

Abstract
Sporulation of Saccharomyces cerevisiae is a developmental process in which a single cell is converted into four haploid spores. GIP1, encoding a developmentally regulated protein phosphatase 1 interacting protein, is required for spore formation. Here we show that GIP1 and the protein phosphatase 1 encoded by GLC7 play essential roles in spore development. The gip1Δ mutant undergoes meiosis and prospore membrane formation normally, but is specifically defective in spore wall synthesis. We demonstrate that in wild-type cells, distinct layers of the spore wall are deposited in a specific temporal order, and that gip1Δ cells display a discrete arrest at the onset of spore wall deposition. Localization studies revealed that Gip1p and Glc7p colocalize with the septins in structures underlying the growing prospore membranes. Interestingly, in the gip1Δ mutant, not only is Glc7p localization altered, but septins are also delocalized. Similar phenotypes were observed in a glc7–136 mutant, which expresses a Glc7p defective in interacting with Gip1p. These results indicate that a Gip1p–Glc7p phosphatase complex is required for proper septin organization and initiation of spore wall formation during sporulation.
Keywords: sporulation; GIP1; GLC7; septin; spore wall
Introduction
Sporulation of Saccharomyces cerevisiae is a developmental process in which a single diploid cell is converted into an ascus containing four haploid spores. Diploid cells subjected to nitrogen starvation in the presence of a nonfermentable carbon source undergo two meiotic nuclear divisions, meiosis I and II, and packaging of the four resulting haploid nuclei into spores. Spore formation occurs in a series of well-defined steps (Lynn and Magee, 1970; Moens, 1971; Moens and Rapport, 1971; Neiman, 1998; Kupiec et al., 1997). Late in meiosis I, the spindle pole body (SPB)* undergoes a structural change and its outer plaque becomes enlarged. As cells enter meiosis II, secretory vesicles coalesce on the cytoplasmic surface of each outer plaque to form a novel intracellular membrane, termed the prospore membrane. The prospore membranes extend out as double membranes from the SPBs and engulf the adjacent nuclear lobes. After the completion of meiosis II, nuclear division gives rise to four haploid nuclei. At the same time, the ends of each double membrane meet and fuse with themselves, encapsulating the haploid nuclei to form prospores. Membrane closure triggers the final step of spore morphogenesis, spore wall formation. Spore wall materials are deposited in the luminal space between the two membranes derived from the prospore membrane.
The spore wall consists of four layers. The two inner layers are composed of glucan and mannan, components of the vegetative cell wall (Klis, 1994). The third layer is composed largely of chitosan, a glucosamine polymer, produced by the combined action of the chitin synthetase, Chs3p, and chitin deacetylases (Briza et al., 1988; Pammer et al., 1992; Christodoulidou et al., 1996; Mishra et al., 1997). The outermost layer consists largely of cross-linked dityrosine molecules (Briza et al., 1986). Dityrosine precursors are synthesized in the spore cytosol by the enzymes encoded by DIT1 and DIT2 (Briza et al., 1990, 1994). These genes are induced specifically around the time of prospore membrane closure. The chitosan and dityrosine layers are specific to spores, and provide much of the spore's resistance to environmental stress. Several mutants and genes related to spore wall formation have been isolated and characterized. Among these are genes coding for protein kinases, SPS1, SMK1, CAK1, and MPS1, and a gene encoding a nuclear protein, SWM1, raising the possibility that signal transduction pathways might regulate the spore wall formation (Friesen et al., 1994; Krisak et al., 1994; Wagner et al., 1997; Ufano et al., 1999; Straight et al., 2000).
Septins are a conserved family of proteins characterized by a central core domain and P-loop nucleotide-binding motif (Longtine et al., 1996; Kartmann and Roth, 2001). Most of them also contain a coiled-coil domain that could be involved in their assembly into filaments. In vegetatively growing yeast cells, septins localize to the bud neck and function as a scaffold interacting with a variety of proteins (DeMarini et al., 1997; Longtine et al., 2000). There are seven septin genes in S. cerevisiae, four of which are transcriptionally upregulated during sporulation: SPR3 and SPR28 are sporulation specific, and CDC3 and CDC10 are induced more than tenfold (Kaback and Feldberg, 1985; Chu et al., 1998; De Virgilio et al., 1996; Fares et al., 1996; Primig et al., 2000). During spore development, septin localization is different than that seen in vegetative cells (Fares et al., 1996). No septin localization to the cell periphery is seen. Rather, the septins appear in early meiosis II as four ring-like structures around each SPB. As the prospore membrane extends, septins disappear from the SPB region and display an extended band-like pattern underlying the prospore membrane. After closure of the prospore membrane, the septins become diffusely localized in the spore periphery (Fares et al., 1996). Surprisingly, disruption studies have revealed only modest sporulation defects in septin mutants; spr3Δ spr28Δ homozygous diploids and cdc10Δ diploids sporulate well (De Virgilio et al., 1996; Fares et al., 1996). Although no strong sporulation phenotype has been observed in septin mutants, their expression profile and specific localization suggest that they have some function in sporulation.
Protein phosphatase type 1 (PP1) is a highly conserved phosphoserine-/phosphothreonine-specific protein phosphatase that plays important roles in a variety of cellular processes including cell cycle progression, glycogen metabolism, glucose repression, and sporulation (Mumby and Walter, 1993; Depaoli-Roach et al., 1994; Shenolikar, 1994). In S. cerevisiae, PP1 is encoded by the essential gene GLC7 (Feng et al., 1991). Consistent with the multiple functions of Glc7p, the localization of this protein is dynamic throughout the cell cycle. In addition to a predominant nuclear localization, it is also observed at the SPB, bud neck, and actomyosin ring at distinct times in the cell cycle (Bloecher and Tatchell, 2000). The localization and substrate specificity of Glc7p are thought to be regulated by interaction with different targeting or regulatory subunits. For instance, bud neck localization of Glc7p requires the septin-binding protein Bni4p (unpublished data), and Gac1p is required to localize Glc7p to glycogen particles (Francois et al., 1992; Stuart et al., 1994). As in vegetative cells, there are multiple points at which GLC7 is required during sporulation, including premeiotic DNA synthesis and passage through meiosis I (Ramaswamy et al., 1998; Bailis and Roeder, 2000).
GIP1 encodes a potential developmentally regulated targeting subunit of Glc7p. GIP1 was isolated as a GLC7 interacting gene in a two-hybrid screen (Tu et al., 1996). This interaction was confirmed by coimmunoprecipitation (Tu et al., 1996). GIP1 is a sporulation-specific gene and diploid strains homozygous for a deletion of this gene are blocked in sporulation and fail to induce SPS100, one of the very late sporulation genes (Tu et al., 1996). Additionally, several glc7 mutant alleles defective in sporulation have been isolated, and in most instances sporulation efficiency correlates with the strength of the two-hybrid interaction between the glc7 mutant protein and Gip1p (Baker et al., 1997; Ramaswamy et al., 1998). These previous reports raise the possibility that GIP1 dependent regulation of Glc7p might be important for sporulation.
In this study, we examine the roles of GIP1 and GLC7 in sporulation. Cytological analyses of mutant strains revealed that GIP1 and GLC7 are required for spore wall formation. Both proteins are found to colocalize with septins and are required for septin assembly during sporulation. These results suggest that GIP1 and GLC7, and perhaps the septins, participate in a signal transduction pathway necessary to monitor prospore membrane growth and initiate the synthesis of the spore wall.
Results
Spore wall formation is blocked in gip1Δ mutant cells
To investigate the essential role of GIP1 during sporulation, we examined where in the process of sporulation gip1Δ cells are defective. First, a homozygous gip1Δ mutant strain, NY501 (Table I), was constructed in the rapidly sporulating SK1 background (Kane and Roth, 1974), and the progression of meiosis was monitored using the DNA-binding dye DAPI. In wild-type and gip1Δ mutant cells, the number of nuclei seen in each cell increased from one to two, then to four, with nearly identical kinetics (Fig. 1). This indicates that meiosis I and II proceed normally in the gip1Δ mutant, and suggests that the sporulation defect in gip1Δ mutants must be due to a failure in some aspect of spore packaging.
