

Abstract
Enhanced formation of reactive oxygen species (ROS), superoxide (O2 ·−), and hydrogen peroxide (H2O2) may result in either apoptosis or other forms of cell death. Here, we studied the mechanisms underlying activation of the apoptotic machinery by ROS. Exposure of permeabilized HepG2 cells to O2 ·− elicited rapid and massive cytochrome c release (CCR), whereas H2O2 failed to induce any release. Both O2 ·− and H2O2 promoted activation of the mitochondrial permeability transition pore by Ca2+, but Ca2+-dependent pore opening was not required for O2 ·−-induced CCR. Furthermore, O2 ·− alone evoked CCR without damage of the inner mitochondrial membrane barrier, as mitochondrial membrane potential was sustained in the presence of extramitochondrial ATP. Strikingly, pretreatment of the cells with drugs or an antibody, which block the voltage-dependent anion channel (VDAC), prevented O2 ·−-induced CCR. Furthermore, VDAC-reconstituted liposomes permeated cytochrome c after O2 ·− exposure, and this release was prevented by VDAC blocker. The proapoptotic protein, Bak, was not detected in HepG2 cells and O2 ·−-induced CCR did not depend on Bax translocation to mitochondria. O2 ·−-induced CCR was followed by caspase activation and execution of apoptosis. Thus, O2 ·− triggers apoptosis via VDAC-dependent permeabilization of the mitochondrial outer membrane without apparent contribution of proapoptotic Bcl-2 family proteins.
Keywords: superoxide anion; mitochondria; VDAC; cytochrome c; apoptosis
Introduction
The superoxide (O2 ·−)*free radical has come to play a pivotal role in a wide variety of pathophysiological conditions. In an aerobic system, sequential reduction of molecular oxygen leads to the generation of O2 ·−, which is rapidly converted to H2O2 by superoxide dismutase (SOD) (Jacobson, 1996). O2 ·− can also rapidly react with an another free radical, nitric oxide (NO), yielding more toxic reactive oxygen species (ROS), such as peroxynitrite (ONOO−). In physiological systems, for example, ROS play an important role in signaling mechanisms, especially in the TNFα-regulated pathways (Hennet et al., 1993). O2 ·− is considered one of the primary ROS generated in cells stimulated by TNFα (Hennet et al., 1993). Furthermore, ROS are known to play a part in proliferation and cell death (Buttke and Sandstrom, 1994; Patel et al., 1996; Fiskum, 2000; Yuan and Yankner, 2000). In several models of apoptosis, increased formation of ROS was described as an early event (Petit et al., 1995; Zamzami et al., 1995a). Most of the ROS have the potential to trigger apoptosis (Buttke and Sandstrom, 1994; Lin et al., 1995; Jacobson, 1996; Hildeman et al., 1999). In addition to enhanced ROS formation, impairment of the cellular antioxidant mechanisms may also lead to apoptosis (Rothstein et al., 1994). Decreased antioxidant profiles were found in epithelial cells that undergo apoptosis (Madesh et al., 1999), and antioxidants have been reported to counter apoptotic cell death (Hockenbery et al., 1993; Zamzami et al., 1995a,b).
Central components of the programmed cell death machinery, which have been conserved throughout evolution, include caspase cascade, Apaf-1 complex formation, and heterodimerization of Bcl-2 family proteins. Cellular organelles, particularly mitochondria, play a crucial role by releasing several apoptotic inducing factors, such as cytochrome c (cyto c), Smac/DIABLO, apoptosis-inducing factor (AIF), and caspases from the intermembrane space into the cytosolic environment in response to a variety of apoptotic stimuli (Liu et al., 1996; Adams and Cory, 1998; Green and Reed, 1998; Krajewski et al., 1999; Susin et al., 1999a,b; Du et al., 2000; Verhagen et al., 2000). During apoptosis, translocation of cyto c to the cytosol determines the assembly of a megaprotein complex (Apaf-1). The Apaf-1 complex initially results in activation of caspase-9 followed by the activation of other caspases. Released Smac/DIABLO interacts with the BIR domain of IAPs (inhibitors of apoptosis) and in turn relieves the inhibition of caspases by IAPs (for review see Green, 2000). Ultimately, caspases chop the cellular proteins resulting in programmed cell death.
Mitochondrial dysfunction is one of the prominent features of ROS-mediated cell death. Mitochondrial depolarization, in association with an increased endogenous production of superoxide anion and subsequent damage to the cardiolipids in the inner mitochondrial membrane, was reported as an early event in dexamethasone-induced apoptosis (Petit et al., 1995; Zamzami et al., 1995a,b). A major pathway leading to mitochondrial damage is based on the amplification of mitochondrial and cytosolic superoxide production in a broad spectrum of inflammatory or ischemia-related conditions. However, the exact mechanism by which ROS, and in particular O2 ·−-mediated apoptosis, establish and utilize CCR remains unclear. Recently, several models have been proposed to elucidate how cyto c and other apoptotic factors are released from mitochondria during apoptosis (for reviews see Green and Reed, 1998; Desagher and Martinou, 2000; Harris and Thompson, 2000; Korsmeyer et al., 2000; Kroemer and Reed, 2000). One paradigm suggests that apoptotic agents trigger mitochondrial permeability transition pore (PTP) opening that results in swelling of the mitochondrial matrix space, causing rupture of the outer mitochondrial membrane (OMM; Marchetti et al., 1996; Zamzami et al., 1996; Kroemer et al., 1998; Bernardi et al., 1999; Crompton, 1999). PTP opening has also been reported to be involved in initiation of the apoptotic machinery without irreversible damage of the mitochondrial membranes (Szalai et al., 1999). ROS and high intramitochondrial Ca2+ may act together to trigger PTP opening, and ROS in interaction with Ca2+ have been proposed to utilize PTP opening to evoke CCR and subsequent activation of caspases (for review see Zoratti and Szabo, 1995; Ankarcrona et al., 1996; Marzo et al., 1998; Crompton, 1999; Duchen, 2000; Fiskum, 2000; Hajnóczky et al., 2000).
The voltage-dependent anion channel (VDAC, or mitochondrial porin) in the outer membrane and the adenine nucleotide translocator (ANT) in the inner membrane have also been proposed to control release of apoptotic factors without opening of the PTP complex. For example, closure of VDAC was reported to establish a defect in ATP/ADP exchange that results in an inhibition of the F1F0-ATPase and, in turn, an initial hyperpolarization of the inner membrane followed by a loss of the outer membrane integrity and CCR (Vander Heiden et al., 1999).
Other models suggest that release of apoptotic factors occurs via large pores assembled from Bcl-2 family proteins (Bax, Bak) in the mitochondrial outer membrane (for review see Green and Reed, 1998; Desagher and Martinou, 2000; Korsmeyer et al., 2000). Insertion of Bax to the outer membrane and formation of Bax oligomers may provide channels conducting large proteins (Gross et al., 1998; Basanez et al., 1999; Antonsson et al., 2000; Saito et al., 2000). Translocation of tBid from cytosol to the mitochondria have also been proposed to induce an allosteric activation and oligomerization of Bak, forming a pore that allows transport of cyto c (Wei et al., 2000). Bid and Bax may induce cyto c efflux through the OMM without affecting the inner mitochondrial membrane (IMM; von Ahsen et al., 2000). Antiapoptotic Bcl-2 family proteins have been shown to regulate mitochondrial ROS formation (Hockenbery et al., 1993; Kane et al., 1993; Gottlieb et al., 2000) and to inhibit ONOO−-induced CCR from isolated mitochondria distal to ONOO− formation (Ghafourifar et al., 1999).
Interactions between Bcl-2 family proteins and components of the PTP (ANT, VDAC) have also been implicated in CCR (Marzo et al., 1998; Shimizu et al., 1999). Interaction with Bax has been reported to facilitate CCR via VDAC and this process was inhibited by Bcl-xL in vitro (Shimizu et al., 1999, 2001). Based on these studies, a number of mechanisms may serve as a potential target of ROS during initiation of the mitochondrial phase of apoptosis. Since mitochondrial ATP production is needed for formation of the apoptosome, it is also necessary to determine whether a damage of the IMM causes impairment in oxidative metabolism before CCR.
