

Abstract
We have shown previously that Ipl1 and Sli15 are required for chromosome segregation in Saccharomyces cerevisiae. Sli15 associates directly with the Ipl1 protein kinase and these two proteins colocalize to the mitotic spindle. We show here that Sli15 stimulates the in vitro, and likely in vivo, kinase activity of Ipl1, and Sli15 facilitates the association of Ipl1 with the mitotic spindle. The Ipl1-binding and -stimulating activities of Sli15 both reside within a region containing homology to the metazoan inner centromere protein (INCENP). Ipl1 and Sli15 also bind to Dam1, a microtubule-binding protein required for mitotic spindle integrity and kinetochore function. Sli15 and Dam1 are most likely physiological targets of Ipl1 since Ipl1 can phosphorylate both proteins efficiently in vitro, and the in vivo phosphorylation of both proteins is reduced in ipl1 mutants. Some dam1 mutations exacerbate the phenotype of ipl1 and sli15 mutants, thus providing evidence that Dam1 interactions with Ipl1–Sli15 are functionally important in vivo. Similar to Dam1, Ipl1 and Sli15 each bind to microtubules directly in vitro, and they are associated with yeast centromeric DNA in vivo. Given their dual association with microtubules and kinetochores, Ipl1, Sli15, and Dam1 may play crucial roles in regulating chromosome–spindle interactions or in the movement of kinetochores along microtubules.
Keywords: chromosome segregation; Ipl1; Sli15; Dam1; kinetochore
Introduction
The Ipl1 protein kinase from budding yeast is a founding member of the aurora/Ipl1-related kinase (AIRK)* family (for review see Bischoff and Plowman, 1999; Giet and Prigent, 1999), and it plays a key role in the regulation of chromosome segregation (Chan and Botstein, 1993). Bipolar mitotic spindle assembly, sister chromatid cohesion and separation, and spindle elongation and disassembly appear relatively normal in the majority of ipl1 mutant cells, but chromosomes become severely missegregated without cell cycle arrest (Francisco et al., 1994; Biggins et al., 1999; Kim et al., 1999). At least part of the chromosome missegregation observed in ipl1 cells may be caused by kinetochore defects, since kinetochores assembled in ipl1 mutant extracts show altered binding to microtubules, and Ipl1 phosphorylates the kinetochore component Ndc10 in vitro (Biggins et al., 1999). Ipl1 is also known to phosphorylate histone H3 at serine 10 in vitro and in vivo. However, this phosphorylation alone is not essential for mitotic or meiotic chromosome segregation in yeast (Hsu et al., 2000).
One protein that appears to be required for Ipl1 function is Sli15, which binds directly to Ipl1 (Kim et al., 1999). Mutations in SLI15 exacerbate the Ts− phenotype of ipl1-2 cells, and Ts− sli15-3 mutant cells have cytological phenotypes very similar to those of ipl1-2 cells. Both proteins also colocalize to spindle microtubules and spindle poles. These observations suggest that Sli15 may be a positive regulator or major physiological target of Ipl1. Recently, a sequence motif known as the IN box (Adams et al., 2000; Kaitna et al., 2000) was found to be present at the COOH termini of both Sli15 and the metazoan “chromosomal passenger” inner centromere protein (INCENP), which localizes along chromosomes, at kinetochores, and at the spindle midzone in a cell cycle stage–specific manner (Cooke et al., 1987). Interestingly, INCENP proteins from Caenorhabditis elegans, Xenopus, and humans bind to aurora-B (AIRK2) and these two proteins colocalize in human cells (Adams et al., 2000; Kaitna et al., 2000). Aurora-B and INCENP, like their counterparts in yeast, are required for chromosome segregation (Mackay et al., 1998; Kaitna et al., 2000; Giet and Glover, 2001).
Dam1 and Duo1 are components of an essential protein complex required for both mitotic spindle integrity and kinetochore function. These two proteins associate with each other and colocalize to spindle poles, spindle microtubules, and kinetochores (Hofmann et al., 1998; Jones et al., 1999; Cheeseman et al., 2001). Although Dam1 by itself can bind microtubules directly in vitro, Dam1 and Duo1 require each other for their localization to the spindle microtubules. Interestingly, in addition to missegregating chromosomes severely, some dam1 mutant cells exhibit premature anaphase events, such as spindle elongation while arrested in metaphase, as well as genetic interactions with a subset of kinetochore components. Similar abnormalities have been observed in some ipl1 mutant cells (Biggins et al., 1999), and they point toward a potential functional connection between Ipl1 and Dam1 in the correct functioning of the kinetochores.
Here we report a molecular characterization of Ipl1 and Sli15 and demonstrate a novel interaction with Dam1. Our results show that, like Dam1, Ipl1 and Sli15 are microtubule-binding proteins associated with yeast kinetochores. Both Sli15 and Dam1 are likely physiological targets of Ipl1, and Sli15 also stimulates the kinase activity of Ipl1 and facilitates its association with the mitotic spindle. The microtubule-binding and Ipl1-stimulating activities of Sli15 reside in different regions of Sli15.
Results
The IN box–containing COOH-terminal region of Sli15 binds to Ipl1
To identify proteins that associate with Ipl1 in vivo, we performed a two-hybrid screen (James et al., 1996) with full-length Ipl1 as the bait. From this screen, we identified prey plasmids that encoded truncated forms of Sli15, the shortest version of which contained the COOH-terminal 88 residues (Sli15611–698; pCC1007). Present within this COOH-terminal domain is the recently described IN box (residues 626–698; Fig. 1), which is conserved between Sli15 and metazoan INCENP (Adams et al., 2000; Kaitna et al., 2000). To find out whether other regions of Sli15 also bind to Ipl1, we tested other prey plasmids that encoded full-length Sli15, the NH2-terminal (Sli151–229 or Sli15-N), the middle (Sli15227–559 or Sli15-M), or the COOH-terminal (Sli15558–698 or Sli15-C) region of Sli15. Only full-length Sli15 and the IN box–containing Sli15-C showed interactions with Ipl1 (Fig. 1; unpublished data).
Figure 1.
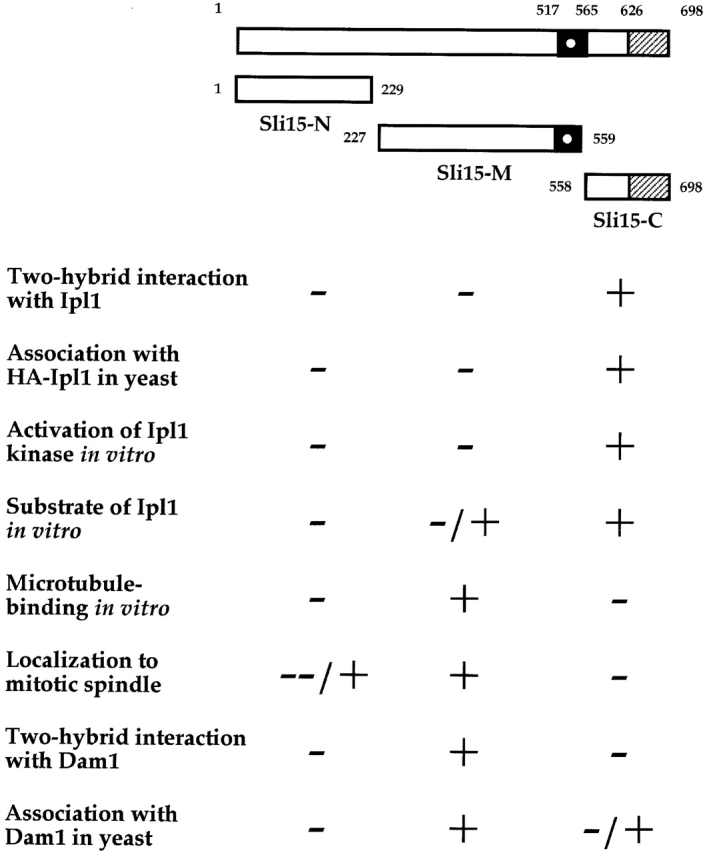
Properties of different regions of Sli15. The region of putative coiled-coil is filled in. The putative nuclear localization signal is shown as a white dot. The conserved IN box is stippled.
To test whether Sli15-C associates with Ipl1 in vivo, we affinity-purified glutathione-S-transferase (GST), GST–Sli15, GST–Sli15-N, GST–Sli15-M, or GST–Sli15-C from yeast cells that expressed a functional version of HA–Ipl1. HA–Ipl1 copurified with GST–Sli15 and GST–Sli15-C, but not with GST, GST–Sli15-N, or GST–Sli15-M (Fig. 2). These two-hybrid interaction and biochemical results, together with our previous demonstration that Ipl1 binds directly to Sli15 (Kim et al., 1999), indicate that the IN box–containing COOH-terminal 141 residues of Sli15 are necessary and sufficient for the direct binding of Sli15 to Ipl1.
Figure 2.