Table I. Strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| NH144 | MATa/MATα ura3/ura3 leu2Δ::hisG/leu2-k his4-x/HIS4 arg4-NspI/ARG4 lys2/lys2 hoΔ::LYS2/hoΔ::LYS2 | Neiman, 1998 |
| AN103 | MATa/MATα his3ΔSK/his3ΔSK ura3/ura3 leu2Δ::hisG/leu2Δ::hisG trp1::hisG/trp1::hisG arg6/ARG6 lys2/lys2 hoΔ::LYS2/hoΔ::LYS2 rme1::LEU2/rme1::LEU2 gip1::HIS3/gip1::HIS3 | This study |
| SB247 | MATa/MATα ura3-52/ura3-52 trp1-1/trp1-1 leu2/leu2 glc7::LEU2/glc7::LEU2 [pNC160-glc7-136 (TRP1)] | Baker et al., 1997 |
| AN117-4B | MATα his3 ura3 trp1::hisG leu2 arg4-NspI lys2 hoΔ::LYS2 rme1::LEU2 | Neiman et al., 2000 |
| AN117-16D | MATa his3 ura3 trp1::hisG leu2 lys2 hoΔ::LYS2 | Neiman et al., 2000 |
| AN120 | MATa/MATα his3/his3 ura3/ura3 trp1::hisG/trp1::hisG leu2/leu2 arg4-NspI/ARG4 lys2/lys2 hoΔ::LYS2/hoΔ::LYS2 rme1::LEU2/RME1 | Neiman et al., 2000 |
| NY1 | MATα his3 ura3 trp1::hisG leu2 arg4-NspI lys2 hoΔ::LYS2 rme1::LEU2 gip1::HIS3 | This study |
| NY2 | MATa his3 ura3 trp1::hisG leu2 lys2 hoΔ::LYS2 gip1::HIS3 | This study |
| NY501 | MATa/MATα his3/his3 ura3/ura3 trp1::hisG/trp1::hisG leu2/leu2 arg4-NspI/ARG4 lys2/lys2 hoΔ::LYS2/hoΔ::LYS2 rme1::LEU2/RME1 gip1::HIS3/gip1::HIS3 | NY1 × NY2 |
| NY13 | MATα his3 ura3 trp1::hisG leu2 arg4-NspI lys2 hoΔ::LYS2 rme1::LEU2 gip1::his5+-spo20pr-HA-GIP1 | This study |
| NY14 | MATa his3 ura3 trp1::hisG leu2 lys2 hoΔ::LYS2 gip1::his5+-spo20pr-HA-GIP1 | This study |
| NY509 | MATa/MATα his3/his3 ura3/ura3 trp1::hisG/trp1::hisG leu2/leu2 arg4-NspI/ARG4 lys2/lys2 hoΔ::LYS2/hoΔ::LYS2 rme1::LEU2/RME1 gip1::his5+-spo20pr-HA-GIP1 / gip1::his5+-spo20pr-HA-GIP1 | NY13 × NY14 |
| NY523 | MATa/MATα his3/his3 ura3/ura3 trp1::hisG/trp1::hisG leu2/leu2 arg4-NspI/ARG4 lys2/lys2 hoΔ::LYS2/hoΔ::LYS2 rme1::LEU2/RME1 glc7::his5+/GLC7 | This study |
| NY525 | MATa/MATα his3/his3 ura3:glc7-136:URA3/ura3 trp1::hisG/trp1::hisG leu2/leu2 arg4-NspI/ARG4 lys2/lys2 hoΔ::LYS2/hoΔ::LYS2 rme1::LEU2/RME1 glc7::his5+/GLC7 | This study |
| NY527 | MATa/MATα his3/his3 ura3:glc7-136:URA3/ura3:glc7-136:URA3 trp1::hisG/trp1::hisG leu2/leu2 arg4-NspI/ARG4 lys2/lys2 hoΔ::LYS2/hoΔ::LYS2 rme1::LEU2/RME1 glc7::his5+/glc7::his5+ | This study |
Figure 1.

Meiotic progression is normal in the gip1 Δ mutant. At each time point, aliquots of strains AN120 (GIP1/GIP1, thin line) and NY501(gip1Δ/gip1Δ, thick line) were removed from sporulation medium, stained with DAPI, and analyzed for meiotic progression. Percentage of mononucleate (▪), binucleate (▴), and tetranucleate (•) cells are shown. At least 200 cells were counted at each time point.
To examine this possibility, EM analysis of sporulating wild-type and the gip1Δ mutant cells was performed. In wild-type cells, prospore membranes capturing each nucleus were observed (Fig. 2 A), and at later stages, spore walls were seen between each of the inner and outer membrane surrounding the spore nucleus (Fig. 2 B). In gip1Δ cells, prospore membranes resembling those in wild-type cells were also observed capturing nuclei (Fig. 2, C and D). Yet strikingly, no spore walls were observed in the mutant cells. The inner and outer membranes derived from each prospore membrane remained closely apposed, suggesting that no spore wall material was deposited. The gip1Δ cells also displayed a proliferation of darkly staining material around the ascal surface (*, Fig. 2, C and D). Accumulation of similar material has been observed in some spore wall defective mutants (Christodoulidou et al., 1999; Wagner et al., 1999). These data suggest that GIP1 is required to initiate spore wall formation.
Figure 2.

The gip1Δ mutant fails to form spore walls. EM analysis of GIP1 (NH144, A and B) and homozygous gip1Δ mutant (AN103, C and D) cells at a late stage of sporulation. Prospore membranes (PrM); spore walls (SW); and nuclei (N) are indicated. *, Darkly staining material formed in the ascal cytoplasm of gip1 mutants. Bar, 500 nm.
DIT1 expression is blocked in gip1Δ mutants
Although the EM study indicated a lack of spore wall material in the gip1 mutant, we could not rule out the possibility that there are some components or layers of spore wall deposited that are not clearly visible by EM. Dityrosine-containing macromolecules can be observed by their natural fluorescence (Briza et al., 1990). Thus, to examine whether the dityrosine layer is formed in gip1Δ mutant, we assayed the sporulated gip1Δ mutant for dityrosine fluorescence. Dityrosine fluorescence was absent in the gip1Δ mutant (unpublished data), indicating that no dityrosine is produced in this mutant.
The DIT1 gene is required for the synthesis of dityrosine and expression of DIT1 is induced in sporulation before SPS100, but after middle genes such as GIP1 or SPR3 (Briza et al., 1990; Chu et al., 1998; Primig et al., 2000). GIP1 is required for expression of the late gene SPS100 (Tu et al., 1996). This previously described effect of gip1Δ mutation on SPS100 expression, as well as the lack of dityrosine fluorescence, led us to examine DIT1 expression in the gip1Δ mutant. A DIT1–lacZ fusion gene was introduced into wild-type and gip1Δ mutant strains. As a control, a lacZ fusion to SPR3 was used. Whereas SPR3–lacZ was induced with the same kinetics in the isogenic wild-type and gip1Δ diploids, no induction of DIT1–lacZ was observed when GIP1 was absent (Fig. 3). Thus, the absence of dityrosine staining in the gip1Δ mutant can be explained by the failure to transcribe DIT1.
Figure 3.
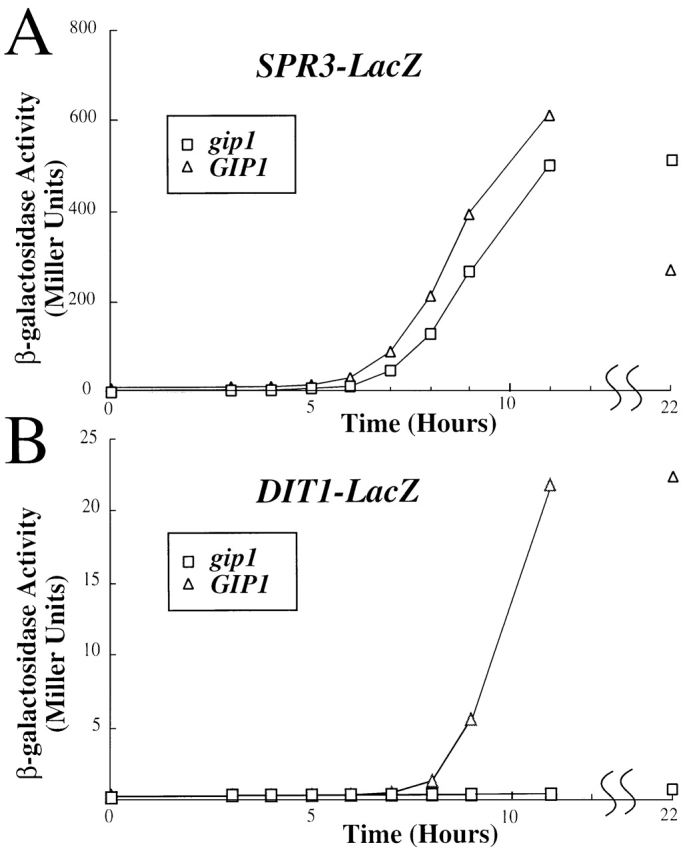
DIT1–lacZ is not induced in gip1 Δ cells. Sporulating cultures of AN120 (GIP1/GIP1, ▵) and NY501 (gip1Δ/gip1Δ, □) carrying either (A) SPR3–lacZ or (B) DIT1–lacZ were analyzed at various time points for β-galactosidase activity. β-galactosidase activity is given in Miller Units.
