

Abstract
Tropomodulin (Tmod) is an actin pointed-end capping protein that regulates actin dynamics at thin filament pointed ends in striated muscle. Although pointed-end capping by Tmod controls thin filament lengths in assembled myofibrils, its role in length specification during de novo myofibril assembly is not established. We used the Drosophila Tmod homologue, sanpodo (spdo), to investigate Tmod's function during muscle development in the indirect flight muscle. SPDO was associated with the pointed ends of elongating thin filaments throughout myofibril assembly. Transient overexpression of SPDO during myofibril assembly irreversibly arrested elongation of preexisting thin filaments. However, the lengths of thin filaments assembled after SPDO levels had declined were normal. Flies with a preponderance of abnormally short thin filaments were unable to fly. We conclude that: (a) thin filaments elongate from their pointed ends during myofibril assembly; (b) pointed ends are dynamically capped at endogenous levels of SPDO so as to allow elongation; (c) a transient increase in SPDO levels during myofibril assembly converts SPDO from a dynamic to a permanent cap; and (d) developmental regulation of pointed-end capping during myofibril assembly is crucial for specification of final thin filament lengths, myofibril structure, and muscle function.
Keywords: actin-capping protein; thin filaments; myofibril; sanpodo; tropomodulin
Introduction
Control of actin polymerization is important to provide architectural support for the cell membrane and to maintain cell shapes, as well as to power motility and contraction (DeRosier and Tilney, 2000). All of these processes depend on the assembly of specialized networks of actin filaments. Actin filaments have a polarized morphology and are built by rapid monomer addition at the barbed end, or by relatively slower monomer addition at the pointed end (for review see Pollard et al., 2000). Many types of actin filaments are constructed in a highly regulated step-wise progression. The first part of this coordinated sequence of events is a nucleation step that requires components to initiate polymerization in the correct cellular location. Next, actin filaments elongate from either the barbed or pointed ends to achieve their final lengths (DeRosier and Tilney, 2000). In many actin cytoskeletal structures, it is known that filaments assemble principally by monomer addition at the barbed end, whereas a role for pointed-end assembly has not been established (Pollard et al., 2000). Further, it is not understood how elongation is limited to produce the precise filament lengths that are characteristic of stable actin cytoskeletal structures such as are present in the sarcomeres of striated muscle, the stereocilia of the hair cells in the inner ear, or the membrane skeleton of the erythroctye (Fowler, 1996, 1997). In this study, we investigate the role of an actin filament pointed-end capping protein, tropomodulin (Tmod),* in the regulation of actin elongation, and the specification of actin filament length during myofibril assembly in development of striated muscle.
Striated muscle is an excellent system to address how actin filaments assemble and how their elongation to a specific length is regulated. The pointed and barbed ends are lined up in regular arrays so that thin filament lengths can be easily visualized and measured. CapZ binds to the barbed ends of the thin filaments (located at the Z-discs), whereas Tmod binds to the pointed ends (located in the middle of the sarcomere) (for review see Littlefield and Fowler, 1998). CapZ is thought to nucleate thin filament assembly early in myofibrillogenesis (Schafer et al., 1995) and to act with α-actinin to anchor thin filaments in the Z-disc (Papa et al., 1999). However, it is not known whether thin filaments elongate from their barbed or pointed ends or how the process of actin filament elongation is regulated to specify length during myofibrillogenesis.
The pointed-end capping protein, Tmod, regulates actin dynamics at pointed ends and is thought to maintain thin filament length in cardiac muscle cells (Fowler, 1996; Littlefield and Fowler, 1998). Changes in Tmod expression levels in cultured cardiac myocytes cause misregulation of thin filament length. Shorter thin filaments are observed when Tmod expression levels are increased (Sussman et al., 1998b; Littlefield et al., 2001); conversely, longer thin filaments are observed when Tmod expression levels or capping activity is reduced (Gregorio et al., 1995; Sussman et al., 1998a). However, these studies were conducted with cells obtained from fully differentiated heart tissue and may not represent mechanisms of de novo myofibril assembly during development in vivo (for review see Gregorio and Antin, 2000). For example, the shorter thin filaments observed in cardiac myocytes overexpressing Tmod may be due to changes in the dynamics of monomer addition in mature thin filaments after assembly is complete, subsequently leading to filament shrinkage, as proposed by Littlefield et al. (2001).
The regulation of thin filament length by Tmod is essential for cardiac muscle function. Cultured cardiac myocytes fail to beat when Tmod capping function is inhibited (Gregorio et al., 1995) or when Tmod levels are altered (Sussman et al., 1998a). Additionally, Tmod-overexpressing transgenic (TOT) (TOT) mouse hearts yield a phenotype reminiscent of dilated cardiomyopathy in humans (Sussman et al., 1998b). However, the molecular mechanisms that cause thin filament length changes and sarcomeric disarray leading to impaired function in cultured cardiac myocytes and TOT mice are not clear. These may be due to defective thin filament assembly during development, or to sarcomeric degeneration in mature muscles after assembly has been completed.
To determine directly whether Tmod regulates the process of actin filament elongation and length specification during de novo myofibril assembly, we developed an in vivo approach to manipulate levels of the Drosophila Tmod homologue, Sanpodo (SPDO), during development of the indirect flight muscles (IFM). Drosophila IFM has several advantages for the purposes of this study. First, myofibril assembly is synchronous and takes place over 4 d of pupation. Second, IFM muscle function is not needed for viability and can be assayed by testing flight. Third, IFM myofibrils are extremely well organized and defects are fairly easy to observe. Finally, muscle sarcomeres undergo dramatic and synchronous changes in length during myofibril assembly in response to induced tension across the developing muscle as a consequence of cuticle expansion (Reedy and Beall, 1993 and references therein). The dorsal longitudinal muscles of the IFM in Drosophila contain ∼310 sarcomeres, ∼1.7 μm long, that span 500 μm of muscle length by day 2 (D2) after pupal formation (APF). By adulthood, the same ∼310 sarcomeres are now 3.2 μm long and span 1,000 μm of muscle length, almost a twofold increase in size. To accommodate these changes in sarcomere size, both the thick and thin filaments increase synchronously in length during myofibril assembly (Reedy and Beall, 1993).
We used a transgenic fly line, hs-43 kD spdo (Dye et al., 1998), to rapidly and transiently overexpress SPDO during different stages of myofibril assembly in IFM development. Surprisingly, we found that transient increases in SPDO at any time in IFM development led to an irreversible block in elongation of preexisting thin filaments. Our results indicate that thin filaments elongate from their pointed ends during myofibril assembly, and that thin filament elongation and final length specification depend on developmental regulation of pointed-end capping by SPDO. Our study is also the first to provide direct evidence that pointed- rather than barbed-end elongation can be important for the assembly of actin filaments into cytoskeletal structures.
Results
Multiple SPDO isoforms are expressed during development
Sanpodo mutations were originally identified from mutagenesis screens designed to uncover genes that control sibling neuron cell fate in the central nervous system (Salzberg et al., 1994). Further studies showed that mutation of spdo affects the developmental patterning of the embryonic peripheral nervous system (Dye et al., 1998; Skeath and Doe, 1998) as well as mesoderm lineage specification (Park et al., 1998). Sequence analysis of genomic and cDNA clones originally revealed only one open reading frame encoding a 367-AA protein with 70% overall similarity to vertebrate Tmods (Dye et al., 1998) (Fig. 1 A, CT41421 mRNA). However, data analysis from the Drosophila genome sequence indicates that alternative promoter usage produces an additional larger SPDO isoform of 402 residues containing a 36-AA extension at the NH2 terminus (Fig. 1 A, CT3919 mRNA). This NH2-terminal extension has no apparent homology to other Tmod isoforms or protein or DNA sequences (http://www.ncbi.nlm.nih.gov/BLAST). Additionally, there is another message that is predicted to code for an SPDO protein differing only by four amino acids at the COOH-terminal end (Chu et al., 2000) (unpublished data). The existence of multiple SPDO isoforms is consistent with previous reports of multiple mRNA species (Dye et al., 1998) and with developmental Western blots (Fig. 1 C).
Figure 1.
