

Abstract
CD44 is a widely distributed cell surface adhesion molecule and is implicated in diverse biological processes. However, the nature of intracellular signaling triggered by CD44 remains to be elucidated. Here, we show that CD44 undergoes sequential proteolytic cleavage in the ectodomain and intracellular domain, resulting in the release of a CD44 intracellular domain (ICD) fragment. Consequently, CD44ICD acts as a signal transduction molecule, where it translocates to the nucleus and activates transcription mediated through the 12-O-tetradecanoylphorbol 13-acetate–responsive element, which is found in numerous genes involved in diverse cellular processes. Expression of an uncleavable CD44 mutant as well as metalloprotease inhibitor treatment blocks CD44-mediated transcriptional activation. In search of the underlying mechanism, we have found that CD44ICD potentiates transactivation mediated by the transcriptional coactivator CBP/p300. Furthermore, we show that cells expressing CD44ICD produce high levels of CD44 messenger RNA, suggesting that the CD44 gene is one of the potential targets for transcriptional activation by CD44ICD. These observations establish a novel CD44 signaling pathway and shed new light on the functional link between proteolytic processing of an adhesion molecule at the cell surface and transcriptional activation in the nucleus.
Keywords: adhesion molecule; CD44; proteolytic cleavage; signal transduction; transcription
Introduction
There is accumulating evidence that adhesion molecules participate not only in mediating cell adhesion but also in a wide variety of processes that transduce signals into the cell (Gumbiner, 1996; Aplin et al., 1999). CD44 is the major adhesion molecule expressed in most human cell types and implicated in a wide variety of physiological and pathological processes, including lymphocyte homing and activation, wound healing, cell migration, and the regulation of tumor cell growth and metastasis (Aruffo et al., 1990; Culty et al., 1990; Günthert et al., 1991; Arch et al., 1992; Lesley and Hyman, 1998). The broad spectrum of functions suggests that CD44 can transduce multiple intracellular signals; however, it remains unclear how CD44 acts as a signal transduction molecule.
We previously presented a proteolysis-based model for the regulation of CD44 function (Fig. 1 A) (Okamoto et al., 1999a,b; Kawano et al., 2000). Our preceding works demonstrated that the ectodomain of CD44 expressed on the surface of various cancer cells undergoes proteolytic cleavage by membrane-associated metalloproteases under physiological conditions, and this cleavage is responsible for dynamic regulation of the interaction between CD44 and the extracellular matrix during cell migration (Okamoto et al., 1999a). Consistent with this notion, membrane type 1 matrix metalloprotease (MT1-MMP) has been shown to cleave CD44 ectodomain at the cell surface and promote cell migration (Kajita et al., 2001). Furthermore, we reported that the CD44 ectodomain cleavage is itself regulated by multiple signaling pathways, for example, the activation of PKC, the influx of extracellular Ca2+, members of the Rho family of small GTPases, and the Ras oncoprotein (Fig. 1 A) (Okamoto et al., 1999b; Kawano et al., 2000). Thus, accumulating evidence indicates that CD44 proteolytic cleavage is emerging as a key regulatory event for CD44 functions besides the initially characterized adhesion-dependent functioning.
Figure 1.

Two-step proteolytic cleavages of CD44 at the ectodomain and intracellular domain. (A) Schematic representation of CD44 proteolytic processing. (B) U251MG cells were left untreated (lane 1) or incubated at 37°C first for 30 min in the absence (lane 2) or presence of 100 μM BB2516 (lane 3) and then for 40 min with TPA (100 ng/ml) (lanes 2 and 3). After TPA treatment, the cells were washed and incubated for an additional 3 h with fresh medium. Blots were probed with anti-CD44cyto Ab. (C) Amino acid sequences in the transmembrane and intracellular domain of human CD44 are shown by the single-letter abbreviations. Numbering of the amino acids is shown above the sequence. Anti-CD44cyto Ab was raised to sequences (boxed) at the COOH terminus of the protein. Peptides of CD44 identified in MALDI-MS and ES-MS analysis are represented by lines spanning the corresponding parts of the sequence.
In this report, we demonstrate for the first time that CD44 ectodomain cleavage is followed by CD44 intracellular cleavage. These sequential proteolytic events result in the release of the CD44 intracellular domain (ICD)* fragment, which translocates to the nucleus and promotes transcription mediated through the 12-O-tetradecanoylphorbol 13-acetate (TPA)-responsive element (TRE). This work demonstrates a novel CD44 signaling pathway linking proteolytic processing at the cell surface to gene expression in the nucleus.