Here, we aimed to delineate the pathway superoxide radicals utilize to evoke the mitochondrial and cellular phases of apoptosis. We examined the relationship between ROS formation and CCR, the loss of mitochondrial membrane potential (ΔΨm), caspase activation, and cell death in HepG2 hepatoma cells. We found that O2 ·−, but not H2O2, induces rapid and massive cyto c release from mitochondria. Although O2 ·− and Ca2+ acted in a coordinated manner in activation of PTP opening, O2 ·−-induced CCR was not affected by Ca2+ or by PTP blockers. In contrast, VDAC blockers or an anti-VDAC antibody (Ab#25) prevented cyto c release induced by O2 ·−. Furthermore, integrity of the IMM was preserved during O2 ·−-mediated CCR. Bcl-2 family proteins did not appear to be essential for the control of O2 ·−-mediated CCR. Thus, this study provides evidence for a novel pathway that links O2 ·− to VDAC-dependent selective permeabilization of the OMM and CCR, and, ultimately leads to activation of the apoptotic cascade.
Results
O2 ·− and H2O2 facilitate Ca2+-induced PTP opening, but only O2 ·− induces rapid release of cyto c
To examine the effect of O2 ·− (generated by xanthine plus xanthine oxidase; X + XO) and H2O2 exposure on PTP, ΔΨm, and [Ca2+]c were monitored in permeabilized HepG2 cells incubated in the presence of extramitochondrial ATP supply. Since thapsigargin was added to abrogate endoplasmic reticulum Ca2+ uptake, changes in [Ca2+]c reflected mitochondrial Ca2+ uptake and release. We have established a Ca2+ pulsing protocol that evoked modest depolarization and rapidly decaying [Ca2+]c transients in naive cells, whereas large and extended depolarization and [Ca2+]c elevation in cells treated with agents that promote the gating of PTP by Ca2+ (Szalai et al., 1999). As shown in Fig. 1 A, upon addition of X + XO the mitochondrial depolarization evoked by Ca2+ pulses was augmented and prolonged. Simultaneously, mitochondria in the X + XO–pretreated cells lost their capacity to keep sequestered the exogenously added Ca2+. Enhanced depolarization and impaired Ca2+ uptake exhibited by X + XO–treated cells reflected opening of PTP as it was inhibited by cyclosporin A (CsA; see Fig. 3 A). The mitochondrial depolarization and [Ca2+]c rise in X + XO–treated cells were also attenuated and shortened by pretreatment with an O2 ·− scavenger, superoxide dismutase mimic (Mn[III]tetrakis [4-benzoic acid] porphyrin, MnTBAP, 20 μM for 5 min; Fig. 1 A), suggesting that O2 ·− formation promoted the activation of PTP by Ca2+.
Figure 1.

Effect of ROS on Ca2+-induced PTP opening and CCR in permeabilized HepG2 cells. (A) O2 ·−-generating system (xanthine [0.1 mM] plus xanthine oxidase [20 mU/ml]) and (B) H2O2 (90 mM) augmented Ca2+-induced depolarization (three pulses, 30 μM CaCl2 each) and decreased mitochondrial Ca2+ uptake. These effects were inhibited by an O2 ·− scavenger, MnTBAP (20 μM; 68 ± 4.5% decrease in depolarization and 78 ± 13% decrease in [Ca2+]c rise at 900 s; n = 3), and catalase (Cat; 2500U/ml), respectively. At the end of the measurements, cells were exposed to FCCP (Unc; 1 μM), a protonophore that caused rapid and complete dissipation of ΔΨm. (C) Induction of CCR from mitochondria by O2 ·− but not H2O2. Cytosolic and membrane fractions were prepared after fluorometric measurements of ΔΨm and [Ca2+]c in permeabilized cells performed as shown in A and B, except that Unc was not added. Released cyto c in the cytosol was visualized by Western blotting with anti–cyto c antibody. (D) Permeabilized cells were incubated with H2O2 at three different concentrations (0.9, 9, and 90 mM) or with X + XO for 375 s. (E) Permeabilized cells were preincubated with an ONOO− scavenger, ebselen, at three different concentrations (100, 50, and 25 μM), before the exposure to X + XO. (F) Quantification of O2 ·−-induced CCR from mitochondria. Cells were exposed to X + XO (0.1 mM plus 20 mU/ml) for 375 s. Various amounts of cytosolic and membrane fraction containing cyto c were compared with cyto c standard.
Figure 3.

PTP blockers fail to inhibit O2 ·− -induced CCR. Permeabilized cells were pretreated with CsA (1 μM) or BA (10 μM) before the addition of X + XO. (A) Mitochondrial depolarization and [Ca2+]c rise evoked by Ca2+ pulses added after the pretreatment with X + XO or CsA + X + XO. (B and C) Released cyto c in the cytosol was visualized by Western blotting. Cytosol samples were prepared after the measurement of ΔΨm but FCCP was not added. Permeabilized cells were pretreated with BA (10 μM) in the presence or absence of ATP-regenerating system before the X + XO treatment. Because ADP supports the binding and inhibitory effect of BA, to prevent extramitochondrial phosphorylation of ADP, ATP-regenerating system was omitted from the medium in some experiments.
Similar to O2 ·−, H2O2 potentiated the Ca2+ pulsing–induced depolarization and [Ca2+]c elevation (Fig. 1 B). These effects of H2O2 were inhibited by an H2O2 scavenger, catalase (Fig. 1 B) and by CsA (unpublished data; n = 2). This finding suggests that H2O2 also promoted Ca2+-dependent PTP opening.
At the end of fluorometric measurements, samples were centrifuged and the cytosol was separated from the membrane fraction. In the control, most of the cyto c was detected only in the mitochondrial pellet (Fig. 1 C). In X + XO–pretreated cells (X + XO for 375 s and Ca2+ for 300 s), less cyto c was in the pellet and a large amount was observed in the cytosol. Importantly, in the presence of MnTBAP, X + XO did not cause cyto c redistribution from mitochondria to cytosol (Fig. 1 C). Similarly, pretreatment with SOD (500 U/ml) also prevented the CCR from mitochondria (unpublished data, n = 2). In contrast to O2 ·−, H2O2 (0.9–90 mM) did not elicit cyto c rise in the cytosol (Fig. 1, C and D). O2 ·− rapidly reacts with NO, yielding ONOO−, which has also been shown to evoke CCR and apoptosis (Lin et al., 1995; Ghafourifar et al., 1999). To evaluate the possibility of involvement of ONOO− in X + XO–dependent CCR, permeabilized cells were pretreated with various concentrations of an ONOO− scavenger, ebselen, and then exposed to O2 ·−. O2 ·−-induced CCR was not inhibited in the concurrent presence of ONOO− scavenger (Fig. 1 E). Together, these results demonstrate that O2 ·− and H2O2 promote Ca2+-dependent PTP opening, whereas O2 ·−, but not H2O2 or the O2 ·− product, ONOO−, selectively induced CCR.
To quantitate O2 ·−-dependent CCR, various amounts of cytosolic and membrane proteins were resolved on SDS-PAGE. The amount of CCR was compared with known quantities of cyto c standard. As shown in Fig. 1 F, the vast majority of mitochondrial cyto c was released to the cytosol in permeabilized cells exposed to X + XO (5 mU/ml) for 375 s. Thus, the loss of cyto c from mitochondria in response to O2 ·− treatment is rapid and almost complete.
O2 ·−-induced CCR is independent of PTP opening
To investigate the mechanism by which O2 ·− induces CCR, simultaneous measurements of ΔΨm and [Ca2+]c were performed at various concentrations of X + XO in the absence and presence Ca2+ pulses (Fig. 2) . In the absence of Ca2+, mitochondrial depolarization was not detectable during 375 s exposure to O2 ·−-generating system (Fig. 2 A). When Ca2+ was added to the permeabilized cells, depolarization was seen in a dose-dependent manner (Fig. 2 B). To determine cyto c distribution, cytosol and membrane fractions were separated as described above. Interestingly, O2 ·− was able to induce CCR in a dose-dependent manner, even in the absence of Ca2+ addition (Fig. 2 C). CCR was observed at ≥4 mU XO and it was almost complete. Addition of Ca2+ did not affect the sensitivity of CCR to X + XO. Collectively, these data show that Ca2+ is not required for O2 ·−-dependent CCR.
Figure 2.

Ca2+-induced mitochondrial depolarization is not essential for O2 ·− -dependent CCR. Permeabilized cells were treated with X (0.1 mM) and different concentrations of XO (0.1, 1, 2, 3, 4, 5, 10, or 20 mU/ml) before addition of Ca2+ pulses (30 μM each) or solvent. (A and B) ΔΨm measured simultaneously with [Ca2+]c. (C and D) cyto c in the cytosol and membrane fractions. Immunoblotting of actin in the cytosolic fraction was used to evaluate whether O2 ·− changed the distribution of nonmitochondrial proteins.
