA molecules per cell was higher at a higher dose of the virus. Once again we found that at higher HA densities, a higher percentage of HA become activated (Fig. 5 C).
As discussed above, one can evaluate the rate and extent of HA activation by measuring its subsequent inactivation. The relationship between HA density and activation established in biochemical assays was confirmed with functional experiments in which we measured HA inactivation. Boosting HA expression by NaBut (a threefold increase in the surface density of HA, as estimated based on HA sensitivity to DTT) notably accelerated Japan HA activation/inactivation (Fig. 6). The HA activation/inactivation was also accelerated in X31 HA–cells with a higher density of trypsin-cleaved HA. For HA-cells pretreated with two different concentrations of trypsin, we studied the effect of an activating pulse (pH 4.9, 2 min) on fusion between HA-cells and bound RBCs after a fusion-triggering pulse (pH 4.9, 5 min). For X31 HA-cells pretreated with 5 μg/ml trypsin (10 min, 22°C), the activating pulse lowered the fusion extent from 48.0 ± 4.7 to 28.4 ± 5.4%, n ≥ 3. In contrast, the same activating pulse did not affect fusion if the cells were treated with only 0.5 μg/ml trypsin (30.9 ± 5.2 vs. 31.6 ± 6.1%, n ≥ 3). Here, as in all other experimental systems we studied, the more HA available for activation, the higher the percentage of activated HA molecules.
Figure 6.

Functional assay confirms the acceleration of activation/inactivation upon increase in the level of HA expression. Japan HA-cells incubated with 0 (closed circles) or 2 mM NaBut (open circles) were pretreated with an activating pulse, pH 4.9. Next, RBCs were added, and fusion was triggered by a 10-min pulse of pH 5.2. As in biochemical experiments, the efficiency of HA activation is higher for the cells with an HA expression level increased by NaBut.
HA activation at lowered surface densities is promoted by the target membrane
Although we performed our biochemical experiments in the absence of RBCs, given that HA-cells form clusters, there was potentially a fraction of HA that could interact with the membrane of an adjacent cell upon low pH application. Thus, one might hypothesize that an increase in the activation efficiency at a high density of HA was mediated by HA interactions with the target membrane. For instance, concerted insertion of fusion peptides of different trimers into the target membrane has been proposed by Danieli et al. (1996) as an explanation for cooperativity of fusion. To test this possibility, we plated Japan HA-cells with different HA concentrations as single cells on poly-l-lysine–treated flasks (0.5 mg/ml, 30 min, 22°C). For such single cells (i.e., in the absence of a target membrane), we again found that the percentage of activated HA molecules was higher at higher HA densities (Fig. 7 A). In a parallel experiment, Japan HA-cells, also plated as single cells, were covered with fluorescently labeled liposomes containing gangliosides (Fig. 7 A). An estimate based on the level of fluorescence associated with each cell after incubation with liposomes at 4°C indicated that the cell surface was saturated with liposomes. The percentage of activated HA molecules under these conditions was independent of HA density and corresponded to that observed in the absence of a target membrane at the highest level of HA expression. Thus, the effect of the target membrane on HA activation was the same as that obtained by increasing HA density.
Figure 7.

A target membrane enhances activation to the level observed at high HA density. (A) Closed circles: Japan HA-cells at different HA densities with saturating concentrations of liposomes. Japan HA-cells with levels of HA expression altered by pretreatment with 0 to 9 mM NaBut were plated as single cells on poly-l-lysine–treated flasks and incubated at 4°C with saturating concentrations of liposomes. HA expression was normalized by that in NaBut-untreated HAb2 cells. After removal of unbound liposomes, cells were treated with a 10-min pulse, pH 4.9 (22°C). Open circles: control, analogous cells in the absence of liposomes. Points are means ± SE, n= 4. (B) The cartoon illustrates HA enrichment in the contact zones between an HA-cell and liposomes. HA1-receptor interaction induces HA concentration in the contact zone. Therefore, as long as the contact area is less than the total area of HA-membrane, effective HA density and hence the level of HA activation in the presence of a target membrane exceed those in the absence of a target membrane. The graph represents a theoretical curve based on the estimate of HA enrichment and activation in the HA-cell–target membrane contact region for cells with low HA density. The expected level of activation, shown as a function of the ratio of the contact zone area to the total HA-membrane area, α, approaches the activation level observed for cells with high HA density in the absence of a target membrane.