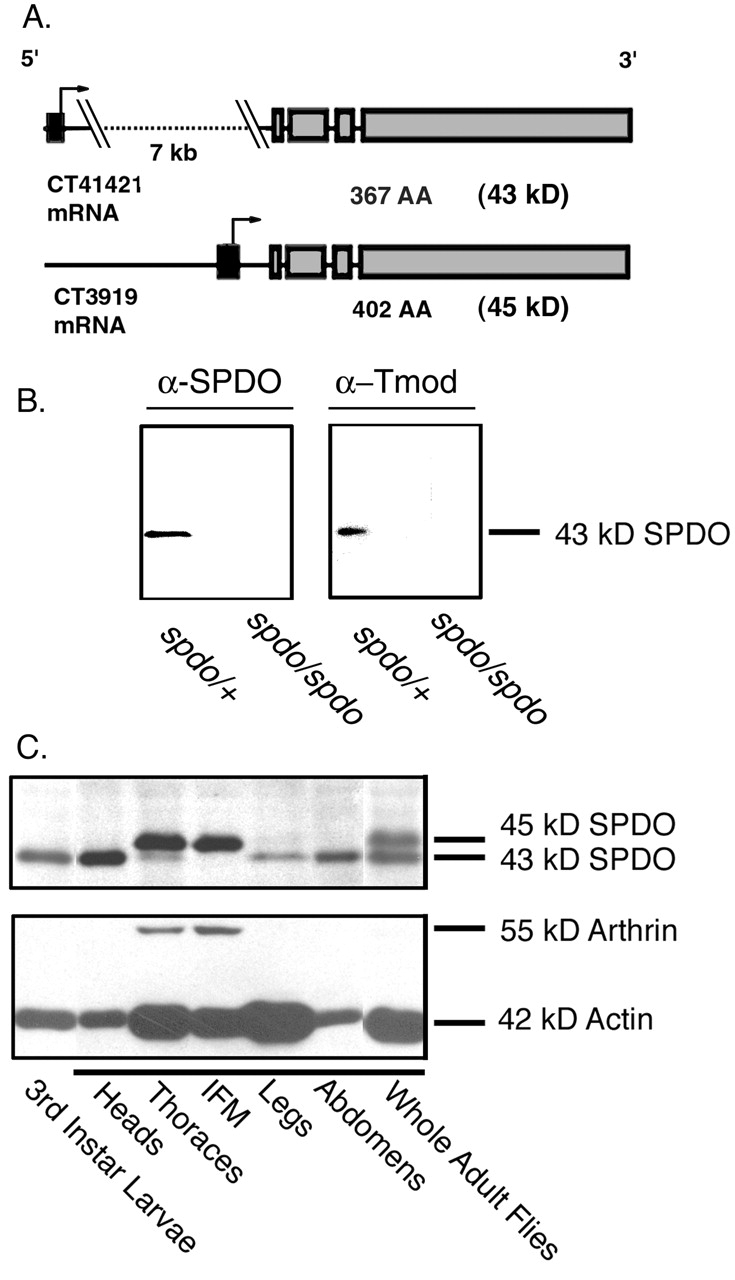
SPDO isoforms are expressed differentially during development and in adult tissues. (A) A data analysis program (GadFly) (http://flybase.bio.indiana.edu:82/) which searches expressed sequence tags (ESTs) from the Drosophila genome project predicts two alternative promoters (black exons) used to generate SPDO isoforms. CT41421 and CT3919 are mRNA transcripts predicted to generate 366 AA (43 kD) and 402 AA (45 kD) protein products, respectively. Arrow represents translational start sites that contribute 2 or 36 AA for CT4121 and CT 3919, respectively. (B) Western Blot prepared from heterozygous (spdo/+) or homozygous (spdo/spdo) spdo embryos probed with mouse anti-SPDO antibody (α-SPDO) (left) or anti–human erythrocyte Tmod antibody (α-Tmod) (right). (C) Western blot prepared from larvae or dissected adult flies probed with anti-SPDO antibodies.
In embryonic extracts, the anti-SPDO antibody recognized a single band migrating at ∼43 kD on an immunoblot of protein homogenates derived from spdo heterozygous embryos, and not in spdo-null embryos (Fig. 1 B). This result agrees with the molecular mass predicted for CT41421 mRNA (43 kD) (Fig. 1 A). In contrast, both this ∼43-kD protein and an additional ∼45-kD protein could be detected in adult tissues (Fig. 1 C). The size of the 45-kD protein is consistent with the molecular mass predicted for CT3919 mRNA (402 AA, 45 kD). The smaller, 43-kD isoform of SPDO was detected at all stages of the Drosophila life cycle (Fig. 1, B, embryos, and C, larvae and adults). In adult flies, the 43-kD SPDO isoform was enriched in the tissues of the head and abdomen, whereas the 45-kD protein was only detected in thoraces and dissected IFM (Fig. 1 C).
The 43-kD SPDO isoform is associated with elongating IFM thin filaments during myofibril assembly
We investigated which SPDO isoform (if any) was associated with the pointed ends of thin filaments during myofibril assembly in IFM development. IFM myofibril assembly begins during early pupation and is completed soon after eclosion in early adulthood (Reedy and Beall, 1993). A schematic of the Drosophila life cycle is depicted in Fig. 2 A (see Fig. 9 , left panel). To determine whether SPDO was associated with assembling IFM myofibrils, whole thoraces from pupae and adults were homogenized and extracted with Triton X-100 and the sarcomeric pellet fraction (P) was subjected to immunoblotting for SPDO. In late pupae on D4 APF and in newly eclosed adults on D1 after eclosion (D1 adults aged 2–3 h), only the 43-kD SPDO isoform was detected in the myofibril fraction (Fig. 2 B). Immunofluorescence staining of myofibrils isolated from D4 APF pupae also revealed that the 43-kD SPDO isoform was associated with the pointed ends of the thin filaments (unpublished data). The 45-kD SPDO isoform was not detected until 8–12 h after eclosion, but was the major isoform in D2 adults (Figs. 1 C and 2 B and see Fig. 7 B, left panel). Immunofluorescence staining of myofibrils from D2 adults demonstrated that the 45-kD SPDO isoform was also associated with the thin filament pointed ends (Fig. 4 A). These results indicate that pointed ends of thin filaments are capped by the 43-kD SPDO isoform during IFM myofibril assembly, and that the 45-kD SPDO isoform does not replace the 43-kD SPDO isoform until well after the majority of myofibril assembly is completed. Because we wanted to study thin filament length regulation during IFM myofibril assembly, we chose to overexpress the endogenous 43-kD isoform during pupation when thin filaments are actively elongating.
Figure 2.
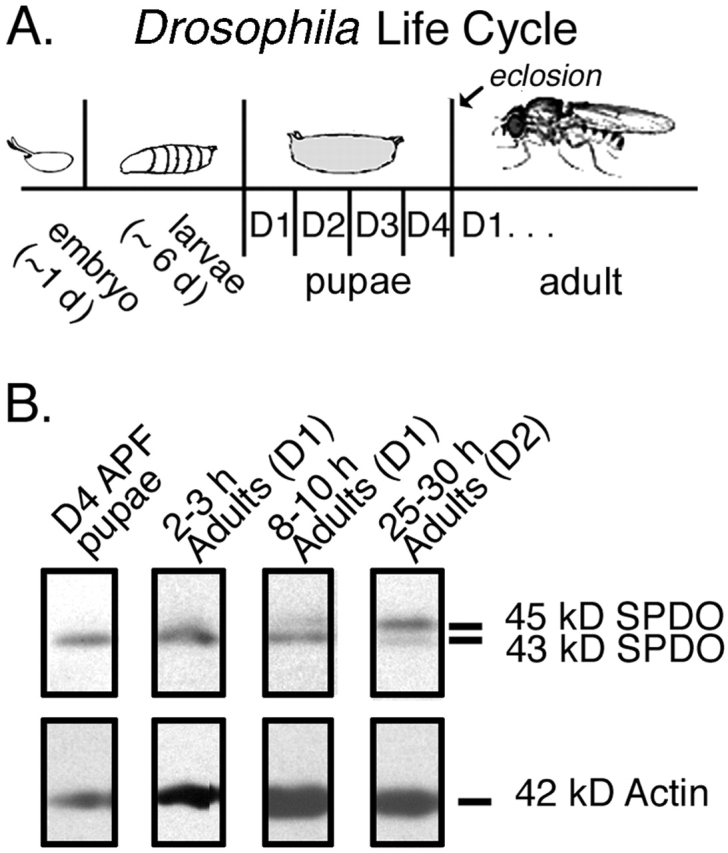
A developmental SPDO isoform switch occurs in the IFM during early adulthood. (A) Schematic of the Drosophila life cycle at 25°C (for review see Ashburner, 1989). After ∼1 d, fertilized embryos hatch to larvae. The larval period lasts ∼6 d before formation of an immobile pupa. Over the next 4 d APF, larval tissues remodel to give rise to a winged adult that emerges from the pupa at eclosion (hatching) (∼10-d cycle). White prepupae formation marks the first day of pupation (D1 APF). Adult flies on the first day after eclosion are indicated as D1 Adults. (B) Whole thoraces were dissected from pupae (12 PM D4 APF), or D1 adult flies aged 2–3 or 8–10 h, or from D2 adults aged 25–30 h after eclosion. Triton X-100 extracted myofibrils from thoraces were isolated and prepared for Western blots. Blots were probed with anti-SPDO (top), and then stripped and reprobed with the C4 anti-actin monoclonal antibody to control for loading (bottom).