To determine whether opening of PTP was involved in O2 ·−-induced CCR, permeabilized cells were treated with inhibitors of the inner membrane components of the PTP, CsA (1 μM), or bongkrekic acid (BA; 10 μM) before the addition of X + XO and Ca2+ pulses (Fig. 3). As shown in Fig. 3 A, CsA that binds to cyclophilin D in the mitochondrial matrix prevented the Ca2+-induced depolarization and sustained [Ca2+]c rise in X + XO–treated permeabilized cells. By contrast, CsA failed to affect the CCR evoked by X + XO both in the presence or absence of Ca2+ (Fig. 3 B). Furthermore, BA, a blocker of the adenine nucleotide translocator that spans the IMM, was also ineffective to attenuate O2 ·−-induced CCR (Fig. 3 C). ADP supports the binding and inhibitory effect of BA, but BA also failed to inhibit O2 ·−-induced CCR when the ATP regenerating system was omitted from the medium to prevent extramitochondrial phosphorylation of ADP (Fig. 3 C, right). Together, these results show that opening of PTP is not required for O2 ·−-induced CCR.
Preservation of ΔΨm requires extramitochondrial ATP supply in cells exposed to O2 ·−
Following release of most of the mitochondrial cyto c (Fig. 2), oxidative metabolism is likely to be impaired. Thus, ΔΨm could be maintained in the presence of X + XO in expense of the extramitochondrial ATP supply (Fig. 2 A). To test whether reversed operation of the F0F1-ATPase allowed extrusion of H+ in mitochondria exposed to O2 ·−, we added an inhibitor of the F0F1-ATPase, oligomycin, and monitored the effect of X + XO on ΔΨm. Under these conditions X + XO caused rapid and complete depolarization (Fig. 4 A). CsA did not affect the X + XO–induced depolarization in oligomycin-pretreated cells (Fig. 4 A). Notably, pretreatment with oligomycin did not change the amount of CCR induced by O2 ·− (unpublished data).
Figure 4.
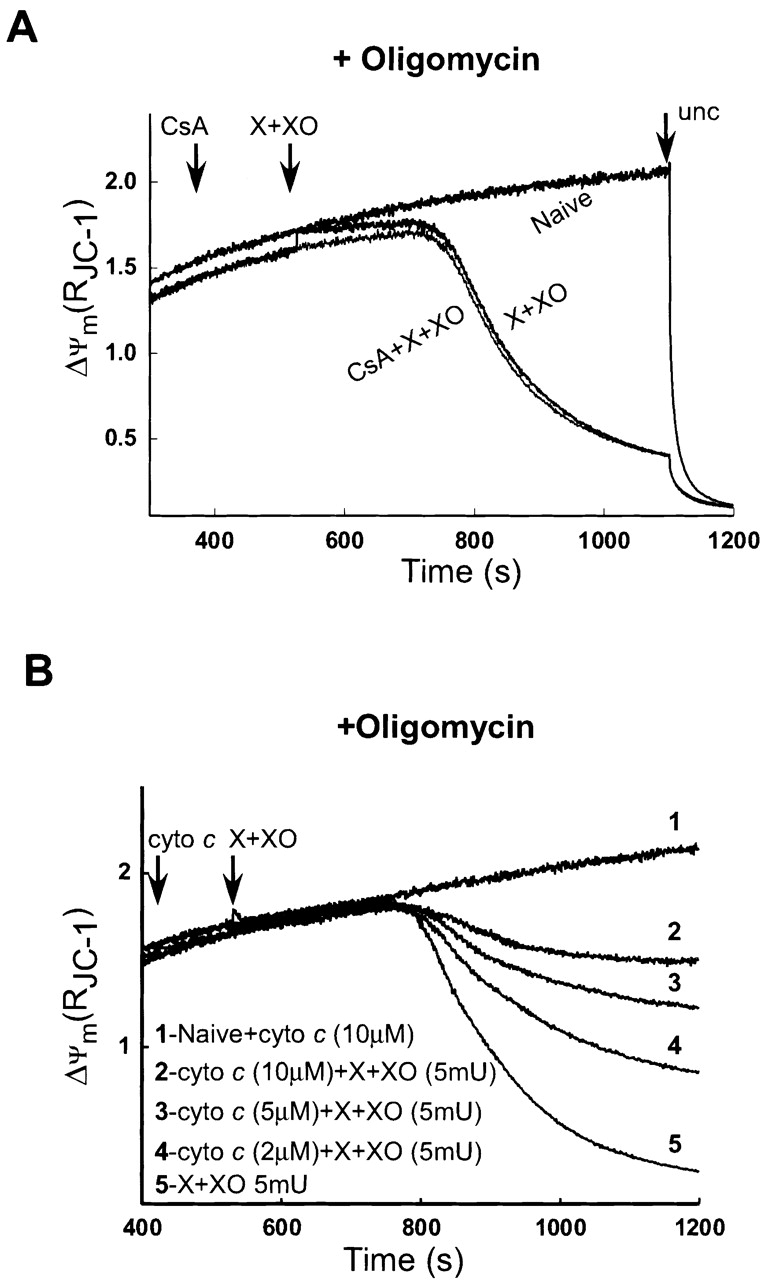
O2 ·− -induced depletion of mitochondrial cyto c yields mitochondrial depolarization in the absence of extramitochondrial ATP supply. (A) Effect of O2 ·− on ΔΨm was monitored in permeabilized cells pretreated with oligomycin (2.5 μg/ml), an inhibitor of the F0F1-ATPase before the addition of X + XO or CsA + X + XO. (B) Rescue of mitochondria from O2 ·−-induced depolarization by exogenously added cyto c. Permeabilized cells were preincubated with cyto c (2, 5, or 10 μM) and oligomycin (2.5 μg/ml) before exposure to X + XO.
O2 ·−-induced CCR precedes the ΔΨm loss
To clarify the temporal relation between CCR and ΔΨm loss, the time-course of CCR and depolarization were studied after exposure to X + XO in oligomycin-pretreated permeabilized cells. As shown in Fig. 5 A, CCR in the cytosol was apparent at 125 s and maximum release was observed at 225 s after X + XO treatment. CCR was associated with a decrease in ΔΨm that started >200 s after O2 ·− addition (Fig. 4 A). Our results suggest that the loss of ΔΨm follows the O2 ·−-induced rapid release of cyto c instantaneously.
Figure 5.
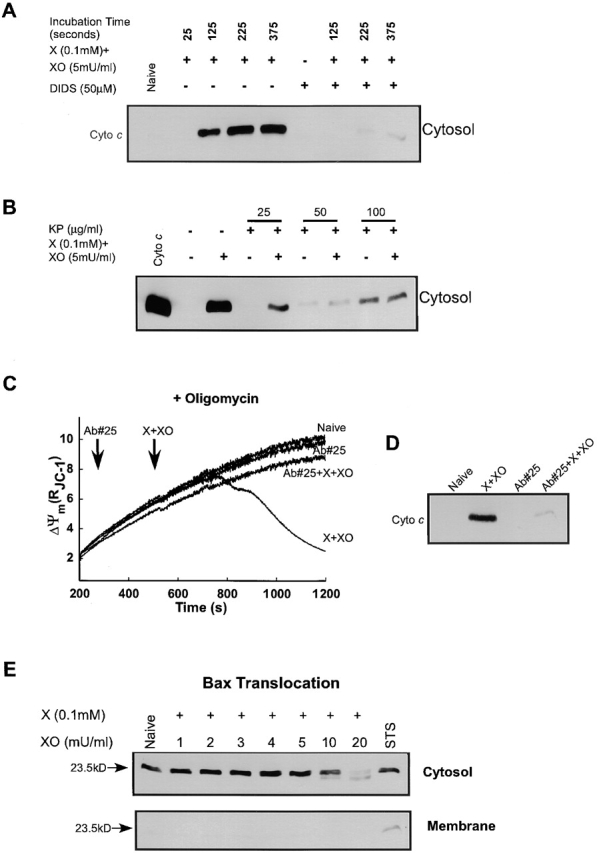
Inhibition of O2 ·− -induced CCR by VDAC blockers and anti-VDAC antibody. Permeabilized cells were exposed to X + XO in the absence of Ca2+ pulses. At various times after X + XO exposure, permeabilized cells were centrifuged for the collection of cytosolic fractions. For inhibitory studies, permeabilized cells were pretreated with (A) DIDS, an inhibitor of VDAC, for 50 s before the addition of X + XO, or (B) with indicated concentrations of KP for 100 s before exposure to X + XO. (C) Prevention of O2 ·− -induced ΔΨm loss by anti-VDAC antibody (Ab#25). Permeabilized cells pretreated with or without Ab#25 (0.56 μg/μl) for 5 min, after which X + XO (0.1 mM plus 5 mU/ml) was added and changes of ΔΨ were monitored for 12 min by spectrofluorimeter. Data presented is representative of one experiment. (D) Inhibition of O2 ·− -induced CCR by anti-VDAC antibody. After the measurement of ΔΨm shown in C, the cells were centrifuged and cyto c was determined in the cytosolic fractions. (E) Translocation of Bax to the mitochondria is absent during exposure to O2 ·− but occurs during staurosporine-mediated CCR. The cytosol and mitochondrial fractions were subjected to 15% SDS-PAGE and transferred to nitrocellulose. Blots were probed with a polyclonal anti-Bax antibody.
