

Abstract
Inhibitory Smads (I-Smads) repress signaling by cytokines of the transforming growth factor-β (TGF-β) superfamily. I-Smads have conserved carboxy-terminal Mad homology 2 (MH2) domains, whereas the amino acid sequences of their amino-terminal regions (N domains) are highly divergent from those of other Smads. Of the two different I-Smads in mammals, Smad7 inhibited signaling by both TGF-β and bone morphogenetic proteins (BMPs), whereas Smad6 was less effective in inhibiting TGF-β signaling. Analyses using deletion mutants and chimeras of Smad6 and Smad7 revealed that the MH2 domains were responsible for the inhibition of both TGF-β and BMP signaling by I-Smads, but the isolated MH2 domains of Smad6 and Smad7 were less potent than the full-length Smad7 in inhibiting TGF-β signaling. The N domains of I-Smads determined the subcellular localization of these molecules. Chimeras containing the N domain of Smad7 interacted with the TGF-β type I receptor (TβR-I) more efficiently, and were more potent in repressing TGF-β signaling, than those containing the N domain of Smad6. The isolated N domain of Smad7 physically interacted with the MH2 domain of Smad7, and enhanced the inhibitory activity of the latter through facilitating interaction with TGF-β receptors. The N domain of Smad7 thus plays an important role in the specific inhibition of TGF-β signaling.
Keywords: TGF-β; bone morphogenetic protein; Smad; receptor; signaling
Introduction
Members of the transforming growth factor-β (TGF-β)* superfamily are multifunctional cytokines that regulate growth, differentiation, motility, adhesion, and morphogenesis of various types of cells (Roberts and Sporn, 1990; Massagué, 1998). The TGF-β superfamily includes TGF-βs, activins, bone morphogenetic proteins (BMPs), growth/differentiation factors, and Müllerian inhibiting substance. Cytokines of the TGF-β superfamily bind to two different types of serine/threonine kinase receptors termed types II and I, and transmit intracellular signals by Smad proteins (Heldin et al., 1997; Massagué and Wotton, 2000; Miyazono et al., 2000). Of the three different subtypes of Smads, receptor-regulated Smads (R-Smads) are directly phosphorylated by type I serine/threonine kinase receptors, and form oligomeric complexes with common mediator Smads (Co-Smads). The R-Smad–Co-Smad complexes then translocate into the nucleus, where they regulate the transcription of target genes.
The third class of Smads comprises the inhibitory Smads (I-Smads), which include Smad6 and Smad7 in mammals and Daughters against Decapentaplegic in Drosophila. Expression of I-Smads is induced by various stimuli, e.g., TGF-β and BMPs (Miyazono, 2000). Smad2 and Smad3, activated by TGF-β, and Smad1 and Smad5, activated by BMPs, bind to the promoter regions and induce the transcription of Smad7 and Smad6 genes, respectively (Nagarajan et al., 1999; Denissova et al., 2000; Ishida et al., 2000). Daughters against Decapentaplegic is also induced by Decapentaplegic signaling (Tsuneizumi et al., 1997). Thus, I-Smads act as components in negative feedback regulation in the Smad signaling pathways. I-Smads stably bind to activated type I receptors, and compete with R-Smads for receptor activation (Hayashi et al., 1997; Imamura et al., 1997; Nakao et al., 1997; Souchelnytskyi et al., 1998). In addition, Smad7, and possibly Smad6, recruit E3 ubiquitin ligases Smurf1 and Smurf2 to type I receptors, leading to ubiquitin-dependent degradation of the TGF-β receptor complexes (Kavsak et al., 2000; Ebisawa et al., 2001). Smad6 has also been reported to form a complex with Smad1, and to compete with Smad4 for oligomer formation (Hata et al., 1998). Moreover, Smad6 has been shown to bind certain transcription factors and repress transcription in the nucleus (Bai et al., 2000).
R-Smads and Co-Smads have highly conserved amino- and carboxy-terminal regions termed Mad homology 1 (MH1) and MH2 domains, respectively, that are linked by linker regions of variable length and sequence. I-Smads have conserved MH2 domains, but their amino-terminal domains (N domains) are highly divergent from the MH1 domains and linker regions of other Smads. Moreover, amino acid sequences of the N domains are only partially conserved between the I-Smads (36.7% between Smad6 and Smad7). Notably, Smad6 and Smad7 have been identified in Xenopus, but their N domains are only 51.3 and 67.4% identical to their mammalian homologues, respectively (Nakayama et al., 1998a,b).
Although I-Smads inhibit signaling by members of the TGF-β superfamily, significant differences have been observed in their biological activities. BMP signaling is antagonized by both Smad6 and Smad7 (Hata et al., 1998; Fujii et al., 1999; Ishisaki et al., 1999), whereas Smad7 is more potent than Smad6 in repressing TGF-β and activin signaling. Thus, Smad7 has been reported to antagonize the growth inhibition, matrix formation, apoptosis induction, and embryonic lung morphogenesis induced by TGF-β or activin (Itoh et al., 1998; Ishisaki et al., 1999; Zhao et al., 2000). Moreover, bleomycin-induced lung fibrosis, which is mediated by endogenous production of TGF-β, is prevented by the administration of adenovirus containing Smad7, but not Smad6 (Nakao et al., 1999).
To elucidate the functional differences between Smad7 and Smad6 in the repression of TGF-β signaling and to determine how Smad7 efficiently inhibits this signaling, we examined the activities of deletion mutants and chimeras of Smad6 and Smad7. Our present findings revealed that the carboxy-terminal MH2 domains of Smad6 and Smad7 are essential for the inhibition of TGF-β and BMP signaling, and the N domain of Smad7 is required for efficient inhibition of TGF-β signaling by Smad7. We also found that the N domains determine the subcellular localization of I-Smads. Physical interaction of Smad7 with TGF-β type I receptor (TβR-I) was found to be one of the most important mechanisms for inhibiting TGF-β signaling. Intriguingly, the isolated N domain of Smad7 physically interacted with the Smad7 MH2 domain, and enhanced the inhibitory activity of the latter through facilitating its interaction with TβR-I.
Results
Smad7 is more potent than Smad6 in inhibiting TGF-β signaling
We examined the inhibition by I-Smads of TGF-β and BMP signaling using transcriptional response assays. We compared the activities of Smad6 and Smad7 using two different promoter–reporter constructs, p3TP–Lux and AR3–Luc, which preferentially respond to TGF-β/activin signaling. Two different types of cells, i.e., R mutant Mv1Lu mink lung epithelial cells and COS7 cells, were used for the transcriptional assays. Smad7 potently inhibited the transcriptional activation of p3TP–Lux induced by a constitutively active form of TβR-I, TβR-I(TD), in both R mutant cells and COS7 cells (Fig. 1 A). In contrast, Smad6 was less potent than Smad7 in inhibiting the transcriptional activation induced by TβR-I(TD). Similar results were obtained using the AR3–Luc promoter–reporter construct (Fig. 1 B), which contains the promoter region of Xenopus Mix.2 and responds to TGF-β/activin signaling in the presence of a forkhead transcription factor, FAST1/FoxH3.
Figure 1.