Results and discussion
CD44 undergoes sequential proteolytic cleavage in the ectodomain and intracellular domain
We previously demonstrated that cleavage of the ectodomain of CD44 generates membrane-tethered fragments that are detectable by immunoblot analysis with antibodies raised to the COOH-terminal region of the protein (anti-CD44cyto antibody [Ab]) (Fig. 1 A) (Okamoto et al., 1999a). Although these cleavage products of CD44 appear to undergo further proteolysis through an intracellular processing pathway (Okamoto et al., 1999b), little is known about its nature or biological significance. To examine the possible intracellular processing of these CD44 fragments, we first incubated human glioma U251MG cells with TPA, a procedure that induces cleavage of the CD44 ectodomain (Okamoto et al., 1999b), and then incubated the cells for an additional 3 h in fresh medium. Immunoblot analysis of cell lysates with anti-CD44cyto Ab revealed the CD44 ectodomain cleavage products as a broad band that migrated at ∼25 kD (Fig. 1 B, lane 2). The ectodomain cleavage products were not detected in TPA-treated cells that had been preincubated with the specific metalloprotease inhibitor BB2516 (Fig. 1 B, lane 3), consistent with our previous demonstration that the ectodomain cleavage is mediated by metalloproteases (Okamoto et al., 1999a,b). Notably, an anti-CD44cyto Ab–specific immunoreactive fragment with a mobility greater than the CD44 ectodomain cleavage products was also apparent in the TPA-treated U251MG cells (Fig. 1 B, lane 2, arrowhead). This fragment was purified by antibody affinity chromatography and subjected to matrix-assisted laser-desorption/ionization mass spectrometry (MALDI-MS) and electrospray (ES)-MS analysis. The mass spectra obtained from MALDI-MS were used for peptide mass database searches which identified CD44 with three peptides matching, covering 80% of CD44 intracellular domain (Fig. 1 C). ES-MS combined with maximum entropy data analysis showed a major mass of 3923.95, which matched the predicted molecular mass of 3923.10 for a peptide composed of amino acid residues 288–324 (Fig. 1 C). These results indicated that the small fragment, designated CD44ICD, is indeed derived from CD44, and the NH2 terminus of CD44ICD begins with amino acid A288, within or very close to the intracellular side of the transmembrane domain (Fig. 1 C) (Lesley and Hyman, 1998).
To further examine the generation of CD44ICD, we treated U251MG cells with TPA and then for various periods with fresh medium in the absence or presence of carbobenzoxy-L-leucyl-L-leucyl-L-leucinal (MG132), an intracellular protease inhibitor which has been shown previously to block γ-secretase–mediated intracellular proteolytic cleavages of Notch and β-amyloid precursor protein (De Strooper et al., 1999). Cells treated with TPA and then immediately lysed revealed a significant amount of CD44 ectodomain cleavage products (Fig. 2 A, lanes 1 and 5). Cells incubated for another 3 h after the removal of TPA resulted in a decrease in the amount of the CD44 ectodomain cleavage fragments and the appearance of CD44ICD (Fig. 2 A, lane 2). The abundance of CD44ICD decreased thereafter (Fig. 2 A, lanes 2–4), likely as a result of further degradation. In contrast, the presence of MG132 prevented both the decrease in abundance of the CD44 ectodomain cleavage products and the concomitant increase in the amount of CD44ICD (Fig. 2 A, lanes 6–8). These data indicate that CD44ICD is generated as a result of proteolytic cleavage from the membrane-tethered products of CD44 ectodomain cleavage.
Figure 2.
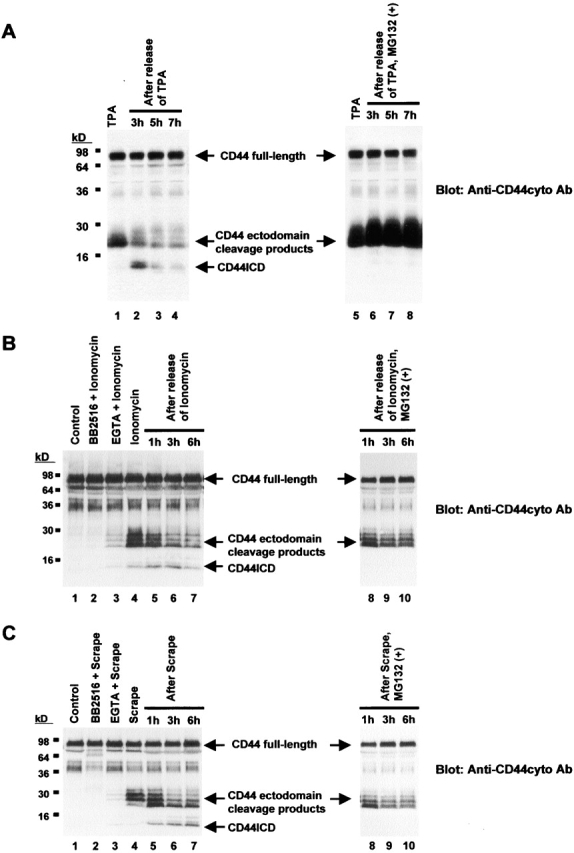
Calcium influx induces CD44 sequential cleavages as well as TPA treatment. (A) Western blot of U251MG cells incubated first for 40 min with TPA (100 ng/ml) and then in the absence (left) or presence (right) of MG132 (15 μM) for the indicated periods. Blots were probed with anti-CD44cyto Ab. (B) U251MG cells were left untreated (lane 1) or treated with ionomycin (10 μM) for 30 min in the presence (lanes 2 and 3) or absence (lanes 4–10) of BB2516 (100 μM) or EGTA (5 mM). After ionomycin treatment, the cells were washed and then incubated in the absence (lanes 5–7) or presence (lanes 8–10) of MG132 (15 μM) for the indicated periods. Blots were probed with anti-CD44cyto Ab. (C) U251MG cells were left untreated (lane 1) or mechanically scraped in the presence (lanes 2 and 3) or absence (lanes 4–10) of BB2516 (100 μM) or EGTA (5 mM). The cells were then incubated in the absence (lanes 5–7) or presence (lanes 8–10) of MG132 (15 μM) for the indicated periods. Blots were probed with anti-CD44cyto Ab.