If depolarization was due to cyto c depletion–induced impairment of ΔΨm generation by the oxidative pathway, supply of exogenous cyto c to the bulk cytosolic buffer may sustain ΔΨm in oligomycin-treated cells (Waterhouse et al., 2001). As shown in Fig. 4 B, exogenously added cyto c (2, 5, and 10 μM) attenuated O2 ·−-induced ΔΨm loss in a dose-dependent fashion. These results show that after O2 ·−-induced CCR, mitochondria can maintain ΔΨm only if ATP was provided by an extramitochondrial source or the decrease in the intramitochondrial concentration of cyto c was compensated by added cyto c. Also, these results suggest that the IMM was intact even after O2 ·−-induced CCR.
Inhibitors of VDAC abrogate O2 ·−-induced CCR
Recent studies have reported that Bax, a proapoptotic Bcl-2 family protein may interact with VDAC to trigger CCR. It is also to note that the antiapoptotic protein, Bcl-xL prevents CCR from mitochondria through the VDAC–Bax complex (Shimizu et al., 1999). The OMM contains a large amount of VDAC, and this protein channel can act as a track for the transport of substances in and out of mitochondria (Colombini et al., 1996). Thus, we next investigated the effect of VDAC blockers, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) (Shafir et al., 1998) and Konig's polyanion (KP), on the O2 ·−-induced CCR. Pretreatment with DIDS (50 μM) for 1 min before the addition of X + XO completely inhibited O2 ·−-induced CCR (Fig. 5 A). Permeabilized cells were also pretreated with different concentrations of KP for 100 s before exposure to X + XO. Treatment with KP also inhibited CCR induced by O2 ·− (Fig. 5 B). At higher concentrations, the KP caused release of a fraction of cyto c by itself. These results indicated that VDAC is involved in the O2 ·−-induced CCR.
Recently, Shimizu et al. (2001) described an anti-VDAC antibody (Ab#25) that inhibits VDAC activity. We used this antibody to further verify the role of VDAC in CCR and ΔΨm loss of permeabilized cells treated with X + XO. In permeabilized cells pretreated with oligomycin, X + XO showed massive amount of CCR and large ΔΨm loss. However, cells pretreated with Ab#25 (0.56 μg/μl) for 5 min prevented the CCR (Fig. 5 D) and the ΔΨm loss (Fig. 5 C).
Involvement of proapoptotic Bcl-2 family proteins is not necessary for O2 ·−-induced CCR
The interaction of Bcl-2 family proteins in the mitochondrial membranes can control the permeability to cyto c. During apoptosis, translocation of Bax from cytosol to mitochondria facilitates the release of cyto c through the OMM (Eskes et al., 1998; Jurgensmeier et al., 1998; Marzo et al., 1998; Narita et al., 1998; Pastorino et al., 1998). Bax has also been reported to interact with VDAC to release cyto c (Shimizu et al., 1999). However, in our study, cytosolic Bax did not translocate to the mitochondrial membrane during O2 ·− exposure (Fig. 5 E). By contrast, cells exposed to a proapoptotic agent, staurosporine (1 μM) for 3 h showed significant amount of mitochondrial anchored Bax. Furthermore, washout of the cytosol after cell permeabilization did not prevent O2 ·−-mediated CCR (unpublished data; n = 3). These data indicate that O2 ·−-induced CCR through VDAC does not require translocation of Bax to the mitochondria.
Another proapoptotic Bcl-2 family protein, Bak, may substitute for Bax in the CCR (Wei et al., 2001). However, no Bak was detected in the lysate of HepG2 cells using a monoclonal antibody (anti-human Bak, clone TC102; Oncogene Research Products) that recognized Bak in the lysate of H9c2 cardiac myotubes (unpublished data). Thus, Bak does not appear to be necessary for the O2 ·−-induced CCR.
Coupling of caspase activation to O2 ·−-mediated CCR
Mitochondrial permeabilization and CCR into the cytosol are essential events for caspase activation (Liu et al., 1996). We next examined whether the O2 ·−-induced release of cyto c and, presumably, other mitochondrial factors, is associated with caspase activation. After a short exposure to O2 ·−, cleavage of procaspase-3 was detected, suggesting caspase-3 activation (Fig. 6 A). The VDAC blocker DIDS completely blocked the O2 ·−-mediated procaspase-3 cleavage, whereas CsA or BA did not prevent O2 ·−-mediated caspase activation (Fig. 6 A). One may speculate that the disappearance of procaspase-3 was due to its degradation by a necrosis-stimulating protease. To evaluate formation of the active caspase-3 enzyme, we also performed measurements of the cleavage product of a fluorogenic caspase substrate, DEVD-AMC (Fig. 6 B). In extracts of X + XO–treated permeabilized cells, the cleavage of DEVD-AMC was augmented. This effect was abolished by DIDS, whereas CsA did not attenuate the X + XO–stimulated DEVD-AMC cleavage (Fig. 6 B). We also observed that a caspase inhibitor, Ac-DEVD-CHO (50 μM), did not affect CCR, but completely blocked caspase 3 activation (unpublished data; n = 2). Thus, O2 ·− resulted in caspase activation which was closely coupled to VDAC-dependent CCR.
Figure 6.
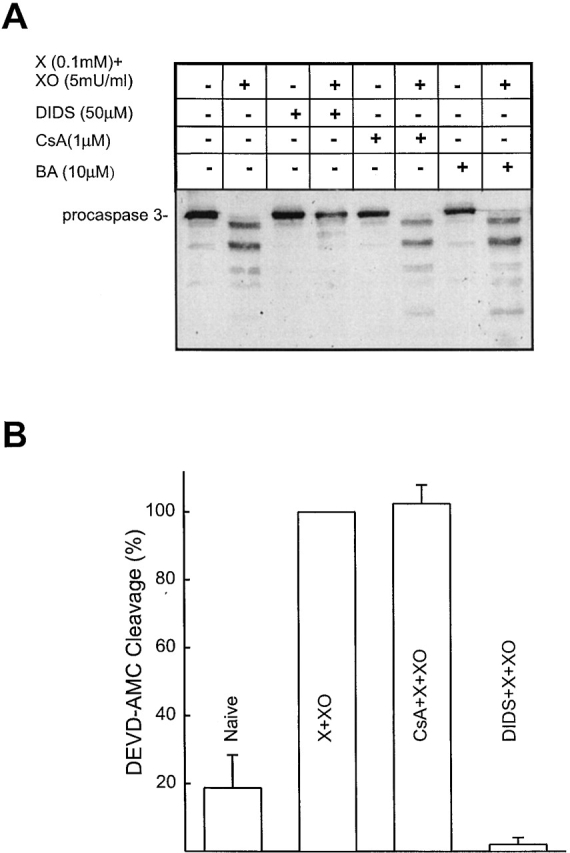
O2 ·− induces caspase-3 activation; prevention by a VDAC blocker but not PTP inhibitors. (A) Cytosolic fractions (prepared as described above) were resolved on 15% SDS-PAGE followed by transfer to nitrocellulose and the blot was probed with polyclonal anticaspase-3 antibody. (B) Fluorometric assay of DEVD-AMC (12.5 μM) cleavage, in cytosol extracts (incubation for 30 min at 35°C). DEVD-AMC cleavage normalized to the activity obtained in the presence of X + XO (0.1 mM plus 5 mU/ml) is shown as mean ± SEM (n = 3).