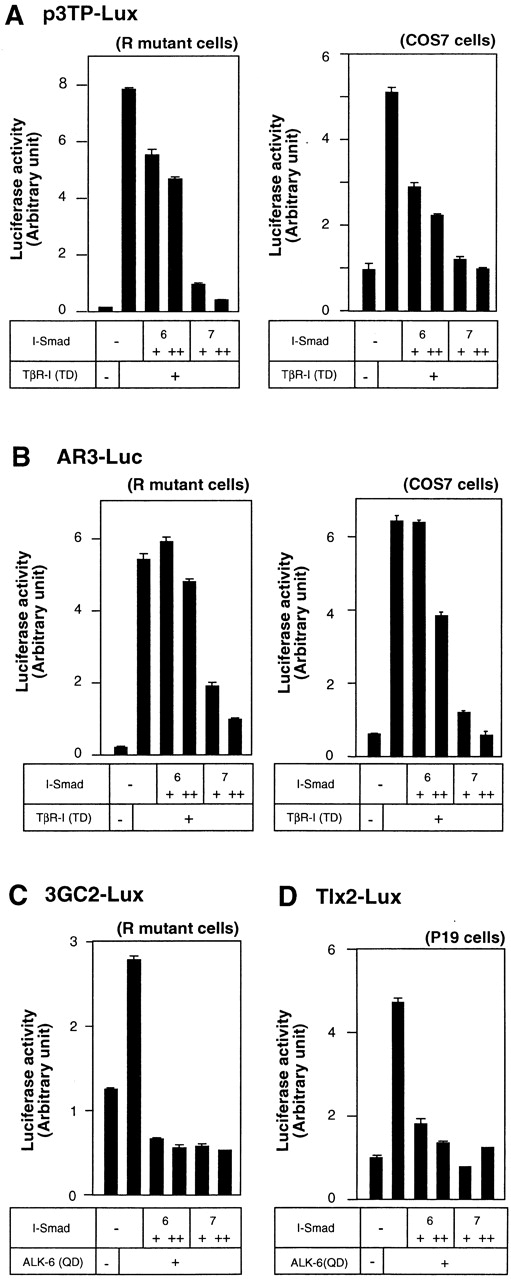
Inhibition of TGF-β and BMP signaling by I-Smads. (A and B) Comparison of the inhibitory effects of Smad6 and Smad7 on transcription from p3TP–Lux (A) and AR3–Luc (B) induced byTβR-I(TD). In A–C, R mutant Mv1Lu cells or COS7 cells were transfected with the indicated plasmids, and luciferase activities were determined as described in the Materials and methods. + and ++ are 0.1 and 0.3 μg of DNA, respectively, transfected in R mutant or COS7 cells. 6 and 7 denote Smad6 and Smad7, respectively. For the analysis using AR3–Luc, FAST1/FoxH3 cDNA was cotransfected. (C) Comparison of the inhibitory effects of Smad6 and Smad7 on 3GC2–Lux transcription induced by a BMPR-I, ALK-6(QD). (D) Inhibitory effects of Smad6 and Smad7 on Tlx2–Lux transcription induced by ALK-6(QD) and BMPR-II. P19 embryonal carcinoma cells were transfected with the indicated plasmids, and luciferase activities were determined as described in the Materials and methods. + and ++ are 0.1 and 0.3 μg of DNA, respectively, transfected in P19 cells.
Transcriptional repression by I-Smads was determined using 3GC2–Lux activated by a constitutively active BMP type I receptor (BMPR-I), ALK-6(QD). In contrast to their differential effects on the inhibition of TGF-β signaling, both Smad6 and Smad7 inhibited BMP signaling induced by ALK-6(QD). Another BMP-responsive luciferase construct, Tlx2–Lux, was also tested to examine the effects of I-Smads on BMP signaling. Again, Smad6 and Smad7 were nearly equal in their inhibition of BMP signaling induced by ALK-6(QD) (Fig. 1 D). Thus, Smad7 is more potent than Smad6 in inhibiting TGF-β signaling, whereas Smad6 and Smad7 were functionally equivalent in inhibiting BMP signaling.
The N domain of Smad7 is important for the inhibition of TGF-β signaling
We next examined which parts of Smad7 are responsible for the inhibition of TGF-β signaling. We prepared deletion mutants of Smad6 and Smad7 (Fig. 2 A). Smad6N and Smad7N have only the N domains of Smad6 and Smad7, respectively, whereas Smad6C and Smad7C contain their MH2 domains. We also generated a chimeric molecule containing the N domain of Smad6 and the MH2 domain of Smad7 (Smad6/7), and one containing the Smad7 N domain and the Smad6 MH2 domain (Smad7/6).
Figure 2.
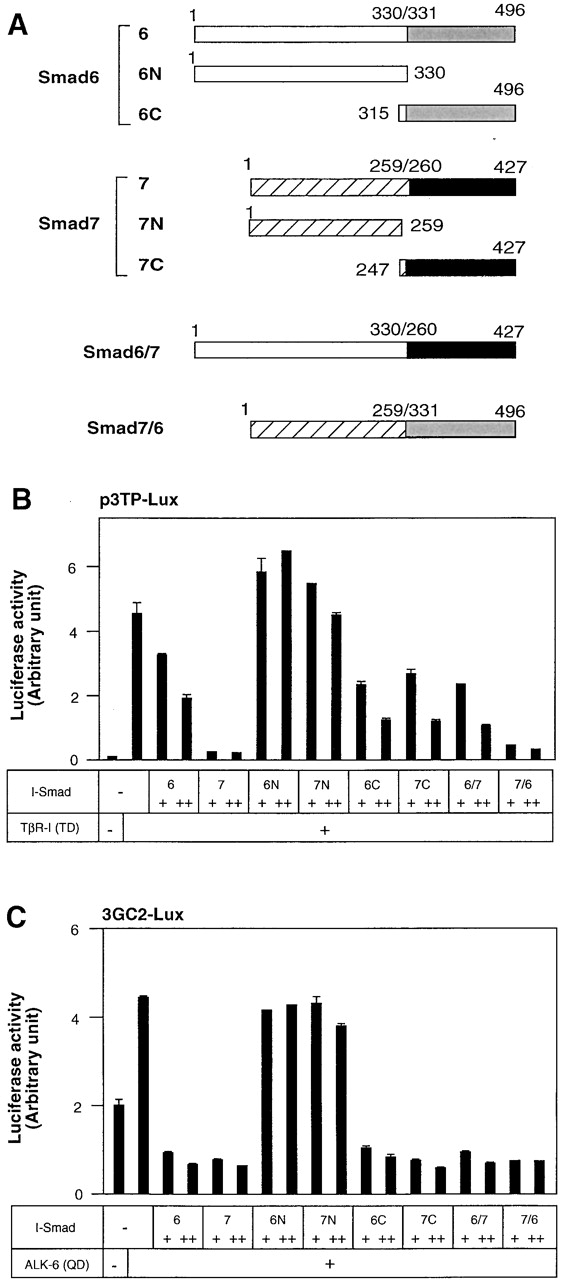
Inhibition of TGF-β or BMP signaling by Smad6 and Smad7 mutants. (A) Structures of Smad6 and Smad7 and their deletion mutants and chimeras. The amino acid numbers of Smad6 and Smad7 are indicated. (B) Inhibitory effects of I-Smads and their deletion mutants and chimeras on p3TP–Lux transcription induced by TβR-I(TD). In B and C, R mutant Mv1Lu cells were transfected with the indicated plasmids, and luciferase activities were determined. + and ++ are 0.1 and 0.3 μg of DNA, respectively, transfected in R mutant cells. (C) Inhibitory effects of I-Smads and their deletion mutants and chimeras on 3GC2–Lux transcription induced by ALK-6(QD).
The isolated N domains of Smad6 and Smad7 (Smad6N and Smad7N) were unable to repress TGF-β signaling induced by TβR-I(TD) (Fig. 2 B). In contrast, the MH2 domains of Smad6 and Smad7 (Smad6C and Smad7C) repressed the transcriptional activity of TβR-I(TD) to extents similar to that induced by the full-length Smad6, but were less potent than the full-length Smad7. Thus, the MH2 domains of Smad6 and Smad7 are essential for repression of TGF-β signaling, but are not sufficient for the efficient transcriptional repression observed with Smad7.
We also examined the two different Smad6 and Smad7 chimeras, i.e., Smad6/7 and Smad7/6. Smad7/6 was as potent as Smad7, whereas Smad6/7 was functionally similar to Smad6 in the inhibition of TGF-β signaling (Fig. 2 B). Thus, the N domain of Smad7 is important for the efficient transcriptional repression induced by Smad7.
Inhibition by Smad6 and Smad7 of BMP signaling was also studied using the 3GC2–Lux promoter–reporter construct. The N domains of Smad6 and Smad7 were not effective in inhibiting BMP signaling (Fig. 2 C). Smad6C and Smad7C were nearly as potent as the full-length Smad6 and Smad7, suggesting that the MH2 domains may be sufficient for repression of BMP signaling. This finding was further confirmed by the observation that the Smad6 and Smad7 chimeras, Smad6/7 and Smad7/6, potently inhibited the signaling activity of the BMPR-I.
Multiple parts of the Smad7 N domain may be responsible for specific inhibition of TGF-β signaling
To determine which parts of the N domain of Smad7 are essential for efficient inhibition of TGF-β signaling, we constructed Smad7 chimeras in which the amino-terminal regions were replaced by the corresponding regions of Smad6 (Fig. 3 A). In the N domain of Smad7, the most amino-terminal region (amino acids 1–17) and the region adjacent to the MH2 domain (amino acids 192–213) are highly conserved between Smad6 and Smad7, whereas the other regions are less conserved. Functional analyses of the Smad7 chimeras by p3TP–Lux and AR3–Luc assays revealed that depending on the length of the N domain of Smad7 replaced by that of Smad6, the inhibitory activity of Smad7 became gradually less potent (Fig. 3, B and C). A significant difference in the inhibition of TGF-β signaling was observed in AR3–Luc when amino acids 32–90 of Smad7 were replaced by the corresponding region of Smad6. The amino acid sequence of this Smad7 region is divergent from that of Smad6. These findings suggest that multiple parts of the Smad7 N domain may contribute to specific inhibition of TGF-β signaling, and that the region containing amino acids 32–90 is one of the most important regions in the Smad7 N domain for repressing TGF-β signaling.
