

Abstract
Using a new assay for membrane fusion between late Golgi/endosomal compartments, we have reconstituted a rapid, robust homotypic fusion reaction between membranes containing Kex2p and Ste13p, two enzymes resident in the yeast trans-Golgi network (TGN). Fusion was temperature, ATP, and cytosol dependent. It was inhibited by dilution, Ca+2 chelation, N-ethylmaleimide, and detergent. Coimmunoisolation confirmed that the reaction resulted in cointegration of the two enzymes into the same bilayer. Antibody inhibition experiments coupled with antigen competition indicated a requirement for soluble NSF attachment protein receptor (SNARE) proteins Tlg1p, Tlg2p, and Vti1p in this reaction. Membrane fusion also required the rab protein Vps21p. Vps21p was sufficient if present on either the Kex2p or Ste13p membranes alone, indicative of an inherent symmetry in the reaction. These results identify roles for a Tlg SNARE complex composed of Tlg1p, Tlg2p, Vti1p, and the rab Vps21p in this previously uncharacterized homotypic TGN fusion reaction.
Keywords: TGN; rab; SNARE; fusion; Kex2p
Introduction
Controlling fusion of intracellular organelles is essential to their functional integrity. This is particularly true for organelles of the secretory pathway that contain both resident enzymes and secretory proteins destined for the cell surface. Identities of secretory organelles, as defined by their enzymatic content, reflect the dynamic and highly regulated processes of selective delivery of lipid and protein by fusion with transport vesicles and selective removal of lipid and protein by budding of transport vesicles. Membrane fusion depends on the complementarity of transmembrane soluble N-ethylmaleimide (NEM)-sensitive factor attachment protein receptor (SNARE)* proteins on opposing bilayers (Sollner et al., 1993; McNew et al., 2000) and is influenced by the action of accessory factors such as Sec1p/unc-18 (Hata et al., 1993; Carr et al., 1999). Formation of a trans-SNARE complex is thought to catalyze actual bilayer fusion (Fukuda et al., 2000). The complex is held together by a parallel four-helix bundle in which a vesicle SNARE (v-SNARE) contributes one helix and two or three target membrane SNAREs (t-SNAREs) contribute the other three (Poirier et al., 1998; Sutton et al., 1998; Antonin et al., 2000). After fusion, the resulting cis-SNARE complexes can be disassembled by the action of NEM-sensitive factor (NSF; Sec18p in yeast), a AAA ATPase (Sollner et al., 1993). Two general types of membrane fusion events can be distinguished (Wickner and Haas, 2000). Homotypic fusion occurs between like membranes having the same sets of v- and t-SNARE proteins and has been observed with endoplasmic reticulum (Latterich and Schekman, 1994), Golgi (Warren and Malhotra, 1998), early and late endosomes (Mills et al., 1999; Antonin et al., 2000), and lysosomal/vacuolar membranes (Wickner and Haas, 2000). Heterotypic fusion occurs between target membranes having a t-SNARE complex and vesicle membranes bearing the cognate v-SNARE and has been studied extensively in the context of transport vesicle fusion with target organelles (Rothman and Wieland, 1996) and secretory vesicle fusion with the plasma membrane (Grote et al., 2000).
The trans-Golgi network (TGN) consists of a complex network of vesicles and tubules in continual communication with Golgi cisternae, early and late endosomes, and the cell surface. Numerous sorting, budding, and fusion events must occur in a coordinated fashion to maintain the functional integrity of the TGN and prevent undesirable mixing of endocytic and secretory cargo. The molecular details of how this is achieved are unclear. One approach toward understanding sorting in this organelle is to reconstitute TGN membrane fusion events.
We have developed an assay to monitor vesicular transport and membrane fusion events between the TGN and endosomal compartments in yeast. Using this assay, we have discovered a vigorous and previously uncharacterized membrane fusion event involving organelles containing the TGN resident enzymes Kex2p and Ste13p (dipeptidyl aminopeptidase [DPAP]A). Membrane fusion was energy and cytosol dependent. It was inhibited by rapid chelation of Ca2+ and the thiol-reactive reagent NEM.
Furthermore, the SNARE proteins Tlg1p, Tlg2p, and Vti1p, the rab5 homologue Vps21p, and the Sec1p homologue Vps45p, all of which have been implicated in late Golgi/endosomal function in vivo, are required for this reaction. NSF-sensitive SNARE complexes containing various combinations of Tlg1p, Tlg2p, and Vti1p have been isolated (Holthuis et al., 1998; Nichols et al., 1998; Coe et al., 1999). TGN homotypic fusion represents an event in which these three SNAREs appear to function together.
Results
A new assay for fusion of membranes containing TGN resident enzymes
The yeast integral membrane protease Kex2p, the prototype of the family of eukaryotic proprotein processing enzymes that includes furin, cleaves pro–α-factor and other precursors of secreted proteins in the late Golgi (Fuller et al., 1989b; Graham and Emr, 1991). After Kex2p cleavage of pro–α-factor, NH2 terminal dipeptides of α-factor intermediates are removed by the Ste13p DPAP (Julius et al., 1983). Kex2p and Ste13p colocalize to the yeast equivalent of the TGN by virtue of signals in their cytosolic tails. These signals regulate their transport to and retrieval from post-Golgi compartments (Redding et al., 1991; Wilcox and Fuller, 1991; Wilcox et al., 1992; Bryant and Boyd, 1993; Nothwehr et al., 1993; Brickner and Fuller, 1997; Bryant and Stevens, 1997). To better understand the trafficking and localization of TGN membrane proteins, we developed a sensitive quantitative assay for fusion of organelles containing Kex2p and Ste13p based on the mixing of their lumenal contents and their enzymatic activities (Fig. 1 A). A Kex2p substrate was created by fusing an α-factor cleavage site followed by the triple hemagglutinin (HA) epitope tag to the lumenal COOH terminus of Ste13p (Ste13αHA). Expression of this modified form of Ste13p complemented a ste13Δ mutation for α-specific mating, indicating that it was functional in vivo (unpublished data). Fusion of membranes containing Ste13αHA with membranes containing Kex2p followed by Kex2p-mediated proteolytic cleavage after the Lys-Arg cleavage site within the α-factor repeat should separate the HA epitope tag from the DPAP domain (Fig. 1 A). The cleaved product could then be quantified by measuring the fraction of DPAP activity that cannot be immunoprecipitated using anti-HA antibody (i.e., the fraction of the Ste13αHA that no longer has the HA tag). To determine if Ste13αHA was localized correctly and available for cleavage, we first examined whether Ste13αHA was processed by Kex2p in vivo. Ste13αHA was processed efficiently when coexpressed with Kex2p; whereas, the molecule was completely unprocessed when expressed in a kex2Δ strain (Fig. 1 B). This indicated that the fusion protein was properly localized and that the α-factor cleavage site was accessible to Kex2p. We next examined Ste13αHA processing in a cell-free system. Cell-free extracts were prepared from semiintact yeast cells by freeze-thaw lysis and centrifugation (Baker et al., 1988; Ruohola et al., 1988) to produce a medium speed supernatant (MSS) fraction containing cytosol along with late Golgi and endosomal membranes but not ER or early Golgi (Fig. 1 C, inset). Efficient Kex2p-dependent processing of Ste13αHA was observed when MSS membranes from a strain expressing Ste13αHA were incubated under reaction conditions with MSS membranes from a strain expressing Kex2p (Fig. 1 C). Processing was blocked at 0°C (Fig. 1 C) and was rapid, proceeding linearly for ∼10 min and reaching at 15 min a maximum level equivalent to cleavage of ∼35–40% of total Ste13αHA (Fig. 1 D). Only a very short lag (<1 min) was observed before processing began in contrast to cell-free vesicular transport reactions between Golgi cisternae (Balch et al., 1984) from the ER to the Golgi (Baker et al., 1988) or from the late endosome/prevacuolar compartment (PVC) to the lysosome (Vida and Gerhardt, 1999), all of which display a pronounced lag (∼10 min).
Figure 1.