Discussion
Activation of influenza HA can cause the membrane fusion step of viral entry only if this activation occurs in the right place and at the right time. Premature activation and discharge of HA trimers, whether in the absence of a suitable target membrane or before assembly of the multi-trimer machine thought to be needed for fusion (Ellens et al., 1990; Gutman et al., 1993; Blumenthal et al., 1996; Danieli et al., 1996; Gaudin et al., 1996; Plonsky and Zimmerberg, 1996; Chernomordik et al., 1998; Markovic et al., 1998; Leikina and Chernomordik, 2000), would be detrimental to viral entry because it would result in HA inactivation and hence depletion of the available pool of fusion-competent proteins. Here, we show that the degree of HA activation rises with increasing HA surface density, and we conclude that HA activation exhibits positive cooperativity. This finding suggests that the mechanism by which adjacent HA molecules effectively synchronize the release of their conformational energy is through positive cooperativity. The involvement of inter-trimer interactions in HA activation is conserved between the fast-activating H3 and the slow-activating H2 influenza subtypes. Below, we discuss the specific stages of activation that are dependent on the trimer–trimer interactions, and the general role of concerted mechanisms in fusion complexes and other multimeric protein machines.
Conformational change in HA depends on trimer–trimer interactions
As shown above, the propensity of HA to restructure into its low pH conformation depends on interactions between cleaved HA trimers capable of undergoing such conformational changes. These interactions may involve different domains of the HA molecule. For instance, low pH forms of the HA ectodomain interact by their fusion peptides, as can be inferred from the formation of rosette structures (Ruigrok et al., 1988). Moreover, fusion peptide interaction among neighboring HAs was hypothesized to be responsible for a measurable decrease in lateral mobility of HA after activation (Gutman et al., 1993). Additional or alternative mechanisms of interaction at low pH might involve the kinked regions of HA2 (residues 106–112), which are responsible for the aggregation of large, membrane-bound polypeptide fragments of HA2 (residues 1–127) at low pH (Kim et al., 1998). It is also possible that the rate of HA refolding does not depend upon direct trimer–trimer interaction but rather depends on a number of adjacent trimers simultaneously interacting with the membrane in which they are anchored. For instance, multiple low pH–activated HAs might act together in inducing local bending of the viral membrane, thus bridging the gap between the virion and the target cell (Kozlov and Chernomordik, 1998). Such HA-generated membrane bending can hypothetically facilitate the activation of yet nonactivated trimers. Since positive cooperativity in activation was observed in the absence of a target membrane, it apparently does not require HA–target membrane interaction.
The effect of the target membrane
Although the presence of a target membrane was not required for cooperative activation of HA, membrane contacts increased the level of low pH–triggered activation at low surface densities of the protein. Since cooperativity was detected in the absence of a target membrane, one may argue that concerted HA activation is involved in the inactivation rather than in the fusion process. This argument would imply that HA–target membrane interaction affects the refolding of individual trimers by lowering the energy barrier of HA activation. For instance, the conformational change in HA may proceed beyond transient exposure of the fusion peptide only if the exposed peptides can interact with each other or if they can insert into the target membrane. This implies that the fusion peptide reaches the target membrane at the early stage of HA refolding (Stegmann et al., 1990), rather than being delivered to the target membrane by extension of the central coiled-coil core of HA in a major and irreversible rearrangement of the protein (Carr et al., 1997). Note, however, that this scenario does not explain why the efficiency of activation at a low density of HAs in the presence of liposomes coincides with that at a high density of HAs in the absence of liposomes.
The simplest and, we believe, the most natural interpretation of the promotion of HA activation in the presence of the target membrane to the level observed at the highest HA densities is an enrichment of HA molecules in the contact zone due to HA1–receptor binding (Mittal and Bentz, 2001). k, the dissociation constant of HA1–sialic acid of ∼3 mM (Sauter et al., 1992), can be renormalized to k′ ≅ 100 μm−2 for HA and receptor concentration in the membrane (Leikina et al., 2000). Even though we know that the entire cell surface was covered with liposomes, the specific geometry of liposome–cell contact, and hence also the percentage of the cell membrane in close contact with liposomes, is not known. If the ratio of the contact zone area to the total HA membrane area equals α, the membrane concentration of receptor-bound HA (C HA-R) can be described by the following equation:
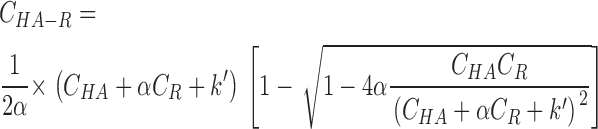 |
where C HA and C R stand for total membrane concentrations of HA (e.g., 2,500 μm−2 in HAb2 cells; Danieli et al., 1996) and receptors (5 mol% or ∼80,000 μm−2). Since the radius of HA trimer is ∼4 nm (Wiley and Skehel, 1987), the maximal membrane concentration of HA in the cell–liposome contact zone was limited to 20,000 μm−2. If more than 15% of the cell membrane is in the close contact zone (α ≥ 0.15), more than 90% of the HA molecules will be assembled in the contact zones, leading to significant enrichment of HA. Taking into account the experimentally observed dependence of activation on the density of HA (Fig. 7 A, open circles), for α varying from 0.1 to 0.9, the percentage of activated HA is expected to be at least twofold higher than in the absence of a target membrane (theoretical curve in Fig. 7 B). Therefore, only at the highest surface densities of HA will the efficiency of its activation in the absence of a target membrane reach the level normally observed in the contact zones.