Figure 3.

Multiple regions in the Smad7 N domain are important for efficient inhibition of TGF-β signaling. (A) Structures of the Smad6 and Smad7 chimeras are shown. The amino acid numbers of Smad6 and Smad7 are indicated. (B and C) Inhibition of TGF-β signaling by the Smad6 and Smad7 chimeras. Effects of the chimeras on p3TP–Lux (B) and AR3–Luc (C) transcription induced by TβR-I(TD) were examined. + and ++ are 0.1 and 0.3 μg of DNA, respectively, transfected in R mutant cells. FAST1/FoxH3 cDNA was cotransfected in C.
N domains determine subcellular localization of I-Smads
Smad7 has been reported to be located in the nuclei of COS7 cells (Itoh et al., 1998). Moreover, Smad7 is exported to the cytoplasm upon stimulation by TGF-β (Itoh et al., 1998), as well as by E3 ubiquitin ligases Smurf1 and Smurf2 (Kavsak et al., 2000; Ebisawa et al., 2001). We examined which parts of I-Smads determine their subcellular localization. As reported previously (Itoh et al., 1998; Kavsak et al., 2000; Ebisawa et al., 2001), Smad7 was predominantly located in the nuclei of transfected COS7 cells, whereas Smad6 was observed in the nuclei as well as in the cytoplasm (Fig. 4 A). The cytoplasmic staining of Smad6 was observed as a dotted pattern. In contrast, Smad6C and Smad7C were detected in the nucleus (unpublished data; see Fig. 8, C and D) . The subcellular localization of the Smad7 chimeras depended on their amino-terminal regions. Thus, S6(31/32)S7 and S6(171/91)S7 were detected in the nucleus, similar to Smad7, whereas S6(225/158)S7 and S6(279/211)S7 were detected as cytoplasmic dotted patterns. These results indicate that the region containing amino acids 91–157 of Smad7 plays an important role in determining the subcellular localization of Smad7 in COS7 cells.
Figure 4.
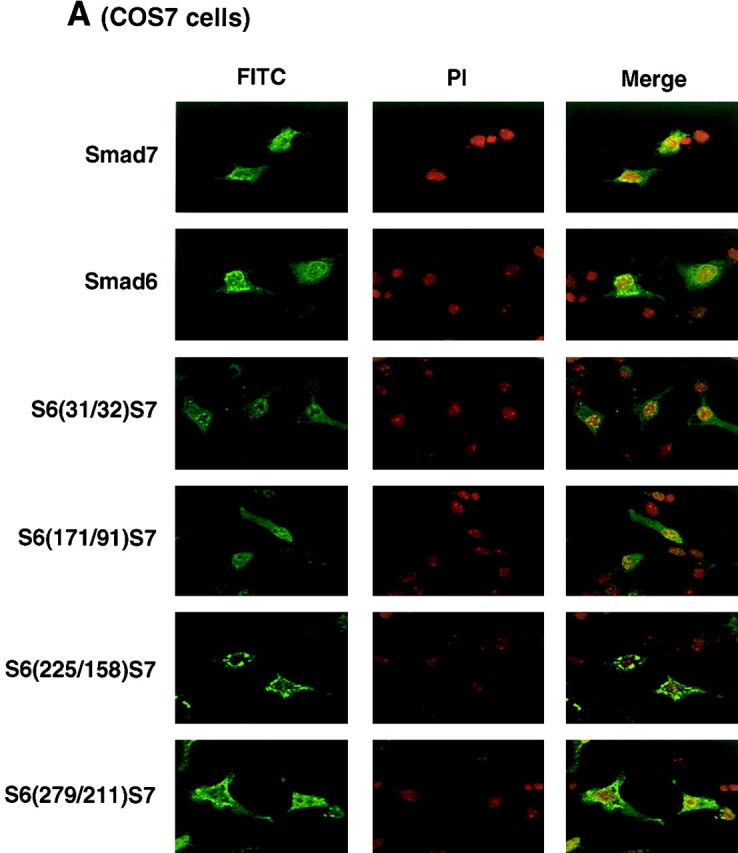
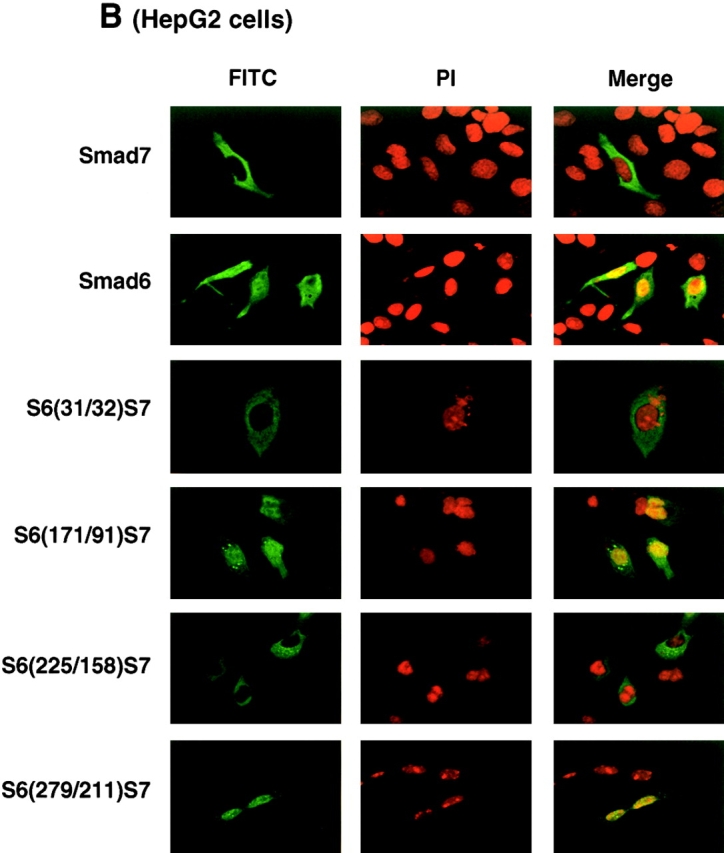
Subcellular localization of I-Smads. Subcellular localization of Smad6, Smad7, and their chimeras was determined in transfected COS7 (A) or HepG2 cells (B). FLAG-tagged I-Smads were transfected in cells and the subcellular localization of I-Smads was demonstrated by FITC. Nuclear staining was performed by 4,6-diamidino-2-phenylindole (PI).
Figure 8.