Next, we investigated the other physiological conditions under which CD44ICD is generated. We have shown previously that a transient increase in intracellular calcium concentration, which can be induced by various physiological events, promotes CD44 ectodomain cleavage (Okamoto et al., 1999b). Accordingly, we examined whether calcium influx subsequently induces the production of CD44ICD. Ionomycin treatment, which induces the calcium influx, of U251MG cells led to the release of CD44 ectodomain cleavage products, whereas this was markedly inhibited in the presence of the metalloprotease inhibitor BB2516 or the calcium chelator EGTA (Fig. 2 B, lanes 1–4). After the induction of CD44 ectodomain cleavage, the generation of CD44ICD was found (Fig. 2 B, lanes 4–7), but not in the presence of MG132 (Fig. 2 B, lanes 8–10), which confirms that the generation of CD44ICD is dependent on the intracellular proteolytic machinery. Furthermore, mechanical scraping of U251MG cells, which mimic cell wounding in vivo and induces the transient influx of calcium (McNeil et al., 1989; Ito et al., 1999; Tran et al., 1999), promoted the sequential CD44 cleavages and production of CD44ICD in a similar manner (Fig. 2 C). These findings indicate that the generation of CD44ICD is part of the diverse cellular signaling events triggered by calcium influx. Thus, our results show that CD44 undergoes two successive proteolytic cleavages in the ectodomain and in the intracellular domain in these physiological processes.
CD44ICD released by sequential proteolytic cleavages translocates into the nucleus
We asked if the intracellular cleavage of CD44 affected its intracellular localization. We transiently transfected COS-7 cells with a plasmid encoding CD44ICD tagged with hemagglutinin (HA) at its NH2 terminus. Immunofluorescence staining of the transfected cells revealed that CD44ICD was localized to the nucleus (Fig. 3 A, left). Identical results were obtained when CD44ICD was epitope-tagged with Myc at its COOH terminus or fused to green fluorescent protein at its NH2 terminus (unpublished data). Next, we examined whether endogenous CD44ICD generated by the sequential cleavage of CD44 also translocates to the nucleus. U251MG cells treated with TPA were lysed and separated into membrane/cytosol and nuclear fractions. Immunoblot analysis with anti-CD44cyto Ab indicated that the CD44 ectodomain cleavage products are present in the membrane/cytosol fraction (Fig. 3 B), as we have shown previously (Okamoto et al., 1999a). In contrast, the CD44ICD proteolytic fragment is found in the nuclear fraction (Fig. 3 B). Both the ectodomain and intracellular cleavage products are not found in cells pretreated with BB2516 (Fig. 3 B). Identical results were obtained from calcium influx–induced CD44 sequential cleavages (Fig. 3 B). Thus, the two sequential proteolytic cleavage of CD44 result in the release of CD44ICD from the plasma membrane and its translocation to the nucleus.
Figure 3.
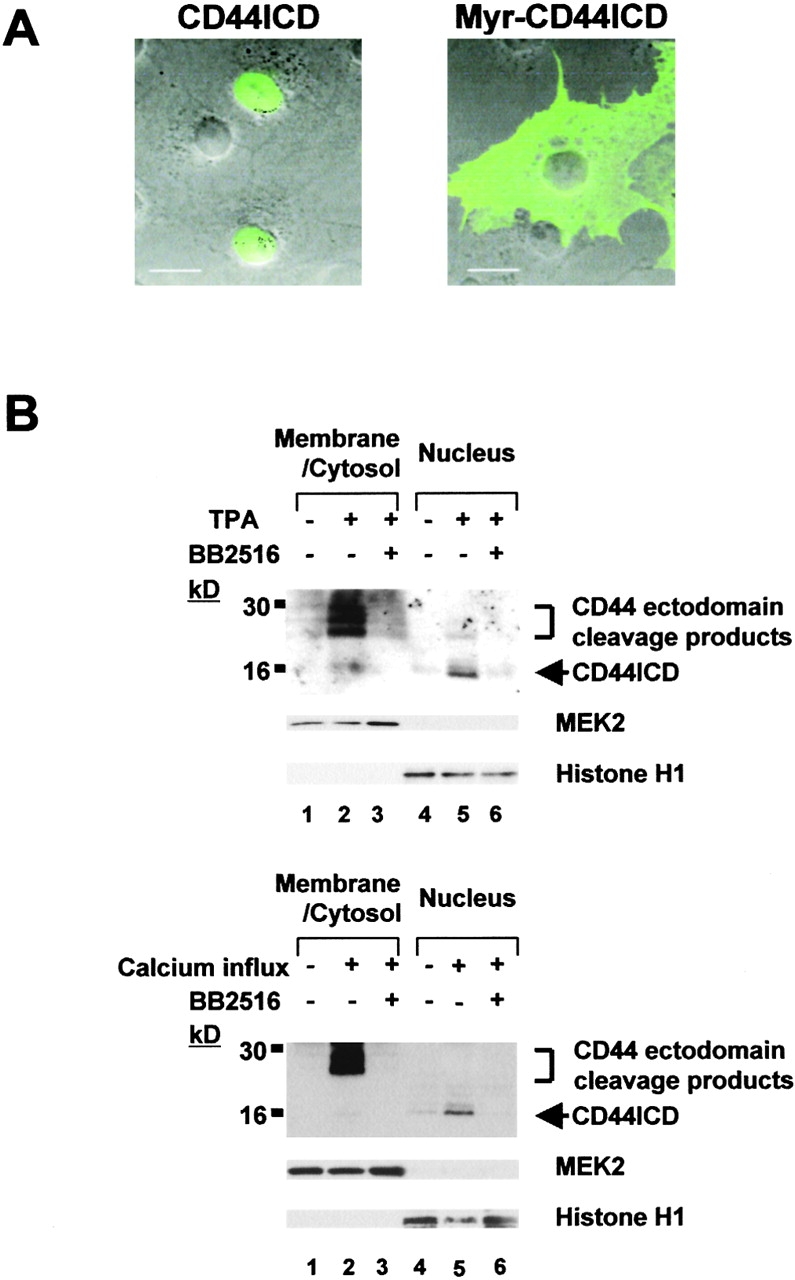
Nuclear translocation of CD44ICD (A) COS-7 cells were transfected with HA-tagged CD44ICD (left) or Myr-CD44ICD (right) expression plasmids. After 24 h, the cells were stained with anti-HA mAb. (B) U251MG cells were treated as described in the legend to Fig. 2, A or B. Membrane/cytosol and nuclear fractions were analyzed by Western blotting with anti-CD44cyto Ab, anti-MEK2 Ab (cytosolic marker), and anti-Histone H1 (nuclear marker). Bars, 20 μm.