Figure 9.

Model of IFM thin filament assembly in wild-type flies and in the presence of excess 43-kD SPDO. Based on the ultrastructural analysis of Reedy and Beall (1993), by the D2 APF, small sarcomeres of ∼1.7 μm in length have already formed with clear interdigitation of thick and thin filaments (a). In wild-type flies (left panel), the sarcomeres continue to develop throughout pupation by elongation of preexisting “core” filaments from their pointed ends (b–c) and by addition of new filaments to the periphery of the myofibril (c–d). The 43-kD SPDO isoform is present throughout thin filament elongation during pupation in noninduced hs = 43 spdo or wild-type flies (a–d), and is replaced by the 45-kD SPDO isoform in early adulthood, after the majority of assembly is completed (e). In contrast (right panel) transient overexpression of the 43-kD SPDO isoform by heat-shock (HS) induction during pupation (c) prevents uncapping and thin filament elongation of the preexisting “core” thin filaments (c–d). Normal replacement of the 43-kD SPDO isoform by the 45-kD isoform at adulthood is also prevented (d–e). However, sarcomere elongation is unaffected as are addition and elongation of peripheral thin filaments, and these peripheral thin filaments appear to be capped by the 45-kD isoform at their pointed ends (c–e) (Fig. 7). Black lines, thin filaments; filled black arrowheads, 43-kD SPDO isoform; hollow arrowheads, 45-kD SPDO isoform; small arrows indicate dynamic exchange of SPDO; Z, Z line.
Figure 7.

Transiently overexpressed 43 kD-SPDO bound to thin filaments irreversibly. (A) Relative amounts of 43-kD or 45-kD SPDO isoforms associated with Triton X-100 extracted pupal myofibrils from whole thoraces (D4 APF), adult myofibrils from thoraces of D3 adult hs-43 kD spdo flies heat shocked (HS) on D4 APF (+), or from untreated D3 adult hs-43 kD spdo flies (−). Samples from soluble (S) and myofibril pellet fractions (P) were electrophoresed on SDS-PAGE followed by immunoblotting with anti-SPDO (top), and then stripped and reprobed with the C4 anti-actin monoclonal antibody to control for loading (bottom). Pupae were dissected immediately after 1 h of heat shock at 12 PM D4 APF, or allowed to develop to adulthood at 25°C. (B) Staged hs-43 kD spdo flies were heat shocked (HS) for 1 h on D4 APF (+) or untreated (−) and allowed to develop to adulthood. Adults were collected and aged up to 10 d. The IFMs were dissected and prepared for SDS-PAGE and immunoblotting. The blots were stripped as above. Both the 43-kD actin and the 55-kD IFM-specific ubiquinated actin (Arthrin) (Ball et al., 1987) were detected at similar levels with or without heat-shock treatment.
Figure 4.

Transient induction of 43-kD SPDO expression during myofibril assembly produces abnormally short core thin filaments. Isolated IFM myofibrils from heat-treated (12 PM D4 APF) yw adult flies (A and C) or heat-treated hs-43 kD spdo adult flies (B and D) were fixed and stained with Bodipy-phallacidin (green) (top panels in A, B) or anti-TM (top panel in C and D) followed by staining with antibodies to SPDO (middle panels). Images were selected from optically sectioned (100 nm increments) myofibrils obtained from deconvolution microscopy and correspond to the center section of the myofibril. White arrowheads, pointed ends; Z, Z-discs. Bar, 5 μm.
Induced expression of the 43-kD SPDO isoform affects IFM function during mid- to late-myofibril assembly
The transgenic fly line hs-43 kD spdo was used to overexpress the 43-kD SPDO isoform during different stages of pupation. The hs-43 kD spdo strain was created by germline transformation with a P element construct containing the heat shock 70 promoter fused with the open reading frame coding for the 43-kD isoform of spdo (Fig. 1 A, CT41421 mRNA) (Dye et al., 1998). To test the effect of transient increases in levels of the 43-kD SPDO isoform on different stages of IFM myofibril assembly, experimental groups of hs-43 kD spdo flies were subjected to 1 h of heat shock during pupation. The time of heat shock treatment during pupation ranged from D2 APF, when striated myofibrils are first evident, to early adulthood, when assembly is complete (Reedy and Beall, 1993). The functional consequences of overexpression of the 43-kD SPDO during IFM assembly were examined by flight testing adult hs-43 kD spdo flies that were heat shocked as pupae and were compared with the flight performance of heat-shocked yw control flies. The effect on IFM muscle function was dramatically different when the 43-kD SPDO was overexpressed on different days of pupation (Fig. 3) .
Figure 3.

Overexpression of the 43-kD SPDO isoform during mid- to late-myofibril assembly during pupation affects adult flight ability. Staged hs-43 kD spdo lines were treated to one 37°C heat-shock treatment for 1 h during pupation (ranging from D2 APF to adult; Fig. 2 A), and then allowed to develop at 25°C. Adults were collected for flight testing. The percentage of 2–3-d-old adult flies that were capable of upward or horizontal flight (% Fliers) was graphed in relation to the timing of heat-shock induction. In the control groups, >70% of adult yw flies that were heat shocked any time between D2 and D4 APF could fly (unpublished data).
Induction of the 43-kD SPDO expression by heat-shock treatment during mid-to late-pupation (D3–D4 APF) interfered with flight, whereas heat shock treatment during early pupation (D2 APF, 12 PM) had no significant effect on flight ability (Fig. 3). The most dramatic change in flight behavior was observed within a 6-h period on D3 APF. The number of flighted adults decreased from 70 to 30% when 43-kD SPDO expression was induced at 12 PM or at 6 PM, respectively. However, heat shock on early D4 APF had the most detrimental effect on adult muscle function. Only 8% of the heat-shocked hs-43 kD spdo flies could sustain some type of flight, whereas the majority completely dropped during flight testing. In control experiments, yw flies that were treated to heat shock conditions during pupation were not flight impaired (unpublished data) and exhibited no changes in myofibril morphology (Fig. 4, A and C and see Fig. 8, D and E) . In contrast to the flight impairment due to overexpression during pupation, newly eclosed flies (heat-shocked D1 adults) were not flight impaired (Fig. 3). Thus, a brief induction of expression of the 43-kD SPDO isoform during the period of mid- to late-myofibril assembly had the most deleterious effect on IFM muscle function.
Figure 8.