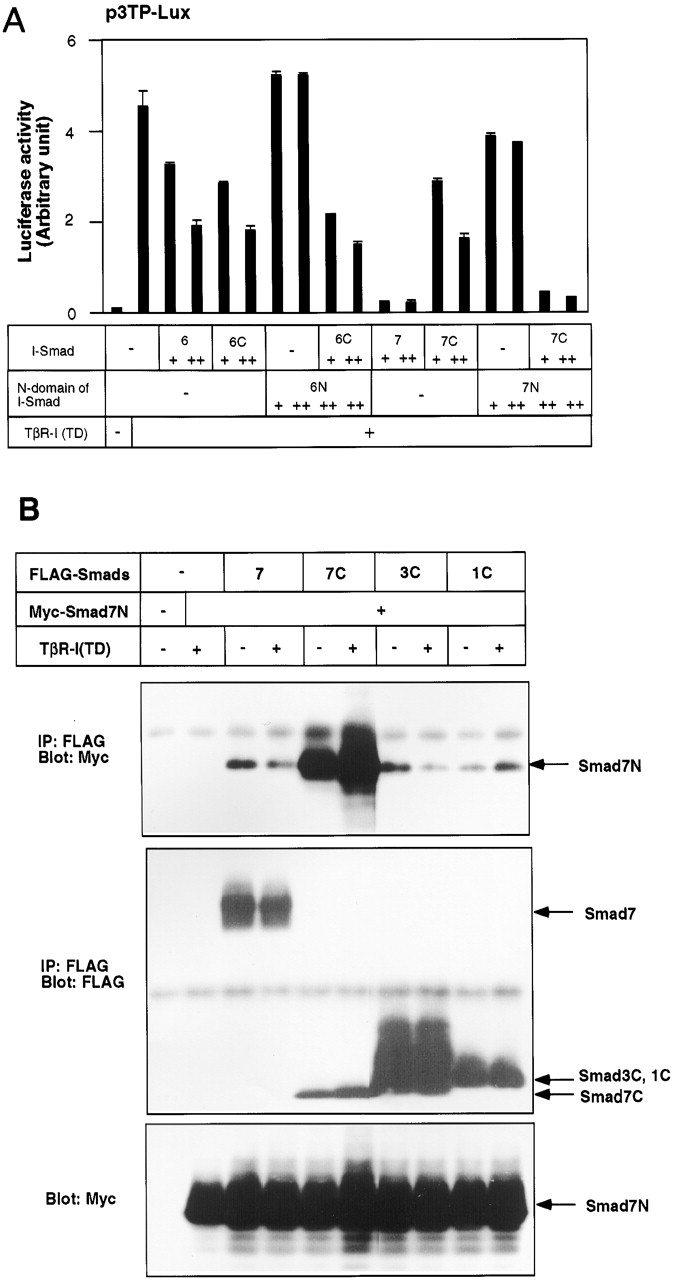
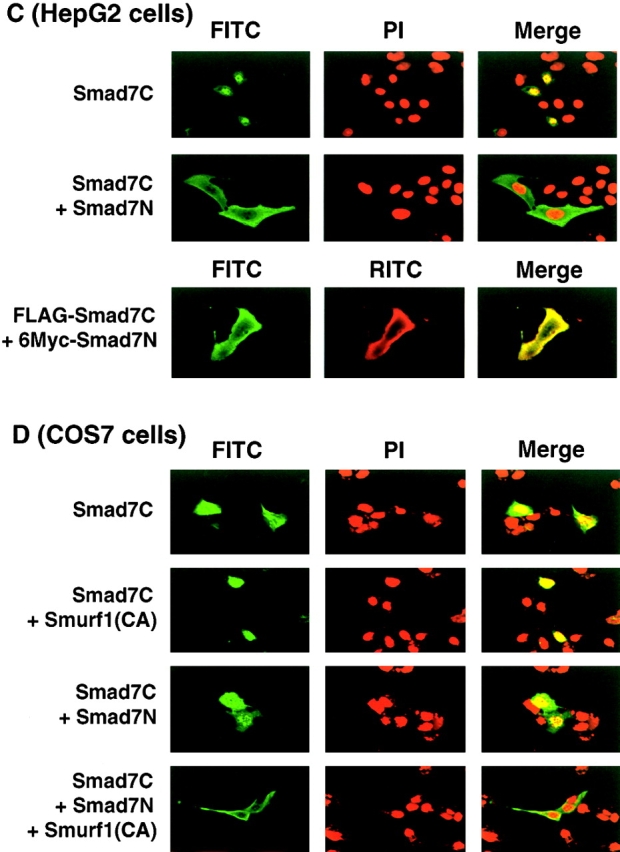
Physical interaction of Smad7N leads to nuclear export and enhancement of the inhibitory activity of Smad7C. (A) Enhancement of the inhibitory activity of Smad7C by Smad7N. Effects of Smad6N and Smad7N were examined in the presence of their MH2 domains (Smad6C and Smad7C) by a p3TP–Lux transcription assay induced by TβR-I(TD). + and ++ are 0.1 and 0.3 μg of DNA, respectively, transfected in R mutant cells. (B) Physical interaction between Smad7N and the MH2 domains of Smad7 (7C), Smad3 (3C), and Smad1 (1C). Interaction of Smad7N with the full-length Smad7 (7) was also tested. Interaction was examined by FLAG immunoprecipitation of FLAG–Smads, followed by Myc immunoblotting of Smad7N using transfected COS7 cells (top). Expression levels of transfected proteins were determined and are shown in the lower two panels. (C and D) Subcellular localization of Smad7C in the presence and absence of Smad7N was examined in transfected HepG2 (C) or COS7 cells (D) using confocal laser scanning microscopy. FLAG–Smad7C was stained by anti-FLAG antibody in the absence and presence of Smad7N and Smurf1(CA), and its subcellular localization was demonstrated by FITC. In HepG2 cells, 6Myc–Smad7N was stained by rhodamine isothiocyanate (RITC; C, bottom). (E) Affinity cross-linking of 125I–TGF-β1 was performed using transfected COS7 cells followed by FLAG immunoprecipitation of FLAG–I-Smads (upper). The lower two panels demonstrate the levels of expression of transfected proteins.
We also examined the subcellular localization of Smad6, Smad7, and their chimeras in other cell types. In contrast to the findings obtained with COS7 cells, Smad7 was predominantly located in the cytoplasm, whereas Smad6 was detected in both the cytoplasm and the nucleus in HepG2 cells (Fig. 4 B). Subcellular localization of the Smad6 and Smad7 chimeras in HepG2 cells also revealed that the localization of I-Smads depended on the amino-terminal region of Smad6 and Smad7, although the chimeras exhibited complicated localization patterns. S6(31/32)S7 and S6(225/158)S7 were detected in the cytoplasm, similar to the full-length Smad7, whereas S6(171/91)S7 and S6(279/211)S7 were in both the nucleus and the cytoplasm. Similar results were obtained when R mutant Mv1Lu cells were used (unpublished data). Thus, the localization of Smad7 in HepG2 and R mutant Mv1Lu cells is different from that in COS7 cells, and multiple parts of the Smad7 N domain appear to determine the subcellular localization of Smad7 in these cells. However, subcellular localization may not primarily determine the potency of Smad7 in inhibiting TGF-β signaling, because the changes in subcellular localization observed with the Smad6 and Smad7 chimeras did not necessarily correlate with the inhibitory activity of Smad7 on TGF-β signaling.
Smurf1 induces nuclear export of both Smad6 and Smad7
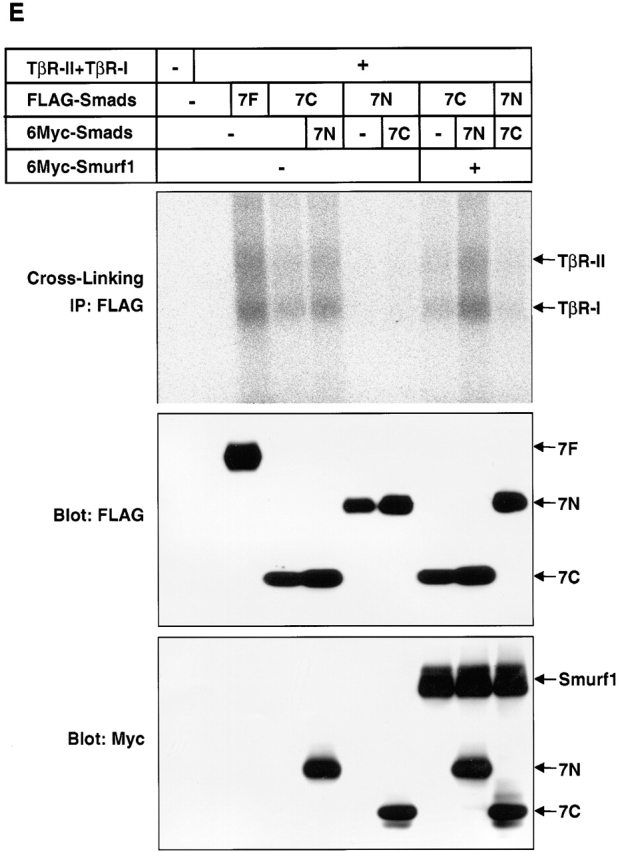
Smurf1 induces the nuclear export of Smad7 through an interaction with the Smad7 PY motif, and induces ubiquitin-dependent degradation of Smad7 and TβR-I. The nuclear export of Smad7 is not dependent on the enzymatic activity of Smurf1, as an enzymatically inactive form of Smurf1, Smurf1(CA), induces the nuclear export of Smad7 (Ebisawa et al., 2001). We examined whether Smurf1 regulates the subcellular localization of Smad6, Smad7, and their mutants. Smurf1(CA) induced cytoplasmic localization of Smad6 and Smad7 in both COS7 cells and HepG2 cells (Fig. 5, A and B) . However, Smad6C and Smad7C, which lack the PY motif, were not exported to the cytoplasm by Smurf1(CA) (Fig. 5 C).
Figure 5.