CD44ICD can activate transcription through the TPA-responsive element
The proteolytic release and nuclear translocation of CD44ICD are reminiscent of those recently described for the signaling proteins Notch and SREBPs (sterol regulatory element–binding proteins) (Sakai et al., 1996; Schroeter et al., 1998; Struhl and Adachi, 1998; Brown et al., 2000). These proteins can be cleaved to liberate cytosolic fragments that enter the nucleus to control gene transcription. We hypothesized that CD44ICD might be able to induce gene transcription. Using a luciferase reporter, we tested the effect of CD44ICD on transcriptional responses from several types of enhancer elements that are the convergent points for many intracellular signal transduction pathways. Remarkably, the reporter under the control of the TRE was strongly transcribed in a dose-dependent manner when cotransfected with CD44ICD (Fig. 4 A). As a control, the mutant TRE reporters showed a reduced response to CD44ICD (Fig. 4, A and B). To determine whether CD44ICD translocation to the nucleus is necessary for the transactivation, we added the myristoylation sequence (MGSSKSKPK) to the NH2 terminus of CD44ICD (myristoylated [Myr]-CD44ICD). Immunofluorescence analysis showed that Myr-CD44ICD fails to translocate to the nucleus (Fig. 3 A, right). Cotransfection of TRE-containing reporter with a plasmid expressing Myr-CD44ICD resulted in no activation of the luciferase reporter (Fig. 4 A). Together, these results strongly suggest that CD44ICD acts as a signaling molecule via translocation to the nucleus and the activation of transcription.
Figure 4.

Transcriptional activation by CD44. (A) COS-7 cells were transfected with plasmids as indicated, and the transcriptional activity from the cotransfected reporter plasmids was measured. The activity of the TRE reporter plasmids was evaluated by transfecting COS-7 cells with RasV12 as a positive control. CD44ICD revealed ∼50% of the TRE-mediated transcriptional activity induced by overexpressed RasV12 protein under the experimental condition (insets). (B) Nucleotides mutated in Mut-TRE reporter plasmids are shown in italics and underlined. (C) COS-7 cells were transfected with a cDNA encoding full-length CD44 (lanes 2 and 3), CD44Δ287–290 constructs (lane 4) or with empty vector (lane 1). 36 h after transfection, the cells were incubated for an additional 12 h in the absence (lanes 1, 2, and 4) or presence of 100 μM BB2516 (lane 3). Blots were probed with anti-CD44cyto Ab. Asterisk indicates endogenous full-length CD44. (D) COS-7 cells were cotransfected with the TRE reporter plasmid together with an expression vector encoding full-length CD44, oncogenic Ras, or the corresponding empty vector. After 32 h, the cells were incubated for an additional 16 h in serum-free medium containing BB2516 (100 μM) or DMSO as a vehicle control before measurement of luciferase activity. Luciferase activity was expressed as fold induction relative to the value for cells transfected with the empty vector. (E) COS-7 cells were cotransfected with TRE reporter plasmid together with full-length CD44, CD44Δ287–290, or the corresponding empty vector. Data are expressed as indicated in Fig. 3 D.
Sequential CD44 cleavage is required for CD44-dependent transcriptional activity
To substantiate these ideas, we investigated the necessity of sequential proteolytic processing for CD44-dependent transcriptional activity. A cDNA encoding the full-length CD44 was transiently transfected into COS-7 cells, and immunoblots of the lysate with anti-CD44cyto Ab revealed, in addition to the overexpressed full-length CD44 bands, bands of ∼25 kD and smaller fragments corresponding to the products of CD44 ectodomain and intracellular cleavage, respectively (Fig. 4 C). The production of both CD44 cleavage products was markedly reduced in cells treated with BB2516 (Fig. 4 C). These results indicate that overexpressed full-length CD44 undergoes two sequential proteolytic cleavages in a manner similar to that observed for endogenous CD44. Expression of full-length CD44 in COS-7 cells significantly increased TRE-dependent transcriptional activity (Fig. 4 D). If sequential proteolytic cleavage of CD44 is necessary for the CD44-dependent transcriptional activity, then manipulations that affect the cleavage of CD44 should also affect the transcriptional activation. We first examined the effects of BB2516 on transcriptional activity in cells transfected with full-length CD44. Treatment with BB2516 blocked any transcriptional activation observed with full-length CD44 (Fig. 4 D). As a control for other potential effects of BB2516, we found that the luciferase activity of cells transfected with oncogenic Ha-Ras did not change on treatment with this inhibitor (Fig. 4 D).