O2 ·− evokes VDAC-dependent CCR in intact cells
To demonstrate whether O2 ·− can also trigger CCR in intact cells, HepG2 cells were treated with X + XO for various time periods. The time-course of the exposure to O2 ·− during treatment with X + XO was evaluated fluorimetrically, using a ROS tracer, 5-(and 6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (Molecular Probes). A substantial increase in ROS was noticed after 5 min and a plateau was reached after 60 min treatment with X + XO (n = 2). To study the cyto c distribution, after treatment with X + XO, cells were harvested, permeabilized and the cytosol was separated from the membrane fraction by centrifugation. Naive cells and cells treated with X + XO for a short duration (15 or 30 min) did not have cyto c in the cytosol (Fig. 7 A). Sometimes, we detected CCR after 45 min but in minute quantities (unpublished data). Treatment with X + XO for 1 h or more caused large release of cyto c (Fig. 7 A). We wished to know whether the effects of PTP blockers or VDAC blocker on the O2 ·−-mediated CCR in intact cells were similar to those of in permeabilized cells. The O2 ·−-dependent release of cyto c into the cytosol was not detected in DIDS treated cells, whereas CsA or BA did not prevent mitochondrial CCR into the cytosol (Fig. 7 B). Thus, O2 ·− produced in the bathing medium evoked CCR in intact cells similar to the effect of cytosolic O2 ·− in permeabilized cells, but the lag time of the CCR was relatively long in intact cells. Several factors may contribute to the delayed onset of the X + XO–induced CCR in intact cells, including the scavenging of ROS in the bathing medium by BSA and in the cytosol by antioxidant mechanisms (e.g., Cu,Zn-SOD). Also O2 ·− does not readily cross the plasma membrane.
Figure 7.

O2 ·− -induced CCR in intact HepG2 cells; time-course, effect of PTP, and VDAC blockers. (A) Cells were grown in 25-cm2 flasks for 48 r and treated with X + XO for different time periods. After treatment, cells were harvested and permeabilized with 40 μg/ml digitonin containing ICM at pH 7.2 for 10 min. The cytosol was then separated from the membrane frac- tion (containing mitochondria) by centrifugation. The cytosolic fractions were resolved on SDS-PAGE and immunoblotted for cyto c. (B) Cells pretreated with DIDS, CsA, or BA for 10 min were exposed to X + XO. After 3 h, cells were isolated, permeabilized, centrifuged, and immunoblot was done as described above. (C) Confocal imaging of O2 ·−-induced CCR using cyto c–GFP. Cyto c-GFP–transfected HepG2 cells were treated with X + XO (0.1 mM and 20 mU/ml, respectively) in the absence or presence of inhibitors. Distribution of cyto c–GFP was imaged by confocal microscopy. The images were taken after different time periods of X + XO exposure with a 60× oil objective. (D) Quantification of cyto c–GFP in the cytosol. Since the nucleus is devoid of mitochondria and released cyto c–GFP entered the nuclear matrix, the amount of cyto c–GFP released into the cytosol was determined by measurements of fluorescence over the nucleus in each cell. f.a.u., fluorescence arbitrary units. To assess the amount of cyto c–GFP release after X + XO treatment, we counted ∼100–200 cells in three independent experiments for each condition. (E) Electron micrographs of control (left) and X + XO–treated (0.1 mM plus 20 mU/ml for 1 h; right) intact cells are shown to illustrate the typical patterns of mitochondrial morphology (>200 mitochondria were evaluated in each condition).
To visualize the effect of O2 ·− induced CCR in individual cells, we transfected HepG2 cells transiently with cyto c–green fluorescent protein (GFP). After transfection, cells were treated with X + XO (0.1 mM and 20 mU/ml) for appropriate time periods. The distribution of cyto c–GFP was visualized using a confocal microscope. In normal cells and at 30 min treatment with X + XO, cyto c–GFP was mostly localized in mitochondria (Fig. 7 C). Cyto c–GFP appeared in the cytosol and in the nuclear matrix after 1 h or longer treatment with X + XO (Fig. 7 C). Cell to cell differences in the lag time of cyto c–GFP redistribution were observed, but once the release started it rapidly involved every mitochondrion. At 3 h of exposure to X + XO, cyto c–GFP distribution was homogeneous in most of the cells (Fig. 7 C). The O2 ·−-induced release of cyto c–GFP was quantitated based on the fluorescence appearing in the nuclear region of the cells (Fig. 7 D). The onset of cyto c–GFP release was similar to that of CCR, whereas the maximal release of cyto c–GFP was obtained 5 h after addition of X + XO (Fig. 7 D). Pretreatment with CsA (5 μM) did not inhibit cyto c–GFP release from mitochondria after O2 ·− exposure. However, DIDS prevented the release of cyto c (Fig. 7 D). Collectively, these studies demonstrated that increased formation of O2 ·− is capable of evoking CCR in intact cells and further supported the role of VDAC in this process.
Selective permeabilization of the OMM by truncated Bid has been described to initiate CCR in the absence of major damage of the mitochondrial structure (von Ahsen et al., 2000). Using transmission electron microscopy, we evaluated the morphology of the mitochondria in intact cells incubated in the absence and presence of X + XO for 1 h (Fig. 7 E). Consistent with previous studies in HepG2 cells (e.g., Lewis et al., 1996), the majority of the mitochondria appeared in close association with the endoplasmic reticulum, was oval shaped, and was packed with cristae (left). Furthermore, 1 h of treatment with X + XO did not cause a major change in the mitochondrial structure (right). Thus, the onset of the CCR was not preceded by X + XO–induced large amplitude swelling of the mitochondria.
O2 ·− evokes cyto c efflux from liposomes reconstituted with VDAC
To further explore the role of VDAC in O2 ·−-mediated apoptosis, we examined the effect of O2 ·− on the permeability of VDAC purified (confirmed by Coomassie staining and immunoblotting; Fig. 8 A) from liver mitochondria and incorporated into liposomes. Plain liposomes and VDAC liposomes loaded with cyto c–FITC were exposed to O2 ·− for 7 min, and the distribution of cyto c–FITC was visualized using confocal microscopy (Fig. 8 B). Plain, cyto c-FITC–loaded liposomes displayed only a small change in fluorescence distribution in response to X + XO (top images; extraliposomic fluorescence: 6.4 ± 0.6 and 11.4 ± 0.3 arbitrary units before and after X + XO addition, respectively; n = 3). By contrast, VDAC reconstituted liposomes allowed for a large increase of extraliposomic fluorescence in response to O2 ·− exposure (middle images; 6.9 ± 0.2 and 33.3 ± 0.7 arbitrary units before and after X + XO addition, respectively; n = 3). Involvement of VDAC in the O2 ·−-induced CCR was further tested using DIDS. Pretreatment with DIDS (100 μM) inhibited the X + XO–induced increase in fluorescence between VDAC liposomes (bottom images; 7.6 ± 0.3 and 15.5 ± 0.5 arbitrary units before and after X + XO addition, respectively; n = 3).
Figure 8.

O2 ·− directly modulates purified VDAC to facilitate cyto c release. (A) Immunoblotting of purified VDAC and VDAC liposomes for VDAC and Bax. (B) Confocal imaging of FITC–cyto c efflux from plain and VDAC liposomes. Cyto c labeled with FITC was loaded into both plain and VDAC-incorporated liposomes. Images were taken before and after 7 min of exposure to X + XO (0.1 mM and 20 mU/ml). Furthermore, the effect of O2 ·− was evaluated on VDAC liposomes incubated in the presence of DIDS (100 μM). (C) Fluorometry of cyto c–FITC release from liposomes. Suspensions of liposomes (4 types; plain, plain loaded with cyto c–FITC, VDAC, and VDAC loaded with cyto c–FITC) were incubated in the absence or presence of X + XO (0.1 mM and 20 mU/ml) and DIDS (100 μM) for 7 min. Subsequently, the suspensions were centrifuged and FITC fluorescence was determined in the supernatants and liposomoic pellets. Cyto c–FITC release is shown by the FITC fluorescence measured in the supernatants. We observed that exposure to X + XO increased the fluorescence of FITC (unpublished data) and this effect could contribute to the increase of FITC in the supernatants of plain as well as VDAC liposomes. However, in response to X + XO, the VDAC liposomes exhibited a several-fold larger increase in the supernatant FITC and a considerably smaller residual pellet FITC than that of in plain liposomes (unpublished data). Data are from two separate experiments; each was performed in duplicates.