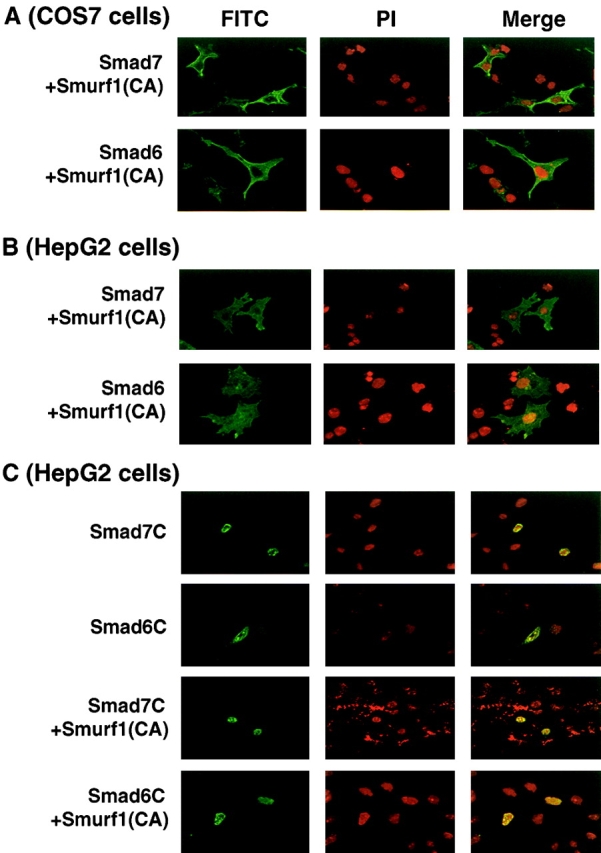

Smurf1 induces nuclear export of Smad6 and Smad7. (A–C) The nuclear export of Smad6, Smad7, and their deletion mutants (Smad6C and Smad7C) induced by Smurf1(CA) was examined in COS7 (A) or HepG2 cells (B and C) as in Fig. 4. (D) The effects of Smad7 amino-terminal deletion mutants on p3TP–Lux transcription induced by TβR-I(TD) were examined. + and ++ are 0.3 and 1.0 μg of DNA, respectively, transfected in R mutant cells. PPPY represents the PY motif.
Functional importance of the I-Smad PY motif was examined by a p3TP–Lux assay. S7Δ191, which lacks the amino-terminal 191 amino acids of Smad7, is less potent than the full-length Smad7 in inhibiting TGF-β signaling. Further deletion including the PY motif of Smad7 (S7Δ210) resulted in a dramatic decrease in the inhibition of TGF-β signaling (Fig. 5 D). In contrast, S6(279/211)S7 was not so weak as S7Δ210 in inhibiting TGF-β signaling (Fig. 3 B), probably because S6(279/211)S7 contains the PY motif of Smad6. These findings suggest that in agreement with a previous report (Ebisawa et al., 2001), the PY motif is required for the nuclear export of I-Smads and the degradation of TβR-I, and the absence of the PY motif leads to a decrease in the inhibition of TGF-β signaling by I-Smads. However, because Smurf1 can interact with Smad6 and Smad7, the interaction of I-Smads with Smurf1 may not be responsible for the differential activity of Smad6 and Smad7.
Repression of Smad2 phosphorylation by I-Smads correlates with their inhibition of transcription induced by TβR-I(TD)
The abilities of the deletion mutants and chimeras of Smad6 and Smad7 to inhibit Smad2 phosphorylation were examined in transfected COS7 cells. Consistent with the results obtained with transcriptional activation assays (Fig. 2 B), phosphorylation of Smad2 was abolished by Smad7 and Smad7/6 (Fig. 6) . Smad6, Smad6/7, Smad6C, and Smad7C were less potent in inhibiting Smad2 phosphorylation. As expected, the N domains of Smad6 and Smad7 did not affect the phosphorylation of Smad2. I-Smads have been reported to regulate TGF-β/BMP signaling through various mechanisms, i.e., competition with R-Smads for receptor activation, ubiquitin-dependent degradation of receptors by Smurfs, prevention of complex formation between R-Smads and Co-Smads, and transcriptional repression in the nucleus. Because the levels of Smad2 phosphorylation correlate with the repression of p3TP–Lux transcription by the I-Smad mutants, Smad7 might regulate TGF-β signaling mainly at the receptor level through competition with R-Smads for receptor activation and degradation by Smurfs.
Figure 6.

Repression of Smad2 phosphorylation by I-Smads, their deletion mutants, and chimeras. COS7 cells were transfected with the indicated plasmids, and phosphorylation of Smad2 was determined by FLAG immunoprecipitation of Smad2, followed by phosphoserine immunoblotting (top). Levels of expression for each protein were determined by immunoblotting using FLAG, HA, or Myc antibodies (lower). + and ++ are 0.1 and 0.3 μg of DNA, respectively, transfected in COS7 cells.
The N domain of Smad7 determines the affinity of Smad7 for TβR-I
Because the interaction between I-Smads and TβR-I appears to be one of the most critical mechanisms in the efficient inhibition of TGF-β signaling, we explored the physical interaction of I-Smads and their mutants with TβR-I. We first examined the interaction between I-Smad constructs with the TβR-I and TβR-II receptor complex cross-linked with 125I–TGF-β1. Fig. 7 A demonstrates that the TβR-II–TβR-I complex was efficiently coimmunoprecipitated by full-length Smad7 and Smad7/6. In contrast, Smad6C and Smad7C weakly interacted with the TGF-β receptor complexes, and the full-length Smad6 and Smad6/7 did so only very weakly. The receptor complexes were not coimmunoprecipitated by the N domains of Smad6 or Smad7. This finding is consistent with those obtained with the transcriptional assays (Fig. 2 B), indicating that the direct association of I-Smads with TGF-β receptor complexes is an important mechanism in the inhibition of TGF-β signaling.
Figure 7.
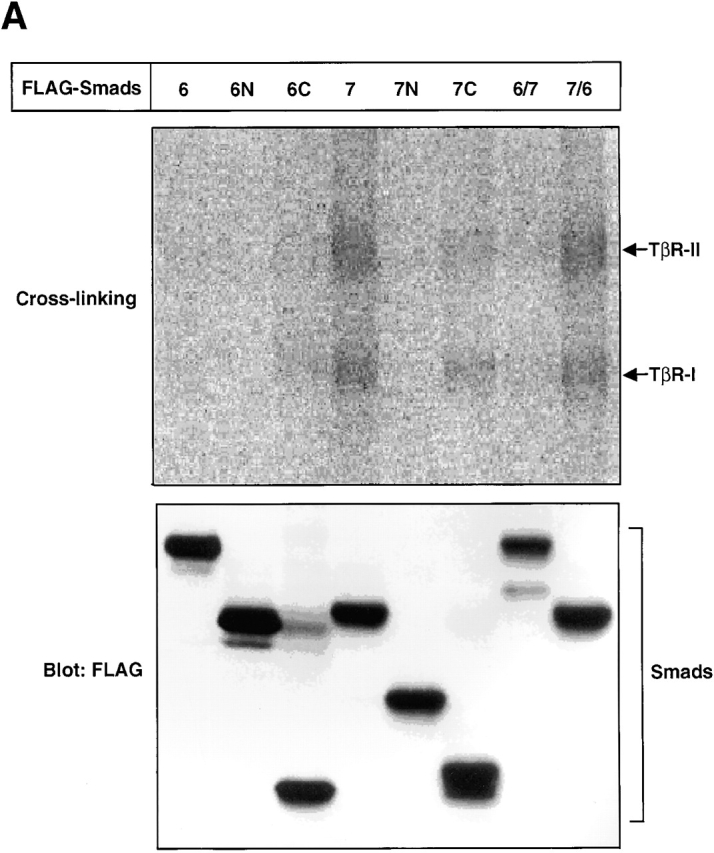

Physical interaction of Smads with TGF-β receptors. (A) Affinity cross-linking using 125I–TGF-β1 followed by immunoprecipitation by anti-FLAG antibody was performed using COS7 cells transfected with the indicated plasmids (top). Levels of expression of I-Smads and their mutants and chimeras were examined by immunoblotting using anti-FLAG antibody (bottom). (B) Interaction of I-Smads with TβR-I(TD) was examined by FLAG immunoprecipitation of I-Smads, followed by HA-immunoblotting of TβR-I(TD) using transfected COS7 cells (top). The Smad6 and Smad7 chimeras shown in Fig. 3 A were used. 6(32), S6(31/32)S7; 6(91), S6(171/91)S7; 6(158), S6(225/158); 6(211), S6(279/211)S7. Note that HA-tagged TβR-I(TD) was comigrated with IgG. Lower panels demonstrate the levels of expression of I-Smads (middle) and TβR-I(TD) (bottom).
As TβR-I acts as a downstream component of TβR-II, we next investigated whether I-Smads and their mutants physically interact with the constitutively active form of TβR-I. Immunoprecipitation of the I-Smad constructs followed by immunoblotting of TβR-I(TD) revealed that the full-length Smad7 and Smad7/6 bound to TβR-I(TD) strongly, whereas Smad6 and Smad6/7 did so only weakly (Fig. 7 B). Of the chimeras between Smad6 and Smad7, S6(31/32)S7 bound to TβR-I(TD) strongly. However, the other chimeras bound to TβR-I(TD) less strongly, depending on the lengths of the N domain of Smad7, and their binding affinity correlated with their potency in inhibiting TGF-β signaling, analyzed by transcriptional assays (Fig. 3). When the interaction of the I-Smad chimeras with a constitutively active BMPR-I, ALK-6(QD), was examined by immunoprecipitation followed by immunoblotting, we observed no differences in affinity with ALK-6(QD) among the I-Smad chimeras (unpublished data).