Based on the intracellular cleavage site (I287 ↓ A288) determined by mass spectrometric analysis (Fig. 1 C), we then eliminated four amino acids (I287–N300) and transfected this mutant construct (CD44Δ287–290) into cells. The CD44ICD proteolytic fragments were barely detectable in cells transfected with CD44Δ287–290 (Fig. 4 C), confirming that this is the essential region for CD44 intracellular cleavage. We then found that CD44Δ287–290 failed to show significant transcriptional enhancement, whereas cells transfected with full-length CD44 exhibited a significant increase in transcriptional activity (Fig. 4 E). We conclude from these analyses that transcriptional activation by CD44 signaling requires the sequential proteolytic processing of this protein and the consequent release of CD44ICD.
CD44ICD potentiates transactivation mediated by transcriptional coactivator CBP/p300
We next set out to investigate how CD44ICD proteolytic fragments are involved in transcriptional activation in the nucleus. We first tested in GAL4 transactivation assays whether CD44ICD itself acts as a transcription factor. CD44ICD was fused to the GAL4 DNA binding domain (GAL4-CD44ICD), and this expression plasmid was transfected into COS-7 cells along with a reporter plasmid containing five tandem GAL4 binding sites. GAL4-CD44ICD had no activity on the GAL4-dependent promoter (Fig. 5 A), suggesting that CD44ICD alone is not sufficient to activate the transcriptional response. We considered the possibility that CD44ICD may modulate transcription by affecting other transcription factors or transcriptional coactivators that are involved in TRE-mediated transcription. To explore this possibility, we assessed the effect of CD44ICD on the activity of transcription factors c-Fos or c-Jun, and the transcriptional coactivators, CBP (CREB-binding protein) and p300 (Goodman and Smolik, 2000). Each of these was fused to the GAL4 DNA binding domain. All of these fusion constructs directed transcriptional activation of the GAL4-dependent promoter (Fig. 5 B). However, the cotransfection of CD44ICD did not affect the transcriptional activity of c-Fos and c-Jun (Fig. 5 B). In contrast, coexpression of CD44ICD did synergistically enhance transcription by GAL4-CBP and GAL4-p300 (Fig. 5 B). The strong synergistic activation was not observed in cells that coexpressed the membrane-bound form of CD44ICD, Myr-CD44ICD (Fig. 5 C). These results suggest that CD44ICD potentiates transcriptional activation through the functional cooperation with CBP/p300. To determine whether CBP/p300 is necessary for CD44-induced transcription, we used the adenoviral E1A protein, which inhibits CBP/p300-dependent transactivation by interacting with CBP/p300 (Arany et al., 1995; Lundblad et al., 1995). E1A clearly inhibited TRE-activated transcription induced by CD44ICD (Fig. 5 D). In contrast, E1AΔ2–36, which has a short NH2-terminal deletion and is defective for CBP/p300 binding (Stein et al., 1990), was impaired in the repression of transactivation by CD44ICD (Fig. 5 D). Together, these data indicate that CBP/p300 plays a role in mediating the transactivation by CD44ICD and support a requirement at least in part for a functional collaboration between CD44ICD and CBP/p300. Since no direct interaction between CBP/p300 and CD44ICD has been found (unpublished data), we propose that CD44ICD might interact with unidentified factors that impinge upon the CBP/p300-mediated transcriptional machinery. In light of several possible ways by which CBP/p300 activates transcription (Giordano and Avantaggiati, 1999), further analysis will be necessary to elucidate the precise mechanism whereby CD44ICD modulates CBP/p300 in transcriptional regulation.
Figure 5.

Functional cooperation between CD44ICD and CBP/p300, and upregulation of CD44 transcript induced by CD44ICD. (A–C) pFR-Luc, which has five GAL4 DNA binding sites, was used as a reporter for transcription mediated by GAL4 fusion proteins. Values are relative to control cells transfected with the same amount of expression vector encoding only the GAL4 DNA-binding domain (GAL4). (A) COS-7 cells were transfected with plasmid encoding GAL4, GAL4-CD44ICD, or GAL4–Elk-1, and the transcriptional activity from the cotransfected pFR-Luc was measured. (B) COS-7 cells were cotransfected with constructs expressing indicated GAL4 fusion proteins combined with CD44ICD or the corresponding empty vector. (C) COS-7 cells were transfected with CD44ICD, Myr-CD44ICD, or the corresponding empty vector and constructs expressing GAL4-p300 or GAL4. (D) COS-7 cells were transfected with TRE reporter plasmid and CD44ICD or the corresponding empty vector. Additionally, some transfections included an expression vector encoding E1A, E1AΔ2–36 or the corresponding empty vector, as indicated. (E) HeLa cells were transfected with a plasmid encoding CD44ICD or the corresponding empty vector together with a puromycin resistance gene plasmid at a molar ratio of 20:1. The cells were fed with fresh complete medium containing puromycin (3.0 μg/ml) 24 h after transfection. 36 h after the addition of puromycin, surviving cells were serum starved for 24 h in DME/F-12 containing 0.5% FCS. Total RNA was isolated and subjected to RT-PCR analysis of the expression of CD44 mRNA. The primers used were derived from sequences in the extracellular domain of human CD44 gene (5′-ATCGATTTGAATATAACCTGCCGC-3′ and 5′-ACAATCTTCTTCAGGTGGAGCTGA-3′). The products from the RT-PCR were normalized by comparison to RT-PCR of glyceraldehyde-3-phosphate dehydrogenase mRNA. The numbers under each lane were expressed as a fold relative to control cells. (F) U251MG cells were mechanically scraped and incubated for various times as indicated in the absence or presence of MG132 (15 μM). At the indicated times, the RT-PCR analyses of the expression of CD44 mRNA were conducted as described in Fig. 4 E.