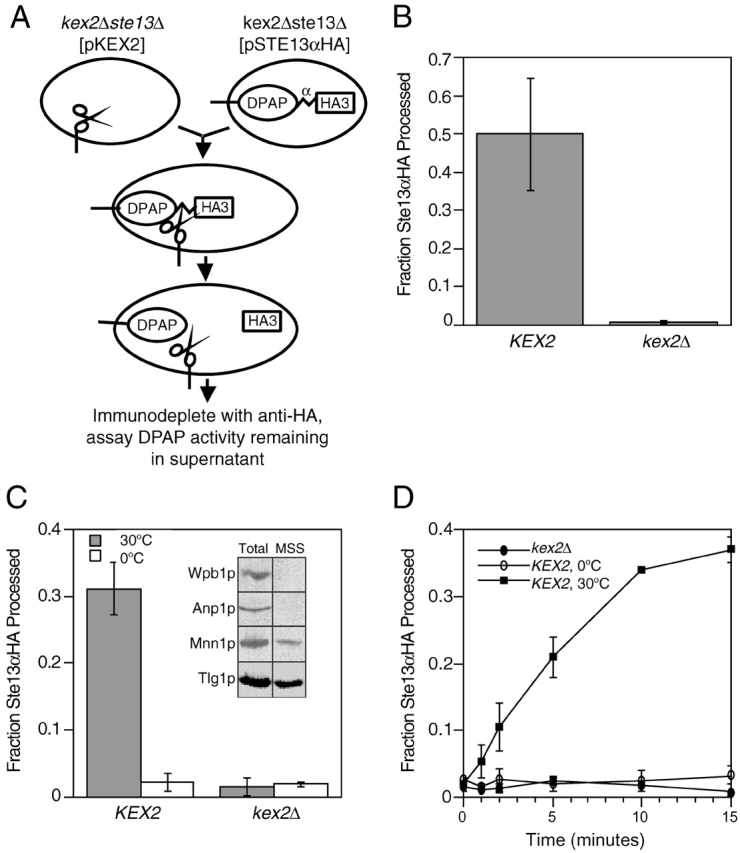
A new assay for fusion of membranes containing TGN resident enzymes. (A) Schematic depiction of cell-free fusion assay. Vesicles isolated from JBY209 (kex2Δ ste13Δ) expressing Kex2p (represented by scissors-shaped protein) fuse with vesicles isolated from JBY209 expressing a Ste13αHA fusion protein containing a Kex2p cleavage site (α) between the lumenal DPAP domain and the HA tag. Cleavage of Ste13αHA by Kex2p separates the HA tag from the DPAP catalytic domain of Ste13. (B) in vivo processing of Ste13αHA by Kex2p. JBY209 expressing Ste13αHA was transformed with either pCWKX10 (KEX2) or a vector control (kex2Δ). Lysates were prepared, and the fraction of Ste13αHA processed was determined as described in Materials and methods. (C) Cell-free Kex2p processing of Ste13αHA. MSS membranes from either Kex2p-expressing cells or kex2Δ cells were combined with MSS membranes from JBY209 expressing Ste13αHA and incubated at either 0 or 30°C. (Inset) Western blot analysis of total lysates before centrifugation (Total) and supernatant fraction obtained by ∼14,000 g centrifugation (MSS) using polyclonal antibodies for organelle marker proteins: Wbp1p, wheat agglutinin binding protein (ER); Anp1p, subunit of cis Golgi mannosyl transferase complex (cis Golgi); Mnn1p, late acting mannosyl transferase (late Golgi); and Tlg1p, t-SNARE of the late Golgi (TGN). Plots in B and C show representative data from many experiments. (D) Time course of Ste13αHA processing. Fourfold scaled up reactions were incubated under reaction conditions. At the indicated times, duplicate 10-μl volumes were removed, and the amount of Ste13αHA processed was determined as described in Materials and methods.
To directly establish that membrane fusion had occurred and Kex2p and Ste13αHA were cointegrated into the same membranes at the end of the reaction, product membranes were immunoisolated using affinity purified antibodies directed against the Kex2p cytosolic tail (Redding et al., 1991) by a method that results in quantitative isolation of membranes containing Kex2p (Sipos and Fuller, 2001). If processing of Ste13αHA by Kex2p resulted from the fusion of compartments containing the two enzymes, then isolation of Kex2p-containing compartments should result in coisolation of DPAP activity. MSS membranes from the Kex2p- and Ste13αHA-expressing strains were incubated under reaction conditions either together (Fig. 2 , Mixed) or separately (Fig. 2, Separate). “Separate” samples were combined at the end of the incubation. Incubation of the membranes together resulted in efficient processing of Ste13αHA (35%; Fig. 2 A). The products of both reactions were then added to magnetic beads coated with affinity purified anti-Kex2p tail antibodies. After immunoisolation, beads were assayed for Kex2p and DPAP activity. In both samples, >90% of Kex2p activity was associated with the beads after immunoisolation (unpublished data). In Kex2p-containing membranes immunoisolated from the “separate” reaction, no measurable DPAP activity was detected, whereas, ∼12% of the total DPAP activity from the “mixed” reaction was associated with the beads (Fig. 2 B). This indicated that the observed Ste13αHA processing correlated with membrane fusion, resulting in membranes containing both Kex2p and DPAP activity.
Figure 2.
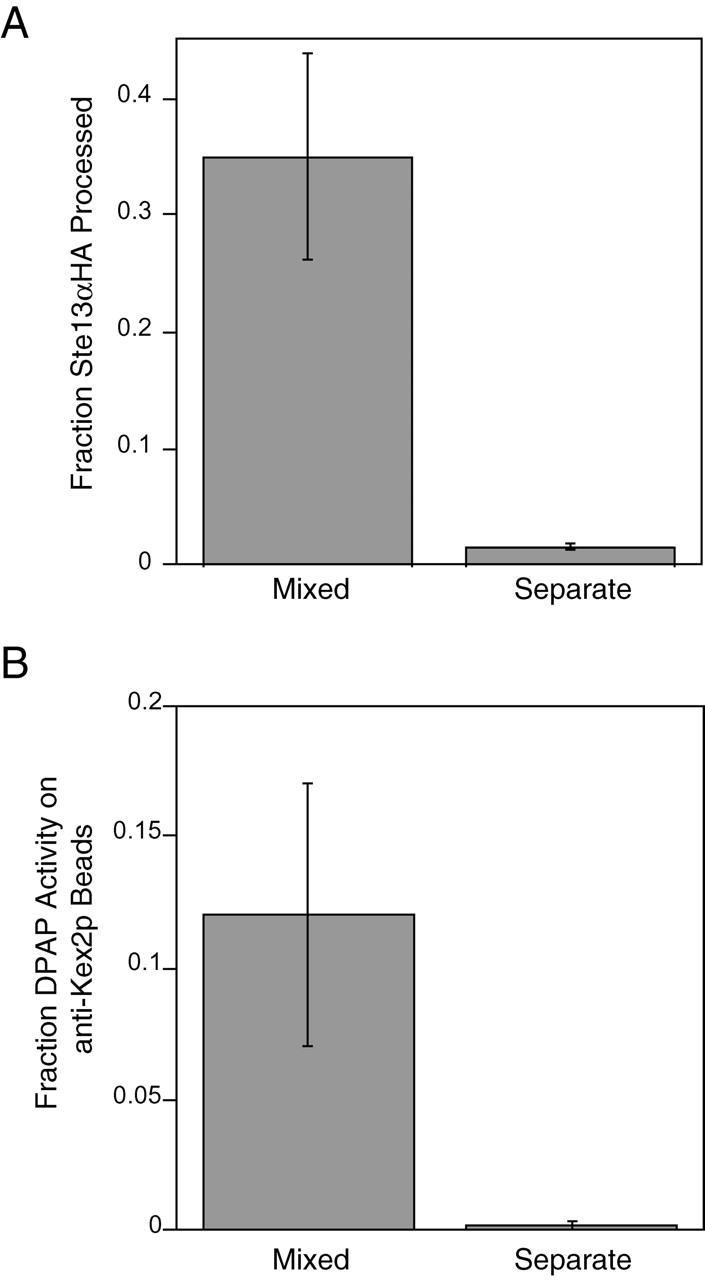
Ste13αHA processing represents a membrane fusion event; DPAP activity is associated with immunopurified Kex2p vesicles. (A) “Mixed” reaction was under standard reaction conditions. Control reaction (Separate) was set up as two half reactions containing MSS membranes from either Ste13αHA- or Kex2p-expressing strains, which were incubated separately at 30°C and then combined on ice. Half of each reaction was used to determine the fraction of Ste13αHA processed. (B) The remainder of the mixed and separate reactions was bound to anti-Kex2p beads, washed, and assayed for the percent of total DPAP activity, which remained bound to the beads. The somewhat lower yield of immunoisolated DPAP activity (12%) compared with the fraction processed (35%) likely resulted from fragmentation of the membranes during the isolation procedure.
Characteristics of the cell-free fusion reaction
The cell-free membrane fusion that we observed exhibited features characteristic of other cell-free biological membrane fusion reactions. First, although Ste13αHA processing in the crude system did not require addition of exogenous ATP (Fig. 3 A), omission of the ATP regeneration system completely blocked processing (Fig. 3 A), indicating that the reaction was ATP dependent. Second, processing of Ste13αHA occurred with maximal efficiency between 25 and 30°C and was inhibited at temperatures below 15°C or above 35°C (Fig. 3 B), similar to the temperature dependence of other cell-free biological membrane fusion reactions (Baker et al., 1988; Latterich and Schekman, 1994). Third, dilution of the MSS membranes progressively inhibited Ste13αHA processing, indicating that membrane concentration was important for efficient fusion (Fig. 3 C). Fourth, addition of Triton X-100 (1% wt vol−1) completely inhibited Kex2p processing of Ste13αHA even though both Kex2p and DPAP were fully active under these conditions (unpublished data), indicating that membrane integrity was essential for efficient processing (Fig. 3 D). Furthermore, this result shows that for processing to be observed in this dilute system Kex2p must be coconcentrated along with the substrate Ste13αHA in the lumenal space of fused late Golgi membrane vesicles. Taken together, the characteristics of the reaction suggested that the observed Kex2p-dependent processing of Ste13αHA represented a biologically relevant vesicular transport or membrane fusion event between membrane compartments containing Kex2p and Ste13αHA.