The N domain of Smad7 potentiates the inhibitory activity of its MH2 domain through physical interaction
To elucidate the mechanism by which the N domain of Smad7 represses TGF-β signaling, we cotransfected Smad7N with Smad7C, and examined the inhibition of TGF-β signaling by p3TP–Lux assays. Although Smad7N alone was unable to inhibit TGF-β signaling, cotransfection of Smad7N with Smad7C led to potent inhibition (Fig. 8 A). In contrast, cotransfection of Smad6N with Smad6C (Fig. 8 A) or Smad7C (unpublished data) did not significantly induce further inhibition of TGF-β signaling compared with transfection of Smad6C alone. When Smad7N was cotransfected with the MH2 domain of Smad3, it did not modulate the transcriptional activity of the latter (unpublished data). These results indicate that the N domain of Smad7 potentiates the inhibitory activity of its MH2 domain in trans.
We next examined whether Smad7N can physically interact with the MH2 domains of Smads. As shown in Fig. 8 B, Smad7N was able to interact with Smad7C. Smad7N also interacted with Smad6C (unpublished data), but failed to associate with the MH2 domains of Smad3 or Smad1 (Fig. 8 B). We also tested the interaction of Smad6N with the MH2 domains of I-Smads. Smad6N weakly interacted with Smad6C, but not with Smad7C (unpublished data).
We examined the localization of Smad7C in the presence and absence of Smad7N in HepG2 and COS7 cells. In HepG2 cells, Smad7 was detected in the cytoplasm (Fig. 4 B), whereas Smad7C was detected in the nucleus (Fig. 8 C). However, when Smad7N was cotransfected, Smad7C was observed in the cytoplasm (Fig. 8 C). Moreover, Smad7C and Smad7N were colocalized in HepG2 cells, consistent with the observation that they physically interact with each other. In COS7 cells, Smad7 and Smad7C were detected in the nucleus (Figs. 8 D and 4 A). Co-transfection of either Smurf1(CA) or Smad7N did not induce the nuclear export of Smad7C. However, Smad7C was detected in the cytoplasm in the presence of both Smad7N and Smurf1(CA) (Fig. 8 D).
We then studied the interaction of Smad7C with the TGF-β receptors in the presence and absence of Smad7N by affinity cross-linking, followed by immunoprecipitation of FLAG–Smads. Smad7C weakly interacted with the TGF-β receptor complex, whereas Smad7N alone failed to do so, as shown in Fig. 7 A. However, strong interaction between Smad7C and the TGF-β receptor complex was detected in the presence of Smad7N (Fig. 8 E). Smurf1 facilitates the interaction of Smad7 with TGF-β receptors by targeting Smad7 to the plasma membrane (unpublished data). In the presence of Smurf1, Smad7C efficiently interacted with TGF-β receptors when cotransfected with Smad7N (Fig. 8 E). By immunoprecipitation of Smad7N, the TGF-β receptor complex was weakly coimmunoprecipitated in the presence of Smurf1 and Smad7C, suggesting that Smad7N interacts with these receptors indirectly through Smad7C. These findings suggest that Smad7N physically interacts with Smad7C, leading to an increase in the affinity between Smad7C and the TGF-β receptors.
Discussion
The N domain of Smad7 is important for repressing TGF-β signaling
We have shown in the present study that Smad7 is more potent than Smad6 in inhibiting TGF-β signaling, whereas Smad6 and Smad7 are functionally similar in repressing BMP signaling. Analyses by p3TP–Lux and AR3–Luc assays revealed that the N domain of Smad7 plays an important role in efficient inhibition of TGF-β signaling. Although multiple regions of the N domain of Smad7 appeared to contribute to the repression of TGF-β signaling, the transcriptional assay using AR3–Luc and the interaction of the I-Smad chimeras with TβR-I(TD) suggested that the region containing amino acids 31–90 of Smad7 is one of the most important regions for the efficient inhibition of TGF-β signaling.
The N domains of Smad6 and Smad7 have thus far been poorly investigated. The amino acid sequences of the N domains are not highly conserved between Smad6 and Smad7 (36.7%). However, certain regions, e.g., the most amino-terminal part (amino acids 1–17) and a region adjacent to the MH2 domain (amino acids 192–213), are relatively well conserved. The region adjacent to the MH2 domain contains a PY motif that is important for interacting with the E3 ubiquitin-ligases Smurf1 and Smurf2 (Kavsak et al., 2000; Ebisawa et al., 2001).
The present findings obtained using Smad chimeras appear to be inconsistent with those described in a recent study using Xenopus assays (Nakayama et al., 2001). However, the Smad6/7 chimera used by Nakayama et al. (2001) contains the amino acids 1–154 of Smad6 and 182–382 of Smad7, which is similar to the S6(225/158)S7 chimera in the present study. It will be interesting to examine whether replacing the rest of the N domain of Smad7 with that of Smad6 results in an alteration of the inhibitory activity of Smad6/7 chimeras in Xenopus assays.
Subcellular localization of I-Smads
Localization in cells is differentially regulated between Smad6 and Smad7. Smad7 has been reported to be located in the nucleus of transfected COS7 cells (Itoh et al., 1998), whereas Smad6 is distributed in both the nucleus and the cytoplasm. Intriguingly, we found that Smad7 was predominantly located in the cytoplasm in HepG2 cells and R mutant Mv1Lu cells, although the reason for the difference in localization of Smad7 between these cell types is unknown. TβR-I(TD) has been shown to induce nuclear export of Smad7 (Itoh et al., 1998), but not of Smad6; however, in the present study we were unable to clearly induce the nuclear export of Smad7 by TβR-I(TD) (unpublished data).
We also found that Smurf1 induces nuclear export of Smad6 and Smad7, whereas Smad6C and Smad7C, which lack the ability to interact with Smurf1, were not exported to the cytoplasm even in the presence of Smurf1. These findings suggest that the weak inhibitory activity of Smad6C and Smad7C may be, at least in part, due to their lack of interaction with Smurfs. However, as the PY motif is present in both Smad6 and Smad7, and Smurf1 can interact equally well with both of them (Ebisawa et al., 2001), this region may not solely account for the functional difference between these I-Smads.
Analyses using Smad6 and Smad7 chimeras revealed that various regions of Smad7 play roles in the subcellular localization of Smad7. Intriguingly, Smad7 is differentially located in either the cytoplasm or the nucleus depending on cell type. It is possible that proteins expressed in only certain types of cells may specifically interact with Smad7 through various regions in the N domain, and thereby induce its nuclear export or import. Although subcellular localization differs between S6(171/91)S7 and S6(225/158)S7 in COS7 cells and between S6(171/91)S7, S6(225/158)S7, and S6(279/211)S7 in HepG2 and R mutant Mv1Lu cells, inhibition of TGF-β signaling differed only slightly between these chimeras. Thus, subcellular localization of I-Smads might not be the major determinant of the differential effects on the inhibition of TGF-β signaling, and other mechanisms may also play important roles in their inhibitory signaling.
Interaction of I-Smads with TβR-I
As levels of Smad2 phosphorylation correlated very well with the inhibitory activity of the I-Smad chimeras, mechanisms mediated upstream of Smad2 phosphorylation, i.e., interaction of I-Smads with type I receptors and ubiquitin-dependent degradation of these receptors, may play important roles in the specific inhibition of TGF-β signaling. By replacing parts of the N domain of Smad7 with the corresponding regions of Smad6, we found that depending on the lengths of the N domain of Smad7, the I-Smad chimeras interacted with TβR-I less efficiently, and their inhibitory activities became less potent. Intriguingly, a significant decrease in the interaction between the I-Smad chimeras and TβR-I(TD) was observed when amino acids 31–90 of Smad7 were replaced, suggesting that this region may be one of the important regions for efficient inhibition of TGF-β signaling by Smad7.
The L45 loop of type I serine/threonine kinase receptors directly interacts with the L3 loop in the MH2 domains of R-Smads, and determines the specificity of inhibitory signals (Chen et al., 1998; Lo et al., 1998). The L45 loops of TβR-I and activin type IB receptor/ALK-4 interact with and activate Smad2 and Smad3, whereas the BMPR-I L45 loops bind to Smad1 and Smad5. The L45 loop of type I receptors may also interact with the L3 loops of I-Smads. However, the amino acid sequences of the L3 loops of I-Smads are divergent from those of R-Smads. Thus, the L3 loops of Smad6 and Smad7 may have low affinity interactions with TβR-I, and the N domain of Smad7, but not Smad6, is required for efficient interaction. In contrast, interaction of ALK-6 with the MH2 domains of Smad6 and Smad7 might occur with high affinity. Thus, the N domain of Smad6 or Smad7 does not appear to be required for the inhibition of BMP signaling. It will be important to determine the modes of interaction between type I receptors and I-Smads by three-dimensional analysis.