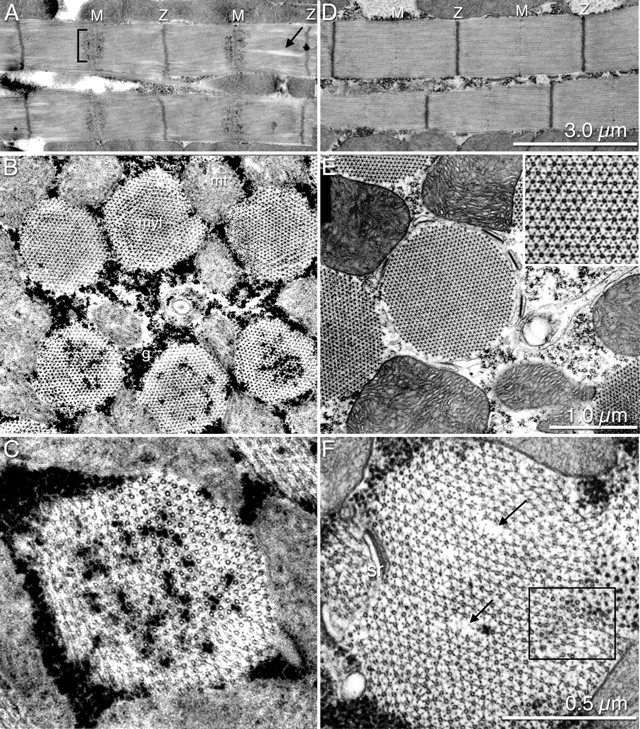
Overexpression of SPDO during assembly affects myofibril morphology. Ultrastructure of IFM from D2 adult flies that had been heat shocked on D4 APF (hs-43 kD spdo (A–C and F) from control yw flies heat shocked on D4 APF (D and E). Longitudinal sections are shown in A and D, cross-sections shown in B, C, E, and F. The ultrastructure is normal for heat-shocked control flies (D and E). Black arrows (A and F), missing thick and thin filaments. g, glycogen granules; M, M-line; MT, mitochondria; myf, myofibril; SR, sarcoplasmic reticulum; Z, Z line. Bars: (A and D) 3.0 μm; (B and E) 1.0 μm; (C, F, and E inset) 0.5 μm.
SPDO overexpression results in shorter thin filaments located in the cores of adult IFM myofibrils
The effect of transient overexpression of 43-kD SPDO during myofibril assembly on thin filament length was monitored by staining isolated adult IFM myofibrils with phallacidin, and then analyzed using deconvolution microscopy. We examined D3 adult IFM myofibrils from hs-43 kD spdo and yw control flies that had been heat shocked on D4 APF (Fig. 4). Phallacidin staining of F-actin revealed that two different populations of thin filaments, based on length, exist in IFM myofibrils from adult flies when expression of 43-kD SPDO was induced on D4 APF. Thin filaments located in the core of the IFM myofibrils were uniformly shorter (1.28 μm) than the thin filaments at the periphery of the myofibril (1.52 μm; P < 0.01) (Table I and Fig. 4). In contrast, all of the IFM thin filaments of non–heat-treated hs-43 kD spdo flies, as well as heat-shocked yw flies were of uniform length (average of 1.52 μm) (Fig. 4 A and Table I), agreeing with previous ultrastructural studies (Reedy and Beall, 1993).
Table I. The effect of 43-kD SPDO overexpression during myofibril assembly.
| Developmental stage when IFM myofibrils fixed | Heat shock | Sarcomere length | Thin filament length | Core thin filament length | Myofibril diameter (DZ) | Core myofibril diameter | Core volume/total | DZ/DH |
|---|---|---|---|---|---|---|---|---|
| time | μm | μm | μm | μm | μm | % | % | |
| yw (controls) | ||||||||
| Adult | − | 3.53 ± .08 | 1.49 ± 0.08 | 1.12 ± 0.09 | 98 ± 7 | |||
| Adult | D4 APF | 3.44 ± 0.1 | 1.45 ± 0.07 | 1.14 ± 0.11 | 98 ± 8 | |||
| hs-43 kD spdo | ||||||||
| Pupal D4 APF | − | 3.01 ± 0.12 | 1.25 ± 0.07 | 0.63 ± 0.14 | ||||
| Pupal D4 APF | D4 APF | 2.91 ± 0.1 | 1.21 ± 0.06 | 0.62 ± 0.03 | ||||
| Adult | − | 3.55 ± 0.10 | 1.52 ± 0.04 | 1.19 ± 0.09 | 98 ± 3 | |||
| Adult | D3 APF | 3.42 ± 0.05 | 1.44 ± 0.04 | 1.06 ± 0.06 | 1.14 ± 0.06 | 0.32 ± 0.05 | 5.3 ± 2 | 98 ± 4 |
| Adult | D4 APF | 3.48 ± 0.12 | 1.52 ± 0.07 | 1.28 ± 0.03 | 1.11 ± 0.04 | 0.62 ± 0.08 | 24.0 ± 6 | 98 ± 5 |
| Adult | D1 Adult | 3.44 ± 0.12 | 1.42 ± 0.06 | 1.23 ± 0.08 | 98 ± 4 | |||
| Adult 1 D | D4 APF | 3.42 ± 0.22 | 1.42 ± 0.10 | 1.16 ± 0.05 | 0.99 ± 0.14 | 0.43 ± 0.15 | ||
| Adult 10 D | D4 APF | 3.50 ± 0.16 | 1.50 ± 0.08 | 1.18 ± 0.04 | 1.06 ± 0.12 | 0.48 ± 0.12 |
Myofibril parameters were measured from deconvolved images. ±, SEM; Dz, diameter measured at the Z-discs; DH, diamerter measured at the H zone; n, 15 myofibrils measured from three separate experiments for 2–3 day adult flies unless otherwise indicated.
IFM myofibrils from both heat-shocked and nontreated hs-43 kD spdo lines were also stained with anti-SPDO antibodies (Fig. 4, middle panels). Deconvolution microscopy showed that SPDO localized to the pointed ends (▵) of the thin filaments located in the core and periphery of myofibrils from both heat-shocked and control hs-43 kD spdo flies. This indicates that both the short and long populations of thin filaments are capped.
To determine whether the changes in actin filament length observed by phallacidin staining were also reflected in other thin filament components, isolated myofibrils from hs-43 kD spdo flies heat-shocked on D4 APF were stained with antibodies specific for tropomyosin (TM) (van Straaten et al., 1999). In control myofibrils from adult flies, TM staining was uniform along the thin filaments, but was excluded from the area of the Z-discs (Fig. 4 C, top), as previously observed (van Straaten et al., 1999). SPDO staining colocalized with TM at the region of the pointed ends (Fig. 4 C, merge, bottom). The pattern of TM staining (Fig. 4 D) was consistent with the phallacidin staining pattern in myofibrils from heat-shocked hs-spdo flies (Fig. 4 B), indicating that TM and SPDO are associated with both the shorter and longer filaments.
43-kD SPDO induction prevented thin filament elongation irreversibly at all stages of myofibril assembly
During IFM myofibril assembly, short thin filaments that are assembled by D2 APF elongate incrementally before reaching a final length shortly after eclosion (see Fig. 9) (Reedy and Beall, 1993). We postulated that overexpression of the 43-kD SPDO isoform during IFM development resulted in a uniformly shorter set of thin filaments in the core of the myofibril (Fig. 4, B and D) by blocking elongation of preexisting thin filaments. Therefore, the dimensions of the affected adult myofibril core (Fig. 5 C) should correspond to the thin filament length and myofibril diameter at the time of 43-kD SPDO induction during pupation.
Figure 5.

Transient induction of 43-kD SPDO during myofibril assembly irreversibly prevents thin filament elongation. Thin filament lengths (A) and myofibril diameters (B) were measured as indicated in the diagram (C) using Deltavision software, from deconvolved images of Bodipy- phallacidin stained IFM myofibrils isolated from either pupal flies (1 PM D4 APF), D1 adult hs-43 kD spdo flies heat-shocked at 12 PM D4 APF, or untreated D2 adult flies. Error bars, SEM. (C) Schematic diagram of longitudinal and cross sections of an IFM myofibil from an adult hs-43 kD spdo fly that was heat shocked during pupation. Light gray shading indicates Bodipy-phallacidin stained “core” of the myofibril with shorter thin filaments and dark gray shading indicates Bodipy-phallacidin stained “peripheral” thin filaments.
To test this hypothesis, we measured thin filament lengths of myofibrils isolated from pupal hs-43 kD spdo flies fixed immediately after the heat-shock treatment, and compared them to thin filament lengths of myofibrils from adult flies subjected to the same heat shock regime. The lengths of the core adult IFM thin filaments (Fig. 5 C, light gray) were 1.28 μm and corresponded closely to the pupal IFM thin filament lengths of 1.25 μm (Fig. 5 A and Table I). In contrast, the lengths of the peripheral IFM thin filaments (Fig. 5 C, dark gray) were similar to filament lengths in control flies (Fig. 5 A and Table I). The 0.62 μm diameter of the core in myofibrils from adult hs-43 kD spdo flies (heat-shocked D4 APF) corresponded to the 0.63-μm diameter of myofibrils isolated from control pupae (Fig. 5 B and Table I). Further, the core of IFM thin filaments from heat-shocked flies did not change any further in length or diameter during adulthood. The parameters measured from D10 adult hs-43 kD spdo flies (heat-shocked on D4 APF) were similar to those obtained from D1 adult hs-43 kD spdo flies treated to the same regime during pupation (Table I). These results support the hypothesis that transient overexpression of 43-kD SPDO during myofibril assembly irreversibly prevented elongation of preexisting thin filaments during IFM development.