To quantitate cyto c–FITC release from the liposomes, we also centrifuged liposome suspensions and measured the fluorescence in the supernatant and in the pellet using a fluorimeter. As shown in Fig. 8 C, the fluorescence in the supernatant was small when liposomes were prepared without cyto c–FITC loading and reconstitution of VDAC, exposure to X + XO or DIDS did not evoke any changes (bars: 1, 3, 5, 7, and 9). When cyto c-FITC–loaded plain liposomes were exposed to X + XO, the fluorescence in the supernatant showed a twofold increase, similar to the data obtained with confocal imaging (bar 4 vs. 2). This increase was due to a direct interaction between the fluorophore and O2 ·−, because a comparable increase was obtained when FITC was incubated with X + XO (unpublished data). However, the supernatant prepared from suspensions of VDAC liposomes exhibited eightfold increase after exposure to X + XO (bar 8 vs. 6). Comparing the plain and VDAC liposomes, incubation with X + XO caused a much larger increase of the fluorescence in the supernatant (bar 8 vs. 4) and this was accompanied with a much larger decrease of the fluorescence in the pellet prepared from VDAC liposomes (unpublished data, n = 2). In addition, X + XO–induced redistribution of the fluorescence was significantly attenuated by pretreatment with DIDS (bar 10 vs. 8). Importantly, Bax was not detected in purified VDAC or in VDAC liposomes, providing further evidence that the effect of O2 ·− on VDAC was not mediated by Bax (Fig. 8 A). Together, these studies show that reconstitution of VDAC in liposomes allows for a O2 ·−-induced increase in cyto c permeability. The lack of O2 ·−-dependent efflux of cyto c–FITC from plain liposomes suggests that O2 ·− does not rupture the lipid domains of the mitochondrial outer membrane. These results further confirmed that O2 ·− modulates opening of VDAC and facilitates CCR. The fact that VDAC blockers and anti-VDAC antibody prevent the CCR in the HepG2 cells and that PTP blockers did not prevent O2 ·−-induced CCR suggested that the ability of O2 ·− to interact with VDAC is sufficient to induce CCR from mitochondria.
Execution of apoptosis in cells exposed to O2 ·−
We have demonstrated CCR and caspase activation in cells exposed to O2 ·−. To determine whether CCR and caspase activation are followed by ordered execution of the apoptotic program, phosphatidylserine (PS) exposure was evaluated by annexin V staining, and membrane permeability was studied by labeling with propidium iodide (Fig. 9) . In X + XO–treated cells, imaging of ΔΨm simultaneously with Alexa 488 annexin showed an early mitochondrial depolarization (Fig. 9 A; decrease in red signal at 1 h). Presumably, extramitochondrial ATP production is relatively small in HepG2 cells and therefore mitochondrial depolarization was closely coupled to cyto c release. After 1 h of X + XO treatment, very few cells exhibited annexin labeling of the plasma membrane, whereas at 3 h most of the cells were positive (green signal). Annexin staining of the cell could occur due to permeabilization of the plasma membrane; however, in cyto c-GFP–expressing cells, the cyto c–GFP released from mitochondria to the cytosol did not leave the cells, suggesting that the plasma membrane barrier was preserved (Fig. 9 B). Also, propidium iodide was initially excluded from the PS-positive cells (Fig. 9 C, see below). Treatment with DIDS (100 μM) attenuated the O2 ·−-induced ΔΨm loss and annexin staining (unpublished data). These observations suggest that execution of the apoptotic program was initiated by O2 ·− through a VDAC-dependent signaling pathway.
Figure 9.
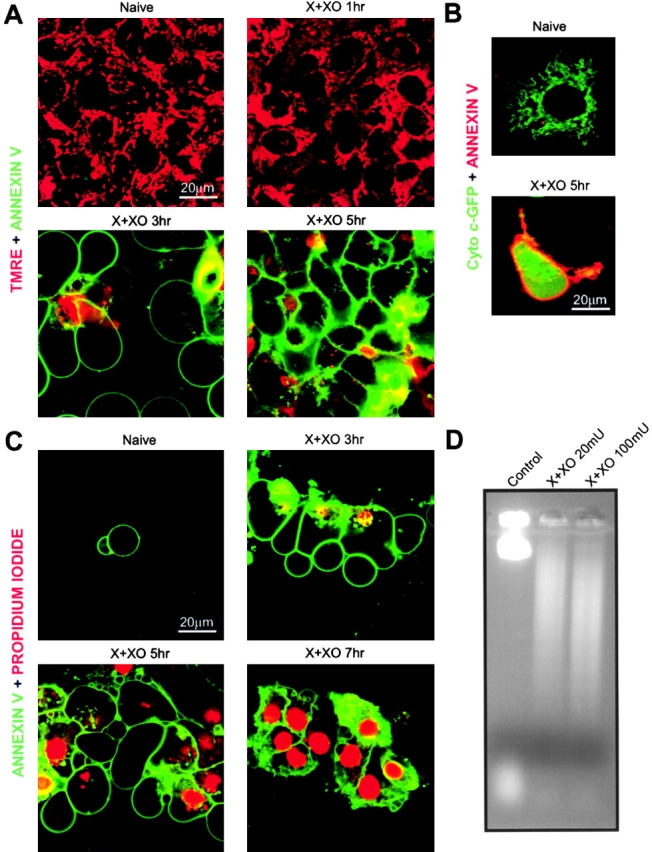
Execution of apoptosis in HepG2 cells exposed to O2 ·− . Mitochondrial depolarization, PS exposure, disruption of plasma membrane integrity, and DNA fragmentation in cells exposed to O2 ·−. Cells were treated with X + XO (0.1 mM plus 20 mU/ml) for various time periods in the absence or presence of DIDS (100 μM). (A) ΔΨm and PS exposure were evaluated with confocal imaging of annexin V Alexa 488 and TMRE. (B) Distribution of cyto c–GFP and annexin staining were visualized. (C) PS exposure and loss of plasma membrane integrity were evaluated with imaging of annexin V Alexa 488 and propidium iodide. (D) DNA fragmentation analysis was performed with cells incubated in the presence or absence of X + XO for 12 h.
Apoptotic cells can be discriminated from necrotic cells based on the fact that apoptotic cells become permeable to propidium iodide (PI) relatively late. To evaluate the contribution of apoptosis and necrosis to cell death, after exposure to the O2 ·−-generating system for 1, 3, 5 and 7 h, incubation of the cells with Alexa 488 annexin and PI was performed. Most of the cells treated with O2 ·− for 1 h were not annexin V– or PI-positive. At 3 h of O2 ·− treatment, cells displayed mostly annexin V staining (i.e., early stage of apoptosis), whereas at 5 and 7 h pretreatment, cells showed both annexin V and PI staining, suggesting the onset of late stage apoptosis (Fig. 9 C). Thus, CCR and mitochondrial depolarization, which occurred at 1–2 h X + XO treatment (Figs. 7 A and 9 A), were followed by phosphatidylserine exposure and permeabilization of the plasma membrane was further delayed. This order of events suggests that the cells went through apoptotic cell death. This conclusion was further supported by the fragmentation of DNA in the cells exposed to O2 ·− (Fig. 9 D).
Discussion
This study describes a novel mechanism underlying activation of CCR by O2 ·−, but not by H2O2 or ONOO−. We showed that O2 ·−-induced CCR takes place quickly and involves most of the cyto c stored in the mitochondria. Although ROS interact with Ca2+ in gating of PTP, we have shown that the ROS-induced CCR pathway is independent of the inner membrane components of the pore. Our data suggest that the release of cyto c by O2 ·− is mainly due to VDAC-dependent selective permeabilization of the mitochondrial outer membrane. Ultimately, these alterations lead to activation of caspases and execution of apoptosis.
We examined the molecular machinery used by ROS to trigger the mitochondrial phase of apoptosis. The first important observation was that O2 ·− generated by the xanthine/xanthine oxidase system caused rapid release of cyto c from mitochondria to the cytosol in intact as well as in permeabilized HepG2 cells. Similar to cyto c, Smac/DIABLO was also rapidly released by O2 ·− (unpublished data). O2 ·−-induced release of cyto c and Smac/DIABLO may have a widespread significance in apoptosis, since in a number of pathological conditions, such as ischemia/reperfusion injury, drug insults, and inflammatory responses, large amounts of O2 ·− are produced by xanthine oxidase–mediated catabolism of purine nucleotides or by increased electron transport chain activity or by activation of NADPH oxidase particularly in neutrophils (Granger et al., 1986; Babior, 1999). Furthermore, increased formation of O2 ·− was described as an early event in several apoptotic paradigms (Petit et al., 1995; Zamzami et al., 1995a).