Figure 3.

Characterization of in vitro TGN membrane fusion. (A) Processing of Ste13αHA requires an ATP regeneration system. Control reactions (±Kex2p) included 1.5 mM exogenous Mg-ATP, 40 mM phosphocreatine, and 0.125 mg ml−1 creatine kinase. Omission of Mg-ATP (−ATP), or phosphocreatine/creatine kinase, (−Regen) is indicated. (B) Temperature dependence of Ste13αHA processing in vitro. Reactions were performed at each temperature using lysates from either KEX2 or kex2Δ strains. (C) Dilution sensitivity of Ste13αHA processing. Reactions were diluted to the indicated extents before (Pre-reaction) or after (Post-reaction) incubation under reaction conditions. (D) Detergent sensitivity. MSS membranes from either KEX2 or kex2Δ strains were combined with MSS membranes from a Ste13αHA-expressing strain, and reactions were performed in the presence or absence of 1% Triton X-100.
Membrane fusion events catalyzed by NSF/Sec18p are sensitive to NEM (Block et al., 1988). We observed partial inhibition of fusion by preincubation of lysates in the presence of NEM (Fig. 4 A). Many membrane fusion events require Ca2+ (Rexach and Schekman, 1991; Bark and Wilson, 1994; Peters and Mayer, 1998). Preincubation of MSS membranes with EGTA had no effect on fusion (unpublished data). In contrast, preincubation with the more rapid chelator BAPTA resulted in strong inhibition of fusion, which could be rescued by addition of millimolar concentrations of Ca2+ but not Mg+2 (Fig. 4 B). This suggested that the Ca2+ required for membrane fusion might be released from a membrane-protected pool immediately before it functions as has been suggested in the case of other fusion events (DeBello et al., 1995; Schiavo et al., 1995; Peters and Mayer, 1998). It was possible that this effect on the processing reaction was due to the Ca2+ dependence of Kex2p proteolytic activity (Fuller et al., 1989a) rather than an effect on the fusion reaction itself, although the resistance to EGTA argued against this interpretation. Furthermore, coimmunoisolation experiments demonstrated that incorporation of Ste13αHA into Kex2p-containing membranes, not processing of Ste13αHA per se, was inhibited by BAPTA (Fig. 4, C and D). Examination of the kinetics of a reaction partially inhibited by BAPTA revealed that both the initial rate and extent of the reaction were reduced (unpublished data).
Figure 4.

TGN membrane fusion is NEM-sensitive and requires Ca 2+. (A) Donor and acceptor MSS membranes were treated separately with NEM or equimolar ratio of NEM + DTT at 30°C for 5 min. NEM was quenched with equimolar concentration of DTT on ice before initiating reactions. (B) MSS membranes were pretreated with 5 mM BAPTA, 7.5 mM CaCl2, or 7.5 mM MgCl2 for 1 h on ice. Higher processing of Ste13αHA was observed when 7.5 mM CaCl2 was added to standard reactions (unpublished data). To confirm that this additional processing was due to Kex2p, control reactions using kex2Δ MSS membranes were performed in the presence of 7.5 mM CaCl2. Complete inhibition could be achieved with higher BAPTA concentrations. However, at these BAPTA concentrations addition of equimolar Ca2+ did not restore more than 50% of the wild-type reaction (unpublished data). (C) BAPTA inhibits membrane fusion and not Kex2p activity. Mixed and separate reactions between MSS membranes pretreated with 5 mM BAPTA or buffer as in B were performed as described in the legend to Fig. 2 A, and the fraction of Ste13αHA processed in each reaction is indicated (C). (D) The remainder of the mixed and separate reactions was bound to anti-Kex2p beads, washed, and assayed for the percent of total DPAP activity that remained bound to the beads as in the legend to Fig. 2 B. Roughly 1% of the DPAP activity from the BAPTA-treated mixed reactions was associated with the anti-Kex2p beads, whereas 7% of the DPAP activity from the control mixed reaction (−BAPTA) coisolated with the beads, demonstrating that cointegration of the Ste13αHA and Kex2p into the same membranes and not simply the processing of the substrate by Kex2p was blocked by BAPTA.
Fusion was cytosol dependent. Membranes pelleted at 200,000 g (P200) to remove cytosol displayed poor processing, which could be stimulated in a concentration-dependent manner by addition of cytosol (Fig. 5 A). The reconstituted processing reaction proceeded with slower kinetics and to a lesser extent, suggesting that the reaction depended on one or more labile components (Fig. 5 B).
Figure 5.
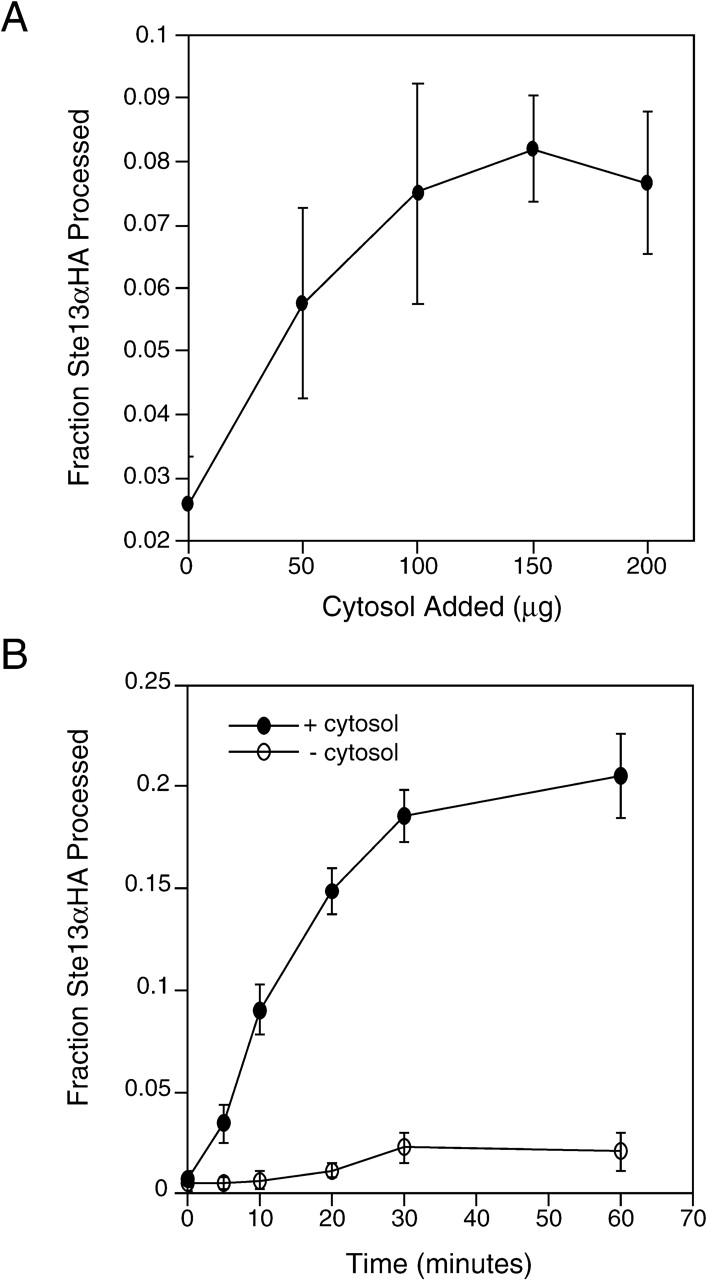
TGN membrane fusion is cytosol dependent. (A) P200 membranes and cytosol were prepared and incubated as described in Materials and methods except that reactions were incubated for 40 min. The average of four experiments is shown, and error bars represent the standard deviation from the mean. (B) Fourfold scaled up reactions containing P200 membranes were incubated under reaction conditions in the presence (660 μg) or absence of cytosol. Time points were collected as in the legend to Fig. 1.