Fluorescence studies reveal the temporal order of spore wall deposition
To examine the formation of spore wall layers other than the dityrosine layer, a series of fluorescent markers was used. The mannan layer was visualized using the mannose-binding lectin concanavalin A coupled to FITC. A monoclonal antibody directed against β1–3Glucan was used to identify the glucan layer, and the chitin-binding dye Calcofluor white was used to visualize the chitosan layer. The use of secondary antibodies conjugated to a red fluor to localize the glucan antibodies allowed all three markers to be used simultaneously. As shown in Fig. 4, the staining patterns of wild-type asci varied according to the stage of spore development. After counting and classifying these asci, we ordered the patterns as follows (Table II): when cells are in the stage of the prospore membrane formation and extension, those membranes are stained only with FITC-ConA (Class 2). Approximately 30% of cells showed this pattern at the seven hour time point. Subsequently, a population of cells appears that is positive for both FITC-ConA and anti-β1–3Glucan (Class 3). At the 7- and 8-h time points, ∼20% of the cells showed staining with anti-β1–3Glucan in the spore precursors. After a transient period of triply positive staining (Class 4), the ascus loses anti-β1–3Glucan staining (Class 5). At this point, the FITC-conA staining pattern changes to stain only the spore periphery. Finally, Calcofluor white no longer stains the spore wall (Class 6). Therefore, these markers allow all the major spore wall polysaccharides to be visualized during the course of spore wall formation and suggest that these components are deposited in a specific sequence: mannan before glucan before chitosan.
Figure 4.
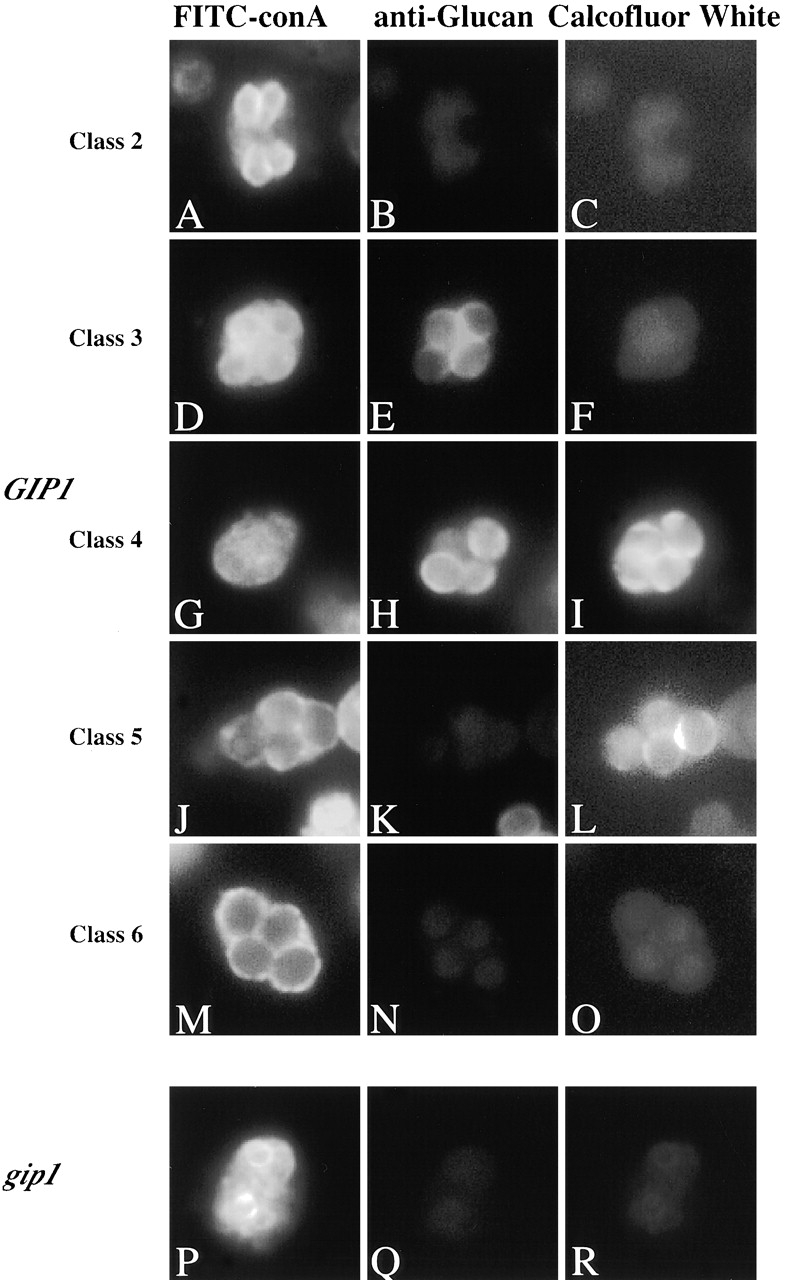
Analysis of spore wall formation in wild type and gip1 Δ mutant. (A–O) Samples were removed from sporulating culture of AN120 (GIP1/GIP1) at 6-, 7-, 8-, and 9-h time points, subjected to the immunofluorescence analysis of spore walls as described in Materials and methods. Classification of the cells are described in Results. (A, D, G, J, and M) FITC-ConA staining; (B, E, H, K, and N) Anti-β-glucan staining; (C, F, I, L, and O) Calcofluor white staining. (P–R) NY501 (gip1Δ / gip1Δ) was sporulated and subjected to the immunofluorescence analysis of spore walls. (P) FITC-ConA staining; (Q) Anti–β-glucan staining; (R) Calcofluor white staining.
Table II. Analysis of spore wall deposition.
| Staining for
|
GIP1
|
gip1
|
||||||
|---|---|---|---|---|---|---|---|---|
| Pattern | Mannan | Glucan | Chitosan | 6 h | 7 h | 8 h | 9 h | 9 h |
| Class 1 | − | − | − | 95.7 | 66.3 | 31.8 | 13 | 52 |
| Class 2 | + | − | − | 4.3 | 22.8 | 17.8 | 4 | 48 |
| Class 3 | + | + | F | 0 | 9.4 | 16.8 | 7.5 | 0 |
| Class 4 | + | + | + | 0 | 0 | 1.4 | 0.5 | 0 |
| Class 5 | + | − | + | 0 | 0.5 | 0.9 | 3.5 | 0 |
| Class 6 | + | − | F | 0 | 1 | 31.3 | 71.5 | 0 |
At each time point, cells were removed from sporulating AN120 (GIP1/GIP1) or NY501 (gip1Δ/gip1Δ) cultures and processed as described in Materials and methods. Cells were placed into six classes based on their staining pattern. +; strong staining, −, no staining; F, faint staining. The percentage of cells in each class is shown. At least 200 cells were counted at each time point.
The gip1Δ mutant fails to form the inner layers of spore wall
To examine the spore wall defect of gip1Δ, we applied this method to gip1Δ mutant cells. As in wild-type cells, the developing prospore membranes stained with FITC-ConA (Fig. 4, P–R). However, the gip1Δ mutant cells never progressed to show anti-β1–3Glucan or Calcofluor white staining. Taken together with the lack of the dityrosine layer, these results indicate that the prospore wall in gip1Δ mutants contains only those mannoproteins present during prospore membrane formation. No additional spore wall material is deposited at the completion of prospore membrane synthesis.
Gip1p colocalizes with Spr28p during spore formation
To examine where in the cell Gip1p functions, the Gip1 protein was localized by immunofluorescence analysis. COOH-terminal–tagged versions of Gip1p were nonfunctional (unpublished data). Therefore, targeted integration was used to fuse the hemagglutinin (HA) epitope to the 5′ end of the GIP1 gene. To preserve the sporulation-specific expression of GIP1, the fusion gene was under the control of SPO20 promoter. SPO20 is a sporulation specific gene (Neiman, 1998) with a similar expression profile to GIP1 (Chu et al., 1998). When NY509 (in which both copies of the genomic GIP1 genes are replaced with the fusion gene) was sporulated, the efficiency of sporulation was similar to wild-type cells (82% asci). However, only 26% of the asci were tetrads, with the remainder predominantly dyads and triads, suggesting that the fusion gene is not completely functional. When the fusion gene was cloned into a multicopy vector and introduced into the homozygous gip1 deletion strain, NY501, NY501 produced tetrads at a comparable level (>60%) to the wild-type strain. This strain carrying multicopy HA-GIP1 was therefore used for examination of Gip1p localization.