Concerted HA activation at the fusion site
We hypothesize that at an acidic pH, individual HA trimers first establish a transient early state (depicted as yellow in Fig. 8 B). This stage might involve a limited relocation of HA1 tops and exposure of the fusion peptide. For individual HA trimers, this state is too short-lived to allow detection by any of our assays. Interaction between adjacent HA trimers increases the lifetime of this early state for trimers physically next to the activated ones and promotes their transition to the irreversible lowest energy state (Fig. 8, C and D, orange). As a consequence of such positive cooperativity, activation spreads among adjacent HAs leading to the synchronized release of HA conformational energy by neighboring trimers assembled around the fusion site. The probability of interaction between two HA trimers, and thus their activation, increases with the increase in the local surface density of HA, for instance in the contact zone or membrane domains (e.g., “rafts;” Simons and Ikonen, 1997) enriched in HA molecules. Note that both the presence of a target membrane and HA enrichment in microdomains are expected to significantly affect HA activation only at low average surface density of HA. In fact, disruption of raft microdomains by cholesterol depletion does not affect the fusion phenotype observed at the relatively high levels of HA expression achieved with either the vaccinia virus or the SV-40 transfection systems (Armstrong et al., 2000; Melikyan et al., 2000).
Figure 8.

Schematic diagram showing the hypothetical mechanism of cooperative activation of HA at low pH. Low pH application triggers restructuring of HA trimers from initial conformation (depicted as blue) to an early, transient activated form (yellow) followed by the final, lowest energy conformation (orange). (A) Membrane-anchored HA trimers before low pH application. (B–D) After acidification, inter-trimer interactions promote transition from a transient activated form of HA to the lowest energy protein conformation, and increases the probability of activation for native HA molecules proximal to activated ones. Activation consequently spreads among neighboring HAs (red arrows). At a high local density of HA, such concerted activation leads to the synchronized release of the conformational energy by multiple trimers assembled around the fusion site.
Our experimental approach detected the interaction of neighboring HA trimers only at low HA densities, when the scarcity of these interactions limit the rate of HA activation. Nonetheless, these results strongly argue that low pH–triggered conformational changes of HA at higher surface densities, such as in the viral envelope, also involve inter-trimer interactions. Concentrating HA molecules in the contact zone and accelerating activation by cooperativity, together ensure that the multiple HA trimers required for a proper fusion complex activate synchronously.
Conclusions
The opening of a fusion pore has been hypothesized to involve the interaction of multiple HA molecules that form a fusion complex. If indeed membrane rearrangements in fusion require simultaneous energy release by the conformational change of several HA trimers, a mechanism that minimizes dissipation of this energy by synchronizing the refolding of HA molecules at the fusion site is needed. Our work suggests that this synchronization of the conformational change of multiple HA trimers involves their concerted activation, such that interaction between adjacent HAs acts to effectively lower the energy barrier separating the initial metastable state from the final low-energy conformation. This mechanism lowers the risk that the first activated trimer at the contact site would already be discharged by the time the next trimer starts its irreversible restructuring. We speculate that this mechanism of concerted activation of individual proteins might optimize the activation potential of viral fusion proteins, lower the probability of their premature activation, and be of crucial importance for the assembly of a functional, multiprotein fusion machine. It is conceivable that the mechanism described here for HA activation applies to other multimeric complexes that operate in the plane of the membrane, such as synaptic signaling (Keleshian et al., 2000) or immune complexes.
Materials and methods
Materials
Japan and X31 viral strains were purchased from Charles River Laboratories, where they were propagated in the allantoic cavity of specific pathogen-free eggs and subsequently purified on a sucrose gradient. SV-40 recombinant virus with the Udorn HA gene was a gift from Dr. R. Lamb (Northwestern University, Evanston, IL). Rabbit polyclonal anti-X31 HA serum was directed toward the COOH-terminal portion of HA1, whereas anti-Japan HA rabbit serum targeted the fusion peptide (Covance Laboratories, Inc.). Monoclonal HC67 antibody (Daniels et al., 1983) was a gift from Dr. J.J. Skehel (The National Institute for Medical Research, London, UK). Anti-HA monoclonal antibody FC-125 was a gift from Dr. Thomas J. Braciale (University of Virginia, Charlottesville, VA). Goat anti–rabbit IgG conjugated with horseradish peroxidase or alkaline phosphatase was purchased from Pierce Chemical Co. ECL and chemifluorescence substrates were obtained from Amersham Pharmacia Biotech. Immobilon-P filters and protease inhibitor cocktail were obtained from Millipore and Boehringer, respectively. DTT was purchased from ICN Biomedicals. The trypsin from a bovine pancreas, neuraminidase from Clostridium perfringens, thermolysin (type X, P1512), lipid-soluble probe, PKH26, and disialoganglioside GD1a were purchased from Sigma-Aldrich. All other lipids were purchased from Avanti Polar Lipids, Inc.