As a further test of this hypothesis, we postulated that induction of expression of 43-kD SPDO at an earlier stage of myofibril assembly should result in a smaller core of even shorter thin filaments. Indeed, adult myofibrils from hs-43 kD spdo flies heat-shocked on D3 APF had a core of 1.06-μm thin filaments (Fig. 6 A), considerably shorter than the 1.28-μm thin filaments present in the IFM myofibril cores of hs-43 kD spdo flies heat shocked on D4 APF (Table I). Further, the measured diameters of the cores of short thin filaments from myofibrils from hs-43 kD spdo flies heat shocked on D3 APF were about half the diameter of the affected cores measured from myofibrils in which 43-kD SPDO expression was induced on D4 APF (Table I). The volume of the core of short filaments as a percentage of the total volume was ∼5% for flies heat shocked on D3 APF, versus ∼24% for D4 APF (Table I). Because the majority of adult hs-43 kD spdo flies that had been heat shocked on D3 APF could still fly (92%; Fig. 3), IFM muscle function was not impaired when 5% or less of the thin filaments of the myofibril core were shorter than controls.
Figure 6.
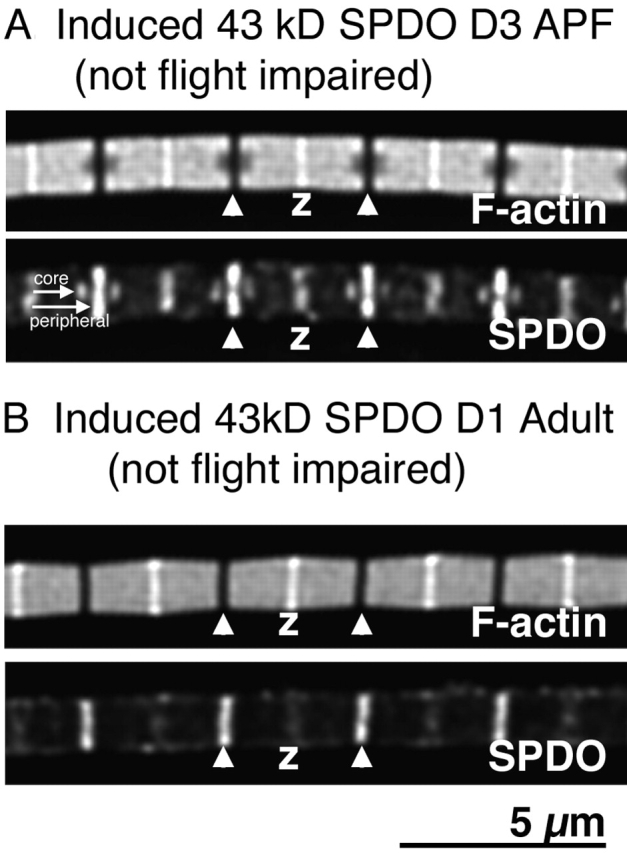
Overexpression of 43-kD SPDO during early- or late-myofibril assembly affects myofibril structure without affecting muscle function. Myofibrils from D2 adult hs-43 kD spdo flies (A) heat shocked at 12 PM D3 APF or (B) 12 PM D1 after eclosion. (Top) Bodipy-phallacidin staining for F-actin; (Bottom) anti-SPDO staining. Short white arrow, short “core” thin filaments; long white arrow, longer “peripheral” thin filaments; white arrowheads, pointed ends; Z, Z-disc. Bar, 5 μm.
We also investigated whether assembly of peripheral thin filaments in newly eclosed adults was sensitive to overexpression of the 43-kD SPDO isoform. However, transient 43-kD overexpression in newly eclosed D1 adults did not result in any discernable core of shorter thin filaments in myofibrils from D3 adult flies (Fig. 6 B). However, phallacidin-stained myofibrils seemed to have a “bow tie” appearance. In control flies, the myofibril diameter measured at the H-zone (DH) was 98% of the myofibril diameter measured at the Z-disc (DZ) (Table I). In contrast, DH of D3 adult hs-43 kD spdo flies heat-shocked as newly eclosed D1 adults was only 82% of the DZ (Table I). The ∼20% reduction in diameter at the H zone is consistent with the possibility that that there were more shorter thin filaments near the Z line versus the H zone. Therefore, the presence of excess 43-kD SPDO during late myofibril assembly most likely interfered with thin filament elongation of newly added peripheral thin filaments at the Z-disc. This result is also consistent with observed posteclosion growth of thoracic muscle of other insects (Houlihan and Brekenridge, 1981). Taken together, these results indicate that transient overexpression of the 43-kD SPDO isoform was capable of preventing thin filament elongation at any stage of myofibril assembly.
Irreversible association of the 43-kD SPDO with IFM thin filaments prevented IFM thin filament elongation
Because the 43-kD SPDO was ordinarily associated with IFM thin filaments during thin filament elongation (Fig. 2 B), we took a biochemical approach to understand how transient overexpression of 43-kD SPDO could irreversibly block thin filament elongation. Immediately after heat-shock, the 43-kD SPDO isoform was present in both the supernatant and myofibril pellet fractions of Triton X-100 extracted pupal hs-43 kD spdo flies that were heat shocked on D4 APF (Fig. 7 A, right). After the flies had been allowed to develop, the 43-kD SPDO isoform was present only in the myofibril pellet fraction of hs-43 kD spdo adult flies heat shocked during pupation, and was not substantially replaced by the 45-kD isoform in adult flies (Fig. 7 A, right). Because the levels of the 43-kD SPDO isoform in homogenates of whole pupae and dissected thoraces returned to normal within 20 h in timed experiments normalized for glycerol-3-phosphate dehydrogenase levels (unpublished data), it appeared that only the 43-kD SPDO that became associated with the myofibril pellet fraction was stable. Further, Coomassie blue staining of samples showed that induction of 43-kD SPDO expression or heat-shock treatment per se did not grossly alter sarcomeric protein levels derived from Triton X-100 extracted thoraces (unpublished data).
To determine whether overexpressed 43-kD SPDO was irreversibly associated with thoracic IFM myofibrils, IFM was dissected from D10 adult flies. Consistent with results obtained with whole thoraces (Fig. 7 A), only the adult hs-43 kD spdo flies heat shocked during pupation had a significant amount of the 43-kD SPDO isoform associated with the myofibril pellet (Fig. 7 B). The levels of the 43-kD SPDO in IFM myofibrils from heat-shocked hs-43 kD spdo flies appeared to be nearly equivalent to the levels of the 45-kD SPDO isoform in myofibrils from control flies. Thus, unlike endogenous 43-kD SPDO, association of the induced 43-kD isoform with developing IFM myofibrils was irreversible and prevented a substantial portion of the 45-kD SPDO isoform from associating with adult IFM myofibrils. These results also indicate that the numbers of capped thin filament pointed ends in myofibrils from control and SPDO-overexpressing flies were roughly equivalent.
The effect of 43kD-SPDO overexpression on IFM myofibril morphology and ultrastructure
We investigated whether the restriction of thin filament lengths in the core of the myofibril led to alterations in sarcomere length or myofibril diameter. Although there was a considerable amount of variation between individual flies, sarcomere lengths and myofibril diameters from myofibrils of heat-treated hs-43 kD spdo flies were similar to these same parameters measured in control flies (Table I). Thus, the presence of short core thin filaments did not appear to grossly impede other aspects of myofibril assembly.
To further investigate the consequences of transient 43-kD SPDO overexpression during myofibrillogenesis, the ultrastructure of heat-treated hs-43 kD spdo flies and control yw flies was analyzed (Fig. 8). In longitudinal sections, sarcomeres were delineated by slightly crooked Z-discs (Fig. 8 A). The most striking feature was the penetration and accumulation of electron dense particles (most likely glycogen) in the core of the myofibril, adjacent to the M line (Fig. 8 A, bracket). This result was consistent with the lack of phallacidin staining causing a “notch-like” appearance in fluorescent images (Figs. 4, B and D, and 6 A). Surprisingly, many myofibrils were also missing a small population of thick filaments (Fig. 8, A and F, arrows).