Gip1p localization during sporulation was examined by immunofluorescence analysis using anti-HA antibodies. HA–Gip1p appears initially in sporulating cells as four rings during meiosis II, and then localizes to the region of extending prospore membranes following the leading edge (Fig. 5). At this stage, parallel bar-like structures were observed. After completion of meiosis II, round structures surrounding the four nuclei were seen. Identical results were obtained when the integrated HA–GIP1 was examined, indicating that the localization is not a consequence of overexpression (unpublished data). This localization pattern of Gip1p resembles the reported localization of septins during sporulation. Therefore, we examined whether Gip1p colocalizes with septins. Gip1p–HA and Spr28p–green fluorescent protein (GFP) were expressed in wild type cells and their localization during sporulation was compared. The distribution of the two proteins overlapped extensively with each other (Fig. 6, A–D). These results indicate that Gip1p colocalizes with septins during spore formation.
Figure 5.

Localization of HA-Gip1p in sporulating cells. NY501 (gip1Δ / gip1Δ) carrying 2 μ HA-GIP1 was sporulated and analyzed by immunofluorescence. (A–E) Anti-HA antibody staining is shown. (F–J) Staining of the same cells with DAPI.
Figure 6.

HA-Gip1p colocalizes with septins and GFP-Glc7p. (A to D) AN120 carrying both pCEN-SPR28-GFP and 2μ HA-GIP1 was sporulated for 7 h and analyzed by staining with anti-HA antibodies. (A) GFP fluorescence. (B) Anti-HA staining. (C) Merge of A and B. (D) DAPI staining of the same cell. (E to H) AN120 carrying both GFP-GLC7 and 2 μ HA-GIP1 was sporulated for 7 h and analyzed by staining with anti-HA antibodies. (E) GFP fluorescence; (F) Anti-HA staining; (G) Merge of E and F; (H) DAPI staining of the same cell.
To determine if the bar-like pattern of Gip1p represents a section through a more complicated structure, image stacks were collected and three dimensional renderings made. Quicktime movies displaying the resulting images are available at http://www.jcb.org/cgi/content/full/jcb.200107008/DC1. The results indicate that the Gip1p-containing structures are actually pairs of parallel sheets that appear as bars when optically sectioned.
Glc7p shows a dynamic localization pattern during sporulation and colocalizes with Gip1p in meiosis II
Because GIP1 encodes a potential Glc7p targeting protein, we examined Glc7p localization in sporulating cells. GFP–GLC7 was introduced into the wild-type strain and the localization of the fusion protein was observed by GFP fluorescence (Fig. 7). Early in sporulation, cells displayed a predominant nuclear staining with a bright nucleolar dot as observed in vegetative cells (Bloecher and Tatchell, 2000). In early meiosis II, in addition to the nuclear staining, four dots were observed near the nuclear periphery suggesting some Glc7p is localized to the SPB. At later stages of meiosis II, Glc7p clearly displayed a bar-like pattern that resembles those of Spr28p and Gip1p. Finally, in postmeiotic cells, Glc7p was again found predominantly in the spore nucleus. These observations indicate that the localization of Glc7p is dynamic during sporulation, and that Glc7p displays a similar localization pattern to Gip1p and Spr28p during meiosis II.
Figure 7.

Localization of GFP-Glc7p during sporulation. AN120 carrying GFP-GLC7 was sporulated and GFP fluorescence was observed. (A–F) GFP fluorescence; (G–L) DAPI staining of corresponding cells in A–F.
To confirm this last point, colocalization of Gip1p and Glc7p was examined directly. GIP1–HA and GFP-GLC7 were expressed in AN120 and the localization of the proteins was analyzed. The distribution of the two proteins showed extensive overlap (Fig. 6, E–H), indicating that Glc7p colocalizes with Gip1p. Taken together with the immunofluorescence analysis of Gip1p and Spr28p, these results demonstrate that Gip1p and Glc7p colocalize with septins during meiosis II.
Glc7p and Septins are not properly localized in the gip1Δ mutant
By analogy to other Glc7p targeting proteins, Gip1p might be a targeting subunit that recruits Glc7p to the septins at the prospore membrane. To test this hypothesis, we examined the localization of Glc7p in the gip1Δ mutant. In this strain, Glc7p did not show a septin-like staining pattern in meiosis II. Instead, it often showed cytosolic staining with four discrete dots, which may indicate SPB localization (Fig. 8 C). Thus, localization of Glc7p to the prospore membrane is dependent on GIP1.
Figure 8.

Localization pattern of GFP-Glc7p and Spr28p-GFP are altered in the gip1 mutant. AN120 (GIP1/GIP1) and NY501 (gip1Δ / gip1Δ) carrying either GFP-GLC7 or pCEN-SPR28-GFP were sporulated and analyzed by fluorescence microscopy. (A) GFP-Glc7p localization in AN120. (B) DAPI staining of cell in A. (C) GFP-Glc7p localization in NY501. (D) DAPI staining of cells in C. (E) Spr28p-GFP localization in AN120. (F) DAPI staining of cells in E. (G and I) Spr28p-GFP localization in NY501 (H and J) DAPI staining of cells in G and I, respectively.
Similar to their function at the bud neck of vegetative cells, the septins may serve as a scaffold to which Gip1p recruits Glc7p. Alternatively, recruitment of Glc7p might be important for the organization of the septins themselves. To address these possibilities, we examined Spr28p localization in the gip1Δ mutant. We found that Spr28p is delocalized in many of the mutant cells (Fig. 8, G and I). In postmeiotic cells, ∼20% of the mutant cells showed a ring-like pattern surrounding four nuclei, indicating localization to the periphery of the prospore. The majority of cells showed only a uniform cytosolic staining pattern. Importantly, in cells in meiosis II, a pattern of bars like those in wild-type cells was only rarely observed (<1%). Similar results were obtained using antibodies to a second sporulation-specific septin Spr3p (unpublished data). Thus, Gip1p does not serve simply as a targeting subunit to connect Glc7p to the septins; rather, GIP1 is required for proper septin organization and the failure of Glc7p to localize to the prospore membrane in gip1Δ cells may reflect the absence of septin structures.
The Don1p ring is not affected by gip1Δ
DON1 encodes a coiled-coil protein that localizes to a ring-like structure at the leading edge of the prospore membrane, and this structure is distinct from the septin bars that underlie the prospore membrane (Knop and Strasser, 2000). To examine whether the effect of gip1 deletion is specific to septins or is perhaps a more general effect on prospore membrane–associated structures, we examined the localization of Don1p in the gip1Δ mutant. DON1-GFP was introduced into wild type and gip1Δ cells, and the localization of the fusion protein was observed using anti-GFP antibodies. In both wild-type and gip1Δ cells, ring-like structures were observed at the lip of the prospore membranes (Fig. 9). Thus, assembly of the Don1p ring is not affected by gip1 deletion, suggesting that the effects of gip1Δ are specific to assembly of the septin complex.
Figure 9.

Don1p ring is formed normally in the gip1 Δ mutant. AN120 (GIP1/GIP1) and NY501 (gip1Δ / gip1Δ) carrying DON1-GFP were sporulated and analyzed by immunofluorescence using anti-GFP antibodies. (A) Don1p-GFP localization in AN120. (B) DAPI staining of cell in A. (C) Don1p-GFP localization in NY501. (D) DAPI staining of cell in C.
The glc7–136 mutant shows similar defect to the gip1Δ mutant
To determine whether the phenotypes of the gip1Δ mutant are caused by a failure to properly localize Glc7p or a second function of the Gip1 protein, we examined the sporulation phenotype of a strain carrying the glc7–136 allele. The glc7–136 mutant causes a sporulation defect (Baker et al., 1997), and the protein is defective in interaction with Gip1p (unpublished data). Thus, if the sporulation defect of the gip1Δ mutant is due to altered localization of Glc7p, the glc7–136 mutant should display similar phenotypes. EM analysis of SB247, a glc7–136 mutant in the JC482 background (Baker et al., 1997), was performed. The cytological phenotype of glc7–136 was more heterogeneous than gip1Δ, probably due to the leaky sporulation defect of the mutant (2% asci). Nonetheless, a significant fraction of the cells (50%) displayed prospores with no apparent deposition of spore wall material (Fig. 10 A), similar to the gip1Δ mutant (Fig. 2). Thus, glc7–136 mutant cells display a block to spore wall deposition similar to that seen in the gip1 deletion strain.
Figure 10.

glc7–136 mutants show a similar phenotype to the gip1 Δ mutant. (A) SB247 (glc7–136/glc7–136) at a late stage of sporulation was analyzed by transmission EM. Prospore membranes (PrM) and nucleus (N) are indicated. (B–I) NY527 (glc7–136/glc7–136) harboring either pCEN-SPR28-GFP or 2 μ HA-GIP1 was sporulated and analyzed by fluorescence microscopy. (B and D) anti-GFP staining; (C and E) DAPI staining of cells in B and D, respectively; (F and H) Anti-HA staining; (G and I) DAPI staining of cells in F and H, respectively. Bar, 500 nm.