The CD44 gene is one of the potential targets for transcriptional activation by CD44ICD
To identify endogenous target genes of CD44ICD-mediated transcription, a plasmid encoding CD44ICD or the corresponding empty vector was transiently transfected into HeLa cells together with a plasmid encoding a puromycin-resistant genes as a selection marker. Following puromycin selection, RNA from these cells was examined for increased expression of candidate target genes by reverse transcript (RT)-PCR. Through a series of RT-PCR analyses, we found that cells expressing CD44ICD express high levels of CD44 mRNA (Fig. 5 E). We have also observed that the generation of endogenous CD44ICD in mechanically scraped cells is accompanied by an increase in the endogenous CD44 transcript (Fig. 5 F). However, this time-dependent induction of CD44 transcript was not observed in the presence of MG132, which blocks intracellular cleavage (Fig. 5 F). These data indicate an important link between the intracellular cleavage of CD44 and the up-regulation of expression of the CD44 transcript. Given that the CD44 gene contains TRE elements within the proximal promoter region (Hofmann et al., 1993), these observations suggest that CD44ICD induces CD44 transcription, thus promoting the rapid turnover of CD44 that is required for efficient cell migration (Lauffenburger and Horwitz, 1996; Okamoto et al., 1999a).
Conclusion
Cell-surface proteins can initiate a variety of intracellular processes. Most of these signal transduction events have been linked to interactions with cytoplasmic proteins which in turn regulate transcription of key genes. The proteolytic cleavage of the ectodomain of a variety of cell surface proteins has recently emerged as a key mechanism for their functional regulation (Hooper et al., 1997; Werb, 1997). Although such proteolysis should produce cleavage products, the subsequent fate or biological effects of these products, especially for adhesion molecules, is poorly understood. Here, we have shown that a fragment of CD44 can directly interact with the transcriptional machinery, resulting in the upregulation of genes containing the TPA-responsive element, including CD44 itself. Our data provide new insights into the functional link between proteolytic processing of adhesion molecules and signal transduction, and suggest that such fragments may directly participate in transcriptional regulation.
Materials and methods
Immunoblot analysis
Cells were directly lysed in 2× Laemmli buffer or cell lysis buffer containing 1% Triton X-100, 0.1% SDS, and 0.5% deoxycholic acid. Equal amounts of protein were separated by SDS-PAGE, transferred onto nitrocellulose membrane and incubated with antibodies. All immunoblots were visualized by enhanced chemiluminescence detection (Amersham Pharmacia Biotech).
Protein purification and mass spectrometry analysis
Purified anti-CD44cyto Ab (1 mg) was coupled to 0.4 ml of ImmunoPure Immobilized ProteinG (Pierce Chemical co.) according to the manufacturer's instruction and then was used as an anti-CD44cyto Ab affinity column. Normal rabbit IgG (1 mg) was separately immobilized, and this column was used for preclearing nonspecific binding proteins. U251MG cells were plated (6 × 106 cells/dish) in 150-mm culture dishes and grown for 16 h. The cells were incubated for 40 min with TPA (100 ng/ml) and then washed, incubated for an additional 3 h with fresh medium. The cells (20 × 150-mm culture dishes) were lysed with PBS/TDS buffer (10 mM Na2HPO4, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 0.2% NaH3, 0.004% NaF, 1 mM NaVO4, 25 mM β-glycerophosphate, 100 μg/ml PMSF, and 1 μg/ml each aprotinin and leupeptin) and centrifuged at 15,000 g to remove insoluble material. The total cell lysates (60 mg) were incubated with Concanavalin A immobilized on 4% cross-linked beaded agarose (Sigma-Aldrich) for 90 min at 4°C with constant rotation to remove the glycosylated full-length CD44. The supernatant was then passed over a normal rabbit IgG column and this precleared lysate was then applied to an anti-CD44cyto Ab affinity column and repassed two additional times. The column was washed with 2 ml of PBS/TDS and then with 1 ml of 10 mM Tris (pH 7.5). The CD44ICD proteolytic fragment was eluted stepwise with 5 passes of 0.5 ml of 100mM triethylamine buffer (pH 11.5). The fractions containing the highest concentrations of CD44ICD were combined, dialyzed against 0.01× PBS, followed by concentration with a centrifugal concentration device. The resultant sample was resolved on 18% SDS-PAGE. The gel was stained with Gel Code Blue stain (Pierce Chemical Co.) following the manufacturer's instruction. The CD44ICD band was excised and subjected to in-gel digestion with Lys-C followed by MALDI-MS and ES-MS analysis. MALDI-MS was performed on a research grade, Micromass Tofspec SE instrument equipped with delayed extraction and a reflectron. ES-MS was performed on a Micromass Q-Tof mass spectrometer. The raw ES-MS spectra were processed using the maximum entropy based approach using MAXENT3 program.