Cells
HAb2 cells constitutively expressing Japan HA (Doxsey et al., 1985) and HA300a cells constitutively expressing X31 HA (Kemble et al., 1993) were cultured as previously described. CV1 cells infected with SV-40 recombinant virus containing the Udorn HA gene were cultured as described in Melikyan et al. (1997b). All HA-cells were prepared for fusion as described in Chernomordik et al. (1998). HA expressed at the cell surface was cleaved from HA0 to the fusion-competent HA1–HA2 form by trypsin (5 μg/ml, 10 min at 37°C, if not stated otherwise). RBCs were labeled with a fluorescent lipid PKH26 (Chernomordik et al., 1998). To modify HA density, the following approaches were used: (1) treatment of HAb2-cells with 0 to 9 mM NaBut 24 h before the experiment, producing an increase in HA expression, in agreement with Danieli et al. (1996); (2) variation of the conditions of trypsinization (1–10 μg/ml), altering the ratio of the HA0 form to the HA1–HA2 form; (3) variation of the multiplicity of infection with SV-40 recombinant virus carrying the Udorn HA gene; and (4) use of reconstituted viral envelopes with different protein to lipid ratios.
Measuring HA activation/inactivation by SDS-PAGE and Western blotting
HA activation was assayed by reducing the HA1–HA2 S–S bond, which is accessible only in the low pH HA conformation (Graves et al., 1983; Wiley and Skehel, 1987). Trypsinized HA-cells were incubated in citric acid–acidified PBS. Next, 20 mM DTT (20 min at 27°C, pH 7.4) was applied to release HA1 from the membrane-anchored HA2 subunit of the low pH HA. The free SH groups were alkylated by a brief wash with 50 mM sodium iodoacetamide in PBS. No additional release of HA1 was observed when the DTT concentration was raised above 20 mM (unpublished data). To study HA activation on viral particles, a viral suspension containing 1 mg/ml total protein was acidified to pH 4.9 at 22°C for 10 min, if not stated otherwise, and then neutralized to pH 7.4 and reduced with DTT. Next, the virus was alkylated and precipitated by centrifugation at 80,000 g for 1 h. In some experiments, acidified HA-cells were incubated with thermolysin (0.05 mg/ml, 10 min at 22°C) to cleave the fusion peptide of low pH HA (Wiley and Skehel, 1987). After treatment, reduced cells and viral particles were lysed in nonreducing SDS-PAGE lysis buffer (50 mM Tris-HCl, pH 7.5; 1.5% SDS; 50 mM sodium iodoacetamide; 5 mM EDTA; 1 mM AEBSF; 100 μM leupeptin; 100 μM 3,4 dichloroisocoumarin; 10% glycerol; 0.01% bromphenol blue) for 5 min with shaking, and then the mixture was boiled for 5 min. Release of the HA1 subunit or the fusion peptide in viral or cellular preparations was detected by SDS-PAGE. In quantitative Western blot analysis, proteins blotted to Immobilon-P filters were incubated in rabbit polyclonal serum (1:500 or 1:2,500) followed by goat anti–rabbit IgG conjugated with alkaline phosphatase (1:14,000). After incubation with enhanced chemifluorescence substrate, dried blots were scanned and quantified on a Molecular Dynamics scanner with the ImageQuant software package (Molecular Dynamics). Japan HA activation is presented as a ratio of HA2 to total band intensity within the sample lane, in which the level of neutral pH cleavage was subtracted from that produced in the low pH treatment. X31 HA activation was calculated as a ratio of the low pH HA0 band to the pH 7.4 HA0 band, which is taken as 100%. Compared with each other for the same strain, either X31 or Japan, the two calculation methods gave statistically indistinguishable results.