To examine the localization of Spr28p and Gip1p in the glc7–136 strain, the glc7–136 mutation was introduced into the same SK1 strains used for the gip1 analysis. As in the JC482 background, the glc7–136 mutant is leaky in SK1, giving 5% asci. SPR28-GFP and HA–GIP1 were introduced into NY527, and the proteins were localized by immunofluorescence. Most of the cells expressing SPR28-GFP displayed uniform cytosolic staining. In ∼30% of the cells, staining of the spore periphery was observed, as is the case with septin localization in the gip1Δ mutant (Fig. 10, B and D). HA–Gip1p displayed similar localization defects to Spr28p-GFP in the glc7–136 strain (Fig. 10, F and H). Thus, as with the spore wall, the glc7–136 mutant shows similar septin organization defects to the gip1Δ mutant. Further, Gip1p and Glc7p are interdependent for localization; interaction of the two proteins is required for either to localize to the prospore membrane. Taken together, these data indicate that Gip1p and Glc7p function together to promote both organization of the septins and spore wall formation.
Discussion
GIP1 and GLC7 are required to signal the onset of spore wall formation
Strains deleted for GIP1 display two clear phenotypes, a block to spore wall formation and delocalization of the septins from the forming prospore membrane. The spore wall phenotype of GIP1 mutants is a discrete arrest at the initiation of spore wall formation. In wild-type cells, deposition of spore wall material is never seen until after the closure of the prospore membrane (Lynn and Magee, 1970). Consistent with this cytological observation, expression of the DIT1 gene appears to occur after prospore closure (Briza et al., 1990). These observations suggest that the cell must have some mechanism to trigger spore wall formation in response to prospore membrane closure. Given their colocalization with septins, the Gip1p–Glc7p holoenzyme would be ideally located to participate in this process. Though we favor the idea that this phosphatase complex acts as a monitor to signal the closure of the prospore membrane, we cannot at present rule out the possibility that the Gip1p–Glc7p phosphatase is required for membrane closure itself. A defect in either membrane closure or the ability to sense that the membrane has closed would result in a failure to initiate spore wall formation.
CHS3 encodes the chitin synthase responsible for the synthesis of the chitosan layer. In contrast to DIT1, the CHS3 gene is not transcriptionally induced during sporulation (Pammer et al., 1992). Therefore, the failure of gip1Δ mutants to produce chitosan suggests that GIP1 may be required for posttranslational regulation of this enzyme. In vegetative cells, Chs3p activity is controlled in part by regulated recycling through an endosomal compartment (Ziman et al., 1998). It will be of interest to examine the localization of Chs3p in sporulating wild-type and gip1Δ cells.
Spore wall formation is an ordered process
Previous studies have shown that Calcofluor white transiently stains the chitosan layer of the developing spore (Briza et al., 1990). The loss of Calcoflour white staining in mature asci is due to maturation of the overlying dityrosine layer blocking access of the dye to its ligand (Briza et al., 1988, 1990). We interpret the transient staining of spores with the anti–β-glucan antibody in the same way. That is, spores are positive for staining until the overlying chitosan layer forms and obscures access of the antibody to the glucan layer. Surprisingly, staining with FITC-ConA is not lost as spores mature, even though the inner mannan layer should also be obscured as the outer layers form. However, at the same time that β-glucan staining is lost, the appearance of the FITC-ConA staining changes markedly. This change probably reflects a loss of access to the mannan layer of the inner spore wall, but residual staining of mannoproteins present in the outer prospore membrane or ascal cytoplasm condensed around the spores. Thus, our immunofluorescence assay of spore wall formation suggests a definitive order of spore wall assembly. Mannoproteins are deposited as the prospore membrane forms. After prospore membrane closure, the glucan layer forms, followed by the chitosan layer and finally the dityrosine layer.
Surprisingly, the layers are deposited from innermost to outermost, with respect to the spore cytoplasm. This suggests that precursors to the chitosan and dityrosine layer must be able to pass through the glucan and mannan layers before assembling. Alternatively, some of the spore wall layers may be constructed from precursors introduced from the ascal cytoplasm rather than the spore cytosol. This possibility has been suggested previously based on specific localization of some sporulation-induced transcripts to the ascal cytoplasm (Kurtz and Lindquist, 1986), and is also consistent with the accumulation of darkly staining vesicles in the region of the ascal cytoplasm surrounding the prospores that is seen in gip1Δ (Fig. 2) and other spore wall–defective mutants (Christodoulidou et al., 1999; Wagner et al., 1999).
Relationship of GIP1 to other spore wall synthesis genes.
Our data indicate that not only is spore wall deposition coordinated with respect to prospore membrane closure, but also a temporal order of deposition, glucan before chitosan before dityrosine, must be maintained. Previously reported spore wall–defective mutants fall broadly into one of two categories: those such as dit1 or chs3 that remove specific spore wall layers (Briza et al., 1990; Pammer et al., 1992), or those such as smk1 and swm1 in which individual spores within an ascus display heterogeneous wall defects (Krisak et al., 1994; Ufano et al., 1999). The heterogeneous spore wall defects seen in these latter mutants could result from an inability to coordinate the timing of deposition within individual spores.
SMK1 is particularly interesting in this regard. Though deletion of SMK1 causes a heterogeneous spore wall phenotype, point mutations produce more homogeneous arrests (Wagner et al., 1999). In the most severe allele, smk1–4, this arrest appears similar to gip1Δ. Further, it has been reported that formation of the glucan, chitosan, and dityrosine layers requires increasing levels of Smk1p activity, respectively (Wagner et al., 1999). As this matches the temporal order of deposition of these layers, SMK1 activity might provide a mechanism to control the timing of synthesis of the different layers. Gip1p–Glc7p phosphatase could control the initiation of spore wall synthesis, at least in part, by regulating the activation of Smk1p.
Gip1p–Glc7p phosphatase regulates septin organization
The Gip1p–Glc7p complex colocalizes with the septins during prospore membrane growth and the first evident phenotype of the gip1Δ mutation is not the spore wall defect, but a failure to organize the septins onto the prospore membrane. However, at times after prospore closure, in a fraction of the gip1Δ mutant cells the septins can be found in the spore periphery as in wild-type cells. These results indicate a specific requirement for the Gip1p–Glc7p phosphatase to organize the septins into structures associated with the growing prospore membrane. Whether this effect is mediated by direct dephosphorylation of the septin proteins or some other modifier of septin organization is not yet known. In vegetative cells, mutation of several different protein kinases has effects on organization of the septin ring (Bouquin et al., 2000; Longtine et al., 2000). For instance, mutation of GIN4 leads to the arrangement of the septins as a series of parallel bars (Longtine et al., 1998a). It has not yet been reported whether any of the septin proteins are direct substrates of any of these kinases. Nonetheless, taken together with our results, it seems likely that phosphorylation and dephosphorylation regulate septin architecture.
The fact that septins are lost from the prospore membrane in the gip1Δ mutant but that the prospore membrane nonetheless forms, indicates that the septins are not required for membrane assembly or growth. One possibility is that the septins function to keep Gip1p–Glc7p associated with the prospore membrane. In this instance, mutation of the septin genes might cause a similar spore wall phenotype to the gip1Δ mutation. However, no significant sporulation phenotype has been reported in septin mutants (De Virgilio et al., 1996; Fares et al., 1996). Therefore, it remains to be determined whether the spore wall and septin phenotypes of the gip1Δ mutant are linked, or whether they represent two independent aspects of Gip1p–Glc7p function.
In higher cells, as in yeast, septins are found at sites of cytokinesis (Neufeld and Rubin, 1994; Kinoshita et al., 1997). However, in contrast to mitotic yeast cells, the septins of higher cells exhibit dynamic localization and are often found in association with regions of active membrane growth (Fares et al., 1995; Kinoshita et al., 1997; Hsu et al., 1998). Thus, the behavior of septins during sporulation is similar to that in higher cells. Our results indicate that the Gip1p–Glc7p complex is required to organize the septins onto the prospore membrane. Given the high degree of conservation of PP1, it will be of interest to learn if septin organization is also regulated by this enzyme in higher cells.