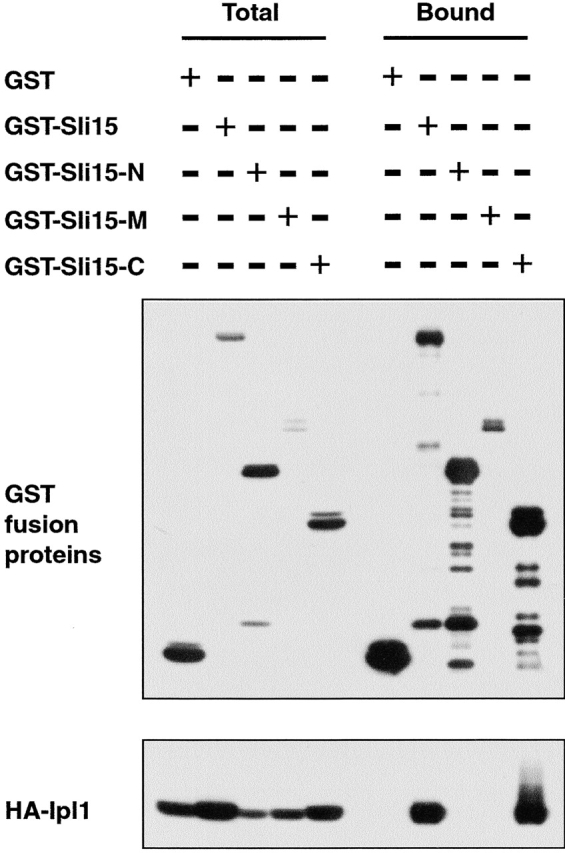
The IN box–containing COOH-terminal region of Sli15 binds Ipl1. GST (pEG[KT]), GST–Sli15 (pCC1061), GST–Sli15-N (pCC1563), GST–Sli15-M (pCC1564), or GST–Sli15-C (pCC1565) was affinity-purified with glutathione-agarose beads from a yeast strain (TD4) that also expressed HA–Ipl1 (pCC1128). Aliquots of proteins from total lysates (Total) or from the affinity-purified (Bound) fractions (after three rounds of washing with a buffer containing 200 mM KCl) were analyzed by immunoblotting using antibodies against GST or the HA epitope. The amount of bound fraction loaded was 4.5-fold that of the total lysates.
The conserved COOH-terminal region of Sli15 activates the in vitro kinase activity of Ipl1
Given the tight association of Sli15 and Ipl1, Sli15 could function as a positive regulator or substrate for Ipl1. We therefore performed in vitro kinase assays with recombinant GST–Ipl1 and GST–Sli15 purified from Escherichia coli. A similar GST–Ipl1 fusion protein expressed in yeast is biologically functional (unpublished data). As reported previously (Biggins et al., 1999), GST–Ipl1 could phosphorylate the artificial substrate myelin basic protein (MBP) and also itself, but not GST (Fig. 3 A, lane 1; unpublished data). The kinase activity observed in vitro was due to GST–Ipl1, and was not due to a contaminating kinase that copurified with GST–Ipl1, since a mutant version of GST–Ipl1 (GST–Ipl1-D245N) could not phosphorylate MBP (Fig. 3 A, lane 6). Strikingly, the ability of GST–Ipl1 to autophosphorylate or phosphorylate MBP was stimulated by the addition of GST–Sli15 but not GST (Fig. 3 A, lane 2; unpublished data). Additional kinase reactions showed that GST–Sli15-C, which can bind to Ipl1, also stimulated the in vitro kinase activity of GST–Ipl1, whereas GST–Sli15-N and GST–Sli15-M, which cannot bind to Ipl1, could not stimulate this kinase activity (Fig. 3 A, lanes 3–5). The extent of stimulation was dependent on the amount of GST–Sli15-C used (Fig. 3 B). Thus, the IN box–containing COOH-terminal region of Sli15 both binds to Ipl1 and stimulates its kinase activity.
Figure 3.
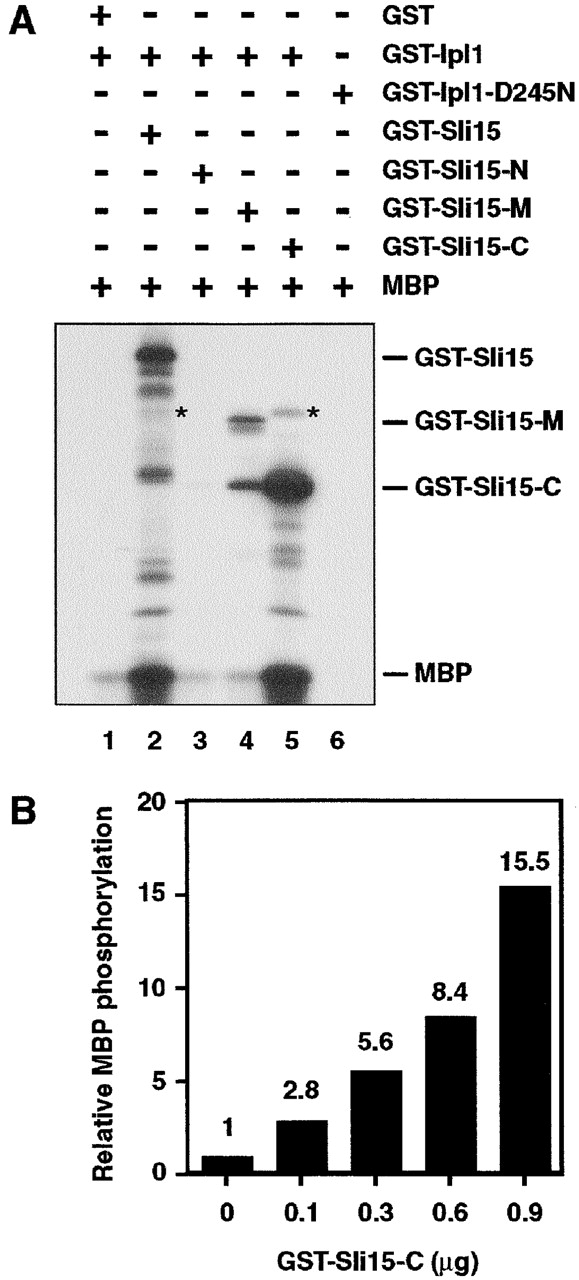

Ipl1 phosphorylates Sli15 and is activated by Sli15. (A) In vitro phosphorylation of Sli15 by Ipl1 and [γ-32P]ATP. With the exception of bovine MBP, all proteins used were purified from E. coli. The asterisks denote labeled GST–Ipl1 bands caused by autophosphorylation. (B) Stimulation of GST–Ipl1 kinase activity by GST–Sli15-C. MBP (1 μg) was phosphorylated by GST–Ipl1 (0.6 μg) in the presence of various amounts of GST–Sli15-C. (C) Phosphorylation of Sli15 is altered in ipl1 mutant cells. Wild-type (WT) (CCY766-9D) or ipl1-2 (CCY915-13C) cells carrying pCC1301 (Sli15–HA) were incubated in supplemented synthetic complete (SC) medium lacking leucine at 26°C and then shifted to 37°C for 2.5 h. Protein extracts from these cells were analyzed by immunoblotting with anti-HA or anti-G6PDH antibodies.
Ipl1 phosphorylates the middle and COOH-terminal regions of Sli15 in vitro
In the in vitro kinase assays described above, GST–Sli15, but not GST, was phosphorylated efficiently by GST–Ipl1 (Fig. 3 A, lanes 1 and 2). Under our reaction conditions, ∼2 mol of phosphate were incorporated per mol of GST–Sli15. When compared with MBP (which was present at a much higher concentration in these reactions), GST–Sli15 was a much better substrate (Fig. 3 A, lanes 1 and 2). When the kinase assays were performed with truncated forms of Sli15, we found that GST–Ipl1 could not phosphorylate GST–Sli15-N, but it could phosphorylate GST–Sli15-M and GST–Sli15-C. Thus, Ipl1 phosphorylation sites exist in the middle and COOH-terminal regions of Sli15.
Sli15 is a phosphoprotein that is under-phosphorylated in ipl1-2 mutant cells
We have previously shown that multiple forms of Sli15 differing in electrophoretic mobility exist in vivo (Kim et al., 1999). Three to four forms that could be resolved by SDS-PAGE were detected in most experiments. The slower-migrating forms represented phosphorylated Sli15, since they could be converted to the fastest migrating form by phosphatase treatment (unpublished data). To find out whether the in vivo phosphorylation of Sli15 is dependent on Ipl1 function, we examined the phosphorylation state of a functional version of Sli15–HA in wild-type and ipl1-2 mutant cells. Strikingly, the abundance of the two slowest-migrating forms of Sli15–HA was greatly reduced in ipl1-2 cells even at the permissive growth temperature of 26°C (Fig. 3 C, lanes 1 and 2). These two forms of Sli15–HA almost totally disappeared in ipl1-2 cells after a 2.5-h incubation at the restrictive growth temperature of 37°C (Fig. 3 C, lanes 3 and 4). These results are consistent with our previous observation that despite a normal growth rate and cell cycle distribution, ipl1-2 cells exhibit an ∼10-fold increase in the frequency of chromosome gain and thus have reduced Ipl1 function at the permissive temperature (Chan and Botstein, 1993). The disappearance of two hyperphosphorylated forms of Sli15 in ipl1-2 cells is also consistent with the observation that Ipl1 could phosphorylate both the middle and COOH-terminal regions of Sli15 in vitro (Fig. 3 A). Together, the in vitro and in vivo results indicate Sli15 most likely functions as a physiological substrate of Ipl1.