Expression plasmids and transfection
Detailed information on the plasmid constructions will be provided upon request. Expression plasmids encoding GAL4-p300 or E1A were gifts from A. Giordano (Jefferson Medical College, Philadelphia, PA). GAL4-CBP was constructed by cloning the BamH1 fragment from CMXCBP kindly provided from R.M. Evans (The Salk Institute for Biological Studies, La Jolla, CA) into pFA-CMV. Transfections were performed using the FuGENE6 transfection reagent (Boehringer).
Immunofluorescence analysis
Cells were fixed with 4% paraformaldehyde for 10 min, exposed to 0.2% (vol/vol) Triton X-100 in phosphate-buffered saline (PBS) for 5 min, washed with PBS, and then incubated for 60 min at room temperature with the 12CA5 mAb directed against HA. After washing three times with PBS, the cells were incubated for 60 min at room temperature with FITC-conjugated secondary antibodies (Biosource International). The cells were again washed with PBS, mounted in 80% glycerol, and examined with a confocal microscope (Fluoview; Olympus).
Cellular subfractionation
Cells were washed and scraped in ice-cold PBS and centrifuged at 1,800 g for 10 min. The pellets were suspended in buffer A (20 mM Hepes-KOH [pH 7.4], 1 mM EDTA, 1 mM EGTA, 1.5 mM MgCl2, 1 mM DTT, and protease inhibitor cocktail) with 0.2% NP-40, disrupted by Dounce homogenization with microscopic monitoring of cell lysis throughout homogenization, and centrifuged at 1,000 g for 5 min; the supernatant was used as soluble fraction. The pellet was washed with and resuspended in buffer A, mixed with 0.34 M sucrose made in buffer A followed by centrifugation at 1,500 g for 5 min, and the pellet was used as the nuclear fraction.
Reporter gene assays
COS-7 cells maintained in culture for <2 wk were seeded into six-well plates (2.5 × 105 cells per well) and grown for 16 h before transfection. Cells were cotransfected with luciferase reporter plasmids and other expression plasmids, as specified in the figure legends. The total amount of DNA transfected was maintained constant by the addition of control DNA. The cells were incubated for 48 h and during the last 16 h they were deprived of serum. Luciferase activity was analyzed in cell lysates (Promega) and normalized to the protein concentration.
Acknowledgments
We thank M. Nakajima and W.G. Stetler-Stevenson for providing metalloprotease inhibitors; R. M. Evans, P. Stiegler, A. Giordano, S. Takeda, M.-C. Hung, and Q. Hu for their gift of expression plasmids; K.K. Tanabe for critical reading; K. Stone of the KECK Facility at Yale University for expert work; and all members of the Wong lab for helpful discussions.
This work was supported by grants CA51093 and CA69495 from the National Institutes of Health (A.J. Wong), the JSPS fellowship for Japanese Biomedical and Behavioral Researchers (to I. Okamoto), “Research for the Future” program of the Japan Society for the Promotion of Science (H. Saya), and a grant for Cancer Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (H. Saya).
I. Okamoto and Y. Kawano contributed equally to this work.
A.J. Wong and H. Saya both served as senior authors for this work.
Footnotes
Abbreviations used in this paper: Ab, antibody; ES, electrospray; ICD, intracellular domain; HA, hemaggulutinin; luc, luciferase; MS, mass spectrometry; Myr, myristoylated; RT, reverse transcript; TPA, 12-O-tetradecanoylphorbol 13-acetate; TRE, TPA-responsive element.