Bailis and Roeder (2000) have reported that GLC7-dependent dephosphorylation of the synaptonemal complex component Red1p is required for progression through the meiotic pachytene checkpoint. In contrast to our findings, this study reported that gip1Δ mutants arrest in pachytene. Further, GIP1 was required for the localization of Glc7p to meiotic chromosomes and overexpression of GLC7 suppressed the gip1Δ sporulation defect. From these results it was suggested that the function of Gip1p is to target Glc7p to meiotic chromosomes. We found no evidence of a meiotic arrest in gip1Δ in two different strain backgrounds, SK1 (Fig. 1) and JC482. Additionally, overexpression of GLC7 does not relieve the spore formation defect of gip1Δ mutants in our strains (unpublished data), as was observed in the BR strain background (Bailis and Roeder, 2000). The reason for the differing phenotypes of gip1Δ in different strain backgrounds remains unclear; however, our results clearly demonstrate roles for GIP1 and GLC7 in postmeiotic events.
In sum, our data suggest a model in which Gip1p is induced during sporulation, binds to Glc7p, and the Gip1p–Glc7p complex then organizes the septins onto the prospore membrane. The Gip1p–Glc7p complex remains associated with the septins during meiosis II, and this places the phosphatase in a position to act as a monitor of prospore membrane growth. Upon closure of the prospore membrane, we propose that Gip1p and Glc7p initiate a signaling cascade that triggers events leading to the synthesis of the spore wall. This includes the induction of DIT1 as well as the activation of chitin and β-glucan synthases. Further work will be necessary to elucidate the nature of this intracellular signaling pathway.
Materials and methods
Yeast strains and media
S. cerevisiae strains used in this work are listed in Table I. AN103 was created by selection of 5-fluoroorotic acid–resistant derivatives of MCY3259 (Tu et al., 1996). To construct NY501, AN117–4B and AN117–16D (Neiman et al., 2000) were transformed with PvuII-digested pJT26-HIS3 (Tu et al., 1996) to produce NY1 and NY2, respectively. For this and all other integrations, proper arrangement of the genomic locus was confirmed by PCR. These strains are mated to create diploid NY501. To create a strain carrying NH2 terminally HA-tagged GIP1 under control of the SPO20 promoter, pFA6a-His3MX6-prSPO20-HA was subjected to PCR with HT1 (5′-CTATTTTCTTCCTCCTTGCCTATTCGTTTGGTGGCTTTCTTTATAATTCGGAATTCGAGCTC-GTTTAAAC-3′) and HT2 (5′-CTTTTCAAAGACTCAAATGGTCTAGCCTTTGGCTGCAAAATAGTTTCCATTTTGTATAGTTCATCCATGC-3′), and the product was used to transform AN117–4B and AN117–16D to obtain NY13 and NY14, respectively. These strains were mated to create diploid NY509. To create the DON1–GFP fusion gene in yeast, pFA6a-GFP (S65T)-His3MX6 (Longtine et al., 1998b) was amplified with HT15 (5′-AAGATGAAAAAAAGCAGGTTCATCCATCTAGCAAGAATTAAGTTTTACGCGGATCCCCGGGT-TAATTAA-3′) and HT16 (5′-AAAGGCAAATAAGCGCAGAAGAAGCAATGGTGGTGTCTGTTCATTGCGAGGAATTCGAGCTCGTTTAAAC-3′), and the product was used to transform AN117–4B to create NY17. To construct NY527, the his5 + gene of Schizosaccharomyces pombe was amplified from pFA6a-His3MX6 (Longtine et al., 1998b) with HT28 (5′-AACTGGGGAGAGGAAAGCAGATTACCACAATATACATTCAAATTAAAGAACGGAT-CCCCGGGTTAATTAA-3′) and HT29 (5′-TAATAAGTATTTTCCTTTTTAAACTTTGATTTAGGACGTGAATCTATTTAGAATTCGAGCTCGTTTAAAC-3′), and the product was used to transform AN120 to create the glc7 heterozygous deletion strain, NY523. NY523 was then transformed with pSB14 to create NY525, which has glc7–136 integrated at the ura3 locus. NY525 was sporulated and dissected. Progeny that had both the glc7 deletion and the glc7–136 integration were identified and mated to produce NY527. Unless otherwise noted, standard media and growth conditions were used (Rose et al., 1990).
Plasmids
To create pFA6a-His3MX6-prSPO20-HA, the promoter sequence of the SPO20 gene was amplified directly from yeast genome using ANO195 (5′-CCTTGAGATCTAAGTCTAGGCGCTTTCAAC-3′) and ANO201 (5′-CCTTGTTAATTAAAGACATTATATATCTAAAAATGGC-3′). The PCR product was then digested with PacI and BglII and used to replace GAL1 promoter region of pFA6a-His3MX6-PGAL1-3HA (Longtine et al., 1998b). To clone the prSPO20-HA-GIP1 fusion gene, genomic DNA from NY13 was subjected to PCR with ANO195 and HT6 (5′-CAGTTGCCTACCAATGTTTC-3′). The product was cloned into SmaI sites of pRS424 and pRS426 (Christianson et al., 1992) to create pSB5 and pSB6, respectively. pNC160-GFP-GLC7 (Bloecher and Tatchell, 2000) was used to express GFP-tagged version of Glc7p. pSB7 was created by excising the SPR28–GFP fusion gene from YEplac181-SPR28-GFP (De Virgilio et al., 1996) with EcoRI and BamHI and cloning it into pRS314 (Sikorski and Hieter, 1989). To clone DON1–GFP into plasmid, genomic DNA prepared from NY17 was subjected to PCR using HT10 (5′-AAACAGATCTATATTACCCTG-3′) and HT19 (5′-GAAGAATTCGATATAGCTCTGAACAATTC-3′). The product was digested with EcoRI and BglII and ligated into similarly digested pRS314 to create pSB8. To integrate glc7–136 at the ura3 locus, the glc7–136 gene was cut out from pSB56 (Baker et al., 1997) with HindIII and SacI, and cloned into pRS306, creating pSB14. pSB14 was linearized with PstI before transformation to target integration to the ura3 locus. The plasmids pHindIII-DIT1-lacZ, a gift of J. Segall (University of Toronto, Toronto, Canada), and pGK16 (Holaway et al., 1987) carrying the DIT1–lacZ and SPR3–lacZ reporters, respectively, were used to monitor gene induction.
Microscopy
For analysis of meiotic progression, cells were sporulated as described (Neiman, 1998). At intervals, aliquots were removed and fixed with 3.7% formaldehyde at 4°C overnight. Cells were collected and mounted in medium containing DAPI (Molecular Probes). EM was performed as described (Neiman, 1998). Samples were analyzed using a JEOL 1200EX transmission electron microscope at the Stony Brook University Microscopy Imaging Center (Stony Brook, NY).
For immunofluorescence analysis, cells were sporulated and fixed with 3.7% formaldehyde for 2 h, collected, resuspended in SHA buffer (1 M Sorbitol, 0.1 M Hepes, pH 7.5, 5 mM NaN3) and stored at 4°C. Immunofluorescence analysis of proteins was performed as described previously (Gao and Dean, 2000). Monoclonal anti-HA antibodies (12CA5) were provided by N. Dean (SUNY Stony Brook, Stony Brook, NY), and affinity-purified anti-GFP antibodies, a gift of N. Davis (Louisiana State University Medical Center, Shreveport, LA), were used at a 1:10 and 1:200 dilution, respectively. Anti-Spr3p antibodies (Fares et al., 1996) were used at a 1:10 dilution. Alexa Fluor 488 goat anti–mouse IgG conjugate, Alexa Fluor 488 goat anti–rabbit IgG conjugate, Alexa Fluor 546 goat anti–rabbit IgG conjugate, and Texas red goat anti–rabbit IgG conjugate (Molecular Probes) were used as secondary antibodies at a 1:400 dilution.
Immunofluorescence analysis of spore walls was performed as follows. Fixed sporulating cells were suspended in Zymolyase solution (27 μg/ml Zymolyase and 0.2% β-mercaptoethanol in SHA buffer) and incubated at 30° for 1 h. Cells were then treated 5 min with SHA buffer containing 0.1% Triton X-100, and 10 min with SHA buffer containing 0.2 mg/ml Calcofluor white (Sigma-Aldrich), washed twice with SHA buffer, and adhered to wells of a polylysine-coated slide. Cells on the slide were treated with methanol for 6 min, followed by acetone for 30 s, and then dried. After blocking with WT buffer (50 mM Hepes, pH 7.5, 150 mM NaCl, 1% Milk, 5 mg/ml bovine serum albumin), cells were incubated with a monoclonal anti–β-1,3 glucan antibody (Biosupplies) (Humbel et al., 2001) at a 1:300 dilution in WT buffer overnight. Wells were washed with PBS-BSA (7 mM Na2HPO4, 3 mM NaH2PO4, 130 mM NaCl, 5 mg/ml bovine serum albumin) and incubated with PBS-BSA containing 1:400 diluted Alexa Fluor 546 goat anti–mouse IgG and 0.1 mg/ml FITC-ConA (Sigma-Aldrich) for 2 h. Finally, wells were washed with PBS-BSA and covered with mounting media. For detection of native GFP fluorescence, cells were fixed with 3.7% formaldehyde for 10 min, washed once with PBS, and mounted with mounting media containing DAPI or processed for immunofluorescence.