In cross-section, individual myofibrils from hs-43 kD spdo flies (heat shocked on D4 APF), exhibited a variety of defects that differed in severity (Fig. 8 B). This diversity appeared to depend on the location and orientation of the cross section. In the affected myofibrils, thin filaments were not visible in the core (Fig. 8 C). Presumably, the absence of distal portions of thin filaments permitted diffusion of electron dense granules around the core thick filaments in the middle of the sarcomere. Individual myofibrils from heat-treated hs-43 kD spdo flies varied greatly in diameter, and we observed examples of normal size myofibril cross-sections (∼1.1 μm; Fig. 8 F) and small myofibrils (∼0.75 μm; Fig. 8 C) measured from different individuals. Further, aberrant thick filament packing was observed, with thick filaments either abnormally packed or loosely spaced (Fig. 8 F, box). These ultrastructural defects resulted from 43-kD SPDO overexpression because heat-shock treatment alone did not perturb myofibril organization in yw flies (Fig. 8, D and E).
To address whether the myofibrils initially assembled correctly and then degenerated in response to exertion during flight, we inspected the ultrastructure of late-pupal IFM. Because the flies have not yet eclosed from the pupal case, these muscles have not been used to attempt flight. It was clear that myofibrils did not assemble correctly in the IFM of heat-treated hs-43 kD spdo lines because similar phenotypes to adult IFM were observed in the pupal IFM (unpublished data).
In comparison to many other flightless mutant flies (for review see Bernstein et al., 1993), the overall myofibrillar organization of heat treated hs-43 kD spdo flies was relatively normal. The most dramatic phenotype was that the distal regions of the thin filaments near the M line were replaced by electron dense granules in the core of the myofibrils.
Discussion
This study provides several novel insights into the process of myofibril assembly and thin filament length specification during muscle development. Our results show that transient overexpression of 43-kD SPDO during myofibril assembly leads to irreversible binding of 43-kD SPDO to the pointed ends of IFM thin filaments. Preventing uncapping at the pointed ends permanently fixes the length of the thin filaments that were preassembled at the developmental period in which 43-kD SPDO was overexpressed (Fig. 9). These observations lead to the following conclusions. First, thin filaments elongate from their pointed ends during de novo myofibril assembly in muscle development. Second, transient increases in SPDO levels convert SPDO from a dynamic pointed end cap into a permanent cap, thus irreversibly blocking elongation and leading to misspecification of final thin filament lengths. This implies that developmental regulation of pointed end capping by SPDO is critical for specification of final thin filament lengths in mature myofibrils. Third, thin filament lengths in Drosophila IFM are not predetermined to a particular length; multiple thin filament lengths are possible. Fourth, prevention of thin filament elongation during myofibril assembly disrupts myofibril structure and affects muscle performance.
In contrast to the rapid polymerization that takes place at the barbed filament ends in the lamellipodia of crawling cells (for review see Pollard et al., 2000), control of actin filament elongation at the pointed end may be a general mechanism for slow assembly (i.e., days) of specialized actin structures (DeRosier and Tilney, 2000). This process may be critical to ensure that actin filaments extend to and are strictly maintained at a given length.
How does SPDO overexpression block thin filament elongation irreversibly?
The ability of excess 43-kD SPDO to irreversibly block thin filament elongation was surprising, based on recent work in which Tmod was shown to rapidly bind to and dissociate from thin filament pointed ends in cardiac myocytes (Littlefield et al., 2001). We had expected that transient overexpression of 43-kD SPDO would merely delay thin filament elongation rather than lead to a complete block. However, excess 43-kD SPDO did not transiently suppress actin monomer addition because the abnormally short core thin filaments never caught up in length, well after SPDO levels had returned to normal. Indeed, the core thin filaments did not increase in length over a 10-d period (Table I). Further, the overexpressed 43-kD SPDO isoform remained irreversibly associated with thin filament pointed ends, as the overexpressed 43-kD SPDO isoform was not replaced by the 45-kD SPDO isoform after eclosion (Fig. 7).
In addition, prevention of thin filament elongation cannot be explained by ectopic or misexpression of the 43-kD SPDO isoform. IFM thin filaments are normally capped by the 43-kD isoform in wild-type flies throughout myofibril assembly during pupation and in early adulthood. The 45-kD isoform does not assemble into myofibrils until many hours after eclosion, well after the majority of myofibril assembly is completed (Fig. 2 B) (Reedy and Beall, 1993). This isoform switch is similar to a Tmod isoform switch in the development of chicken skeletal muscle in which E-Tmod (Tmod1), the predominant isoform in embryonic pectoralis major muscle, is replaced by Sk-Tmod (Tmod4) after hatching (Almenar-Queralt et al., 1999a, 1999b).
Because the endogenous 43-kD SPDO isoform is present throughout myofibril assembly in Drosophila IFM, it must function as a dynamic cap to allow continuous elongation from the pointed ends during normal myofibril assembly. Thus, a transient increase in the level of 43-kD SPDO appears to convert SPDO into a permanent cap, irreversibly blocking elongation. These results may indicate that the dynamics of Tmod capping are more highly regulated in developing IFM as compared with cultured cardiac myocytes (Littlefield et al., 2001). We propose that actin filaments are only able to elongate during certain discrete and periodic steps of myofibril assembly in the IFM. Thin filament elongation may result upon a signal that encourages 43-kD SPDO uncapping and subsequent actin monomer addition to the pointed ends of thin filaments. Providing excess 43-kD SPDO could override this regulation, leading to a permanent pointed end cap, thus resulting in an undersized thin filament.
How could pointed end capping be regulated? The signal for pointed end capping proteins to uncap and permit thin filaments to elongate may be initiated specifically near the M line by concurrent changes in the thick filament length. The thick filament may regulate and coordinate thin filament elongation via actomyosin interactions. For example, actomyosin binding near the pointed ends of thin filaments may cause movement of TM on the thin filament leading to changes in the relative position of TM with respect to actin at the pointed end. This conformational change could alter 43-kD SPDO binding sites and cause the thin filament to uncap, as proposed by Littlefield and Fowler (1998). One way to test a requirement for actomyosin interactions in thin filament uncapping would be to determine whether transgenic flies that express myosin isoforms lacking the actin binding domain (Cripps et al., 1999) can suppress the 43-kD SPDO overexpression phenotype.
What specifies the final length after thin filament elongation is terminated?
Our data and that of others do not support the idea that a ruler protein, such as nebulin, acts as a molecular template to dictate changes in actin thin filament lengths during IFM myofibril assembly, as has been proposed for vertebrate skeletal muscle (for review see Littlefield and Fowler, 1998). First, like vertebrate cardiac muscle, no nebulin homologue has been found in Drosophila. Second, the gradual and continuous changes in sarcomere and thin filament lengths during myofibril assembly are inconsistent with a ruler mechanism (Reedy and Beall, 1993). Third, our data is not consistent with a ruler protein to specify length, as transient overexpression of 43-kD SPDO has irreversible effects on thin filament length specification. Finally, if rulers were to dictate thin filament length, then multiple stretch-activated rulers would be necessary to act as templates during myofibril assembly.
We favor the idea that thick filament length is a key determinant and coordinator of thin filament lengths (as discussed in the previous section). In mutations in which the thick filament length is affected, such as in IFM (2)2 heterozygotes (Beall et al., 1989) or flightin homozygotes (Reedy et al., 2000), thin filament lengths are changed correspondingly. For example, in flightin mutants, the IFM sarcomeres and thick and thin filaments in pupal IFM are 25–30% longer than in wild-type sarcomeres, whereas the sarcomeric structure is otherwise normal (Reedy et al., 2000).
Control of thick filament length also appears to dictate the length of a sarcomere. In flightin nulls, sarcomeres are longer than wild-type sarcomeres, reflecting the longer lengths of thick filaments (Reedy et al., 2000). In addition, sarcomere length is also sensitive to the stability of the thin filament. Sarcomeres are shorter in actin and TM mutants (Kreuz et al., 1996; Beall et al., 1989). In contrast, sarcomere length was not affected by excess 43-kD SPDO (Table I). However, the lack of effect on sarcomere length when core thin filaments are abnormally short may indicate that only a fraction of full-length thin filaments are needed to provide the stability required for longitudinal sarcomeric growth induced by stretch.