In the cells, O2 ·− can be converted to other ROS (H2O2, ONOO−), which have been reported to evoke CCR from isolated mitochondria (Yang and Cortopassi, 1998; Ghafourifar et al., 1999). However, in our studies, H2O2 (90 mM) did not establish rapid CCR and a ONOO− scavenger, ebselen did not attenuate the O2 ·−-induced CCR. Thus, we conclude that O2 ·− triggered CCR by itself.
A principal mechanism of the CCR could involve ROS-induced promotion of Ca2+-dependent PTP opening. Permanent PTP opening causes CCR via matrix swelling and rupture of the mitochondrial membranes (for review see Kroemer et al., 1998; Bernardi et al., 1999; Crompton, 1999; Desagher and Martinou, 2000), but reversible PTP opening was also shown to set up partial CCR without irreversible damage of mitochondrial function (Szalai et al., 1999). We also observed that Ca2+-induced PTP opening was amplified in permeabilized cells exposed to O2 ·− or H2O2, and simultaneous presence of either O2 ·− scavengers SOD or SOD mimic, MnTBAP, or H2O2 scavenger catalase attenuated this effect. However, O2 ·−-induced CCR was insensitive to inhibitors of PTP (CsA, BA) and was also observed in the absence of Ca2+. Thus, PTP opening is not essential for O2 ·−-induced CCR. In addition, we demonstrated that O2 ·−-induced CCR does not yield ΔΨm loss if the mitochondria were provided with exogenous cyto c or ATP. These data suggest that integrity of the mitochondrial inner membrane and matrix space was preserved during O2 ·−-induced CCR. It is important to note that in intact cells, mitochondrial depolarization was closely coupled to CCR, providing evidence that CCR caused impairment in the function of the electron transport chain and extramitochondrial ATP production could not maintain mitochondrial proton extrusion. Thus, O2 ·−-induced CCR is mediated by selective permeabilization of the OMM and the O2 ·−-induced mitochondrial depolarization was secondary to the loss of mitochondrial cyto c.
In regard to the changes in mitochondrial function, the origin of O2 ·− is likely to be of great significance. O2 ·− produced by the respiratory chain has been reported to cause cardiolipid destruction in the IMM and dissipation of the ΔΨm (Zamzami et al., 1995a). However, based on our data, O2 ·− produced by X + XO in the cytosol or in the extracellular medium elicits selective permeabilization of the OMM and the damage of the IMM may occur after impairment of the OMM barrier. Presumably, slow permeation of O2 ·− through the OMM and the ROS scavengers in the mitochondria may attenuate exposure of the IMM to O2 ·− produced outside the mitochondria. Protection of the IMM may be particularly effective when a small amount of O2 ·− is produced, whereas a robust increase in extramitochondrial O2 ·− may cause an early damage of both the OMM and IMM and, in turn, necrosis (Higuchi et al., 1998). Along this line, HepG2 cells are expected to exhibit necrosis when exposed to a high dose of O2 ·−. Interestingly, 100 mU XO plus 0.1 mM X was not sufficient to cause necrosis in all HepG2 cells, since DNA fragmentation was also noticed in this condition. Notably, cultured cells grown in 20% oxygen are essentially preadapted to conditions of oxidative stress (e.g., Davies, 1999).
Selective permeabilization of mitochondrial outer membrane may be established by pores formed by Bcl-2 family proteins (for review see Green and Reed, 1998; Desagher and Martinou, 2000; Korsmeyer et al., 2000) or by pores produced by the interaction of VDAC and Bax (Shimizu et al., 1999, 2001). Here, we showed that O2 ·−-induced CCR was inhibited by VDAC blockers (KP, DIDS) as well as by an antibody that interferes with the function of VDAC (#25; Shimizu et al., 2001). Furthermore, liposomes reconstituted with purified VDAC displayed O2 ·−-induced release of FITC–cyto c. Together, these data demonstrate that VDAC plays a central role in O2 ·−-induced CCR. In the studies of Shimizu et al. (1999), permeation of cyto c by VDAC liposomes required addition of Bax. By contrast, in our studies, O2 ·−-induced release of cyto c from the liposomes did not depend on the presence of Bax. The present study also showed that in normal HepG2 cells, the monomeric Bax form was present in the cytosol and translocated into the mitochondrial membrane upon staurosporine treatment. In contrast, cells exposed to the O2 ·−-generating system did not show any translocation of Bax into mitochondria, even at high concentrations of X + XO, but almost complete release of cyto c was still observed. On the same line, washout of the cytosol did not attenuate O2 ·−-induced CCR in the permeabilized cell experiments. Collectively, these data suggest that O2 ·− directly targets VDAC or a factor that is tightly coupled to VDAC in the process that established CCR and Bax does not mediate the effect of O2 ·− on VDAC.
In the cytosol, released cyto c can bind to Apaf-1 and upstream caspases to form a complex, where they are cleaved into their active forms (Zou et al., 1997). Activated caspase-9 can process inactive procaspase-3 into active caspase-3 and thereby triggers intracellular disintegration (Li et al., 1997). Our studies demonstrated that O2 ·− triggered caspase-3 activation and similar to O2 ·−-induced CCR, caspase activation was not inhibited by PTP blockers (CsA or BA), whereas it was prevented by DIDS. In intact cells, O2 ·−-induced CCR was followed by mitochondrial depolarization, PS exposure and finally the cells become propidium iodide positive. This sequence of events suggests that O2 ·−-induced execution of the apoptotic death program. The apoptotic process exhibits rapid progression and takes place in most of the cells exposed to O2 ·−, suggesting that O2 ·− may play a fundamental role in apoptosis caused by enhanced ROS production.
Materials and methods
Cell culture and transfections
HepG2, human hepatocellular carcinoma cells were grown in minimum essential medium (MEM; GIBCO-BRL) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin, and 2 mM glutamine at 37°C in 5% CO2. For imaging experiments, cells were plated onto poly-d-lysine–treated glass coverslips (2–3 × 104/cm2) and were grown for 72 h. For cell suspension studies, cells were cultured for 4–5 d in 75-cm2 flasks, and were harvested by brief treatment with trypsin-EDTA. For transient transfections, cells were transfected using plasmid DNA (cyto c–GFP; Heiskanen et al., 1999) keeping total DNA at 2 μg/ml and cationic lipid Lipofecta–AMINE 2000 according to the manufacturer's instructions. After 5 h, the cells were placed in a normal growth medium. Imaging was performed 48 h after transfection.
Measurement of ΔΨm and [Ca2+]c in suspension of permeabilized cells
Cells were washed in ice cold Ca2+-free extracellular buffer containing 120 mM NaCl, 5 mM KCl, 1 mM KH2PO4, 0.2 mM MgCl2, 0.1 mM EGTA, 20 mM Hepes-NaOH, pH 7.4. An equal amount of cells (9–10 × 106 cells, 7 mg/ml protein) were resuspended and permeabilized with 40 μg/ml digitonin in 1.8 ml of intracellular medium (ICM) composed of 120 mM KCl, 10 mM NaCl, 1 mM KH2PO4, 20 mM Hepes-Tris, pH 7.2, supplemented with 1 μg/ml of each of antipain, leupeptin, and pepstatin. ICM was passed through a Chelex column before addition of protease inhibitors to lower the ambient Ca2+. All the measurements were performed in the presence of 2 mM MgATP, 2 mM succinate, and ATP-regenerating system composed of 5 mM phosphocreatine and 5 U/ml creatine kinase.
For the simultaneous measurements of ΔΨm and mitochondrial Ca2+ uptake, the permeabilized cells were supplemented with 800 nM JC-1 and 0.5 μM fura2FF/FA. Fluorescence was monitored in a multiwavelength excitation dual wavelength emission fluorimeter (Delta RAM, PTI) using 340 and 380 nm excitation and 535 nm emission for fura2FF, whereas 490 nm excitation/535 nm emission and 570 nm excitation/595 nm emission were used for JC-1. Cytosolic Ca2+ is shown as the excitation ratio (340 nm/380 nm) of fura2FF/FA fluorescence, whereas ΔΨm is shown as the ratio of the fluorescence of J-aggregate (570 nm excitation/595 nm emission) and monomer (490 nm excitation/535 nm emission) forms of JC-1 (Szalai et al., 1999). All the experiments were performed at 35°C with constant stirring.