Ipl1 and Sli15 bind to microtubules in vitro
Because Ipl1 and Sli15 both localize along the length of the mitotic spindle (Biggins et al., 1999; Kim et al., 1999), we tested whether either of these proteins can bind to microtubules directly by examining the ability of GST and GST fusion proteins (that were purified from E. coli) to cosediment with taxol-stabilized microtubules in vitro. GST did not sediment with microtubules (Fig. 4 A). In contrast, whereas only a small fraction of GST–Ipl1 sedimented in the absence of microtubules, the bulk of GST–Ipl1 did cosediment with microtubules in a microtubule concentration–dependent manner (Fig. 4 B). The estimated dissociation constant of the GST–Ipl1–microtubule interaction is ∼0.5 μM, which represents a slightly lower affinity than that between tau and microtubules (Goode and Feinstein, 1994).
Figure 4.
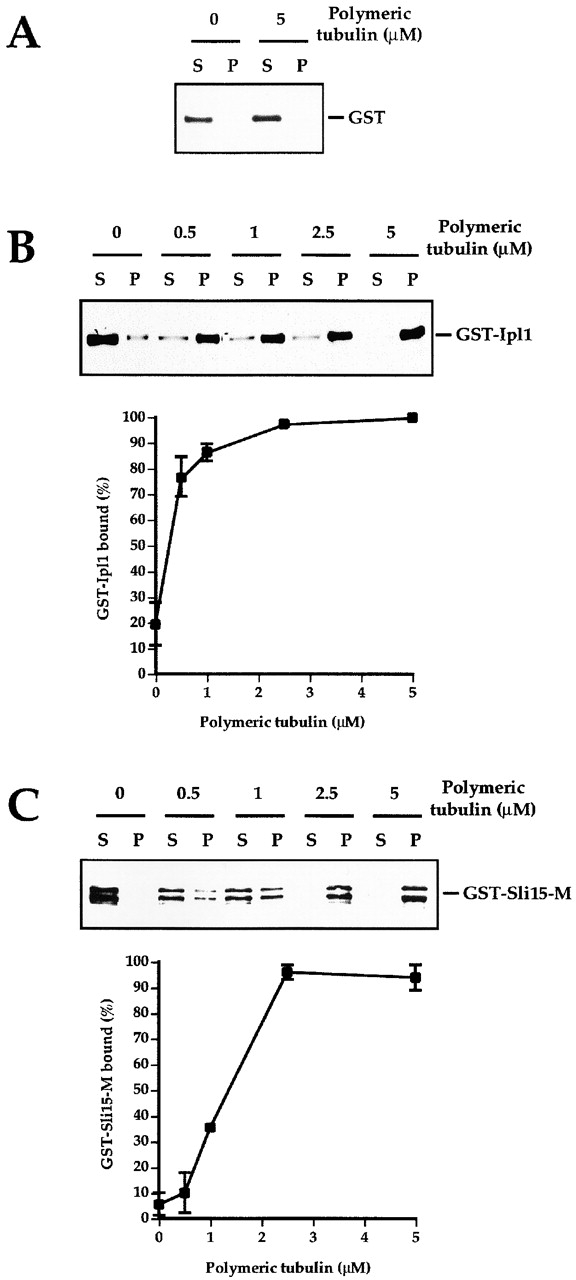
GST–Ipl1 and GST–Sli15-M cosediment with microtubules. Different concentrations of taxol-stabilized microtubules were incubated with GST (A), GST–Ipl1 (B), or GST–Sli15-M (C). The reactions were then centrifuged at high speed to generate the supernatant (S) and pellet (P) fractions. The presence of GST or GST fusion protein in either fraction was determined by immunoblotting with anti-GST antibodies. The percentage of GST–Ipl1 or GST–Sli15-M that was bound to and copelleted with the microtubules in each reaction was quantitated by densitometry of the immunoblots. The binding curves display the average and range of values obtained from duplicate experiments.
When different regions of Sli15 were tested, GST–Sli15-M (Fig. 4 C), but not GST–Sli15-N or GST–Sli15-C (unpublished data), showed microtubule concentration–dependent binding. The estimated dissociation constant of the GST–Sli15-M–microtubule interaction is ∼1.5 μM. Consistent with the ability of Sli15-M to bind to microtubules in vitro, green fluorescent protein (GFP)–Sli15-M, like GFP–Sli15, was localized along the length of the mitotic spindle in wild-type cells (Fig. 5 A). In contrast, the concentrations of GFP–Sli15-N and GFP–Sli15-C in the cytoplasm were much higher, and these proteins were often found to be concentrated around the nuclear periphery, suggesting a problem with the import of these proteins into the nucleus. In a small fraction of cells that expressed GFP–Sli15-N, this protein also could be detected at a low level along the mitotic spindle or spindle poles (unpublished data). Together, these results indicate that the Ipl1 binding (involving residues 558–698) and localization (involving residues 227–559) activities of Sli15 are separable. Thus, Sli15 is unlikely to be targeted to the mitotic spindle solely through its interaction with Ipl1. Consistent with this conclusion, the mitotic spindle localization of GFP–Sli15 was unaffected in ipl1-2 cells even at a restrictive temperature (unpublished data).
Figure 5.
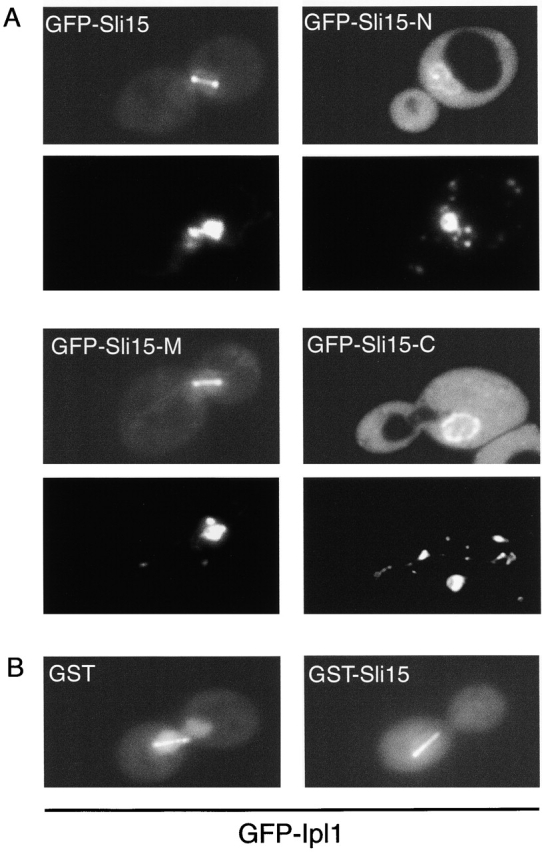
Subcellular localization of GFP–Sli15 and GFP–Ipl1. (A) Subcellular localization of full-length and truncated forms of Sli15. Wild-type diploid yeast cells (DBY1830) expressing GFP–Sli15 (pCC1060), GFP–Sli15-N (pCC1533), GFP–Sli15-M (pCC1534), or GFP–Sli15-C (pCC1566) were stained with DAPI and examined. For each pair of images: top, GFP fusion protein image; bottom, DAPI-stained image. (B) Subcellular localization of GFP–Ipl1. Wild-type haploid yeast cells (TD4) carrying pCC1584 (GFP–Ipl1) and pEG(KT) (GST) or carrying pCC1584 and pCC1061 (GST–Sli15) were cultured for 2 h in medium containing 4% galactose to induce expression of GST or GST–Sli15, respectively. Images of GFP–Ipl1 are shown.
Sli15 facilitates the association of Ipl1 with the mitotic spindle
We demonstrated above that both Ipl1 and Sli15 are capable of binding to microtubules individually. Because Ipl1 and Sli15 seem to be tightly associated, this suggests that in vivo, their binding to microtubules may be cooperative. To test this hypothesis, we examined whether Sli15 is able to facilitate the association of Ipl1 with the mitotic spindle. We first examined GFP–Ipl1 localization in sli15-3 mutant cells and found it to be normal (unpublished data). We also tested whether the subcellular localization of GFP–Ipl1 was affected by an increase in the expression level of Sli15. In a previous study of GFP–Ipl1 and GFP–Sli15, we found that a significantly higher fraction of GFP–Ipl1 was found throughout the interior of the nucleus, although both proteins were concentrated at the spindle poles and on the mitotic spindle (Fig. 5, A and B; Kim et al., 1999). In this study, both fusion proteins were expressed under the control of the relatively strong ACT1 promoter. Thus, GFP–Ipl1 and GFP–Sli15 were probably present at levels higher than those of endogenous Sli15 and Ipl1, respectively. If Sli15 were required for the efficient localization of GFP–Ipl1 to the mitotic spindle and spindle poles, the overabundance of GFP–Ipl1 relative to Sli15 would explain why a higher level of GFP–Ipl1 (relative to GFP–Sli15) was found throughout the nucleoplasm. This hypothesis predicted that more GFP–Ipl1 would be targeted to the mitotic spindle and spindle poles in cells that overexpress Sli15. We found this to be true since GFP–Ipl1 was much more restricted to the mitotic spindle and spindle poles in cells that expressed GST–Sli15 under the control of the strong GAL1/10 promoter (Fig. 5 B). This effect was not caused by a reduction in the level of GFP–Ipl1, since expression of GST–Sli15 is known to increase the abundance of Ipl1 (Fig. 2; Kim et al., 1999). Thus, Sli15 facilitates the localization of Ipl1 to the mitotic spindle.