Immunofluorescence images were acquired using a Zeiss Axioskop with attached SPOT camera (Diagnostic Instruments) and Adobe Photoshop 5.0, or on a Zeiss Axioplan 2 microscope with a Zeiss Axiocam and Zeiss Axiovision 3.0.6 software. Figures were prepared using Adobe Photoshop 6.0 and Canvas 5.0 (Deneba Software).
β-Galactosidase and dityrosine assays
Natural fluorescence of the dityrosine molecules was observed as described previously (Briza et al., 1990). DIT1–lacZ and SPR3–lacZ were separately introduced into both AN120 and NY501. The resulting cells were sporulated and aliquots were collected at each time point and subjected to β-galactosidase assay as described (Stern et al., 1984).
Online supplemental material
To create three-dimensional projections of Gip1 immunofluorescence, cells carrying HA–GIP1 were prepared after 7 h in sporulation medium, and stacks of images were collected with a Bio-Rad Radiance 2000 confocal microscroscope (25mW Arlaser, Nikon TE300, 100 X Plan Apo objective 1.4 NA). Images were collected at 0.2-μm intervals in the Z-axis at 1.5% laser power, pinhole aperture at 2.6, Kalman filtration of four images. Three-dimensional projections were created using Bio-Rad Lasersharp2000 software. Projected images were tilted at angles of −45°–45° at 1°-intervals.
To create three-dimensional projects of HA-Gip1p and DAPI fluorescence, stacks of images were collected through an Olympus UPlanFl 100 × 1.3 NA objective with a Roper Coolmax HQ CCD Camera using Scanalytics IPlab Spectrum software. Images were collected at 0.2-μm intervals in the Z-axis, and three-dimensional projections were created using Iplab software. Projected images were tilted at angles of −45°–45° at 5°-intervals. Both projections were saved as Apple QuickTime movies, available at http://www.jcb.org/cgi/content/full/jcb.200107008/DC1
Supplemental Material
Acknowledgments
The authors are grateful to J. Pringle (University of North Carolina, Chapel Hill, NC), N. Dean (SUNY Stony Brook, Stony Brook, NY), and N. David (Louisiana State University Medical Center, Shreveport, LA) for antibodies, and to H. Friesen and J. Segall (University of Toronto, Toronto, Canada) and N. Hollingsworth (SUNY Stony Brook) for reagents. A.M. Neiman is grateful to R. Sternglanz (SUNY Stony Brook) for material support during the early phases of this work. The authors are also grateful to N. Hollingsworth and J. Engebrecht for comments on the manuscript, and to members of the Neiman Lab for discussions during the course of this work.
This work was supported by National Institutes of Health grants GM62184 to A.M. Neiman and GM47789 to K. Tatchell.
The online version of this article contains supplemental material.
A. Bloecher's present address is Division of Basic Sciences, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. North, Seattle, WA 98109.
Footnotes
Abbreviations used in this paper: GFP, green fluorescent protein; HA, hemagglutinin; PP1, protein phosphatase type 1; SPB, spindle pole body.
References
- Bailis, J.M., and G.S. Roeder. 2000. Pachytene exit controlled by reversal of Mek1-dependent phosphorylation. Cell. 101:211–221. [DOI] [PubMed] [Google Scholar]
- Baker, S.H., D.L. Frederick, A. Bloecher, and K. Tatchell. 1997. Alanine-scanning mutagenesis of protein phosphatase type 1 in the yeast Saccharomyces cerevisiae. Genetics. 145:615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloecher, A., and K. Tatchell. 2000. Dynamic localization of protein phosphatase type 1 in the mitotic cell cycle of Saccharomyces cerevisiae. J. Cell Biol. 149:125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouquin, N., Y. Barral, R. Courbeyrette, M. Blondel, M. Snyder, and C. Mann. 2000. Regulation of cytokinesis by the Elm1 protein kinase in Saccharomyces cerevisiae. J. Cell Sci. 113:1435–1445. [DOI] [PubMed] [Google Scholar]
- Briza, P., M. Breitenbach, A. Ellinger, and J. Segall. 1990. Isolation of two developmentally regulated genes involved in spore wall maturation in Saccharomyces cerevisiae. Genes Dev. 4:1775–1789. [DOI] [PubMed] [Google Scholar]
- Briza, P., M. Eckerstorfer, and M. Breitenbach. 1994. The sporulation-specific enzymes encoded by the DIT1 and DIT2 genes catalyze a two-step reaction leading to a soluble LL-dityrosine-containing precursor of the yeast spore wall. Proc. Natl. Acad. Sci. USA. 91:4524–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briza, P., A. Ellinger, G. Winkler, and M. Breitenbach. 1988. Chemical composition of the yeast ascospore wall. The second outer layer consists of chitosan. J. Biol. Chem. 263:11569–11574. [PubMed] [Google Scholar]
- Briza, P., G. Winkler, H. Kalchhauser, and M. Breitenbach. 1986. Dityrosine is a prominent component of the yeast ascospore wall. A proof of its structure. J. Biol. Chem. 261:4288–4294. [PubMed] [Google Scholar]
- Christianson, T.W., R.S. Sikorski, M. Dante, J.H. Shero, and P. Hieter. 1992. Multifunctional yeast high-copy-number shuttle vectors. Gene. 110:119–122. [DOI] [PubMed] [Google Scholar]
- Christodoulidou, A., V. Bouriotis, and G. Thireos. 1996. Two sporulation-specific chitin deacetylase-encoding genes are required for the ascospore wall rigidity of Saccharomyces cerevisiae. J. Biol. Chem. 271:31420–31425. [DOI] [PubMed] [Google Scholar]
- Christodoulidou, A., P. Briza, A. Ellinger, and V. Bouriotis. 1999. Yeast ascospore wall assembly requires two chitin deacetylase isozymes. FEBS Lett. 460:275–279. [DOI] [PubMed] [Google Scholar]
- Chu, S., J. DeRisi, M. Eisen, J. Mulholland, D. Botstein, P.O. Brown, and I. Herskowitz. 1998. The transcriptional program of sporulation in budding yeast. Science. 282:699–705. [DOI] [PubMed] [Google Scholar]
- De Virgilio, C., D.J. DeMarini, and J.R. Pringle. 1996. SPR28, a sixth member of the septin gene family in Saccharomyces cerevisiae that is expressed specifically in sporulating cells. Microbiology. 142:2897–2905. [DOI] [PubMed] [Google Scholar]
- DeMarini, D.J., A.E. Adams, H. Fares, C. De Virgilio, G. Valle, J.S. Chuang, and J.R. Pringle. 1997. A septin-based hierarchy of proteins required for localized deposition of chitin in the Saccharomyces cerevisiae cell wall. J. Cell Biol. 139:75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaoli-Roach, A.A., I.K. Park, V. Cerovsky, C. Csortos, S.D. Durbin, M.J. Kuntz, A. Sitikov, P.M. Tang, A. Verin, and S. Zolnierowicz. 1994. Serine/threonine protein phosphatases in the control of cell function. Adv. Enzyme Regul. 34:199–224. [DOI] [PubMed] [Google Scholar]
- Fares, H., L. Goetsch, and J.R. Pringle. 1996. Identification of a developmentally regulated septin and involvement of the septins in spore formation in Saccharomyces cerevisiae. J. Cell Biol. 132:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares, H., M. Peifer, and J.R. Pringle. 1995. Localization and possible functions of Drosophila septins. Mol. Biol. Cell. 6:1843–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, Z.H., S.E. Wilson, Z.Y. Peng, K.K. Schlender, E.M. Reimann, and R.J. Trumbly. 1991. The yeast GLC7 gene required for glycogen accumulation encodes a type 1 protein phosphatase. J. Biol. Chem. 266:23796–23801. [PubMed] [Google Scholar]
- Francois, J.M., S. Thompson-Jaeger, J. Skroch, U. Zellenka, W. Spevak, and K. Tatchell. 1992. GAC1 may encode a regulatory subunit for protein phosphatase type 1 in Saccharomyces cerevisiae. EMBO J. 11:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen, H., R. Lunz, S. Doyle, and J. Segall. 1994. Mutation of the SPS1-encoded protein kinase of Saccharomyces cerevisiae leads to defects in transcription and morphology during spore formation. Genes Dev. 15:2162–2175. [DOI] [PubMed] [Google Scholar]
- Gao, X.D., and N. Dean. 2000. Distinct protein domains of the yeast Golgi GDP-mannose transporter mediate oligomer assembly and export from the endoplasmic reticulum. J. Biol. Chem. 275:17718–17727. [DOI] [PubMed] [Google Scholar]