However, thick filaments do not assemble correctly without some type of “feedback signaling” from thin filaments. Although thick filaments can assemble in the absence of thin filaments, they are extremely disorganized (for review see Bernstein et al., 1993). Our results also show that the presence of abnormally short thin filaments affects the packing and lattice of thick filaments (Fig. 8, A and F, arrows). This is consistent with other IFM studies indicating that actomyosin interactions are required to regulate and constrain myofilament lengths (Reedy and Beall, 1993). In summary, it is likely that thin and thick filaments support the growth of one another. Perturbations in either type of myofilament will affect the stability and growth of the others proportionately, as well as influence the length of the sarcomere.
Effects of Tmod overexpression on thin filament length in vertebrate cardiac cells
It is interesting to reinterpret previous overexpression data in vertebrates based on the phenotype reported here. Overexpression of GFP-Tmod by transient transfection in cultured embryonic chick cardiomyocytes prevents rhodamine-actin incorporation at the pointed ends, and results in thin filament lengths that are uniformly shorter by ∼10% (Littlefield et al., 2001). Based on the dynamic capping properties of Tmod, we had interpreted these results to mean that filaments were shorter due to a net loss of actin monomers (i.e., filament shrinkage). However, the reduction in length may have also resulted if thin filaments failed to elongate during myofibril assembly as described here. Indeed, Rhee et al. (1994) have proposed that mature myofibrils develop from “mini-sarcomeres” that contain short actin thin filaments and elongate over time in cultured cardiomyocytes.
Overexpression of Tmod using expression vectors that drive sustained, high levels of expression caused severe myofibril perturbations and degeneration in TOT mouse hearts (Sussman et al., 1998b). However, our observations showing that increased SPDO levels perturbed thin filament assembly during Drosophila muscle development suggests instead that overexpression of Tmod may have disrupted cardiac thin filament assembly during heart development. We propose that a primary defect of misassembled thin filaments resulted in a decrease in heart contractile function, which then led to myofibril degeneration and the pathogenic state of dilated cardiomyopathy reported by Sussman et al. (1998b).
The dramatic elongation of actin filaments during myofibril assembly in Drosophila IFM development may be used as a model for understanding the events that occur during myofibril assembly in vertebrate cardiac muscle. In particular, the thin filaments of both systems appear to elongate during development without the presence of a ruler protein. Indeed, during early myofibrillogenesis in the chick heart sarcomeres grow from ∼1.4 to 1.8–2.5 μm (Gregorio and Antin, 2000), indicating that in cardiac tissue, thin filaments may elongate correspondingly during their development similar to IFM. Preliminary measurements from chick precardiac mesoderm explants also suggest that thin filament lengths increase during vertebrate heart muscle development (D. Rudy and C. Gregorio, personal communication). Our genetic system provides a novel developmental window in which discrete requirements and events that occur during myofibril assembly can be studied in vivo.
Materials and methods
Fly stocks
Gene and chromosome symbols are as described by Lindsley and Zimm (1992). The transgenic line hs-43 kD spdo was supplied by Dr. Hugo Bellen (Baylor University, Houston Texas). Because the P element vector construct (P[hs-43 kD spdo]) was originally injected into yw flies, the same genotype was used for all the control experiments. Flies were grown routinely at 25°C (± 1°C) as described in Cripps et al. (1994). Flies were staged for heat-shock treatment by allowing caged flies to lay eggs on grape agar plates for 2–3 h. 50 eggs were transferred with a camel hair paintbrush onto small strips of paper towels and placed in fresh vials of food. Pupae formed between 6.5–7.5 d after egg collection and adults eclosed after 110–125 h of pupation. Transient heat-shock treatments were performed by placing vials containing staged pupae in a 37°C incubator for 1 h followed by culture at 25°C.
Flight tests
Adult flies aged 2–3 d after eclosion were collected and tested for flight as described (Cripps et al., 1994). Briefly, flies were released inside a clear plastic box that was illuminated from the top. Flies were scored as flyers or nonflyers. Flyers were categorized by their ability to fly upward toward the light source or achieve horizontal flight. Nonflyers flew downward or simply fell down.
Immunofluorescence microscopy
IFM myofibrils were isolated from dissected thoraces that were homogenized in a relaxing buffer (Ringers + EGTA) and allowed to adhere to coverslips for 20 min, and then fixed with 3.7% formaldehyde (Sigma-Aldrich) in relaxing buffer for 10 min and washed with PBS (Fowler et al., 1993). The myofibrils were extracted with 0.2% Triton X-100 in PBS and blocked with 1% normal goat serum in PBS. Myofibrils were double stained with 1/200 of Bodipy-phallicidin (Molecular Probes) and 1:200 of anti-SPDO (polyclonal mouse antiserum provided by Dr. Chris Doe, University of Oregon, Eugene, OR), or anti-tropomyosin (1:50) and anti–α-actinin (1:50) (rat monoclonal antisera to Lethocerus proteins provided by Dr. Belinda Bullard, European Molecular Biology Laboratory, Heidleberg, Germany) (Lakey et al., 1990). Dilutions of secondary antibodies were rabbit anti–rat IgG-TRITC (Sigma-Aldrich) (1:50) and donkey anti–mouse IgG-Rhodamine red (1:200) (Accurate Chemical and Scientific Corp.). Stained myofibrils were imaged by deconvolution microscopy using a 60× (N.A. 1.40) Plan Apochromat objective lens with a 1.5× optivar on an Olympus IX-70 inverted microscope. Data (1050 × 1050 pixels) from 100-nm optical sections was acquired by a Photometrics CCD camera and deconvolved using DeltaVision software version 2.1. Digital images were also processed for presentation using Adobe Photoshop 5.5.
Immunoblots
Samples were prepared from homogenates of dissected tissues or from whole flies in 2× SDS sample buffer and were run out on 12% SDS-PAGE gels to separate the 43- and 45-kD isoforms of SPDO. In some experiments, dissected thoraces were extracted with Triton X-100 before solubilization in SDS (Cripps and Sparrow, 1992). SDS-PAGE was performed as described by Laemmli (1970), using a Bio-Rad Mini-Protean II minigel format (Fig. 1 B) or a large format Hoeffer vertical gel apparatus (Figs. 1 C, 2 B, and 7). Gels were transferred to nitrocellulose (Schelicher and Schuell) and transfer of proteins to nitrocellulose was performed as described by the manufacturers. Immunoblots were blocked with 5% nonfat milk in TBS (with 0.1% Tween-20) and incubated with a primary antibody at room temperature for 1 h. The mouse anti-SPDO antibody was diluted to (1/5,000). Primary antibodies were detected by secondary antibodies conjugated to horseradish peroxidase (Sigma-Aldrich) and were developed by standard chemiluminescence detection methods.
Transmission electron microscopy
Control yw and hs-43 kD spdo flies were heat shocked for 1 h on D4 of pupation and collected just before eclosion or as 1–3 d old adults. Dissected thoraces were prepared for electron microscopy as described in Cripps et al. (1994). Gold thin sections were visualized on a Philips 410 (San Diego State University EM Facility, San Diego, CA) or on a Philips 210 (The Scripps Research Institute Microscopy Facility, La Jolla, CA) at 80 kV.
Acknowledgments
We wish to thank members of the Fowler lab for critical comments, especially Robert Fischer for help with initial deconvolution microscopy studies, and Angels Almenar-Queralt and Ryan Littlefield for help with Adobe Photoshop.
This work was supported by an American Heart Association-Western Affiliate postdoctoral fellowship to M. Mardahl-Dumesnil, and grants to V.M. Fowler from the National Institutes of Health (GM34225), and the Human Frontiers in Science Program (RG0242).
Footnotes
Abbreviations used in this paper: APF, after pupal formation; DH, H zone; DZ, Z-disc; IFM, indirect flight muscle; SPDO, sanpodo; TM, tropomyosin; Tmod, tropomodulin; TOT, Tmod-overexpressing transgenic.