Ipl1–Sli15 can associate with Dam1–Duo1 in vivo
To identify additional proteins that associate with Sli15 (and possibly Ipl1) in vivo, we performed a two-hybrid screen (James et al., 1996) with full-length Sli15 as the bait. From this screen, we identified a prey plasmid (pCC1428) that encoded a truncated form of Duo1 lacking the first nine residues (Duo110–247). Because Duo1 is known to bind to Dam1 (Hofmann et al., 1998; Cheeseman et al., 2001), we tested whether Dam1 also could interact with Sli15 in the two-hybrid assay. Our results indicated that Sli15 interacted with full-length Dam1 even more strongly than with full-length Duo1 (unpublished data).
To confirm the in vivo association of Dam1–Duo1 with Sli15 and possibly Ipl1, we purified GST, GST–Dam1, or GST–Duo1 (which were expressed under the control of the GAL1/10 promoter) from yeast cells that also expressed functional HA–Ipl1 or Sli15–Myc. Our results showed that HA–Ipl1 and Sli15–Myc could be copurified with GST–Dam1 (Fig. 6 A, lanes 6 and 11), whereas HA–Ipl1 but not Sli15–Myc could be copurified with GST–Duo1 (Fig. 6 A, lanes 5 and 12). The association of HA–Ipl1 with GST–Dam1 appeared to be stronger than that with GST–Duo1 since repeated rounds of washing led to the dissociation of most HA–Ipl1 from GST–Duo1 but not from GST–Dam1. These results are consistent with the two-hybrid results described above, which suggested that Sli15 interacted more strongly with Dam1 than with Duo1. One possible interpretation of these results is that Dam1 may mediate the association between Duo1 and Ipl1–Sli15.
Figure 6.

Dam1 binds to Ipl1 and Sli15. (A) In vivo association of Ipl1 or Sli15 with Dam1 or Duo1. GST (pEG[KT]), GST–Dam1 (pDD1017), or GST–Duo1 (pDD475) were affinity-purified with glutathione-agarose beads from a yeast strain (TD4) that also expressed HA–Ipl1 (pCC1128) or Sli15–Myc (pCC1173). Aliquots of proteins from total lysates (total) or from the affinity-purified (bound) fractions (after three rounds of washing with a buffer containing 200 mM KCl) were analyzed by immunoblotting using anti-GST, anti-HA, or anti-Myc antibodies. (B) Direct binding of Dam1 to Ipl1 and Sli15. GST (pGEX-2T), GST–Sli15 (pCC1062), and GST–Ipl1 (pCC669) were affinity-purified with glutathione-agarose beads from E. coli and tested for their ability to bind His6–Dam1 (pDD884). Proteins were detected by immunoblotting with anti-GST or anti-Dam1 antibodies. (C) The middle and COOH-terminal regions of Sli15 bind Dam1. The ability of GST (pEG[KT]), GST–Sli15 (pCC1061), GST–Sli15-N (pCC1563), GST–Sli15-M (pCC1564), or GST–Sli15-C (pCC1565) to associate with Dam1 (pCC1575) was determined as in A. The amount of bound fraction loaded in A and C was 4.5-fold that of the total lysates.
To determine whether Dam1 can bind to Ipl1 or Sli15 directly, we expressed GST, GST–Ipl1, GST–Sli15 and His6–Dam1 separately in E. coli. As shown in Fig. 6 B, the His6–Dam1 that was present in an E. coli crude extract associated with GST–Ipl1 and GST–Sli15, but not GST, which were immobilized on glutathione-agarose beads. Similar results were obtained using Dam1 translated in vitro (unpublished data). Together, these results demonstrate that, at least under conditions of protein overproduction, Dam1 binds Ipl1 and Sli15 in vivo, most likely through direct protein–protein interactions.
Dam1 binding primarily involves the middle region of Sli15
To determine whether different regions of Sli15 are responsible for its binding to Dam1–Duo1 and Ipl1, we repeated the two-hybrid assays with truncated versions of Sli15. Our results showed that Sli15-M, but not Sli15-N and Sli15-C, interacted with Dam1 and Duo1 in this assay (Fig. 1; unpublished data). To confirm the in vivo association of Sli15-M with Dam1 or Duo1, we affinity-purified GST, GST–Sli15, GST–Sli15-N, GST–Sli15-M, and GST–Sli15-C from yeast. Duo1 did not copurify with any GST fusion proteins (unpublished data). For reasons that we do not understand, Dam1 also did not copurify with GST–Sli15 (Fig. 6 C, lane 7). However, Dam1 was readily copurified with GST–Sli15-M and, to a lesser degree, with GST–Sli15-C, but not with GST or GST–Sli15-N (Fig. 6 C, lanes 6–10). Interestingly, the Dam1 species that copurified with GST–Sli15-C had a slower electrophoretic mobility than the Dam1 species that copurified with GST–Sli15-M. Because reduced electrophoretic mobility of Dam1 is caused by its hyperphosphorylation (see below) and since Dam1 is known to bind directly to Ipl1 and Sli15 (Fig. 6 B), these results suggest that Dam1 associates directly with the middle region of Sli15 (Sli15-M) and indirectly with the COOH-terminal region of Sli15 (Sli15-C) through Ipl1.
Because Sli15-M binds to Dam1 and is localized to the mitotic spindle, we tested whether the localization of Ipl1–Sli15 and Dam1 to the mitotic spindle is interdependent. Our results showed that GFP–Sli15 and GFP–Ipl1, unlike Dam1 and Duo1 (Cheeseman et al., 2001), were properly localized to the mitotic spindle in dam1-1, dam1-9, and dam1-11 cells at 25°C as well as 37°C (unpublished data). Similarly, Dam1 and Duo1 were properly localized in ipl1-2 and sli15-3 cells at both temperatures (unpublished data). Thus, the localization of Ipl1–Sli15 and Dam1 to the mitotic spindle does not appear to be interdependent.
Dam1, but not Duo1, is an in vitro substrate of Ipl1
Because Dam1 and Duo1 can associate with Ipl1–Sli15, this raised the possibility that Dam1 or Duo1 may be substrates of Ipl1. Therefore, we performed in vitro kinase assays with His6–Dam1, GST–Dam1-C (containing the COOH-terminal residues 175–343 of Dam1), GST–Sli15, and GST–Ipl1 purified from E. coli, as well as GST–Duo1 that was purified from yeast. As shown in Fig. 7 A (lanes 6–8), GST–Duo1 was not phosphorylated by GST–Ipl1 even in the presence of GST–Sli15. In contrast, His6–Dam1 and GST–Dam1-C were readily phosphorylated by GST–Ipl1 (Fig. 7 A, lanes 2 and 3). Furthermore, the addition of GST–Sli15 to the kinase reaction greatly stimulated the ability of GST–Ipl1 to phosphorylate these two proteins (Fig. 7 A, lanes 4 and 5). GST–Dam1-C could be phosphorylated almost quantitatively, resulting in the conversion of the majority of GST–Dam1-C to an electrophoretically slower–migrating form (unpublished data). Thus, Dam1 is a very good in vitro substrate of Ipl1–Sli15. Because the amount of GST–Sli15 used in these experiments was much less than that of His6–Dam1 or GST–Dam1-C, the relatively low level of Sli15 phosphorylation (Fig. 7 A, lanes 4 and 5) did not necessarily mean that Dam1 was a much better in vitro substrate than Sli15.
Figure 7.

Ipl1 phosphorylates Dam1. (A) In vitro phosphorylation of Dam1 by Ipl1 and [γ-32P]ATP. With the exception of bovine MBP and GST–Duo1 (from yeast), all proteins used were purified from E. coli. (B) Dam1 is phosphorylated in vivo. Dam1, immunoprecipitated from yeast cells (DDY904), was treated with λ phosphatase and analyzed by immunoblotting with anti-Dam1 antibodies. (C) Phosphorylation of Dam1 is altered in ipl1 and sli15 mutant cells. Wild-type (WT) (DDY904), ipl1-2 (CCY915-13C), or sli15-3 (CCY1124-4B) cells were incubated in YEPD medium at 25°C and then shifted to 37°C for 3 h. Protein extracts from these cells were analyzed by immunoblotting with anti-Dam1 antibodies.
To find out whether Dam1 also could stimulate the kinase activity of Ipl1, we examined the effect of His6–Dam1 on the ability of GST–Ipl1 to phosphorylate MBP. Our results indicated that His6–Dam1, unlike GST–Sli15, could not stimulate the kinase activity of GST–Ipl1 (Fig. 7 A, lanes 9 and 10).
Dam1 is a phosphoprotein that is underphosphorylated in ipl1 and sli15 mutant cells
Previous studies using anti-Dam1 antibodies indicated that Dam1 exists as multiple forms that differ in electrophoretic mobility (Cheeseman et al., 2001). On a 10% polyacrylamide gel, as many as five different forms of Dam1 were visible (Fig. 7 C, lane 1). The slower-migrating forms were caused by phosphorylation since phosphatase treatment of immunoprecipitated Dam1 led to the disappearance of the slower-migrating forms (Fig. 7 B). The abundance of the different forms of Dam1 does not change over the cell cycle (unpublished data).