HA expressed at the cell surface is accessible to trypsin cleavage (Clague et al., 1991). Thus, to determine the ratio of surface HA to total HA we treated HA-cells with trypsin (5 μg/ml, 10 min, 37°C; our standard trypsinization protocol, see above) and measured the loss of the uncleaved HA0 form by Western blotting. The percentage of cleaved, and thus surface-expressed, HA (65–80% and 90% of the total HA for X31 HA-cells and Japan HA-cells, respectively) did not increase when the trypsin concentration was increased to 10 μg/ml. Thus, we assume that all the surface HA is in the HA1–HA2 form under the conditions of our experiments (if not stated otherwise). An alternative way to determine surface density of HA was by cell surface biotinylation. Cell surface labeling of 0–9 mM NaBut-treated HA-cells was performed with 0.5 mg/ml EZ-Link Sulfo NHS-Biotin (Pierce Chemical Co.) for 30 min at 22°C. Labeled cells were lysed in the buffer containing 20 mM sodium phosphate buffer, pH 7.5, 500 mM NaCl, 0.1% SDS, 1.5% Triton X-100. The lysate was incubated with the UltraLink Immobilized Streptavidin Plus (Pierce Chemical Co.) for 1 h at 4°C. Immobilized Streptavidin with bound proteins was washed four times in lysis buffer, and then biotin-labeled proteins were liberated from Streptavidin by boiling in SDS-containing sample buffer and analyzed for HA content by Western blotting. In parallel, we also determined the amount of nonbiotinylated, Streptavidin-unbound HA in the supernatant to estimate the percentage of surface HA among total cellular HA. To minimize the NaBut effect of slowing the rate of cell division (a 1.5-fold higher cell count in 0 mM vs. 9 mM NaBut 24 h after the treatment), each SDS-PAGE sample was normalized by the total protein content.
For Japan HA-expressing cells, the percentage of surface HA among the total cellular HA did not vary when the level of HA expression was boosted by NaBut. Therefore, the intensity of the HA1–HA2 band in lysates of ∼5 × 105 HAb2 cells treated with different concentrations of NaBut, normalized by that of untreated HAb2 cells, was used in Fig. 4, B and C and Fig. 7 A as a measure of the increase in HA surface density relative to the level of HA expression in untreated HAb2 cells: ∼2,500 trimers per μm2 (Danieli et al., 1996).
Surface HA expression for different NaBut concentrations was also evaluated by means of flow cytometry. Cells were labeled with the anti-HA monoclonal antibody FC-125 with Cy5 tag (a gift from Dr. Mukesh Kumar, National Institutes of Health [NIH], Bethesda, MD). The FACSCalibur® with the CELLQuest software package (Becton Dickinson) was used to record the mean fluorescence of Cy5-positive cells with different HA expression levels.
Measuring HA activation/inactivation by CELISA
The high degree of sequence homology (i.e., 74%) between X31 and Japan HA fusion peptides allowed the use of rabbit serum raised against the Japan HA fusion peptide on both strains in a CELISA. The CELISA assay was performed as described in Leikina and Chernomordik (2000). In brief, surface HA in HA300a or HAb2 cells was reacted with a 1:100 dilution of the antiserum for 1 h at 22°C.
Measuring HA activation/inactivation by cell–cell fusion
Fusion was assayed by fluorescence microscopy as the PKH26 transfer from RBCs to unlabeled HAb2 or HA300a cells, as previously described (Chernomordik et al., 1998). Data were quantified as the ratio of dye-redistributed–bound RBCs to the total number of bound RBCs. HA activation was initiated by a pH 4.9 pulse (the activating pulse) to HAb2 or HA300a cells in the absence of RBCs. Next, RBCs were added and allowed to bind to HA-cells; the second low pH pulse (the fusion-triggering pulse) followed. Fusion was measured 20 min after the triggering pulse. Longer incubations at low pH (i.e., 30 min) did not increase the extent of fusion. In general, a higher degree of HA activation after the activating pulse resulted in a greater inactivation and lower fusion after the fusion-triggering pulse.
In Fig. 1 B, HA-cells with bound RBCs were treated with thermolysin (0.05 mg/ml) for 20 min at 22°C at the LPC-arrested fusion stage (Chernomordik et al., 1997). Cells were triggered to fuse by application of a low pH medium at 22°C in the presence of 285 μM lauroyl LPC, which reversibly blocked fusion. LPC removal 30 min after the low pH pulse gave the full extent of fusion.
Udorn HA expression
Infection with SV-40 recombinant virus carrying the Udorn HA gene (Melikyan et al., 1997b) was performed on confluent monolayers of CV1 cells at 0.01, 0.1, and 1 mg/ml total viral protein. After a 1-h viral adsorption period, the viral suspension was diluted at a 1:5 ratio, and cells were incubated at 37°C for the next 48 h. Expression of HA at the surfaces of uninfected (i.e., control) CV1 cells and CV1 cells infected with SV-40 at different levels was evaluated by flow cytometry with HA-specific HC67x monoclonal primary antibodies.