References
- Aplin, A.E., A.K. Howe, and R. Juliano. 1999. Cell adhesion molecules, signal. transduction and cell growth. Curr. Opin. Cell Biol. 11:737–744. [DOI] [PubMed] [Google Scholar]
- Arany, Z., D. Newsome, E. Oldread, D.M. Livingston, and R. Eckner. 1995. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 374:81–84. [DOI] [PubMed] [Google Scholar]
- Arch, R., K. Wirth, M. Hofmann, H. Ponta, S. Matzku, P. Herrlich, and M. Zoller. 1992. Participation in normal immune responses of a metastasis-inducing splice variant of CD44. Science. 257:682–685. [DOI] [PubMed] [Google Scholar]
- Aruffo, A., I. Stamenkovic, M. Melnick, C.B. Underhill, and B. Seed. 1990. CD44 is the principal cell surface receptor for hyaluronate. Cell. 61:1303–1313. [DOI] [PubMed] [Google Scholar]
- Brown, M.S., J. Ye, R.B. Rawson, and J.L. Goldstein. 2000. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 100:391–398. [DOI] [PubMed] [Google Scholar]
- Culty, M., K. Miyake, P.W. Kincade, E. Sikorski, E.C. Butcher, C. Underhill, and E. Sikorski. 1990. The hyaluronate receptor is a member of the CD44 (H-CAM) family of cell surface glycoproteins. J. Cell Biol. 111:2765–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper, B., W. Annaert, P. Cupers, P. Saftig, K. Craessaerts, J.S. Mumm, E.H. Schroeter, V. Schrijvers, M.S. Wolfe, W.J. Ray, et al. 1999. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 398:518–522. [DOI] [PubMed] [Google Scholar]
- Giordano, A., and M.L. Avantaggiati. 1999. p300 and CBP: partners for life and death. J. Cell Physiol. 181:218–230. [DOI] [PubMed] [Google Scholar]
- Goodman, R.H., and S. Smolik. 2000. CBP/p300 in cell growth, transformation, and development. Genes Dev. 14:1553–1577. [PubMed] [Google Scholar]
- Gumbiner, B.M. 1996. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 84:345–357. [DOI] [PubMed] [Google Scholar]
- Günthert, U., M. Hofmann, W. Rudy, S. Reber, M. Zoller, I. Haussmann, S. Matzku, A. Wenzel, H. Ponta, and P. Herrlich. 1991. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 65:13–24. [DOI] [PubMed] [Google Scholar]
- Hofmann, M., W. Rudy, U. Günthert, S.G. Zimmer, V. Zawadzki, M. Zoller, R.B. Lichtner, P. Herrlich, and H. Ponta. 1993. A link between ras and metastatic behavior of tumor cells: ras induces CD44 promoter activity and leads to low-level expression of metastasis-specific variants of CD44 in CREF cells. Cancer Res. 53:1516–1521. [PubMed] [Google Scholar]
- Hooper, N.M., E.H. Karran, and A.J. Turner. 1997. Membrane protein secretases. Biochem. J. 321:265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, K., I. Okamoto, N. Araki, Y. Kawano, M. Nakao, S. Fujiyama, K. Tomita, T. Mimori, and H. Saya. 1999. Calcium influx triggers the sequential proteolysis of extracellular and cytoplasmic domains of E-cadherin, leading to loss of beta-catenin from cell-cell contacts. Oncogene. 18:7080–7090. [DOI] [PubMed] [Google Scholar]
- Kajita, M., Y. Itoh, T. Chiba, H. Mori, A. Okada, H. Kinoh, and M. Seiki. 2001. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 153:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano, Y., I. Okamoto, D. Murakami, H. Itoh, M. Yoshida, S. Ueda, and H. Saya. 2000. Ras oncoprotein induces CD44 cleavage through phosphoinositide 3-OH kinase and the rho family of small G proteins. J. Biol. Chem. 275:29628–29635. [DOI] [PubMed] [Google Scholar]
- Lauffenburger, D.A., and A.F. Horwitz. 1996. Cell migration: a physically integrated molecular process. Cell. 84:359–369. [DOI] [PubMed] [Google Scholar]
- Lesley, J., and R. Hyman. 1998. CD44 structure and function. Front. Biosci. 3:616–630. [DOI] [PubMed] [Google Scholar]
- Lundblad, J.R., R.P. Kwok, M.E. Laurance, M.L. Harter, and R.H. Goodman. 1995. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature. 374:85–88. [DOI] [PubMed] [Google Scholar]
- McNeil, P.L., L. Muthukrishnan, E. Warder, and P.A. D'Amore. 1989. Growth factors are released by mechanically wounded endothelial cells. J. Cell Biol. 109:811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto, I., Y. Kawano, H. Tsuiki, J. Sasaki, M. Nakao, M. Matsumoto, M. Suga, M. Ando, M. Nakajima, and H. Saya. 1999. a. CD44 cleavage induced by a membrane-associated metalloprotease plays a critical role in tumor cell migration. Oncogene. 18:1435–1446. [DOI] [PubMed] [Google Scholar]
- Okamoto, I., Y. Kawano, M. Matsumoto, M. Suga, K. Kaibuchi, M. Ando, and H. Saya. 1999. b. Regulated CD44 cleavage under the control of protein kinase C, calcium influx, and the Rho family of small G proteins. J. Biol. Chem. 274:25525–25534. [DOI] [PubMed] [Google Scholar]
- Sakai, J., E.A. Duncan, R.B. Rawson, X. Hua, M.S. Brown, and J.L. Goldstein. 1996. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell. 85:1037–1046. [DOI] [PubMed] [Google Scholar]
- Schroeter, E.H., J.A. Kisslinger, and R. Kopan. 1998. Notch-1 signalling requires ligand- induced proteolytic release of intracellular domain. Nature. 393:382–386. [DOI] [PubMed] [Google Scholar]
- Stein, R.W., M. Corrigan, P. Yaciuk, J. Whelan, and E. Moran. 1990. Analysis of E1A- mediated growth regulation functions: binding of the 300-kilodalton cellular product correlates with E1A enhancer repression function and DNA synthesis-inducing activity. J. Virol. 64:4421–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl, G., and A. Adachi. 1998. Nuclear access and action of notch in vivo. Cell. 93:649–660. [DOI] [PubMed] [Google Scholar]
- Tran, P.O., L.E. Hinman, G.M. Unger, and P.J. Sammak. 1999. A wound-induced [Ca2+]i increase and its transcriptional activation of immediate early genes is important in the regulation of motility. Exp. Cell Res. 246:319–326. [DOI] [PubMed] [Google Scholar]
- Werb, Z. 1997. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 91:439–442. [DOI] [PubMed] [Google Scholar]