Because Ipl1 can phosphorylate Dam1 in vitro (Fig. 7 A), we sought to determine whether Dam1 is an in vivo target of Ipl1. As shown in Fig. 7 C (lanes 1 and 4), Dam1 existed as multiple phosphorylated forms in wild-type cells incubated at either 25°C or 37°C. In contrast, the abundance of the three slowest-migrating forms of Dam1 was greatly reduced in ipl1-2 and sli15-3 mutant cells incubated at the permissive temperature of 25°C (Fig. 7 C, lanes 2 and 3), and these phosphorylated forms of Dam1 were absent in mutant cells that had been incubated at 37°C for 3 h (lanes 5 and 6). These results are very similar to those observed for Sli15–HA (Fig. 3 C). The in vitro and in vivo results together indicate that Ipl1 is required for Dam1 phosphorylation in yeast, most likely because Dam1 functions as a physiological target of Ipl1. The disappearance of three hyperphosphorylated forms of Dam1 in ipl1–2 cells suggests that there are at least three Ipl1 phosphorylation sites within Dam1, or that phosphorylation of Dam1 by Ipl1 is required for its subsequent phosphorylation by other kinases. At least one of the Ipl1 phosphorylation sites most likely resides within the COOH-terminal 169 residues of Dam1 (Dam1-C; Fig. 7 A). Furthermore, these results support the idea that Sli15 functions in vivo as a positive regulator of Ipl1.
Genetic links between IPL–SLI15, DUO1-DAM1, and GLC7
In addition to the physical and biochemical interactions observed between Ipl1–Sli15 and Duo1-Dam1, we sought to determine whether there was any genetic evidence to support the importance of such interactions in vivo. Crosses were conducted between ipl1 or sli15 mutants and duo1 or dam1 mutants. A synthetic lethal interaction was observed at 25°C (a permissive temperature for all single mutant cells) in dam1-1 ipl1-2 and dam1-1 sli15-3 double mutants. In contrast, no genetic interactions were observed in crosses between ipl1-2 or sli15-3 and duo1-2, dam1-9, or dam1-11 mutants.
Previous studies have indicated that the Ipl1 protein kinase activity is opposed by the action of the Glc7 type-1 protein phosphatase (Francisco et al., 1994; Tung et al., 1995). In fact, ipl1 mutant cells cannot tolerate a large increase in the dosage of GLC7 (Francisco et al., 1994). Wild-type cells carrying a multicopy GLC7 plasmid did not show a major growth defect when compared with cells carrying an empty vector (unpublished data). In contrast, duo1-2 and dam1-9 mutants carrying the GLC7 plasmid showed a significant lowering of the restrictive growth temperature when compared with mutant cells carrying the empty vector (Fig. 8). Thus, increased Glc7 type-1 protein phosphatase activity exacerbates the phenotype of not only ipl1 but also dam1 and duo1 mutant cells. Together, these genetic interactions highlight the in vivo importance of Dam1 as a physiological target for Ipl1–Sli15.
Figure 8.

Exacerbation of temperature sensitivity of dam1 and duo1 mutants by increased dosage of GLC7. Suspensions of dam1-9 (DDY1906) and duo1-2 (DDY1525) cells carrying the control plasmid pRS316 (−) or the GLC7-2μ plasmid pCC510 (+) were diluted 10-fold serially, spotted on SC medium lacking uracil, and allowed to grow at the indicated temperatures.
Ipl1 and Sli15 associate with centromeric DNA
Biggins et al. (1999) have shown that Ipl1 regulates microtubule binding to kinetochores that are assembled in vitro, most probably through the phosphorylation of the kinetochore protein Ndc10. The extensive biochemical and genetic interactions observed between Ipl1–Sli15 and Dam1–Duo1, which are present at kinetochores (Cheeseman et al., 2001), also suggest that Ipl1–Sli15 may function at least partly at kinetochores in vivo. To address this possibility, we used the chromatin immunoprecipitation method (Orlando and Paro, 1993; Meluh and Koshland, 1997) to determine whether these two proteins are associated with centromeric DNA. Extracts prepared from formaldehyde-fixed yeast cells that did or did not express HA–Ipl1 or Sli15–Myc were sonicated to produce DNA fragments having an average length of ∼300–700 bp. These extracts were used for immunoprecipitation with anti-HA or anti-Myc antibodies. Coprecipitated DNA fragments were used as templates for polymerase chain reactions, using primers flanking CEN3 or CEN16, or primers from a region located ∼1 kb from CEN16. Anti-HA antibodies specifically immunoprecipitated CEN3 and CEN16 sequences from cells that expressed HA–Ipl1 but not from cells that did not express this protein (Fig. 9, lanes 2, 5 and 6). Similarly, anti-Myc antibodies specifically immunoprecipitated CEN3 and CEN16 sequences from cells that expressed Sli15–Myc (Fig. 9, lanes 3, 8, and 9). The immunoprecipitated DNA fragments were specific for centromeres since a DNA sequence that was located ∼1 kb from CEN16 was not precipitated in these experiments (Fig. 9, lanes 5 and 8). These results together demonstrate that Ipl1 and Sli15 associate directly or indirectly with centromeric DNA. Thus, these two proteins are localized in vivo at kinetochores, where they can function with Dam1–Duo1 to regulate chromosome segregation.
Figure 9.

Association of Ipl1 and Sli15 with centromeric DNA. Extracts prepared from formaldehyde-fixed yeast cells (CCY776-5D) that did not (untagged) or did express HA–Ipl1 (pCC1128) or Sli15–Myc (pCC1193) were prepared. These extracts were left untreated (WCE), mock-precipitated without antibodies (No Ab), or immunoprecipitated with anti-HA (α HA) or anti-Myc (α Myc) antibodies. PCR was performed to amplify DNA fragments from WCE or precipitated samples, using primers that flank CEN3 or CEN16, or primers from a region located ∼1 kb from CEN16.
To determine whether the kinetochore localization of Dam1 is dependent on Ipl1 or Sli15 function, we performed immunofluorescent staining of chromosome spreads with ipl1-2 and sli15-3 cells. Our results showed that Dam1 staining in ipl1-2 and sli15-3 cells was similar to that in wild-type cells (unpublished data).
Discussion
Sli15 facilitates Ipl1 protein kinase function in multiple ways
Because protein kinases are notoriously promiscuous in their specificity, mechanisms must operate in cells to insulate them from working in an inappropriate place, at an inappropriate time, or on inappropriate targets. We showed here that Sli15 functions in both the activation and targeting of the Ipl1 protein kinase. In vitro, GST–Sli15 and GST–Sli15-C both stimulate the ability of Ipl1 to phosphorylate a variety of substrates. The stimulation of Ipl1 kinase activity by GST–Sli15-C was as much as 15-fold. The effect of Sli15 on the in vivo Ipl1 kinase activity may be much greater since the in vivo abundance of Ipl1 and its phosphorylation state appear to depend on the abundance of Sli15, at least under conditions where Ipl1 is overproduced (Kim et al., 1999).
In addition to its stimulatory function, Sli15 also facilitates the association of Ipl1 with the mitotic spindle, as Ipl1 becomes more concentrated on the mitotic spindle in cells with elevated levels of Sli15. Although Ipl1 can bind to microtubules in vitro, very little, if any, Ipl1 is found on cytoplasmic microtubules in vivo (Biggins et al., 1999; Kim et al., 1999). Thus, Ipl1 may be prevented from acting in the cytoplasm through its nuclear import. Sli15, which contains a putative nuclear localization signal in its middle region, potentially may target Ipl1 to the nucleus. The finding that the middle region of Sli15 also binds directly to microtubules in vitro suggests that once inside the nucleus, Ipl1 and Sli15 may bind cooperatively to microtubules. Together, the targeting and stimulatory functions of Sli15 should ensure that only correctly targeted Ipl1 can phosphorylate its mitotic spindle–associated physiological substrates. This is clearly true in the case of Dam1, which is hypophosphorylated in ipl1-2 as well as sli15-3 mutant cells. Interestingly, Dam1 can bind to Ipl1 and Sli15 directly in vitro. Thus, Sli15 may enhance the association of Ipl1 with at least one of its substrates. This may represent yet another means by which Sli15 may facilitate the ability of Ipl1 to phosphorylate its physiological substrates.
Ipl1–Sli15 and their homologues in other eukaryotes
Our finding that the COOH-terminal IN box–containing region of Sli15 binds to Ipl1 is consistent with the recent report that the IN box–containing COOH-terminal region of INCENP (ICP-1) from C. elegans is sufficient for its binding to aurora-B (AIRK2) (Kaitna et al., 2000). The observation that this region of Sli15 functions as a stimulator of Ipl1 suggests that the IN box in INCENP also may stimulate AIRK. This idea is further supported by our recent finding that a fusion protein containing essentially only the IN box of Sli15 can stimulate the kinase activity of Ipl1 (unpublished results). Our finding that Sli15 facilitates the association of Ipl1 with the mitotic spindle is also consistent with the recent reports that the mitotic spindle association of aurora-B (AIRK2) is dependent on INCENP in humans and C. elegans (Adams et al., 2000; Kaitna et al., 2000). The middle region of Sli15 can bind to microtubules directly and is localized to the mitotic spindle. The equivalent region of chicken INCENP also localizes to microtubules in mammalian cells (Mackay et al., 1993, 1998). Because the sequence homology between Sli15 and metazoan INCENP is largely restricted to the COOH-terminal IN box domain, it remains to be determined whether a less conserved sequence motif is responsible for the binding of Sli15 to microtubules.
Sli15 and Dam1 as physiological targets of Ipl1
Two lines of evidence suggest that Sli15 and Dam1 are physiological targets of Ipl1. First, they are very good in vitro substrates of Ipl1. In fact, Sli15 is by far the best in vitro substrate of Ipl1 that we have identified. It is a better substrate than the previously identified in vitro (and possibly in vivo) substrate Ndc10 (Biggins et al., 1999) and the physiological target histone H3 (Hsu et al., 2000). Second, both Sli15 and Dam1, like histone H3, are hypophosphorylated in ipl1-2 cells even at a permissive growth temperature. Even though the significance of the phosphorylation of these two proteins by Ipl1 is not yet known, the binding or phosphorylation of Dam1 by Ipl1–Sli15 is likely to be biologically important since certain combinations of ipl1, sli15, and dam1 mutations show synthetic lethal genetic interactions (Kim et al., 1999; this study).