Physical interaction between the N and MH2 domains of Smad7
Interestingly, Smad7N alone did not inhibit TGF-β signaling, but Smad7N and Smad7C together potently repressed it, as reported by Nakayama et al. (2001). We showed in the present study that Smad7N physically interacts with its MH2 domain, and suggested that its ability to do so is important for its inhibition of TGF-β signaling. Smad7N induced the nuclear export of Smad7C in HepG2 cells. In COS7 cells, Smad7N required Smurf1 for the nuclear export of Smad7C. Finally, we demonstrated that Smad7N enhances the interaction of Smad7C with the TGF-β receptor complex. Thus, Smad7N has the ability to physically interact with Smad7C, which might result in a conformational change of the MH2 domain and enhancement of its affinity for the receptor complex.
The amino-terminal MH1 domains of R-Smads have been shown to physically interact with their MH2 domains (Hata et al., 1997). However, the MH1 domains of R-Smads repress the activity of their MH2 domains through physical interaction, and this interaction is released upon receptor activation and phosphorylation of R-Smads. In contrast, the N domain of Smad7 physically interacts with the MH2 domain, resulting in enhancement of the inhibitory activity of Smad7 through facilitation of the interaction with TGF-β receptors (Fig. 9) . Our present study also revealed that Smad7N specifically interacts with the MH2 domains of I-Smads, but not with those of R-Smads. Thus, the present findings suggest a new mode of action for the Smad7 N domain in supporting the inhibitory activity of the MH2 domain in TGF-β signaling.
Figure 9.

Modes of interaction between the amino-terminal regions and the MH2 domains of R-Smads and I-Smads. The amino-terminal regions and the MH2 domains physically interact with each other in both R-Smads and I-Smads. The amino-terminal MH1 domains and the MH2 domains of R-Smads repress each other's activity, and this physical interaction is released upon receptor activation. In contrast, the N domain of Smad7 enhances the inhibitory activity of its MH2 domain through physical interaction.
Materials and methods
Construction of plasmids
The original constructions of constitutively active forms of TβR-I and ALK-6 (TβR-I[TD] and ALK-6[QD], respectively), TβR-II, BMPR-II, Smad1, Smad2, Smad3, Smad6, Smad7, Smurf1(CA), and FAST1/FoxH3 cDNAs were created as previously described (Imamura et al., 1997; Kawabata et al., 1998; Hanai et al., 1999; Ebisawa et al., 2001). FLAG–pcDNA3, 6Myc–pcDNA3, and pcDNA3–HA were created as described previously (Inoue et al., 1998; Kawabata et al., 1998). Deletion mutants and chimeras of Smad6 and Smad7 were prepared by a PCR-based approach. To obtain efficient levels of protein expression, some constructs, including all the FLAG-tagged Smad constructs, were subcloned into pcDEF3 (Goldman et al., 1996). Expression levels of transfected Smad proteins were similar when they were examined by immunoblotting. All of the PCR products were sequenced before use.
Cell culture and cDNA transfection
R mutant Mv1Lu mink lung epithelial cells (gift of J. Massagué, Memorial Sloan-Kettering Cancer Center, New York, NY, and M. Laiho, Haartman Institute, University of Helsinki, Finland), COS7 cells, HepG2 cells (American Type Culture Collection accession number HB-8065), and P19 embryonal carcinoma cells (gift of T. Momoi, National Institute of Neuroscience, Kodaira, Tokyo, Japan) were cultured in DME containing 10% fetal bovine serum and antibiotics. Cells were transiently transfected using FuGENE6 (Roche Diagnostics) following the manufacturer's recommendations.
Luciferase assay
R mutant mink lung epithelial cells, COS7 cells, or P19 embryonal carcinoma cells were transiently transfected with an appropriate combination of p3TP–lux (Cárcamo et al., 1994), AR3–Luc (Hayashi et al., 1997), 3GC2–Lux (Ishida et al., 2000), or Tlx2–Lux (Tang et al., 1998) promoter–reporter constructs, expression plasmids, and pcDNA3. Total amounts of transfected DNAs were the same in each experiment, and values were normalized using Renilla luciferase activity. All the Smad constructs used in luciferase assays were subcloned in pcDEF3, and their protein expression levels were verified by immunoblotting before use or in parallel with luciferase assays.
Immunoprecipitation and immunoblotting
COS7 cells, transfected with expression constructs, were solubilized in a buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 1% aprotinin, and 1 mM phenylmethyl sulfonyl fluoride. The cell lysates were precipitated by centrifugation and the supernatants were incubated with anti-FLAG M2 (Eastman Kodak Co.) antibody for 1 h, followed by incubation with protein A– or G–Sepharose beads. The beads were washed four times with the buffer used for cell solubilization. The immune complexes were then eluted by boiling for 3 min in SDS sample buffer (100 mM Tris-HCl, pH 8.8, 0.01% bromophenol blue, 36% glycerol, 4% SDS, 10 mM DTT) and applied to SDS-PAGE. Aliquots of the cell lysates were directly subjected to SDS-PAGE without immunoprecipitation. Proteins were electrotransferred to ProBlott membranes (Applied Biosystems) and immunoblotted with anti-HA 3F10 (Boehringer), anti-phosphoserine (Zymed Laboratories), anti-FLAG M2, or anti-myc 9E10 (PharMingen) antibodies and detected using an ECL detection system (Amersham Pharmacia Biotech). For reblotting, the membranes were stripped in accordance with the manufacturer's protocol.
Affinity cross-linking analysis
Recombinant TGF-β1 (R&D Systems) was iodinated using the chloramine T method (Frolik et al., 1984). COS7 cells were transfected with expression vectors, and affinity cross-linked using 125I–TGF-β1. Subsequent immunoprecipitation and analysis by SDS-PAGE were performed as previously described (Imamura et al., 1997).
Immunofluorescence labeling
Immunohistochemical staining of Smad6 and Smad7 was performed using transfected COS7, HepG2, or R mutant cells. Eight-well Lab-Tek chamber slides (Nalge Nunc International) were treated with 0.03 mg/ml Cellmatrix type IV (Nitta Gelatin Co., Ltd.) at 37°C for 30 min. Cells were seeded in the chambers, transfected with indicated cDNAs, and incubated for an additional 24 h. Cells were then fixed and treated with mouse anti-FLAG antibody and biotinylated horse anti–mouse IgG (affinity purified IgG [H+L]; Vector Laboratories), followed by incubation with FITC-labeled Avidin DCS (Vector Laboratories). For double staining of FLAG- and 6Myc-tagged Smads, 6Myc–Smads were stained using rabbit anti-Myc antibody (gift of C.-H. Heldin, Ludwig Institute for Cancer Research, Uppsala, Sweden), followed by incubation with rhodamine isothiocyanate–labeled goat anti–rabbit IgG (Cappel). The nuclei were stained by 4,6-diamidino-2-phenylindole. Intracellular localization was determined by confocal laser scanning microscopy.
Acknowledgments
We are grateful to Y. Inada and A. Nishitoh-Sakai for technical help.
This study was supported by Grants-in-Aid for Scientific Research and Special Coordination Funds for Promoting Science and Technology of the Ministry of Education, Culture, Sport, Science, and Technology of Japan. This study was also supported by a Ludwig Institute for Cancer Research Fund.
Footnotes
Abbreviations used in this paper: BMP, bone morphogenetic protein; BMPR-I, BMP type I receptor; Co-Smad, common mediator Smad; I-Smad, inhibitory Smad; MH, Mad homology; R-Smad, receptor-regulated Smad; TβR-I, TGF-β type I receptor; TGF-β, transforming growth factor-β.