Virosomes
Virosomes from X31 influenza virus were prepared as described in Bron et al. (1993). In brief, viral particles were solubilized in 100 mM C12E8 and reconstituted by detergent removal with BioBeads SM2. Approximately 55% of the viral HA and 42% of the phospholipid, relative to the starting material, were recovered in the virosomes, as evaluated by means of quantitative immunoblotting and measuring the fluorescence of virosomes formed from viral particles prelabeled with rhodamine dipalmitoyl phosphatidylethanolamine (Rho-PE, at a final concentration of 0.7 mol%). This recovery efficiency was in agreement with that reported in Bron et al. (1993). To lower HA density, C12E8-solubilized virus was supplemented with different amounts of the lipid mixture, egg phosphatidylethanolamine/egg phosphatidylcholine/cholesterol (1:1:1) including 1.4 mol% Rho-PE. Influenza virus is estimated to have 500–1,000 trimers per viral particle of ∼100-nm diameter (Taylor et al., 1987), i.e., 100–200 lipid molecules per trimer. Assuming that virosomes without exogenous lipids have a HA trimer to lipid ratio of 1:200, which corresponds to 15,000 HA trimers per μm2, the ratios for the two preparations of “diluted” virosomes formed with exogenous lipid were 1:3,650 and 1:1,100, which correspond to densities of 820 and 2,800 trimers per μm2, respectively. HA incorporation in reconstituted vesicles was readily assessed by means of equilibrium density-gradient analysis using a 2.5–40% linear sucrose gradient. Fractions were collected and analyzed for protein and lipid content by means of Western blotting and fluorescence scanning. The amount of HA in the bottom fraction never exceeded 3.5% of all HA in the sample, indicating that the amount of nonreconstituted HA (HA rosettes) in our preparations was negligible.
Liposomes
Liposomes made of distearoylphosphatidylcholine/cholesterol/Rho-PE/gangliosides GD1a (49.5:40.5:5:5 mol%) were prepared by extrusion through a 100-nm Nucleopore filter. The size of the extruded liposomes (i.e., less than 100 nm in diameter) was verified by means of quasi-elastic light scattering. Liposomes (∼0.5-μmole total lipid) were incubated with HA-cells (106 cells) at 4°C for 60 min. After the removal of unbound liposomes, cells were treated with a 10-min, pH 4.9, pulse at 22°C, and the percentage of activated HA was assayed by measurement of protein sensitivity to DTT.
The degree of HA activation, the extent of NaBut-induced promotion of HA expression, and the extent of fusion varied from day to day, possibly because of variation in the level of HA expression. Each experiment presented here was repeated several times, and all functional dependencies reported were observed in each experiment. The data shown in the figures are for the representative experiment or, if shown with error bars, for results averaged over at least three experiments.
Acknowledgments
We are grateful to Drs. A. Kajava (NIH), M. Kozlov (Tel Aviv University, Tel Aviv, Israel), K. Melikov (NIH), A. Sokoloff (University of Wisconsin, Madison, WI), T. Stegmann (CNRS, Toulouse, France), A. Walter (St. Olaf College, Northfield, MN), and J. Wilschut (University of Groningen, Groningen, Netherlands) for friendly and helpful advice and discussions on different aspects of this work. We are indebted to Drs. T.J. Braciale, R. Lamb, and J.J. Skehel for their gifts of antibody FC-125, Udorn HA construct, and antibody HC67, respectively, and to Dr. M. Kumar for labeled FC-125 antibody. We also wish to thank Dr. J.Ch. Grivel (NIH) for his expert advice and help with flow cytometry experiments.
I. Markovic's current address is Division of Monoclonal Antibodies, Office of Therapeutics Research and Review, Center for Biologics Evaluation and Research, FDA, Bethesda, MD 20892.
Footnotes
Abbreviations used in this paper: CELISA, cell surface enzyme-linked immunosorbent assay; DTT, dithiothreitol; HA, influenza hemagglutinin; HA-cell, HA-expressing cell; HA-membrane, HA-expressing membrane; LPC, lysophosphatidylcholine; NaBut, sodium butyrate; RBC, red blood cell; Rho-PE, rhodamine dipalmitoyl phosphatidylethanolamine.