Tlg SNARE complex components are required for cell-free membrane fusion
To better define the nature of the reaction, we examined the role of t-SNARE proteins known to function late in the secretory pathway in the fusion reaction by observing the effects of treating the MSS membranes with specific antisera. Preincubation of membranes with antisera against the t-SNAREs Tlg1p, Tlg2p, Vti1p, and the Sec1p homologue, Vps45p, completely inhibited membrane fusion (Fig. 6 A). In contrast, antiserum against Pep12p or the lumenal domain of Kex2p had no effect (Fig. 6 A). Finally, consistent with the partial inhibition observed upon addition of NEM, preincubation with antiserum against Sec18p partially inhibited the reaction (Fig. 6 A). To verify the specificity of inhibition by the various antisera, antigens in the form of purified tagged recombinant cytosolic domains (indicated as Δ–transmembrane domain [TMD]) of the SNARE proteins were used to compete with the inhibitory activity of the antisera. Addition of excess purified Tlg2ΔTMDp blocked inhibition by anti-Tlg2p serum (Fig. 6 B), confirming the specificity of antibody inhibition. At levels of anti-Tlg1p antisera that inhibited ∼60% of fusion, preincubation of serum with 10 μg of purified Tlg1ΔTMDp blocked the majority of anti-Tlg1p inhibition (Fig. 6 B). The antigen competition was specific for Tlg1p because an equivalent molar concentration of Vti1ΔTMDp (also 10 μg; the proteins have nearly identical molecular weights) was ineffective at blocking anti-Tlg1p inhibition (Fig. 6 B). Antigen competition experiments with anti-Vti1p serum at levels that reduced membrane fusion by ∼60% required high levels of antigen (>20 μM). These levels of Vti1ΔTMDp (antigen) inhibited fusion, presumably by a competition mechanism (Bennett et al., 1993; Hua and Scheller, 2001) even in the absence of antiserum (Fig. 6 B). Significantly, addition of Vti1ΔTMDp to the anti-Vti1p–inhibited reaction restored membrane fusion to the same level observed in the presence of Vti1ΔTMDp antigen alone (Fig. 6 B). As an additional control for the specificity of SNARE function in this reaction, we raised a new antiserum against the Tlg2p cytosolic domain (see Materials and methods) and compared the inhibitory effects of IgG purified from this new serum with IgG purified from antiserum raised against the ER/Golgi t-SNARE, Bet1p (Stone et al., 1997) (Fig. 6 C). After preincubation of MSS membranes with IgG levels roughly equivalent to 1 μl of unfractionated antiserum (∼6 μg), anti-Tlg2p IgG inhibited membrane fusion, whereas anti-Bet1 IgG had no effect. Taken together, these results argue for specific roles for Tlg1p, Tlg2p, and Vti1p and a potential role for Vps45p in the fusion reaction.
Figure 6.
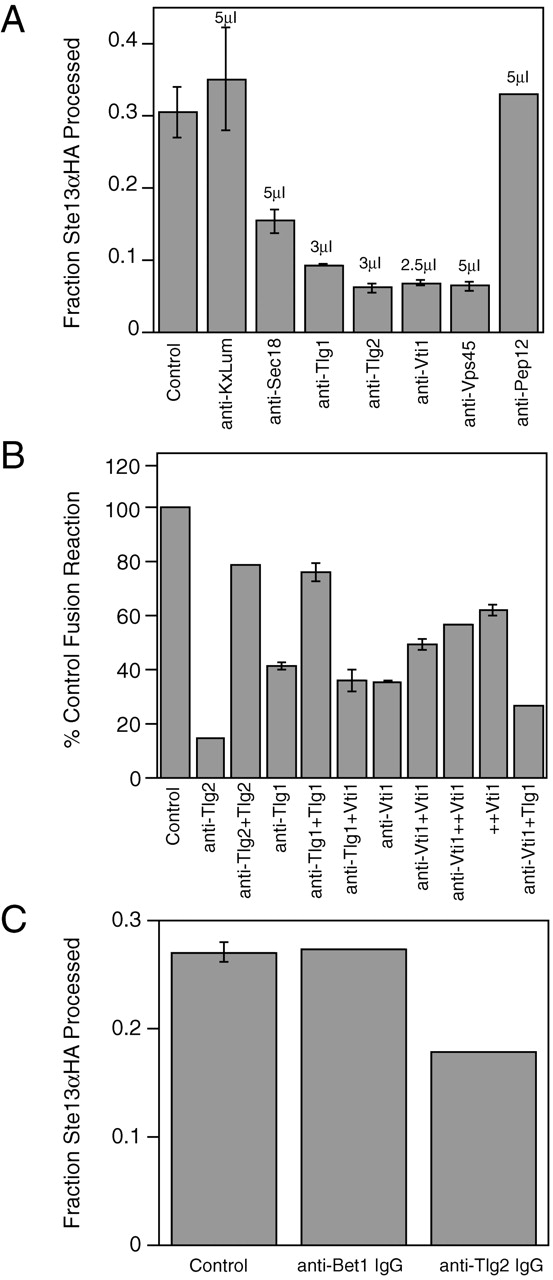
TGN SNARE complex is required for TGN membrane fusion. (A) MSS membranes were combined on ice and incubated 1 h with 3–5μL of antisera (indicated above the appropriate bar). Reaction mix was then added and fusion was initiated at 30°C. Except for anti-Pep12p, all preincubations were performed in duplicate. Control represents the average of control reactions for each of the antiserum inhibition experiments. The data presented for anti-Pep12p and are representative of three separate experiments. (B) Inhibition by anti-Tlg2p, anti-Tlg1p, and anti-Vti1p is antigen specific. Anti-Tlg2p anti- serum was preincubated with 20 μg purified recombinant His6-Tlg2p lacking the COOH-terminal TMD (Tlg2) or with buffer for 1 h. Anti-Tlg1p antiserum was preincubated with 10 μg purified recombinant His-tagged Tlg1p lacking the COOH-terminal TMD (Tlg1) or with 10 μg purified recombinant His6-Vti1 lacking the COOH-terminal TMD (Vti1) as a control. Anti-Vti1p antiserum was preincubated with either 10 μg His6-Vti1ΔTMD (+Vti1), 25 μg His6-Vti1ΔTMD (++Vti1), or 10 μg His6-Tlg1ΔTMD (Tlg1). MSS membranes were then added to anti- serum and incubated on ice for 1 h before initiating fusion reactions at 30°C. (C) IgG purified from anti-Tlg2 antiserum inhibits membrane fusion, whereas IgG purified from anti-Bet1 antiserum does not. IgG was fractionated from serum proteins as described in Materials and methods and concentrated on microcon-10 spin columns to ∼1–2mg/ml. MSS membranes were combined on ice and incubated for 1 h with ∼6 μg anti-Tlg2p or anti-Bet1p IgG as indicated. After incubation under standard reaction conditions (described in Materials and methods), reactions were stopped by addition of Triton X-100 to 1% and precleared of IgG by 30 min incubation with pansorbin. Resulting IgG-depleted reactions were then subjected to anti-HA IP and mock IP as described in Materials and methods.
Vps21p rab protein is required on one of the two participating membranes for cell-free fusion to proceed
All known membrane fusion events in the secretory pathway require a Ypt/rab protein (Lazar et al., 1997). To examine the role of the rab5 homologue Vps21p in this reaction, we prepared membranes from vps21 mutant strains expressing either Kex2p or Ste13αHA. We observed an elevated fraction of DPAP activity in MSS membranes prepared from the vps21 mutant strain that could not be precipitated by anti-HA (10%; as opposed to ≤2% observed in the case of wild-type strains), indicating that this fraction of the Ste13αHA had undergone Kex2-independent cleavage of the HA tag in vivo. Further Kex2-independent processing was not observed upon incubation of the membranes under reaction conditions (Fig. 7 A). When Kex2p- and Ste13αHA-containing membranes prepared from vps21 mutant strains were incubated together under reaction conditions, no additional processing of Ste13αHA above the elevated background level was observed (Fig. 7 B, column 4 compared with 6). In contrast, normal levels of processing of Ste13αHA were displayed by reactions in which either the Kex2p- or the Ste13αHA-containing membranes were prepared from a wild-type VPS21 strain (Fig. 7 B). These results suggested that Vps21p was essential for the membrane fusion reaction and could be donated by either MSS fraction.
Figure 7.