References
- Almenar-Queralt, A., C.C. Gregorio, and V.M. Fowler. 1999. a. Tropomodulin assembles early in myofibrillogenesis in chick skeletal muscle: Evidence that thin filaments rearrange to form striated myofibrils. J. Cell Sci. 112:1112–1123. [DOI] [PubMed] [Google Scholar]
- Almenar-Queralt, A., A. Lee, C.A. Conley, L. Ribas de Pouplana, and V.M. Fowler. 1999. b. Identification of a novel tropomodulin isoform, skeletal tropomodulin, that caps actin filament pointed ends in fast skeletal muscle. J. Biol Chem. 274:28466–28475 (erratum published 275[17]:13164). [DOI] [PubMed] [Google Scholar]
- Ashburner, M. 1989. Drosophila, a Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Ball, E., C.C. Karlik, C.J. Beall, D.L. Saville, J.C. Sparrow, B. Bullard, and E.A. Fyrberg. 1987. Arthrin, a myofibrillar protein of insect flight muscle, is an actin-ubiquitin conjugate. Cell. 51:221–228. [DOI] [PubMed] [Google Scholar]
- Beall, C.J., M.A. Sepanski, and E.A. Fyrberg. 1989. Genetic dissection of Drosophila myofibril formation: effects of actin and myosin heavy chain null alleles. Genes Dev. 3:131–140. [DOI] [PubMed] [Google Scholar]
- Bernstein, S.I., P.T. O'Donnell, and R.M. Cripps. 1993. Molecular genetic analysis of muscle development, structure, and function in Drosophila. Int. Rev. Cytol. 143:63–152. [DOI] [PubMed] [Google Scholar]
- Chu, X., D. Thompson, L.J. Yee, and L.A. Sung. 2000. Genomic organization of mouse and human erythrocyte tropomodulin genes encoding the pointed end capping protein for the actin filaments. Gene. 256:271–281. [DOI] [PubMed] [Google Scholar]
- Cripps, R.M., and J.C. Sparrow. 1992. Polymorphism in a Drosophila indirect flight muscle-specific tropomyosin isozyme does not affect flight ability. Biochem. Genet. 30:159–168. [DOI] [PubMed] [Google Scholar]
- Cripps, R.M., K.D. Becker, M. Mardahl, W.A. Kronert, D. Hodges, and S.I. Bernstein. 1994. Transformation of Drosophila melanogaster with the wild-type myosin heavy-chain gene: rescue of mutant phenotypes and analysis of defects caused by overexpression. J. Cell Biol. 126:689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cripps, R.M., J.A. Suggs, and S.I. Bernstein. 1999. Assembly of thick filaments and myofibrils occurs in the absence of the myosin head. EMBO J. 18:1793–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosier, D.J., and L.G. Tilney. 2000. F-actin bundles are derivatives of microvilli: what does this tell us about how bundles might form? J. Cell Biol. 148:1–6. [PMC free article] [PubMed] [Google Scholar]
- Dye, C.A., J.K. Lee, R.C. Atkinson, R. Brewster, P.L. Han, and H.J. Bellen. 1998. The Drosophila sanpodo gene controls sibling cell fate and encodes a tropomodulin homolog, an actin/tropomyosin-associated protein. Development. 125:1845–1856. [DOI] [PubMed] [Google Scholar]
- Fowler, V.M. 1996. Regulation of actin filament length in erythrocytes and striated muscle. Curr. Opin. Cell Biol. 8:86–96. [DOI] [PubMed] [Google Scholar]
- Fowler, V.M. 1997. Capping actin filament growth: tropomodulin in muscle and nonmuscle cells. Soc. Gen. Physiol. Ser. 52:79–89. [PubMed] [Google Scholar]
- Fowler, V.M., M.A. Sussmann, P.G. Miller, B.E. Flucher, and M.P. Daniels. 1993. Tropomodulin is associated with the free (pointed) ends of the thin filaments in rat skeletal muscle. J. Cell Biol. 120:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio, C.C., and P.B. Antin. 2000. To the heart of myofibril assembly. Trends Cell Biol. 10:355–362. [DOI] [PubMed] [Google Scholar]
- Gregorio, C.C., A. Weber, M. Bondad, C.R. Pennise, and V.M. Fowler. 1995. Requirement of pointed-end capping by tropomodulin to maintain actin filament length in embryonic chick cardiac myocytes. Nature. 377:83–86. [DOI] [PubMed] [Google Scholar]
- Houlihan, D., and L. Brekenridge. 1981. Stretch-induced growth of blowfly muscle. J. Insect Physiol. 27:521–525. [Google Scholar]
- Kreuz, A.J., A. Simcox, and D. Maughan. 1996. Alterations in flight muscle ultrastructure and function in Drosophila tropomyosin mutants. J. Cell Biol. 135:673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227:680–685. [DOI] [PubMed] [Google Scholar]
- Lakey, A., C. Ferguson, S. Labeit, M. Reedy, A. Larkins, G. Butcher, K. Leonard, and B. Bullard. 1990. Identification and localization of high molecular weight proteins in insect flight and leg muscle. EMBO J. 9:3459–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley, D.L., and G. Zimm. 1992. The Genome of Drosophila melanogaster. Academic Press, San Diego, CA.
- Littlefield, R., and V.M. Fowler. 1998. Defining actin filament length in striated muscle: rulers and caps or dynamic stability? Annu. Rev. Cell Dev. Biol. 14:487–525. [DOI] [PubMed] [Google Scholar]
- Littlefield, R., A. Almenar-Queralt, and V.M. Fowler. 2001. Actin dynamics at pointed ends regulates thin filament length in striated muscle. Nat. Cell Biol. 3:544–551. [DOI] [PubMed] [Google Scholar]
- Papa, I., C. Astier, O. Kwiatek, F. Raynaud, C. Bonnal, M.C. Lebart, C. Roustan, and Y. Benyamin. 1999. Alpha actinin-CapZ, an anchoring complex for thin filaments in Z-line. J. Muscle Res. Cell Motil. 20:187–197. [DOI] [PubMed] [Google Scholar]
- Park, M., L.E. Yaich, and R. Bodmer. 1998. Mesodermal cell fate decisions in Drosophila are under the control of the lineage genes numb, Notch, and sanpodo. Mech. Dev. 75:117–126. [DOI] [PubMed] [Google Scholar]
- Pollard, T.D., L. Blanchoin, and R.D. Mullins. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29:545–576. [DOI] [PubMed] [Google Scholar]
- Reedy, M.C., and C. Beall. 1993. Ultrastructure of developing flight muscle in Drosophila. I. Assembly of myofibrils. Dev. Biol. 160:443–465. [DOI] [PubMed] [Google Scholar]
- Reedy, M.C., B. Bullard, and J.O. Vigoreaux. 2000. Flightin is essential for thick filament assembly and sarcomere stability in Drosophila flight muscles. J. Cell Biol. 151:1483–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, D., J.M. Sanger, and J.W. Sanger. 1994. The premyofibril: evidence for its role in myofibrillogenesis. Cell Motil. Cytoskeleton. 28:1–24. [DOI] [PubMed] [Google Scholar]
- Salzberg, A., D. D'Evelyn, K.L. Schulze, J.K. Lee, D. Strumpf, L. Tsai, and H.J. Bellen. 1994. Mutations affecting the pattern of the PNS in Drosophila reveal novel aspects of neuronal development. Neuron. 13:269–287. [DOI] [PubMed] [Google Scholar]
- Schafer, D.A., C. Hug, and J.A. Cooper. 1995. Inhibition of CapZ during myofibrillogenesis alters assembly of actin filaments. J. Cell Biol. 128:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeath, J.B., and C.Q. Doe. 1998. Sanpodo and Notch act in opposition to Numb to distinguish sibling neuron fates in the Drosophila CNS. Development. 125:1857–1865. [DOI] [PubMed] [Google Scholar]
- Sussman, M.A., S. Baque, C.S. Uhm, M.P. Daniels, R.L. Price, D. Simpson, L. Terracio, and L. Kedes. 1998. a. Altered expression of tropomodulin in cardiomyocytes disrupts the sarcomeric structure of myofibrils. Circ. Res. 82:94–105. [DOI] [PubMed] [Google Scholar]
- Sussman, M.A., S. Welch, N. Cambon, R. Klevitsky, T.E. Hewett, R. Price, S.A. Witt, and T.R. Kimball. 1998. b. Myofibril degeneration caused by tropomodulin overexpression leads to dilated cardiomyopathy in juvenile mice. J. Clin. Invest. 101:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Straaten, M., D. Goulding, B. Kolmerer, S. Labeit, J. Clayton, K. Leonard, and B. Bullard. 1999. Association of kettin with actin in the Z-disc of insect flight muscle. J. Mol. Biol. 285:1549–1562. [DOI] [PubMed] [Google Scholar]