References
- Bai, S., X. Shi, X. Yang, and X. Cao. 2000. Smad6 as a transcriptional corepressor. J. Biol. Chem. 275:8267–8270. [DOI] [PubMed] [Google Scholar]
- Cárcamo, J., F.M.B. Weis, F. Ventura, R. Wieser, J.L. Wrana, L. Attisano, and J. Massagué. 1994. Type I receptors specify growth-inhibitory and transcriptional responses to transforming growth factor β and activin. Mol. Cell. Biol. 14:3810–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y.G., A. Hata, R.S. Lo, D. Wotton, Y. Shi, N. Pavletich, and J. Massagué. 1998. Determinants of specificity in TGF-β signal transduction. Genes Dev. 12:2144–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denissova, N.G., C. Pouponnot, J. Long, D. He, and F. Liu. 2000. Transforming growth factor β-inducible independent binding of SMAD to the Smad7 promoter. Proc. Natl. Acad. Sci. USA. 97:6397–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebisawa, T., M. Fukuchi, G. Murakami, T. Chiba, K. Tanaka, T. Imamura, and K. Miyazono. 2001. Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 276:12477–12480. [DOI] [PubMed] [Google Scholar]
- Frolik, C.A., L.M. Wakefield, D.M. Smith, and M.B. Sporn. 1984. Characterization of a membrane receptor for transforming growth factor-β in normal rat kidney fibroblasts. J. Biol. Chem. 259:10995–11000. [PubMed] [Google Scholar]
- Fujii, M., K. Takeda, T. Imamura, T.K. Sampath, S. Enomoto, M. Kawabata, M. Kato, H. Ichijo, and K. Miyazono. 1999. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol. Biol. Cell. 10:3801–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman, L.A., E.C. Cutrone, S.V. Kotenko, C.D. Krause, and L.A. Langer. 1996. Modifications of vectors pEF-BOS, pcDNA1 and pcDNA3 result in improved convenience and expression. Biotechniques. 21:1013–1015. [DOI] [PubMed] [Google Scholar]
- Hanai, J.-i., L.F. Chen, T. Kanno, N. Ohtani-Fujita, W.Y. Kim, W.-H. Guo, T. Imamura, Y. Ishidou, M. Fukuchi, M.-J. Shi, et al. 1999. Interaction and functional cooperation of PEBP2/CBF with Smads: synergistic induction of the immunoglobulin germline Cα promoter. J. Biol. Chem. 274:31577–31582. [DOI] [PubMed] [Google Scholar]
- Hata, A., R.S. Lo, D. Wotton, G. Lagna, and J. Massagué. 1997. Mutations increasing autoinhibition inactivate tumour suppressors Smad2 and Smad4. Nature. 388:82–87. [DOI] [PubMed] [Google Scholar]
- Hata, A., G. Lagna, J. Massagué, and A. Hemmati-Brivanlou. 1998. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 12:186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, H., S. Abdollah, Y. Qiu, J. Cai, Y.Y. Xu, B.W. Grinnell, M.A. Richardson, J.N. Topper, M.A. Gimbrone, Jr., J.L. Wrana, and D. Falb. 1997. The MAD-related protein Smad7 associates with the TGFβ receptor and functions as an antagonist of TGFβ signaling. Cell. 89:1165–1173. [DOI] [PubMed] [Google Scholar]
- Heldin, C.-H., K. Miyazono, and P. ten Dijke. 1997. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 390:465–471. [DOI] [PubMed] [Google Scholar]
- Imamura, T., M. Takase, A. Nishihara, E. Oeda, J.-i. Hanai, M. Kawabata, and K. Miyazono. 1997. Smad6 inhibits signalling by the TGF-β superfamily. Nature. 389:622–626. [DOI] [PubMed] [Google Scholar]
- Inoue, H., T. Imamura, Y. Ishidou, M. Takase, Y. Udagawa, K. Tsuneizumi, T. Tabata, K. Miyazono, and M. Kawabata. 1998. Interplay of signal mediators of Decapentaplegic (Dpp): molecular characterization of Mothers against Dpp, Medea, and Daughters against Dpp. Mol. Biol. Cell. 9:2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida, W., K. Hamamoto, K. Kusanagi, K. Yagi, M. Kawabata, K. Takehara, T.K. Sampath, M. Kato, and K. Miyazono. 2000. Smad6 is a Smad1/5-induced Smad inhibitor: characterization of bone morphogenetic protein-responsive element in the mouse Smad6 promoter. J. Biol. Chem. 275:6075–6079. [DOI] [PubMed] [Google Scholar]
- Ishisaki, A., K. Yamato, S. Hashimoto, A. Nakao, K. Tamaki, K. Nonaka, P. ten Dijke, H. Sugino, and T. Nishihara. 1999. Differential inhibition of Smad6 and Smad7 on bone morphogenetic protein- and activin-mediated growth arrest and apoptosis in B cells. J. Biol. Chem. 274:13637–13642. [DOI] [PubMed] [Google Scholar]
- Itoh, S., M. Landström, A. Hermansson, F. Itoh, C.-H. Heldin, N.-E. Heldin, and P. ten Dijke. 1998. Transforming growth factor β1 induces nuclear export of inhibitory Smad7. J. Biol. Chem. 273:29195–29201. [DOI] [PubMed] [Google Scholar]
- Kavsak, P., R.K. Rasmussen, C.G. Causing, S. Bonni, H. Zhu, G.H. Thomsen, and J.L. Wrana. 2000. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell. 6:1365–1375. [DOI] [PubMed] [Google Scholar]
- Kawabata, M., H. Inoue, A. Hanyu, T. Imamura, and K. Miyazono. 1998. Smad proteins exist as monomers in vivo and undergo homo- and hetero-oligomerization upon activation by serine/threonine kinase receptors. EMBO J. 17:4056–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, R.S., Y.G. Chen, Y. Shi, N.P. Pavletich, and J. Massagué. 1998. The L3 loop: a structural motif determining specific interactions between SMAD proteins and TGF-β receptors. EMBO J. 17:996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué, J. 1998. TGF-β signal transduction. Annu. Rev. Biochem. 67:753–791. [DOI] [PubMed] [Google Scholar]
- Massagué, J., and D. Wotton. 2000. Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 19:1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono, K. 2000. Positive and negative regulation of TGF-β signaling. J. Cell Sci. 113:1101–1109. [DOI] [PubMed] [Google Scholar]
- Miyazono, K., P. ten Dijke, and C.-H. Heldin. 2000. TGF-β signaling by Smad proteins. Adv. Immunol. 75:115–157. [DOI] [PubMed] [Google Scholar]
- Nagarajan, R.P., J. Zhang, W. Li, and Y. Chen. 1999. Regulation of Smad7 promoter by direct association with Smad3 and Smad4. J. Biol. Chem. 274:33412–33418. [DOI] [PubMed] [Google Scholar]
- Nakao, A., M. Afrakhte, A. Morén, T. Nakayama, J.L. Christian, R. Heuchel, S. Itoh, M. Kawabata, N.-E. Heldin, C.-H. Heldin, and P. ten Dijke. 1997. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature. 389:631–635. [DOI] [PubMed] [Google Scholar]
- Nakao, A., M. Fujii, R. Matsumura, K. Kumano, Y. Saito, K. Miyazono, and I. Iwamoto. 1999. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J. Clin. Invest. 104:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama, T., H. Gardner, L.K. Berg, and J.L. Christian. 1998. a. Smad6 functions as an intracellular antagonist of some TGF-β family members during Xenopus embryogenesis. Genes Cells. 3:387–394. [DOI] [PubMed] [Google Scholar]
- Nakayama, T., M.A. Snyder, S.S. Grewal, K. Tsuneizumi, T. Tabata, and J.L. Christian. 1998. b. Xenopus Smad8 acts downstream of BMP-4 to modulate its activity during vertebrate embryonic patterning. Development. 125:857–867. [DOI] [PubMed] [Google Scholar]
- Nakayama, T., L.K. Berg, and J.L. Christian. 2001. Dissection of inhibitory Smad proteins: both N- and C-terminal domains are necessary for full activities of Xenopus Smad6 and Smad7. Mech. Dev. 100:251–262. [DOI] [PubMed] [Google Scholar]
- Roberts, A.B., and M.B. Sporn. 1990. The transforming growth factor-βs. Peptide Growth Factors and Their Receptors, Part I. M.B. Sporn and A.B. Roberts, editors. Springer-Verlag New York Inc., New York. 419–472.
- Souchelnytskyi, S., T. Nakayama, A. Nakao, A. Morén, C.-H. Heldin, J.L. Christian, and P. ten Dijke. 1998. Physical and functional interaction of murine and Xenopus Smad7 with bone morphogenetic protein receptors and transforming growth factor-β receptors. J. Biol. Chem. 273:25364–25370. [DOI] [PubMed] [Google Scholar]
- Tang, S.J., P.A. Hoodless, Z. Lu, M.L. Breitman, R.R. McInnes, J.L. Wrana, and M. Buchwald. 1998. The Tlx-2 homeobox gene is a downstream target of BMP signalling and is required for mouse mesoderm development. Development. 125:1877–1887. [DOI] [PubMed] [Google Scholar]
- Tsuneizumi, K., T. Nakayama, Y. Kamoshida, T.B. Kornberg, J.L. Christian, and T. Tabata. 1997. Daughters against dpp modulates dpp organizing activity in Drosophila wing development. Nature. 389:627–631. [DOI] [PubMed] [Google Scholar]
- Zhao, J., W. Shi, H. Chen, and D. Warburton. 2000. Smad7 and Smad6 differentially modulate transforming growth factor β-induced inhibition of embryonic lung morphogenesis. J. Biol. Chem. 275:23992–23997. [DOI] [PubMed] [Google Scholar]