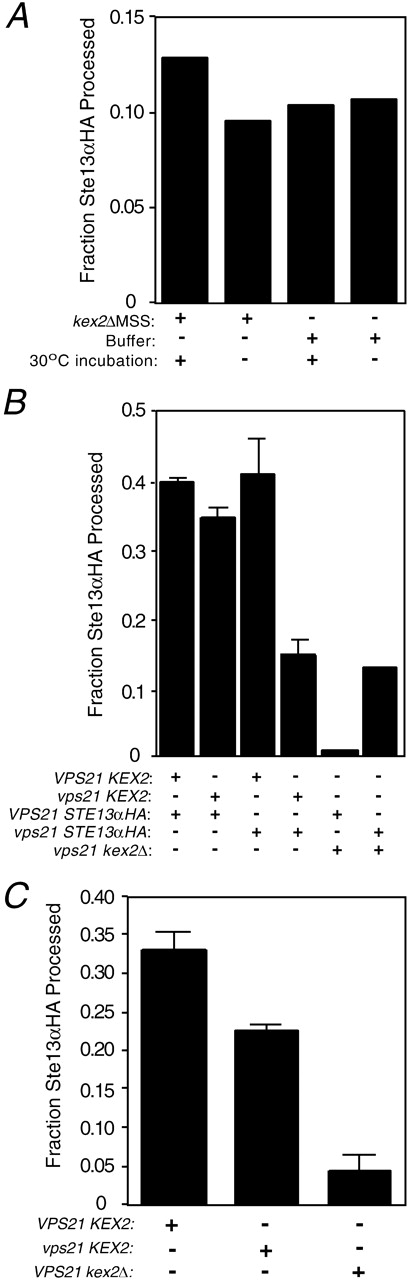
Vps21p function is required on only one membrane for TGN membrane fusion to proceed. (A) MSS membranes from Ste13αHA-expressing vps21 strain BLY9 were combined with either MSS from kex2Δ strain (JBY209) or buffer on ice. The samples were immunoprecipitated with anti-HA after incubations at either 30 or 0°C. (B) MSS membranes from Kex2p- or Ste13αHA-expressing vps21 strain BLY9 (or JBY209 as VPS21 kex2Δ control) were combined as indicated under standard reaction conditions. Data are representative of three separate experiments performed using two different MSS preparations. (C) P200 membranes from JBY209 expressing Ste13αHA (VPS21 Ste13αHA) were combined with MSS membranes from JBY209 (pCWKX10) (VPS21 KEX2), BLY9 (pCWKX10) (vps21 KEX2), or BLY9 (vps21 kex2Δ) under standard reaction conditions. Reactions in B and C were performed in duplicate.
Based on these data, it was possible that Vps21p function was required on only one of the two participating membranes. However, because Vps21p is found both on membranes and in the cytosol and presumably cycles between the two (Horazdovsky et al., 1994), it was possible that the cytosolic pool of wild-type Vps21p supplied by the VPS21 MSS could bind to the membranes from the vps21 strain, providing functional Vps21p on both membranes. If this were the case, then mixing Ste13αHA-containing VPS21 P200 membranes, from which the cytosolic pool of Vps21p had been removed, with Kex2p-containing vps21 MSS (i.e., membranes plus cytosol) should not result in membrane fusion. However, as shown in Fig. 7 C reactions containing P200 membranes from a Ste13αHA-expressing VPS21 strain and MSS membranes from a Kex2p-expressing vps21 strain supported a substantial level of fusion, two thirds as much as seen in a reaction containing P200 membranes from the Ste13αHA-containing VPS21 strain and MSS from the Kex2p-containing VPS21 strain. These data argue that the rab5 homologue Vps21p is required for the reaction and rab activity on either of the two participating membranes will suffice to support fusion.
Discussion
We report here the reconstitution and characterization of a membrane fusion event involving organelles enriched in TGN resident enzymes. This reaction displayed characteristics of other biological membrane fusion reactions: it depended on ATP and cytosol, and it was inhibited by NEM and rapid Ca2+ chelation. Sensitivity of the reaction to BAPTA but not EGTA is consistent with a requirement for a transient utilization of Ca2+ during fusion as has been suggested for homotypic fusion of vacuoles (Peters and Mayer, 1998) and fusion of synaptic vesicles at the nerve terminus (DeBello et al., 1995; Schiavo et al., 1995). The reaction was blocked by antisera against components of a late Golgi Tlg SNARE complex consisting of Tlg1p, Tlg2p, and Vti1p. Antisera against Vps45p also inhibited fusion, suggesting that Vps45p may function as the Sec1p component of this complex. Vps45p interacts both physically and functionally with Tlg2p and plays a role in multiple protein trafficking events originating at the TGN (Cowles et al., 1994; Bryant et al., 1998; Nichols et al., 1998; Abeliovich et al., 1999; Bassham et al., 2000). Vps21p rab function was required on one of the two membranes for fusion to proceed. Finally, we observed a partial requirement for Sec18p/NSF.
Mutations in the genes encoding the components of the Tlg SNARE complex give rise to distinguishable phenotypes, suggesting that each of these proteins catalyzes an unique set of fusion events (von Mollard et al., 1997; Abeliovich et al., 1999; Coe et al., 1999). The fusion event we measure here appears to represent one reaction in which these three proteins participate together. Consistent with our data, a t-SNARE complex of Tlg1p, Tlg2p, and Vti1p has been found to pair specifically with Snc1p to catalyze fusion when reconstituted in liposomes (Paumet et al., 2001). Preliminary evidence suggests that Snc1p is likely to function as the v-SNARE in conjunction with the Tlg SNARE complex in homotypic TGN fusion (unpublished data); however, this remains to be conclusively demonstrated. This study provides a biochemical assay for the coordinate function of all of these proteins in a single fusion event involving physiological membranes.
Steady-state localization of transmembrane proteins to the TGN in yeast depends on continuous cycles of transport between TGN and post-Golgi endosomal compartments (Vida et al., 1993; Cereghino et al., 1995; Piper et al., 1995; Cooper and Stevens, 1996; Brickner and Fuller, 1997; Bryant and Stevens, 1997). Thus, this reaction could hypothetically represent vesicular transport events between the TGN and the PVC. However, our data suggest that the reaction described here does not correspond to a vesicular transport event. First, the reaction did not display the extended lag phase typical of cell-free vesicular transport (Balch et al., 1984; Baker et al., 1988; Vida and Gerhardt, 1999). Second, whereas the PVC t-SNARE Pep12p is required for TGN to PVC transport and the class E Vps protein Vps27p is required for PVC to TGN transport, the reaction we observed required neither protein based on the following observations. Membranes prepared from a pep12 temperature-sensitive mutant strain (Burd et al., 1997) were able to support processing of Ste13αHA after a 5-min preincubation at the restrictive temperature to inactivate the protein (unpublished data). Moreover, anti-Pep12p antiserum failed to inhibit the reaction. Membranes prepared from vps27-null or temperature-sensitive mutants were fully competent to support processing of Ste13αHA (unpublished data). Finally, there was no requirement for the trans-Golgi localization signal (TLS)1 (Brickner and Fuller, 1997) in the cytosolic tail of Kex2p, which would be required if the reaction involved transport of Kex2p from the PVC to the TGN (unpublished data).
Several lines of evidence argue that the reaction described here measures a homotypic fusion event. Homotypic fusion involves fusion of like organelles through the pairing of t-SNAREs and v-SNAREs distributed comparably onto the fusing membranes. In the case of homotypic vacuolar fusion, v-SNAREs can be genetically ablated from one membrane and t-SNAREs from the other and fusion can still proceed (Wickner and Haas, 2000). In contrast, heterotypic fusion requires pairing of a v-SNARE on a vesicle with t-SNAREs on the target membrane. The absence of either blocks the reaction. Ideally, mutations of SNARE genes therefore can allow the classification of a fusion reaction as homotypic or heterotypic as in the case of vacuolar fusion (Wickner and Haas, 2000). However, because the existence of the compartments to which our reporter proteins are localized depends on the components of the Tlg SNARE complex, such an experiment would be difficult to interpret. An alternative way to distinguish homotypic and heterotypic fusion is the sensitivity of the reaction to pretreatment of one of the membranes with antiserum against one of the participating SNARE proteins. We have found that in the reconstituted reaction involving pelleted membranes and cytosol pretreatment of either membrane before centrifugation with antisera against Tlg1p or Tlg2p was sufficient to completely inhibit the fusion reaction (unpublished data). Although this result is somewhat different from what was found in vacuolar fusion (see above), it argues that identical v- and t-SNAREs are present and are functional in the reaction on both of the fusing membranes, a necessary characteristic of homotypic fusion. Vps21p rab function was required, but on only one of the two membranes, to support membrane fusion. The symmetry inherent in the fact that rab activity on either membrane was sufficient supports the conclusion that the reaction occurs between two identical membranes.
Because both Kex2p and Ste13p cycle between the TGN and late endosomal compartments and possibly through early endosomal compartments (unpublished data) the fusion reaction between Kex2p- and Ste13αHA-containing membranes could represent homotypic fusion of TGN or early or late endosomal membranes. It seems unlikely that this reaction involves late endosomal/PVC membranes because (a) antibodies against Pep12p, the major late endosomal t-SNARE, did not inhibit the fusion reaction and (b) MSS membranes from vps27-null mutants, which accumulate aberrant late endosomal membranes, exhibited normal fusion activity (unpublished data). Subsets of components of the Tlg SNARE complex have been implicated in endocytosis (Tlg1p, Tlg2p, and Vps21p [Abeliovich et al., 1998; Seron et al., 1998; Gerrard et al., 2000]), biosynthetic traffic to the vacuole (Vti1p and Vps21p [Horazdovsky et al., 1994; von Mollard et al., 1997]), and early endosome homotypic fusion in mammalian cells (rab5/Vps21p; [Gorvel et al., 1991]). These observations could implicate the involvement of early endosomal membranes in this reaction. Although this possibility cannot be excluded at this time, we believe it is more likely that the homotypic fusion reaction described here represents fusion of TGN membranes. First, up to 45% of Ste13αHA was processed in this cell-free reaction, suggesting that at least half of the Ste13αHA is localized to the membrane compartment that participates in the reaction. Ste13p has been shown to possess a strong TGN retention signal that slows its delivery to the late endosome, presumably by retarding its exit from the TGN (Bryant and Stevens, 1997). Therefore, it is unlikely that the majority of Ste13αHA is localized to the endocytic compartments. Second, Tlg1p and Tlg2p have been colocalized with Kex2p in the late Golgi and have been shown to be required for its proper localization and activity (Holthuis et al., 1998). These SNAREs do not colocalize extensively with Pep12p, a late endosomal marker, or with Sec7p, an early Golgi marker (Lewis et al., 2000).