References
- Armstrong, R.T., A.S. Kushnir, and J.M. White. 2000. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol. 151:425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal, R., D.P. Sarkar, S. Durell, D.E. Howard, and S.J. Morris. 1996. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell–cell fusion events. J. Cell Biol. 135:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron, R., A. Ortiz, J. Dijkstra, T. Stegmann, and J. Wilschut. 1993. Preparation, properties, and applications of reconstituted influenza virus envelopes (virosomes). Methods Enzymol. 220:313–331. [DOI] [PubMed] [Google Scholar]
- Bullough, P.A., F.M. Hughson, J.J. Skehel, and D.C. Wiley. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 371:37–43. [DOI] [PubMed] [Google Scholar]
- Carr, C.M., and P.S. Kim. 1993. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell. 73:823–832. [DOI] [PubMed] [Google Scholar]
- Carr, C.M., C. Chaudhry, and P.S. Kim. 1997. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. USA. 94:14306–14313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik, L.V., E. Leikina, V. Frolov, P. Bronk, and J. Zimmerberg. 1997. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J. Cell Biol. 136:81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik, L.V., V.A. Frolov, E. Leikina, P. Bronk, and J. Zimmerberg. 1998. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 140:1369–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague, M.J., C. Schoch, and R. Blumenthal. 1991. Delay time for influenza virus hemagglutinin-induced membrane fusion depends on hemagglutinin surface density. J. Virol. 65:2402–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danieli, T., S.L. Pelletier, Y.I. Henis, and J.M. White. 1996. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol. 133:559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels, R.S., A.R. Douglas, J.J. Skehel, and D.C. Wiley. 1983. Analyses of the antigenicity of influenza haemagglutinin at the pH optimum for virus-mediated membrane fusion. J. Gen. Virol. 64:1657–1662. [DOI] [PubMed] [Google Scholar]
- Doxsey, S.J., J. Sambrook, A. Helenius, and J. White. 1985. An efficient method for introducing macromolecules into living cells. J. Cell Biol. 101:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellens, H., J. Bentz, D. Mason, F. Zhang, and J.M. White. 1990. Fusion of influenza hemagglutinin-expressing fibroblasts with glycophorin-bearing liposomes: role of hemagglutinin surface density. Biochemistry. 29:9697–9707. [DOI] [PubMed] [Google Scholar]
- Gaudin, Y., R.W.H. Ruigrok, and J. Brunner. 1995. Low-pH induced conformational changes in viral fusion proteins: implications for the fusion mechanism. J. Gen. Virol. 76:1541–1556. [DOI] [PubMed] [Google Scholar]
- Gaudin, Y., H. Raux, A. Flamand, and R.W. Ruigrok. 1996. Identification of amino acids controlling the low-pH-induced conformational change of rabies virus glycoprotein. J. Virol. 70:7371–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves, P.N., J.L. Schulman, J.F. Young, and P. Palese. 1983. Preparation of influenza virus subviral particles lacking the HA1 subunit of hemagglutinin: unmasking of cross-reactive HA2 determinants. Virology. 126:106–116. [DOI] [PubMed] [Google Scholar]
- Gunther-Ausborn, S., P. Schoen, I. Bartoldus, J. Wilschut, and T. Stegmann. 2000. Role of hemagglutinin surface density in the initial stages of influenza virus fusion: lack of evidence for cooperativity. J. Virol. 74:2714–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman, O., T. Danieli, J.M. White, and Y.I. Henis. 1993. Effects of exposure to low pH on the lateral mobility of influenza hemagglutinin expressed at the cell surface: correlation between mobility inhibition and inactivation. Biochemistry. 32:101–106. [DOI] [PubMed] [Google Scholar]
- Keleshian, A.M., R.O. Edeson, G.J. Liu, and B.W. Madsen. 2000. Evidence for cooperativity between nicotinic acetylcholine receptors in patch clamp records. Biophys. J. 78:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble, G.W., D.L. Bodian, J. Rose, I.A. Wilson, and J.M. White. 1992. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J. Virol. 66:4940–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemble, G.W., Y.I. Henis, and J.M. White. 1993. GPI- and transmembrane-anchored influenza hemagglutinin differ in structure and receptor binding activity. J. Cell Biol. 122:1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, C.H., J.C. Macosko, and Y.K. Shin. 1998. The mechanism for low-pH-induced clustering of phospholipid vesicles carrying the HA2 ectodomain of influenza hemagglutinin. Biochemistry. 37:137–144. [DOI] [PubMed] [Google Scholar]
- Korte, T., K. Ludwig, M. Krumbiegel, D. Zirwer, G. Damaschun, and A. Herrmann. 1997. Transient changes of the conformation of hemagglutinin of influenza virus at low pH detected by time-resolved circular dichroism spectroscopy. J. Biol. Chem. 272:9764–9770. [DOI] [PubMed] [Google Scholar]