Although it may not be apparent in Fig. 7 C, the abundance of at least one slower-migrating and presumably phosphorylated form of Dam1 remains unchanged in ipl1-2 and sli15-3 cells, thus indicating that Dam1 may be the target of one additional protein kinase. Dam1 is known to interact genetically with the Mps1 protein kinase (Jones et al., 1999). However, the electrophoretic mobility of Dam1 appears unaffected in mps1-1 cells (unpublished results), thus it is not yet possible to conclude whether Dam1 is a physiological target for Mps1. Nevertheless, this observation argues that Ipl1 protein kinase activity is not greatly altered in mps1-1 cells.
Ipl1, Sli15, and Dam1 as microtubule-binding proteins that associate with centromeres
For chromosome segregation to occur, spindle microtubules from opposite poles must attach to the kinetochores of sister chromatids. Microtubule-binding proteins that are found at kinetochores potentially may play important roles in the poorly understood process of kinetochore–microtubule attachment. The finding that Ipl1, Sli15, and Dam1 associate with both microtubules and kinetochores suggests that these proteins may connect kinetochores to spindle microtubules (this study; Hofmann et al., 1998; Cheeseman et al., 2001). The phosphorylation of Sli15 and Dam1 by Ipl1 may modulate their ability to associate with each other, the kinetochores, or microtubules, thus affecting kinetochore–microtubule attachments or the movement of kinetochores along kinetochore microtubules. Consistent with the idea that Ipl1 function is required for normal kinetochore–microtubule attachment or movement, Biggins et al. (1999) have shown that Ipl1 phosphorylates the kinetochore protein Ndc10 and inhibits the binding of kinetochores to microtubules in vitro. We have also shown that the kinetochore protein Nuf2 (Janke et al., 2001; Wigge and Kilmartin, 2001) is found along the length of mitotic spindles much more frequently in ipl1 and sli15 mutant cells than in wild-type cells (Kim et al., 1999). This pattern of Nuf2 localization suggests that kinetochores are attached to spindle microtubules in ipl1 and sli15 cells but these kinetochores do not move efficiently toward the opposite spindle poles. Interestingly, recent live-cell analysis of chromosome dynamics in strains carrying mutant kinetochore proteins revealed that ipl1 and dam1 mutants are similar in that sister chromatids show close association with a single pole, presumably reflecting monopolar microtubule attachment (He et al., 2001).
In addition to serving as a physical link between kinetochores and microtubules, Ipl1, Sli15, and Dam1 may play a role in monitoring the attachment of kinetochores to microtubules or their function may be regulated by this attachment. In this regard, it is interesting to note that one human homologue of Ipl1 (aurora-A or AIRK1) is known to bind to Cdc20, a key activator of the anaphase-promoting complex, which is the target for the kinetochore attachment checkpoint control (Farruggio et al., 1999).
Materials and methods
Strains, plasmids, media, and genetic techniques
The yeast strains and plasmids used in this study are listed in Table I. The E. coli strain DB1142 (leu pro thr hsdR hsdM recA) was used routinely as a host for plasmids, except in experiments involving recombinant protein expression, where the E. coli strains BL21 (F− ompT hsdS [r B− mB−] gal dcm) (for tac promoter–regulated expression) and BL21(DE3) (F− ompT hsdS [r B− mB−] gal dcm [DE3]) (for T7 promoter–regulated expression) were used. Yeast cells were grown at 26°C unless otherwise specified.
Table I. Plasmids and yeast strains used in this study.
| Name | Relevant features | Source |
|---|---|---|
| pCC510 | URA3, 2m, GLC7 | Francisco et al., 1994 |
| pCC669 | lacIq, GST-IPL1 under tac promoter control (in pGEX-3X) | This study |
| pCC929 | TRP1, CYH2, 2μ, GAL4BD-IPL1 under ADH1 promoter (in pAS2) | This study |
| pCC1007 | LEU2, 2μ, GAL4AD-SLI15611-698 under ADH1 promoter (in pGAD-C2) | This study |
| pCC1053 | LEU2, 2μ, GAL4AD-IPL1 under ADH1 promoter (in pGAD-C2) | This study |
| pCC1060 | URA3, CEN, GFPS65T-SLI15 under ACT1 promoter (in pRB2138) | Kim et al., 1999 |
| pCC1061 | URA3, leu2-d, 2μ, GST-SLI15 under GAL1/10 promoter (in pEG(KT)) | Kim et al., 1999 |
| pCC1062 | lacIq, GST-SLI15 under tac promoter control (in pGEX-2T) | Kim et al., 1999 |
| pCC1128 | LEU2, 2μ, HA-IPL1 | Kim et al., 1999 |
| pCC1173 | LEU2, 2μ, SLI15-Myc | Kim et al., 1999 |
| pCC1193 | URA3, 2μ, SLI15-Myc | Kim et al., 1999 |
| pCC1212 | TRP1, 2μ, GAL4BD-SLI15 under ADH1 promoter (in pGBD-C2) | This study |
| pCC1271 | lacIq, GST-IPL1D245N under tac promoter control (in pGEX-3X) | This study |
| pCC1301 | LEU2, 2μ, SLI15-HA | This study |
| pCC1309 | URA3, 2μ, GAL4BD-SLI15 under ADH1 promoter (in pGBDU-C2) | This study |
| pCC1428 | LEU2, 2μ, GAL4AD-DUO1110-247 under ADH1 promoter (in pGAD-C3) | This study |
| pCC1469 | lacI q, GST-SLI151-229 under tac promoter control (in pGEX-2T) | This study |
| pCC1470 | lacI q, GST-SLI15227-559 under tac promoter control (in pGEX-2T) | This study |
| pCC1471 | lacI q, GST-SLI15558-698 under tac promoter control (in pGEX-1) | This study |
| pCC1533 | URA3, CEN, GFPS65T-SLI151-229 under ACT1 promoter (in pRB2138) | This study |
| pCC1534 | URA3, CEN, GFPS65T-SLI15227-559 under ACT1 promoter (in pRB2138) | This study |
| pCC1560 | TRP1, 2μ, GAL4BD-SLI151-229 under ADH1 promoter (in pGBD-C1) | This study |
| pCC1561 | TRP1, 2μ, GAL4BD-SLI15227-559 under ADH1 promoter (in pGBD-C1) | This study |
| pCC1562 | TRP1, 2μ, GAL4BD-SLI15558-698 under ADH1 promoter (in pGBD-C1) | This study |
| pCC1563 | URA3, leu2-d, 2μ, GST-SLI151-229 under GAL1/10 promoter (in pEG(KT)) | This study |
| pCC1564 | URA3, leu2-d, 2μ, GST-SLI15227-559 under GAL1/10 promoter (in pEG(KT)) | This study |
| pCC1565 | URA3, leu2-d, 2μ, GST-SLI15558-698 under GAL1/10 promoter (in pEG(KT)) | This study |
| pCC1566 | URA3, CEN, GFPS65T-SLI15558-698 under ACT1 promoter (in pRB2138) | This study |
| pCC1575 | LEU2, 2μ, DAM1 | This study |
| pCC1584 | TRP1, CEN, GFPS65T-IPL1 under ACT1 promoter (in pRS314) | This study |
| pDD475 | URA3, leu2-d, 2μ, GST-DUO1 under GAL1/10 promoter (in pEG(KT)) | Hofmann et al., 1998 |
| pDD884 | lacIq, His6-DAM1 under T7 promoter (in pRSETa) | Cheeseman et al., 2001 |
| pDD1017 | URA3, leu2-d, 2μ, GST-DAM1 under GAL1/10 promoter (in pEG(KT)) | This study |
| pDD1018 | LEU2, 2μ, GAL4AD-DAM1 under ADH1 promoter (in pACT2) | This study |
| pDD1019 | lacIq, His6-GST-DAM1175-343 under T7 promoter (in pGAT2) | This study |
| pDD1020 | LEU2, 2μ, GAL4AD-DUO1 under ADH1 promoter (in pACT2) | This study |
| pEG(KT) | URA3, leu2-d, 2μ, GST under GAL1/10 promoter | Mitchell et al., 1993 |
| pGEX-2T | lacIq, GST under tac promoter | Smith and Johnson, 1988 |
| pRS316 | URA3, CEN | Sikorski and Hieter, 1989 |
| CCY766-5D | α lys2-801 his3-Δ200 ura3-52 leu2-3,112 trp1-1 | This study |
| CCY766-9D | a lys2-801 his3-Δ200 ura3-52 leu2-3,112 trp1-1 | Kim et al., 1999 |
| CCY915-13C | a lys2-801 his3-Δ200 ura3-52 leu2-3,112 trp1-1 ipl1-2 | Kim et al., 1999 |
| CCY1124-4B | α lys2-801 his3-Δ200 ura3-52 leu2-3,112 sli15-3 | This study |
| DBY1830 | a /α ade2/1 lys2-801/1 his3-Δ200/his3-Δ200 ura3-52/ura3-52 leu2-3,112/leu2-3,112 trp1-1/trp1-1 | Kim et al., 1999 |
| DDY904 | α lys2-801 his3-Δ200 ura3-52 leu2-3,112 | D. Drubin |
| DDY1906 | α lys2-801 his3-Δ200 ura3-52 leu2-3,112 dam1-9::KanMX | Cheeseman et al., 2001 |
| DDY1910 | α lys2-801 his3-Δ200 ura3-52 leu2-3,112 dam1-11::KanMX | Cheeseman et al., 2001 |
| DDY1914 | α lys2-801 his3-Δ200 ura3-52 leu2-3,112 dam1-1::KanMX | Cheeseman et al., 2001 |
| DDY1525 | α lys2-801 his3-Δ200 ura3-52 leu2-3,112 duo1-2::LEU2 | Hofmann et al., 1998 |
| PJ69-4A | a trp1-901 leu2-3,112 ura3-52 his3-Δ200 gal4Δ gal80Δ LYS2::GAL1→HIS3 GAL2→AΔE2 met2::GAL7-lacZ | James et al., 1996 |
| TD4 | a ura3-52 leu2-3,112 trp1-289 his4-519 | Kim et al., 1999 |
| Y190 | a gal4 gal80 his3 trp1-901 ade2-101 ura3-52 leu2-3,112 URA3::GAL→lacZ LYS2::GAL→HIS3 cyhr | Harper et al., 1993 |
All plasmids contain the ampicillin-resistance marker for selection in E. coli.