We hope to use the cell-free fusion assay described here to analyze the function of the Tlg SNARE complex and identify the cytosolic components required for the reaction. The basic assay we have described may also be applied to reconstitute other transport and fusion events in the TGN/endosomal system by exploiting the use of distinct localization signals or accumulation in specific compartments to achieve differential localization of the enzyme (Kex2p) and substrate (Ste13αHA). Finally, these results call attention to the need for developing assays for TGN homotypic fusion in vivo in order to understand its importance for TGN membrane protein localization and TGN function.
Materials and methods
Antibodies and reagents
Monoclonal antihemagglutinin (HA) antibody 12CA5 was from Roche, and rabbit anti–mouse IgG and affinity purified goat anti–rabbit IgG/Fc fragment-specific were from Jackson ImmunoResearch Laboratories. Antisera against Pep12p/Vps45p, Vti1p, Tlg1p/Tlg2p, Bet1p, and Sec18p were provided by Drs. Scott Emr (University of California, San Diego, CA), Tom Stevens (University of Oregon, Eugene, OR), Hugh R.B. Pelham (Medical Research Council, Cambridge, UK), Randy Schekman (University of California, Berkeley, CA), and William T. Wickner (Dartmouth University, Hanover, NH), respectively. Polyclonal rabbit anti-Tlg2p antiserum used in Fig. 6 C was raised against Tlg2p cytosolic domain purified from Escherichia coli as described for anti-Tlg2p antiserum obtained from Hugh R.B. Pelham (Medical Research Council) (Holthuis et al., 1998). Polyclonal antisera were heat treated at 70°C for 45 min in a PerkinElmer 9600 thermocycler to inactivate serum DPAP.
FCS was from GIBCO BRL. Boc-Leu-Lys-Arg-7-amino-4-methylcoumarin (LKR-AMC) was from Bachem and Ala-Pro-AMC (AP-AMC) was from Enzyme Systems Products. Restriction endonucleases and DNA modification enzymes were from New England Biolabs, Inc. Unless indicated otherwise, all other chemicals and reagents were from Sigma-Aldrich.
Strains and plasmids
The following strains were used: JBY209 (ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 kex2Δ::hisG dap2Δ::kan r pep4Δ::HIS3 ste13Δ::LEU2 MATα), JBY209a (isogenic with JBY209, except that it is MATa), and BLY9 (his3 leu2 trp1 ura3 kex2Δ::hisG dap2Δ::kan r pep4Δ::HIS3 ste13Δ::LEU2 vps21). BLY9 was constructed by crossing the vps21 allele from strain 0587-21 (provided by Dr. Scott Emr, University of California, San Diego, CA) into JBY209a and isolating His+, Leu+, G418r, Vps−, and kex2Δ segregants after sporulation. To create Kex2p- or Ste13αHA-expressing strains, plasmids pCWKX10 or pSTE13αHA, respectively, were introduced by transformation. Untransformed JBY209 was used as the kex2Δ control.
Plasmids pCWKX10 (Wilcox et al., 1992), containing the KEX2 gene under its own promoter, pTLG2HISN1 and pTLG1HISC1 (Holthuis et al., 1998), and pET28aHis6VTI1ΔTMD (Fukuda et al., 2000) have been described. Plasmid pSTE13αHA was constructed as follows. The STE13 gene was amplified by PCR from pBRSTE13 (Jeremy Thorner, University of California, Berkeley, CA) using the following primers (5′ to 3′): GTTATTCGTGTAAAAAATCTAGAAAGCCCCTA, which anneals directly upstream of the start codon and introduces an XbaI site and CAATCATCCATAAAGAATTCTAAATGCAAA, which anneals at the end of STE13 and both introduces an EcoRI site and changes the termination codon to GAA. The PCR product was ligated between the XbaI and EcoRI sites of p416TEF (Mumberg et al., 1995). The resulting plasmid, pTEF-STE13, was digested with EcoRI and SalI and ligated to the following phosphorylated oligonucleotides, which had been annealed to each other (5′ to 3′): AATTGGATGCATCGGAATTCAGCGGCCGCTTG and TCGACAAGCGGCCGCTGAATTCCGATGCATCC. The resulting plasmid, pTEF-STE13 linker, was digested with NsiI and ligated to the following phosphorylated and annealed oligonucleotides (5′ to 3′): TGGCATTGGTTGCAACTAAAACCTGGCCAACCAATGTACAAGAGAGATGCA and TCTCTCTTGTACATTGGTTGGCCAGGTTTTAGTTGCAACCAATGCCATGCA, encoding the following amino acid sequence from prepro–α-factor: Trp His Leu Gln Leu Lys Pro Gly Gln Pro Met Tyr Lys Arg Asp Ala, confirmed by DNA sequencing. The resulting plasmid, pSTE13α, was digested with NotI and ligated to NotI-digested triple HA tag (Tyers et al., 1992), giving pSTE13αHA.
Preparation of membranes and cytosol
Permeabilized cells were prepared from JBY209 containing pCWKX10 or pSTE13αHA as described (Baker et al., 1988). Frozen spheroplasts (200 μl) were thawed (25°C, 2 min) and centrifuged (5 min, 14,000 rpm), and MSS fraction (containing microsomes and cytosol) was collected. For assays in which membranes were separated from cytosol, MSS membranes were diluted fourfold with lysis buffer and centrifuged through a 250-μl band of 12.5% ficoll in a TLS55 rotor (Beckman Coulter) at 55,000 rpm (200,000 g at ravg) for 1 h at 4°C to generate a high speed pellet fraction (P200). The P200 was resuspended in lysis buffer to 15% of the MSS volume and assayed for Kex2p or DPAP activity. P200 fractions were diluted to the same specific activity as the MSS membranes; 10 μl were used per reaction. Cytosol was prepared by centrifugation of MSS from JBY209 in a TLS55 rotor at 55,000 rpm for 1 h at 4°C and was concentrated on microcon-10 columns (Millipore) to a final protein concentration of 20–25 mg/ml as determined by the Bradford assay (Bio-Rad Laboratories).
Cell-free fusion
10 μl of MSS membranes from both the Kex2p- and Ste13αHA-expressing strains were added to 10 μl of 3× reaction mix (0.2 M sorbitol, 10 mM Hepes, pH 7, 75 mM KOAc, 4 mM MgOAc, 0.25 mM EGTA, 140 mM phosphocreatine, 0.375 mg ml−1 creatine kinase, and 9 mM CaCl2) on ice. Reactions were started by shifting to 30°C and unless indicated otherwise were incubated for 20 min. 10 μl were then removed and added to tubes containing either immunoprecipitation (IP) mix (1% Triton X-100, 1 mM EDTA, 20 μl pansorbin, 1 μl 12CA5 monoclonal anti-HA, and 1 μl rabbit anti–mouse IgG) or mock IP mix (1% Triton X-100, 1 mM EDTA, 20 μl pansorbin, and 2 μl water). IPs were incubated at RT with gentle agitation for 30 min. Pansorbin was pelleted, and 30 μl of each supernatant fraction were assayed for DPAP activity. The fraction of Ste13αHA processed was calculated as the ratio of DPAP activity in the supernatant fraction (i.e., the activity that was immunodepletion resistant) to the DPAP activity in the mock IP reaction (i.e., the total). Error bars represent the standard deviation of the average of at least two reactions.
Enzymatic assays
DPAP assays were performed as described (Julius et al., 1983). The supernatant fraction from the IP reactions (30 μl) was added to 30 μl 2× DPAP reaction mix (240 mM Hepes, pH 8, 1% Triton X-100, 200 μM AP-AMC), and reactions were incubated in 96-well plates at 30°C in an f max plate fluorimeter (360 nm excitation/460 nm emission filter pair; Molecular Devices). Data were collected approximately every 20 s for 20 min. Kex2p was assayed as described (Brenner and Fuller, 1992) except that LKR-AMC was used as substrate and 5 mM o-phenanthroline was included to inhibit a membrane-associated Kex2p-independent activity present in crude lysates (Sipos and Fuller, 2001).
Immunoisolation of Kex2p membranes
Anti-Kex2p cytosolic tail antibodies were affinity purified (Redding et al., 1991). Dynabeads (Dynal) were covalently coated with affinity purified goat anti-rabbit IgG/Fc fragment-specific antibodies and bound to affinity purified anti-Kex2p tail antibodies as recommended by the manufacturer. Beads were washed three times with 50 mM Hepes, pH 7.0, 200 mM KOAc, 2 mM EDTA, and 5% FCS before use.