- Holaway, B.L., G. Kao, M.C. Finn, and M.J. Clancy. 1987. Transcriptional regulation of sporulation genes in yeast. Mol. Gen. Genet. 210:449–459. [DOI] [PubMed] [Google Scholar]
- Hsu, S.C., C.D. Hazuka, R. Roth, D.L. Foletti, J. Heuser, and R.H. Scheller. 1998. Subunit composition, protein interactions and structures of the mammalian brain sec6/8 complex and septin filaments. Neuron. 20:1111–1122. [DOI] [PubMed] [Google Scholar]
- Humbel, B.M., M. Konomi, T. Takagi, N. Kamasawa, S.A. Ishijima, and M. Osumi. 2001. In situ localization of beta-glucans in the cell wall of Schizosaccharomyces pombe. Yeast. 18:433–444. [DOI] [PubMed] [Google Scholar]
- Kaback, D.B., and L.R. Feldberg. 1985. Saccharomyces cerevisiae exhibits a sporulation-specific temporal pattern of transcript accumulation. Mol. Cell. Biol. 5:751–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane, S., and R. Roth. 1974. Carbohydrate metabolism during ascospore development in yeast. J. Bacteriol. 118:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartmann, B., and D. Roth. 2001. Novel roles for mammalian septins: from vesicle trafficking to oncogenesis. J. Cell Sci. 114:839–844. [DOI] [PubMed] [Google Scholar]
- Kinoshita, M., S. Kumar, A. Mizoguchi, C. Ide, A. Kinoshita, T. Haraguchi, Y. Hiraoka, and M. Noda. 1997. Nedd5, a mammalian septin, is a novel cytoskeletal component interacting with actin-based structures. Genes Dev. 11:1535–1547. [DOI] [PubMed] [Google Scholar]
- Klis, F.M. 1994. Review: cell wall assembly in yeast. Yeast. 10:851–869. [DOI] [PubMed] [Google Scholar]
- Knop, M., and K. Strasser. 2000. Role of the spindle pole body of yeast in mediating assembly of the prospore membrane during meiosis. EMBO J. 19:3657–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krisak, L., R. Strich, R.S. Winters, J.P. Hall, M.J. Mallory, D. Kreitzer, R.S. Tuan, and E. Winter. 1994. SMK1, a developmentally regulated MAP kinase, is required for spore wall assembly in Saccharomyces cerevisiae. Genes Dev. 8:2151–2161. [DOI] [PubMed] [Google Scholar]
- Kupiec, M., B. Byers, R.E. Esposito, and A.P. Mitchell. 1997. Meiosis and sporulation in Saccharomyces cerevisiae The Molecular and Cellular Biology of the Yeast Saccharomyces. Vol. 3. J.R. Pringle, J.R. Broach, and E.W. Jones, editors. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY 889–1036.
- Kurtz, S., and S. Lindquist. 1986. Subcellular differentiation in sporulating yeast cells. Cell. 45:771–779. [DOI] [PubMed] [Google Scholar]
- Longtine, M.S., D.J. DeMarini, M.L. Valencik, O.S. Al-Awar, H. Fares, C. De Virgilio, and J.R. Pringle. 1996. The septins: roles in cytokinesis and other processes. Curr. Opin. Cell Biol. 8:106–119. [DOI] [PubMed] [Google Scholar]
- Longtine, M.S., H. Fares, and J.R. Pringle. 1998. a. Role of the yeast Gin4p protein kinase in septin assembly and the relationship between septin assembly and septin function. J. Cell Biol. 143:719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine, M.S., A. McKenzie III, D.J. Demarini, N.G. Shah, A. Wach, A. Brachat, P. Philippsen, and J.R. Pringle. 1998. b. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 14:953–961. [DOI] [PubMed] [Google Scholar]
- Longtine, M.S., C.L. Theesfeld, J.N. McMillan, E. Weaver, J.R. Pringle, and D.J. Lew. 2000. Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol. Cell. Biol. 20:4049–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn, R.R., and P.T. Magee. 1970. Development of the spore wall during ascospore formation in Saccharomyces cerevisiae. J. Cell Biol. 44:688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra, C., C.E. Semino, K.J. McCreath, H. de la Vega, B.J. Jones, C.A. Specht, and P.W. Robbins. 1997. Cloning and expression of two chitin deacetylase genes of Saccharomyces cerevisiae. Yeast. 13:327–336. [DOI] [PubMed] [Google Scholar]
- Moens, P.B. 1971. Fine structure of ascospore development in the yeast Saccharomyces cerevisiae. Can. J. Microbiol. 17:507–510. [DOI] [PubMed] [Google Scholar]
- Moens, P.B., and E. Rapport. 1971. Spindles, spindle plaques, and meiosis in the yeast Saccharomyces cerevisiae (Hansen). J. Cell Biol. 50:344–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumby, M.C., and G. Walter. 1993. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol. Rev. 73:673–699. [DOI] [PubMed] [Google Scholar]
- Neiman, A.M. 1998. Prospore membrane formation defines a developmentally regulated branch of the secretory pathway in yeast. J. Cell Biol. 140:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiman, A.M., L. Katz, and P.J. Brennwald. 2000. Identification of domains required for developmentally regulated SNARE function in Saccharomyces cerevisiae. Genetics. 155:1643–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld, T.P., and G.M. Rubin. 1994. The Drosophila peanut gene is required for cytokinesis and encodes a protein similar to yeast putative bud neck filament proteins. Cell. 77:371–379. [DOI] [PubMed] [Google Scholar]
- Pammer, M., P. Briza, A. Ellinger, T. Schuster, R. Stucka, H. Feldmann, and M. Breitenbach. 1992. DIT101 (CSD2, CAL1), a cell cycle-regulated yeast gene required for synthesis of chitin in cell walls and chitosan in spore walls. Yeast. 8:1089–1099. [DOI] [PubMed] [Google Scholar]
- Primig, M., R.M. Williams, E.A. Winzeler, G.G. Tevzadze, A.R. Conway, S.Y. Hwang, R.W. Davis, and R.E. Esposito. 2000. The core meiotic transcriptome in budding yeasts. Nat. Genet. 26:415–423. [DOI] [PubMed] [Google Scholar]
- Ramaswamy, N.T., L. Li, M. Khalil, and J.F. Cannon. 1998. Regulation of yeast glycogen metabolism and sporulation by Glc7p protein phosphatase. Genetics. 149:57–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, M.D., F. Winston, and P. Hieter. 1990. Methods in yeast genetics. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, NY. 198 pp.
- Shenolikar, S. 1994. Protein serine/threonine phosphatases—new avenues for cell regulation. Annu. Rev. Cell Biol. 10:55–86. [DOI] [PubMed] [Google Scholar]
- Sikorski, R.S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern, M., R. Jensen, and I. Herskowitz. 1984. Five SWI genes are required for expression of the HO gene in yeast. J. Mol. Biol. 178:853–868. [DOI] [PubMed] [Google Scholar]
- Straight, P.D., T.H. Giddings, Jr., and M. Winey. 2000. Mps1p regulates meiotic spindle pole body duplication in addition to having novel roles during sporulation. Mol. Biol. Cell. 11:3525–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart, J.S., D.L. Frederick, C.M. Varner, and K. Tatchell. 1994. The mutant type 1 protein phosphatase encoded by glc7-1 from Saccharomyces cerevisiae fails to interact productively with the GAC1-encoded regulatory subunit. Mol. Cell. Biol. 14:896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, J., W. Song, and M. Carlson. 1996. Protein phosphatase type 1 interacts with proteins required for meiosis and other cellular processes in Saccharomyces cerevisiae. Mol. Cell. Biol. 16:4199–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ufano, S., P. San-Segundo, F. del Rey, and C.R. Vazquez de Aldana. 1999. SWM1, a developmentally regulated gene, is required for spore wall assembly in Saccharomyces cerevisiae. Mol. Cell. Biol. 19:2118–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, M., P. Briza, M. Pierce, and E. Winter. 1999. Distinct steps in yeast spore morphogenesis require distinct SMK1 MAP kinase thresholds. Genetics. 151:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, M., M. Pierce, and E. Winter. 1997. The CDK-activating kinase CAK1 can dosage suppress sporulation defects of smk1 MAP kinase mutants and is required for spore wall morphogenesis in Saccharomyces cerevisiae. EMBO J. 16:1305–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziman, M., J.S. Chuang, M. Tsung, S. Hamamoto, and R. Schekman. 1998. Chs6p-dependent anterograde transport of Chs3p from the chitosome to the plasma membrane in Saccharomyces cerevisiae. Mol. Biol. Cell. 9:1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.