Two-hybrid screens with Ipl1 and Sli15
Yeast genes encoding proteins that bind Ipl1 were identified in a two-hybrid screen (Harper et al., 1993), using Gal4BD–Ipl1 (pCC929) as bait. Y190 cells containing pCC929 were transformed with a Gal4AD fusion genomic library constructed in pGAD-C (James et al., 1996). Transformants were selected on SC medium lacking histidine, leucine, and tryptophan but containing 20 mM 3-amino-1,2,4-triazole (3-AT; Sigma-Aldrich). Fast-growing transformant colonies were tested for their β-galactosidase activity using an X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; US Biological) colony filter lift assay. pGAD-C plasmids were recovered from 3-AT–resistant transformants that had higher than background levels of β-galactosidase activity. Screening of ∼7.3 × 106 transformants led to the identification of 28 plasmids that encoded Gal4AD fusions to three different proteins. 12 of these plasmids encoded Gal4AD–Sli15 fusion proteins. Yeast genes encoding proteins that bind Sli15 were identified similarly, except that Gal4BD–Sli15 (encoded by pCC1309) was used as bait in yeast strain PJ69–4A, and transformants were selected on SC medium lacking histidine, leucine, and uracil but containing 3 mM 3-AT. Screening of ∼3.6 × 106 transformants led to the identification of 17 plasmids that encoded Gal4AD fusions to three different proteins. One such plasmid encoded Gal4AD–Duo1.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as previously described (Saitoh et al., 1997), with some minor modifications. Approximately 4 × 108 yeast cells growing exponentially at 30°C in SC medium lacking leucine or uracil were fixed at room temperature with 1% formaldehyde for 15 min and then with 125 mM glycine for 5 min. Cells were harvested and washed three times with TBS (20 mM Tris-HCl [pH 7.6], 200 mM NaCl), and then resuspended in 400 μl of lysis buffer D (50 mM Hepes-KOH [pH 7.5], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100 [vol/vol], 0.1% SDS [wt/vol]) that contained protease inhibitors (complete mini from Roche Diagnostics GmbH). Cell lysis and fragmentation of chromosomes were achieved by sonicating the samples for 14 rounds of 20 s each, using a Tekmar sonic disruptor with microtip and set at 60% duty cycle. This yielded chromatin fragments with average lengths of ∼300–700 bp. Cell debris was removed by centrifuging in a microcentrifuge at 15,000 rpm for 5 min and then for 15 min at 4°C. The volume of the supernatant (whole cell extracts [WCE]) was adjusted to 450 μl with lysis buffer D. 400 μl of each WCE was used for immunoprecipitation and the rest was stored at −20°C as untreated WCE. WCE was incubated for 4 h at 4°C with 4 μg of antibodies (9E10 in the case of anti-Myc; 16B12 in the case of anti-HA; both from BAbCO). 60 μl of protein A–Sepharose CL-4B beads (Amersham Pharmacia Biotech) was added and incubated for 1 h at 4°C. Beads were harvested by centrifugation, followed by five rounds of washing with 1.5 ml of lysis buffer D. Washed beads were resuspended in 200 μl of TE (10 mM Tris-HCl [pH 7.0], 1 mM EDTA). Immunoprecipitated sample and untreated WCE (see above) were adjusted to give final concentrations of 0.25% SDS (wt/vol) and 250 μg/ml proteinase K (Promega). These samples were incubated at 37°C for at least 8 h. Cross-linking was reversed by incubating the samples at 65°C for 6 h, followed by phenol/chloroform extraction. DNA was precipitated and resuspended in 50 μl of TE. 1 μl of each sample was used as template for 30 cycles of PCR reactions with Taq DNA polymerase (Roche Diagnostics GmbH). CEN3 and CEN16 primers were described by Meluh and Koshland (1997). CEN16 proximal primers had the following sequences (5′-ACACCATGGTAGCGGTTCTA-3′ and 5′- GGTAGAAGCCTTTGTACCAT-3′).
Purification and analysis of proteins
GST or GST fusion proteins were purified from yeast essentially as previously described (Kim et al., 1999), except that the lysis buffer contained 50 mM Hepes-KOH (pH 7.4), 200 mM KCl, 10% glycerol (vol/vol), 1% NP-40 (vol/vol), 10 mM EGTA, 2 mM DTT, 50 mM NaF, 0.1 mM Na3VO4, 5 mM β-glycerophosphate, and the following protease inhibitors (Sigma-Aldrich): 2 μg/ml each of antipain, leupeptin, pepstatin A, chymostatin, and aprotinin; 10 μg/ml of phenanthroline; 16 μg/ml of benzamidine-HCl; and 1 mM PMSF. Cell lysis was performed in glass tubes (13 × 100 mm), each containing 0.1 ml of cell suspension.
E. coli cells (BL21 or BL21[DE3]) expressing GST or GST fusion proteins were harvested and rinsed once in 10 ml of ice cold washing buffer, which consisted of 50 mM Hepes-KOH (pH 7.4) and 200 mM KCl. Cells were resuspended in 1.5 ml of buffer B, which consisted of 50 mM Hepes-KOH (pH 7.4), 200 mM KCl, 1% NP-40 (vol/vol), 1 mM EDTA, 2 mM DTT, and the protease inhibitors listed above. Cells were resuspended in 1.5 ml of the same buffer and lysed in a French press at 16,000 psi. Cell debris was removed by a 10-min centrifugation at 20,000 g, thus generating the supernatant fraction. The supernatant was added to a 1.5-ml microcentrifuge tube that contained 0.1 ml of a 50% slurry (vol/vol) of glutathione-agarose beads. After a 2-h incubation at 4°C with constant agitation, the glutathione-agarose beads were harvested, followed by two 5-min washes with 1 ml of lysis buffer B. The proteins bound on the glutathione-agarose beads were eluted by the addition of 0.1 ml of elution buffer (50 mM Tris-HCl [pH 8.0], 50 mM KCl, 15 mM reduced glutathione; Sigma-Aldrich). The binding of His6–Dam1 to GST–Ipl1 and GST–Sli15 was performed essentially as described previously (Kim et al., 1999).
Protein samples were electrophoretically separated in SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with 2,000-fold diluted rabbit anti-GST antibodies (Molecular Probes, Inc.), 1,000-fold diluted mouse anti-HA or anti-Myc ascites fluid (both from BAbCO), 100,000-fold diluted anti-G6PDH antibodies (Sigma-Aldrich), 1,000-fold diluted affinity-purified guinea pig anti-Dam1 antibodies (Cheeseman et al., 2001), or 2,000-fold diluted affinity-purified rabbit anti-Duo1 antibodies (Hofmann et al., 1998). Proteins recognized by primary antibodies were visualized by the ECL chemiluminescent system (Amersham Pharmacia Biotech).
Dam1 immunoprecipitation and phosphatase treatment
Cells from 50 ml of a log phase culture were harvested and washed with sorbitol buffer (1.3 M sorbitol, 0.1 M KPO4, pH 7.5) and incubated for 50 min with lyticase. Cells were pelleted gently and resuspended in lysis buffer E (1% SDS [wt/vol], 10 mM EDTA, 50 mM Tris-HCl, pH 8.0) with protease inhibitors, 1 mM PMSF, and phosphatase inhibitors (10 mM sodium pyrophosphate, 10 mM sodium azide, 10 mM NaF, 0.4 mM Na3VO4). The resuspended cells were sonicated 10 times for 10 s each and pelleted at maximum speed in a microcentrifuge. The supernatant was diluted 10-fold with dilution buffer (1% Triton [vol/vol], 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.0). Approximately 15 μl of anti-Dam1 antibody precoupled to 20 μl of protein A Affi-gel beads (Bio-Rad Laboratories) was added. The beads were incubated overnight at 4°C and then washed three times with dilution buffer. Sample buffer was added directly to half of the sample. The remaining sample was treated with 800 u of λ phosphatase (New England Biolabs, Inc.) for 2 h at 30°C, according to the manufacturer's guidelines, and sample buffer was then added. Both samples were then run on a 12% polyacrylamide gel.