Reactions (scaled up twofold) were performed as described above. In control reactions, Kex2p- and Ste13αHA-expressing membranes were incubated separately in reaction mix and combined after incubation. At the end of the incubation, half of each reaction was processed for IP or mock IP to determine processing efficiency. The remainder (30 μl) was adjusted to 50 mM Hepes, pH 7.0, 200 mM KOAc, 2 mM EDTA, 5% FCS, and 0.8 M sorbitol in a final volume of 50 μl and added to 150 μg Dynabeads. Binding was performed for 2 h with slow rotation at RT. Beads were isolated magnetically, washed three times with cold 50 mM Hepes, pH 7, 200 mM KOAc, and 0.8 M sorbitol and assayed for Kex2p and DPAP activity.
Preparation of soluble domains of Tlg2p, Tlg1 p, and Vti1p
His6-Tlg2ΔTMDp was expressed in E. coli BL21 (DE3) and purified using ProBond™ (Invitrogen) according to manufacturer's denaturing protocol. Recombinant protein was eluted with 20 mM NaPO4, pH 4.0, and 1 M urea. Fractions enriched for recombinant protein were pooled, dialyzed in 20 mM NaPO4, pH 7.0, 1 M urea, and concentrated to 10–20 mg ml−1 on microcon-10 columns. Tlg1-His6ΔTMDp and His6-Vti1ΔTMDp were expressed and purified as described above except that denaturants were omitted during lysis and purification and elution was performed with 400 mM imidazole-HCl in 20 mM NaPO4, pH 7.4, and 500 mM NaCl.
Antigen competition
Polyclonal antiserum (∼100 μg total protein) was incubated with 10–40 μg recombinant protein on ice for 1 h. Antigen-saturated serum was then incubated with MSS membranes on ice for 1 h, and the membranes were tested for fusion competence.
IgG purification
Antisera were dialyzed overnight in 500-vol 20 mM KPO4, pH 7.2. Dialyzed serum was loaded onto a column containing equal volumes of CM- and DEAE-Sepharose. Flow through was collected and concentrated on microcon-10 columns. IgG purity and concentration were determined by SDS-PAGE and the Bradford assay.
Acknowledgments
We are grateful to the following for generously providing reagents, strains, and antisera and for discussing unpublished data: Scott Emr, Jeffrey Gerst, Todd Graham, Sean Munro, Hugh R.B. Pelham, James E. Rothman, Randy Schekman, Tom Stevens, and William T. Wickner. We also thank Scott Emr, Daniel J. Klionsky, Hugh R.B. Pelham, Gustavo Pesce, James E. Rothman, Ursula Rüegsegger, and members of the Fuller lab for helpful discussions and comments on the article.
This work was supported in part by National Institutes of Health grant GM50915 to R.S. Fuller and by Genetics Training grant GM07544 to J.M. Blanchette.
J.H. Brickner and J.M. Blanchette contributed equally to this work.
J.H. Brickner's present address is Dept. of Biochemistry and Biophysics, University of California, San Francisco, CA 94143.
Footnotes
Abbreviations used in this paper: DPAP, dipeptidyl aminopeptidase; HA, hemagglutinin; IP, immunoprecipitation; MSS, medium speed supernatant; NEM, N-ethylmaleimide; NSF, NEM-sensitive fusion protein; P200, 200,000 g pellet; PVC, prevacuolar compartment; SNARE, soluble NSF attachment protein receptor; TGN, trans-Golgi network; TLS, trans-Golgi localization signal; TMD, transmembrane domain; t-SNARE, target membrane SNARE; v-SNARE, vesicle SNARE.
References
- Abeliovich, H., E. Grote, P. Novick, and S. Ferro-Novick. 1998. Tlg2p, a yeast syntaxin homolog that resides on the Golgi and endocytic structures. J. Biol. Chem. 273:11719–11727. [DOI] [PubMed] [Google Scholar]
- Abeliovich, H., T. Darsow, and S.D. Emr. 1999. Cytoplasm to vacuole trafficking of aminopeptidase I requires a t-SNARE-Sec1p complex composed of Tlg2p and Vps45p. EMBO J. 18:6005–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonin, W., C. Holroyd, D. Fasshauer, S. Pabst, G. Fischer Von Mollard, and R. Jahn. 2000. A SNARE complex mediating fusion of late endosomes defines conserved properties of SNARE structure and function. EMBO J. 19:6453–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, D., L. Hicke, M. Rexach, M. Schleyer, and R. Schekman. 1988. Reconstitution of SEC gene product-dependent intercompartmental protein transport. Cell. 54:335–344. [DOI] [PubMed] [Google Scholar]
- Balch, W.E., W.G. Dunphy, W.A. Braell, and J.E. Rothman. 1984. Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell. 39:405–416. [DOI] [PubMed] [Google Scholar]
- Bark, I.C., and M.C. Wilson. 1994. Regulated vesicular fusion in neurons: snapping together the details. Proc. Natl. Acad. Sci. USA. 91:4621–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassham, D.C., A.A. Sanderfoot, V. Kovaleva, H. Zheng, and N.V. Raikhel. 2000. AtVPS45 complex formation at the trans-Golgi network. Mol. Biol. Cell. 11:2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M.K., J.E. Garcia-Arraras, L.A. Elferink, K. Peterson, A.M. Fleming, C.D. Hazuka, and R.H. Scheller. 1993. The syntaxin family of vesicular transport receptors. Cell. 74:863–873. [DOI] [PubMed] [Google Scholar]
- Block, M.R., B.S. Glick, C.A. Wilcox, F.T. Wieland, and J.E. Rothman. 1988. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc. Natl. Acad. Sci. USA. 85:7852–7856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, C., and R.S. Fuller. 1992. Structural and enzymatic characterization of a purified prohormone-processing enzyme: secreted, soluble Kex2 protease. Proc. Natl. Acad. Sci. USA. 89:922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner, J.H., and R.S. Fuller. 1997. SOI1 encodes a novel, conserved protein that promotes TGN-endosomal cycling of Kex2p and other membrane proteins by modulating the function of two TGN localization signals. J. Cell Biol. 139:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, N.J., and A. Boyd. 1993. Immunoisolation of Kex2p-containing organelles from yeast demonstrates colocalisation of three processing proteinases to a single Golgi compartment. J. Cell Sci. 106:815–822. [DOI] [PubMed] [Google Scholar]
- Bryant, N.J., and T.H. Stevens. 1997. Two separate signals act independently to localize a yeast late Golgi membrane protein through a combination of retrieval and retention. J. Cell Biol. 136:287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant, N.J., R.C. Piper, S.R. Gerrard, and T.H. Stevens. 1998. Traffic into the prevacuolar/endosomal compartment in Saccharomyces cerevisiae: a VPS45-dependent intracellular route and a VPS45-independent endocytic route. Eur. J. Cell Biol. 76:43–52. [DOI] [PubMed] [Google Scholar]
- Burd, C.G., M. Peterson, C.R. Cowles, and S.D. Emr. 1997. A novel Sec18p/NSF-dependent complex required for Golgi-to-endosome transport in yeast. Mol. Biol. Cell. 8:1089–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr, C.M., E. Grote, M. Munson, F.M. Hughson, and P.J. Novick. 1999. Sec1p binds to SNARE complexes and concentrates at sites of secretion. J. Cell Biol. 146:333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereghino, J.L., E.G. Marcusson, and S.D. Emr. 1995. The cytoplasmic tail domain of the vacuolar protein sorting receptor Vps10p and a subset of VPS gene products regulate receptor stability, function, and localization. Mol. Biol. Cell. 6:1089–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe, J.G., A.C. Lim, J. Xu, and W. Hong. 1999. A role for Tlg1p in the transport of proteins within the Golgi apparatus of Saccharomyces cerevisiae. Mol. Biol. Cell. 10:2407–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, A.A., and T.H. Stevens. 1996. Vps10p cycles between the late-Golgi and prevacuolar compartments in its function as the sorting receptor for multiple yeast vacuolar hydrolases. J. Cell Biol. 133:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles, C.R., S.D. Emr, and B.F. Horazdovsky. 1994. Mutations in the VPS45 gene, a SEC1 homologue, result in vacuolar protein sorting defects and accumulation of membrane vesicles. J. Cell Sci. 107:3449–3459. [DOI] [PubMed] [Google Scholar]
- DeBello, W.M., V. O'Connor, T. Dresbach, S.W. Whiteheart, S.S. Wang, F.E. Schweizer, H. Betz, J.E. Rothman, and G.J. Augustine. 1995. SNAP-mediated protein-protein interactions essential for neurotransmitter release. Nature. 373:626–630. [DOI] [PubMed] [Google Scholar]
- Fukuda, R., J.A. McNew, T. Weber, F. Parlati, T. Engel, W. Nickel, J.E. Rothman, and T.H. Sollner. 2000. Functional architecture of an intracellular membrane t-SNARE. Nature. 407:198–202. [DOI] [PubMed] [Google Scholar]