- Korte, T., K. Ludwig, F.P. Booy, R. Blumenthal, and A. Herrmann. 1999. Conformational intermediates and fusion activity of influenza virus hemagglutinin. J. Virol. 73:4567–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov, M.M., and L.V. Chernomordik. 1998. A mechanism of protein-mediated fusion: coupling between refolding of the influenza hemagglutinin and lipid rearrangements. Biophys. J. 75:1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikina, E., and L.V. Chernomordik. 2000. Reversible merger of membranes at the early stage of influenza hemagglutinin-mediated fusion. Mol. Biol. Cell. 11:2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikina, E., I. Markovic, L.V. Chernomordik, and M.M. Kozlov. 2000. Delay of influenza hemagglutinin refolding into a fusion-competent conformation by receptor binding: a hypothesis. Biophys. J. 79:1415–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic, I., H. Pulyaeva, A. Sokoloff, and L.V. Chernomordik. 1998. Membrane fusion mediated by baculovirus gp64 involves assembly of stable gp64 trimers into multiprotein aggregates. J. Cell Biol. 143:1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan, G.B., S.A. Brener, D.C. Ok, and F.S. Cohen. 1997. a. Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol-anchored influenza hemagglutinin-induced hemifusion to full fusion. J. Cell Biol. 136:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan, G.B., H. Jin, R.A. Lamb, and F.S. Cohen. 1997. b. The role of the cytoplasmic tail region of influenza virus hemagglutinin in formation and growth of fusion pores. Virology. 235:118–128. [DOI] [PubMed] [Google Scholar]
- Melikyan, G.B., R.M. Markosyan, M.G. Roth, and F.S. Cohen. 2000. A point mutation in the transmembrane domain of the hemagglutinin of influenza virus stabilizes a hemifusion intermediate that can transit to fusion. Mol. Biol. Cell. 11:3765–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal, A., and J. Bentz. 2001. Comprehensive kinetic analysis of influenza hemagglutinin-mediated membrane fusion: role of sialate binding. Biophys. J. 81:1521–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plonsky, I., and J. Zimmerberg. 1996. The initial fusion pore induced by baculovirus GP64 is large and forms quickly. J. Cell Biol. 135:1831–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri, A., F.P. Booy, R.W. Doms, J.M. White, and R. Blumenthal. 1990. Conformational changes and fusion activity of influenza virus hemagglutinin of the H2 and H3 subtypes: effects of acid pretreatment. J. Virol. 64:3824–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho-Santos, J., S. Nir, N. Duzgunes, A.P. de Carvalho, and M.D.C. de Lima. 1993. A common mechanism for influenza virus fusion activity and inactivation. Biochemistry. 32:2771–2779. [DOI] [PubMed] [Google Scholar]
- Ruigrok, R.W., A. Aitken, L.J. Calder, S.R. Martin, J.J. Skehel, S.A. Wharton, W. Weis, and D.C. Wiley. 1988. Studies on the structure of the influenza virus haemagglutinin at the pH of membrane fusion. J. Gen. Virol. 69:2785–2795. [DOI] [PubMed] [Google Scholar]
- Sauter, N.K., J.E. Hanson, G.D. Glick, J.H. Brown, R.L. Crowther, S.J. Park, J.J. Skehel, and D.C. Wiley. 1992. Binding of influenza virus hemagglutinin to analogs of its cell-surface receptor, sialic acid: analysis by proton nuclear magnetic resonance spectroscopy and X-ray crystallography. Biochemistry. 31:9609–9621. [DOI] [PubMed] [Google Scholar]
- Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- Skehel, J.J., and D.C. Wiley. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69:531–569. [DOI] [PubMed] [Google Scholar]
- Stegmann, T., J.M. White, and A. Helenius. 1990. Intermediates in influenza induced membrane fusion. EMBO J. 9:4231–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian, S.A., P. Hinterdorfer, G. Baber, and L.K. Tamm. 1995. Influenza hemagglutinin assumes a tilted conformation during membrane fusion as determined by attenuated total reflection FTIR spectroscopy. EMBO J. 14:5514–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, H.P., S.J. Armstrong, and N.J. Dimmock. 1987. Quantitative relationships between an influenza virus and neutralizing antibody. Virology. 159:288–298. [DOI] [PubMed] [Google Scholar]
- Wharton, S.A., L.J. Calder, R.W. Ruigrok, J.J. Skehel, D.A. Steinhauer, and D.C. Wiley. 1995. Electron microscopy of antibody complexes of influenza virus haemagglutinin in the fusion pH conformation. EMBO J. 14:240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, J. 1996. Membrane fusion: the influenza paradigm. Cold Spring Harb. Symp. Quant. Biol. 60:581–588. [DOI] [PubMed] [Google Scholar]
- White, J.M., and I.A. Wilson. 1987. Anti-peptide antibodies detect steps in a protein conformational change: low-pH activation of the influenza virus hemagglutinin. J. Cell Biol. 105:2887–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, D.C., and J.J. Skehel. 1987. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 56:365–394. [DOI] [PubMed] [Google Scholar]