Ipl1 kinase assay
Unless otherwise stated, the reaction mixture consisted of 5 μl kinase buffer (60 mM MgCl2, 60 mM MnCl2, 6 mM DTT, 0.6 mM Na3VO4, 30 mM β-glycerophosphate), 5 μl [γ-32P]ATP (160 μM), 10 μl GST–Ipl1 or GST–Ipl1D245N (∼0.3 μg) in elution buffer (see above), and 10 μl substrate (∼5 μg of bovine MBP [Sigma-Aldrich] or ∼1 μg of GST fusion protein) in elution buffer. After 15 min at 30°C, reactions were stopped by adding 10 μl of 4× sample buffer. Protein samples were electrophoretically separated in SDS-PAGE. The stoichiometry of phosphate incorporated into GST–Sli15 was determined by scintillation counting of the phosphorylated GST–Sli15 band that had been excised from the dried SDS-PAGE.
Microtubule binding assay
Purified bovine brain tubulin (60 μM) in PME buffer (80 mM K-Pipes, pH 6.8, 1 mM EGTA, 1 mM MgCl2) was thawed and centrifuged in a microcentrifuge for 5 min at 4°C to remove insoluble protein. To assemble microtubules, GTP was added to a final concentration of 1 mM, glycerol was added to a final concentration of 25% (vol/vol), and the reaction was incubated at 35°C for 30 min. After assembly, taxol was added to a final concentration of 20 μM to stabilize microtubules. Microtubules were then diluted in PME buffer containing 1 mM GTP and 10 μM taxol.
For cosedimentation assays, GST and GST fusion proteins were purified from E. coli and dialyzed into PME buffer, diluted fivefold with PME buffer, and then centrifuged for 20 min at 60,000 rpm and 25°C in a TLA100 rotor (Beckman Coulter). 20 μl of soluble protein was added to variable concentrations of taxol-stabilized microtubules (0–5 μM) in 40-μl reactions. The reactions were incubated for 20 min at 25°C to allow binding to occur, and then centrifuged as above to pellet the microtubules. Pellets and supernatants were fractionated on SDS-PAGE gels and probed by immunoblotting with anti-GST antibodies.
Acknowledgments
We thank Phil James (University of Wisconsin, Madison, WI) for the supply of plasmid libraries; Makkuni Jayaram (University of Texas, Austin, TX) for support (of S. Velmurugan); David Drubin (University of California, Berkeley, CA) for comments on the manuscript; Maria Enquist-Newman (University of California) for helpful discussions and assistance with experiments; and Shelly Jones (University of Colorado, Boulder, CO) and Mark Winey (University of Colorado) for discussions.
This work was supported in part by National Institutes of Health grants to C.S.M. Chan (GM45185) and G. Barnes (GM47842) and a National Science Foundation Graduate Research fellowship to I.M. Cheeseman.
Footnotes
Abbreviations used in this paper: 3-AT, 3-amino-1,2,4-triazole; AIRK, aurora/Ipl1-related kinase; GFP, green fluorescent protein; GST, glutathione-S-transferase; INCENP, inner centromere protein; MBP, myelin basic protein; SC, synthetic complete; WCE, whole cell extracts.
References
- Adams, R.R., S.P. Wheatley, A.M. Gouldsworthy, S.E. Kandels-Lewis, M. Carmena, C. Smythe, D.L. Gerloff, and W.C. Earnshaw. 2000. INCENP binds the Aurora-related kinase AIRK2 and is required to target it to chromosomes, the central spindle and cleavage furrow. Curr. Biol. 10:1075–1078. [DOI] [PubMed] [Google Scholar]
- Biggins, S., F.F. Severin, N. Bhalla, I. Sassoon, A.A. Hyman, and A.W. Murray. 1999. The conserved protein kinase Ipl1 regulates microtubule binding to kinetochores in budding yeast. Genes Dev. 13:532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff, F.R., and G.D. Plowman. 1999. The Aurora/Ipl1p kinase family: regulators of chromosome segregation and cytokinesis. Trends Cell Biol. 9:454–459. [DOI] [PubMed] [Google Scholar]
- Chan, C.S.M., and D. Botstein. 1993. Isolation and characterization of chromosome-gain and increase-in-ploidy mutants in yeast. Genetics. 135:677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman, I.M., M. Enquist-Newman, T. Müller-Reichert, D.G. Drubin, and G. Barnes. 2001. Mitotic spindle integrity and kinetochore function linked by the Duo1p/Dam1p complex. J. Cell Biol. 152:197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke, C.A., M.M.S. Heck, and W.C. Earnshaw. 1987. The inner centromere protein (INCENP) antigens: movement from inner centromere to midbody during mitosis. J. Cell Biol. 105:2053–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farruggio, D.C., F.M. Townsley, and J.V. Ruderman. 1999. Cdc20 associates with the kinase aurora2/Aik. Proc. Natl. Acad. Sci. USA. 96:7306–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco, L., W. Wang, and C.S.M. Chan. 1994. Type-1 protein phosphatase acts in opposition to the Ipl1 protein kinase in regulating yeast chromosome segregation. Mol. Cell. Biol. 14:4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giet, R., and D.M. Glover. 2001. Drosophila Aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J. Cell Biol. 152:669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giet, R., and C. Prigent. 1999. Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J. Cell Sci. 112:3591–3601. [DOI] [PubMed] [Google Scholar]
- Goode, B.L., and S.C. Feinstein. 1994. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of Tau. J. Cell Biol. 124:769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper, J.W., G.R. Adami, N. Wei, K. Keyomarsi, and S.J. Elledge. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 75:805–816. [DOI] [PubMed] [Google Scholar]
- He, X., D.R. Rines, C.W. Espelin, and P.K. Sorger. 2001. Molecular analysis of kinetochore-microtubule attachment in budding yeast. Cell. 106:195–206. [DOI] [PubMed] [Google Scholar]
- Hofmann, C., I.M. Cheeseman, B.L. Goode, K.L. McDonald, G. Barnes, and D.G. Drubin. 1998. Saccharomyces cerevisiae Duo1p and Dam1p, novel proteins involved in mitotic spindle function. J. Cell Biol. 143:1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, J.-Y., Z.-W. Sun, X. Li, M. Reuben, K. Tatchell, D.K. Bishop, J.M. Grushcow, C.J. Brame, J.A. Caldwell, D.F. Hunt, R. Lin, M.M. Smith, and C.D. Allis. 2000. Mitotic phosporylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 102:279-291. [DOI] [PubMed] [Google Scholar]
- James, P., J. Halladay, and E.A. Craig. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 144:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke, C., J. Ortiz, J. Lechner, A. Shevchenko, A. Shevchenko, M.M. Magiera, C. Schramm, and E. Schiebel. 2001. The budding yeast proteins Spc24p and Spc25p interact with Ndc80p and Nuf2p at the kinetochore and are important for kinetochore clustering and checkpoint control. EMBO J. 20:777–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, M.H., J.B. Bachant, A.R. Castillo, T.H. Giddings, Jr., and M. Winey. 1999. Yeast Dam1p is required to maintain spindle integrity during mitosis and interacts with the Mps1p kinase. Mol. Biol. Cell. 10:2377–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaitna, S., M. Mendoza, V. Jantsch-Plunger, and M. Glotzer. 2000. Incenp and an Aurora-like kinase form a complex essential for chromosome segregation and efficient completion of cytokinesis. Curr. Biol. 10:1172–1181. [DOI] [PubMed] [Google Scholar]
- Kim, J.-H., J.-S. Kang, and C.S.M. Chan. 1999. Sli15 associates with the Ipl1 protein kinase to promote proper chromosome segregation in Saccharomyces cerevisiae. J. Cell Biol. 145:1381–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, A.M., D.M. Eckley, C. Chue, and W.C. Earnshaw. 1993. Molecular analysis of the INCENPs (inner centromere proteins): separate domains are required for association with microtubules during interphase and with the central spindle during anaphase. J. Cell Biol. 123:373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, A.M., A.M. Ainsztein, D.M. Eckley, and W.C. Earnshaw. 1998. A dominant mutant of inner centromere protein (INCENP), a chromosomal protein, disrupts prometaphase congression and cytokinesis. J. Cell Biol. 140:991–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meluh, P.B., and D. Koshland. 1997. Budding yeast centromere composition and assembly as revealed by in vivo cross-linking. Genes Dev. 11:3401–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, D.A., T.K. Marshall, and R.J. Deschenes. 1993. Vectors for the inducible overexpression of glutathione S-transferase fusion proteins in yeast. Yeast. 9:715–723. [DOI] [PubMed] [Google Scholar]
- Orlando, V., and R. Paro. 1993. Mapping polycomb-repressed domains in the bithorax complex using in vivo formaldehyde cross-linked chromatin. Cell. 75:1187–1198. [DOI] [PubMed] [Google Scholar]
- Saitoh, S., K. Takahashi, and M. Yanagida. 1997. Mis6, a fission yeast inner centromere protein, acts during G1/S and forms specialized chromatin required for equal segregation. Cell. 90:131–143. [DOI] [PubMed] [Google Scholar]
- Sikorski, R.S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, D.B., and K.S. Johnson. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 67:31–40. [DOI] [PubMed] [Google Scholar]
- Tung, H.Y.L., W. Wang, and C.S.M. Chan. 1995. Regulation of chromosome segregation by Glc8p, a structural homolog of mammalian inhibitor 2 that functions as both an activator and an inhibitor of yeast protein phosphatase 1. Mol. Cell. Biol. 15:6064–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigge, P.A., and J.V. Kilmartin. 2001. The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J. Cell Biol. 152:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]