- Fuller, R.S., A. Brake, and J. Thorner. 1989. a. Yeast prohormone processing enzyme (KEX2 gene product) is a Ca2+-dependent serine protease. Proc. Natl. Acad. Sci. USA. 86:1434–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller, R.S., A.J. Brake, and J. Thorner. 1989. b. Intracellular targeting and structural conservation of a prohormone-processing endoprotease. Science. 246:482–486. [DOI] [PubMed] [Google Scholar]
- Gerrard, S.R., N.J. Bryant, and T.H. Stevens. 2000. VPS21 controls entry of endocytosed and biosynthetic proteins into the yeast prevacuolar compartment. Mol. Biol. Cell. 11:613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvel, J.P., P. Chavrier, M. Zerial, and J. Gruenberg. 1991. rab5 controls early endosome fusion in vitro. Cell. 64:915–925. [DOI] [PubMed] [Google Scholar]
- Graham, T.R., and S.D. Emr. 1991. Compartmental organization of Golgi-specific protein modification and vacuolar protein sorting events defined in a yeast sec18 (NSF) mutant. J. Cell Biol. 114:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote, E., C.M. Carr, and P.J. Novick. 2000. Ordering the final events in yeast exocytosis. J. Cell Biol. 151:439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata, Y., C.A. Slaughter, and T.C. Sudhof. 1993. Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature. 366:347–351. [DOI] [PubMed] [Google Scholar]
- Holthuis, J.C., B.J. Nichols, S. Dhruvakumar, and H.R. Pelham. 1998. Two syntaxin homologues in the TGN/endosomal system of yeast. EMBO J. 17:113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horazdovsky, B.F., G.R. Busch, and S.D. Emr. 1994. VPS21 encodes a rab5-like GTP binding protein that is required for the sorting of yeast vacuolar proteins. EMBO J. 13:1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua, Y., and R.H. Scheller. 2001. Three SNARE complexes cooperate to mediate membrane fusion. Proc. Natl. Acad. Sci. USA. 98:8065–8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius, D., L. Blair, A. Brake, G. Sprague, and J. Thorner. 1983. Yeast alpha factor is processed from a larger precursor polypeptide: the essential role of a membrane-bound dipeptidyl aminopeptidase. Cell. 32:839–852. [DOI] [PubMed] [Google Scholar]
- Latterich, M., and R. Schekman. 1994. The karyogamy gene KAR2 and novel proteins are required for ER-membrane fusion. Cell. 78:87–98. [DOI] [PubMed] [Google Scholar]
- Lazar, T., M. Gotte, and D. Gallwitz. 1997. Vesicular transport: how many Ypt/Rab-GTPases make a eukaryotic cell? Trends Biochem. Sci. 22:468–472. [DOI] [PubMed] [Google Scholar]
- Lewis, M.J., B.J. Nichols, C. Prescianotto-Baschong, H. Riezman, and H.R. Pelham. 2000. Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol. Biol. Cell. 11:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNew, J.A., F. Parlati, R. Fukuda, R.J. Johnston, K. Paz, F. Paumet, T.H. Sollner, and J.E. Rothman. 2000. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407:153–159. [DOI] [PubMed] [Google Scholar]
- Mills, I.G., A.T. Jones, and M.J. Clague. 1999. regulation of endosome fusion. Mol. Membr. Biol. 16:73–79. [DOI] [PubMed] [Google Scholar]
- Mumberg, D., R. Muller, and M. Funk. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 156:119–122. [DOI] [PubMed] [Google Scholar]
- Nichols, B.J., J.C. Holthuis, and H.R. Pelham. 1998. The Sec1p homologue Vps45p binds to the syntaxin Tlg2p. Eur. J. Cell Biol. 77:263–268. [DOI] [PubMed] [Google Scholar]
- Nothwehr, S.F., C.J. Roberts, and T.H. Stevens. 1993. Membrane protein retention in the yeast Golgi apparatus: dipeptidyl aminopeptidase A is retained by a cytoplasmic signal containing aromatic residues. J. Cell Biol. 121:1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumet, F., B. Brügger, F. Parlati, J.A. McNew, T.H. Söllner, and J.E. Rothman. 2001. A t-SNARE involved in endocytosis must be activated for fusion. J. Cell Biol. 155:961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, C., and A. Mayer. 1998. Ca2+/calmodulin signals the completion of docking and triggers a late step of vacuole fusion. Nature. 396:575–580. [DOI] [PubMed] [Google Scholar]
- Piper, R.C., A.A. Cooper, H. Yang, and T.H. Stevens. 1995. VPS27 controls vacuolar and endocytic traffic through a prevacuolar compartment in Saccharomyces cerevisiae. J. Cell Biol. 131:603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier, M.A., W. Xiao, J.C. Macosko, C. Chan, Y.K. Shin, and M.K. Bennett. 1998. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat. Struct. Biol. 5:765–769. [DOI] [PubMed] [Google Scholar]
- Redding, K., C. Holcomb, and R.S. Fuller. 1991. Immunolocalization of Kex2 protease identifies a putative late Golgi compartment in the yeast Saccharomyces cerevisiae. J. Cell Biol. 113:527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach, M.F., and R.W. Schekman. 1991. Distinct biochemical requirements for the budding, targeting, and fusion of ER-derived transport vesicles. J. Cell Biol. 114:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman, J.E., and F.T. Wieland. 1996. Protein sorting by transport vesicles. Science. 272:227–234. [DOI] [PubMed] [Google Scholar]
- Ruohola, H., A.K. Kabcenell, and S. Ferro-Novick. 1988. Reconstitution of protein transport from the endoplasmic reticulum to the Golgi complex in yeast: the acceptor Golgi compartment is defective in the sec23 mutant. J. Cell Biol. 107:1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo, G., M.J. Gmachl, G. Stenbeck, T.H. Sollner, and J.E. Rothman. 1995. A possible docking and fusion particle for synaptic transmission. Nature. 378:733–736. [DOI] [PubMed] [Google Scholar]
- Seron, K., V. Tieaho, C. Prescianotto-Baschong, T. Aust, M.O. Blondel, P. Guillaud, G. Devilliers, O.W. Rossanese, B.S. Glick, H. Riezman, et al. 1998. A yeast t-SNARE involved in endocytosis. Mol. Biol. Cell. 9:2873–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipos, G., and R.S. Fuller. 2001. Separation of Golgi and endosomal compartments. Methods Enzymol. In press. [DOI] [PubMed] [Google Scholar]
- Sollner, T., S.W. Whiteheart, M. Brunner, H. Erdjument-Bromage, S. Geromanos, P. Tempst, and J.E. Rothman. 1993. SNAP receptors implicated in vesicle targeting and fusion. Nature. 362:318–324. [DOI] [PubMed] [Google Scholar]
- Stone, S., M. Sacher, Y. Mao, C. Carr, P. Lyons, A.M. Quinn, and S. Ferro-Novick. 1997. Bet1p activates the v-SNARE Bos1p. Mol. Biol. Cell. 8:1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, R.B., D. Fasshauer, R. Jahn, and A.T. Brunger. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 395:347–353. [DOI] [PubMed] [Google Scholar]
- Tyers, M., G. Tokiwa, R. Nash, and B. Futcher. 1992. The Cln3-Cdc28 kinase complex of S. cerevisiae is regulated by proteolysis and phosphorylation. EMBO J. 11:1773–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida, T., and B. Gerhardt. 1999. A cell-free assay allows reconstitution of Vps33p-dependent transport to the yeast vacuole/lysosome. J. Cell Biol. 146:85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vida, T.A., G. Huyer, and S.D. Emr. 1993. Yeast vacuolar proenzymes are sorted in the late Golgi complex and transported to the vacuole via a prevacuolar endosome-like compartment. J. Cell Biol. 121:1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mollard, G.F., S.F. Nothwehr, and T.H. Stevens. 1997. The yeast v-SNARE Vti1p mediates two vesicle transport pathways through interactions with the t-SNAREs Sed5p and Pep12p. J. Cell Biol. 137:1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, G., and V. Malhotra. 1998. The organisation of the Golgi apparatus. Curr. Opin. Cell Biol. 10:493–498. [DOI] [PubMed] [Google Scholar]
- Wickner, W., and A. Haas. 2000. Yeast homotypic vacuole fusion: a window on organelle trafficking mechanisms. Annu. Rev. Biochem. 69:247–275. [DOI] [PubMed] [Google Scholar]
- Wilcox, C.A., and R.S. Fuller. 1991. Posttranslational processing of the prohormone-cleaving Kex2 protease in the Saccharomyces cerevisiae secretory pathway. J. Cell Biol. 115:297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox, C.A., K. Redding, R. Wright, and R.S. Fuller. 1992. Mutation of a tyrosine localization signal in the cytosolic tail of yeast Kex2 protease disrupts Golgi retention and results in default transport to the vacuole. Mol. Biol. Cell. 3:1353–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]