Analysis of cyto c distribution and caspase-3 activation by immunoblotting
At the end of the ΔΨm and [Ca2+]c measurements in permeabilized cell suspensions, cells were immediately centrifuged at 14,000 g for 10 min and the supernatant collected as cytosol. The amount of protein present in the cytosol was measured using the Bradford method.
In studies with intact HepG2 cells, cells were grown in 25-cm2 flasks for 48 h and were later treated with either xanthine 0.1 mM plus xanthine 20 mU/ml or in combination with DIDS (100 μM), CsA (5 μM), and BA (10 μM). At appropriate time periods, cells were harvested and washed in PBS. Cells were then resuspended in and permeabilized with 40 μg/ml digitonin in 1.0 ml of ICM supplemented with 1 μg/ml of each of antipain, leupeptin, and pepstatin for 15 min. The cytosol (14,000 g supernatant) and mitochondria (pellet) were stored at −80°C for Western blot analysis.
For Western blot experiments, proteins (20–25 μg) were loaded onto each lane of a 15% SDS-PAGE and electrophoretically transferred to nitrocellulose filters. Filters were blocked with blocking buffer (Pierce Chemical Co.) overnight, followed by incubation with anti–cyto c (7H8.2C12; BD PharMingen) and anticaspase-3 antibody (CPP32, YAMA, APOPAIN; BD PharMingen) at a dilution of 1:500 and 1:1,000, respectively. After incubation with the primary antibody, bound antibodies were visualized using horseradish peroxidase–coupled secondary antibody and Dura Signal chemiluminescence–developing kit (Pierce Chemical Co.).
Fluorimetric measurement of caspase activity
DEVD-AMC cleavage was measured in cell extracts as described previously (Szalai et al., 1999).
DNA fragmentation analysis
Cells grown in 100-mm culture dishes (3 × 107 cells) were collected in their culture medium, pelleted at 125 g for 5 min, and pellets were washed twice with ice-cold PBS. Pellets were digested with 400 μl of digestion buffer (100 mM NaCl, 100 mM Tris-Cl, pH 8.0, 25 mM EDTA, pH 8.0, 0.5% SDS, 20 ug/ml DNase-free RNase final concentration) for 60 min at 37°C. These homogenates were further incubated with proteinase K (0.1 mg/ml) for 15 min at 50°C. Supernatants were collected to a new tube, in which they were extracted with phenol:chloroform:isoamyl alcohol (25:24:1). DNA was precipitated with 2 vol of cold ethanol and DNA was recovered by centrifugation at 500 g for 2 min. Precipitated DNA was washed once with 70% ethanol and resuspended in 1 mM EDTA, 10 mM Tris-HCl, pH 8.0. Isolated genomic DNA was resolved on a 1.5% agarose gel and visualized by ethidium bromide staining.
Purification and incorporation of VDAC into liposomes and confocal imaging of cyto c translocation
Mitochondria isolated from rat liver were used for purification of VDAC as described previously (de Pinto et al., 1987). The purity of VDAC was assessed by SDS-PAGE electrophoresis and confirmed by Western blotting (antiporin 31HL antibody Calbiochem). Liposomes were prepared by sonicating 500 mg of phospholipid mixture (soybean Type II-S) in 10 ml of buffer containing 50 mM KCl, 20 mM KH2PO4, 20 mM Hepes and 1 mM EDTA, pH 7.0. After sonication, incorporation of purified VDAC and loading of cyto c–FITC (cyto c–FITC was purified as described in the labeling kit protocol [Molecular Probes] and purity was assessed by phosphor imager and Western blotting) was performed by subjecting these vesicles for two freeze–thaw cycles. For imaging of cyto c efflux, fluorescence of cyto c–FITC was monitored using a confocal system (BioRad-1024/2P, excitation 488 nm) fitted to an Olympus IX70 inverted microscope (60× objective). Images were taken from plain and VDAC incorporated liposomes before and 7 min after addition of X (100 μM) and XO (20 mU/ml). For quantitative analysis, liposomes were centrifuged and FITC was released to the supernatant and residual FITC in the liposomoic fraction were determined fluorometrically (490 nm excitatation/510 nm emission).
Electron microscopy
Naive and X + XO (0.1 mM plus 20 mU/ml) –treated cells were fixed in 2% glutaraldehyde, 1% tannic acid, 0.1 M sodium cacodylate at pH 7.4 for 2 h at room temperature. Processing, embedding of the fixed cells, cutting, staining, and examination of the sections (80-nm thick) were performed as described recently (Pacher et al., 2000).
Confocal imaging analysis of CCR and apoptotic markers in intact individual cells
For imaging of cyto c distribution, cells transfected with cyto c–GFP construct were treated with either X (0.1 mM) plus XO (20 mU) or in combination with DIDS (100 μM) or CsA (1 μM). After 3 h, cells were washed twice with extracellular medium (2.0% BSA/ECM) consisting of 121 mM NaCl, 5 mM NaHCO3, 10 mM NaHepes, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, 10 mM glucose, and 2% BSA, pH 7.4. The coverslips were transferred to the microscope stage and cyto c–GFP distribution was monitored with confocal imaging (488 nm excitation).
For imaging of phosphatidylserine exposure and ΔΨm, HepG2 cells were exposed to X + XO for various time periods in the absence or presence of DIDS (100 μM), then incubated with annexin V Alexa 488 for 15 min using 1× binding buffer (Molecular Probes). During the annexin incubation period, TMRE (50 nM) was also loaded (10 min) for the measurement of ΔΨm or propidium iodide (0.5 μg/ml) to evaluate the plasma membrane integrity.
Fluorometry recordings and confocal images are representative of three to six independent experiments. The data combined from separate experiments are shown as mean ±SE. Significance of differences from the relevant controls was calculated by Student's t test.
Acknowledgments
We would like to thank Dr. György Csordás and Timothy Schneider for their help with the electron microscopy. We thank Drs. M. Colombini, A.L. Nieminen, and Y. Tsujimoto for providing us with polyanion, cyto c–GFP plasmid DNA, and anti-VDAC antibody (Ab#25), respectively, and C.J. Buzas for his assistance in liposome preparation.
This work was supported by an American Cancer Society Research Project Grant (G. Hajnoczky). G. Hajnoczky is a recipient of a Burroughs Wellcome Fund Career Award in the Biomedical Sciences.
Footnotes
Abbreviations used in this paper: BA, bongkrekic acid; CCR, cytochrome c release; CsA, cyclosporin A; cyto c, cytochrome c; DIDS, 4,4′-diisothiocyanatostilbene-2,29-disulfonic acid; GFP, green fluorescent protein; IMM, inner mitochondrial membrane; KP, Konig's polyanion; NO, nitric oxide; OMM, outer mitochondrial membrane; ONOO−, peroxynitrite; O2 ·−, superoxide; PI, propidium iodide; PS, phosphatidylserine; PTP, permeability transition pore; ROS, reactive oxygen species; SOD, superoxide dismutase; VDAC, voltage-dependent anion channel; X + XO, xanthine plus xanthine oxidase.
References
- Adams, J.M., and S. Cory. 1998. The Bcl-2 protein family: arbiters of cell survival. Science. 281:1322–1326. [DOI] [PubMed] [Google Scholar]
- Ankarcrona, M., J.M. Dypbukt, S. Orrenius, and P. Nicotera. 1996. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Lett. 394:321–324. [DOI] [PubMed] [Google Scholar]
- Antonsson, B., S. Montessuit, S. Lauper, R. Eskes, and J.C. Martinou. 2000. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cyto c release from mitochondria. Biochem. J. 345:271–278. [PMC free article] [PubMed] [Google Scholar]
- Babior, B.M. 1999. NADPH oxidase: an update. Blood. 93:1464–1476. [PubMed] [Google Scholar]
- Basanez, G., A. Nechushtan, O. Drozhinin, A. Chanturiya, E. Choe, S. Tutt, K.A. Wood, Y. Hsu, J. Zimmerberg, and R.J. Youle. 1999. Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc. Natl. Acad. Sci. USA. 96:5492–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi, P., L. Scorrano, R. Colonna, V. Petronilli, and F. Di Lisa. 1999. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur. J. Biochem. 264:687–701. [DOI] [PubMed] [Google Scholar]
- Buttke, T.M., and P.A. Sandstrom. 1994. Oxidative stress as a mediator of apoptosis. Immunol. Today. 15:7–10. [DOI] [PubMed] [Google Scholar]
- Colombini, M., E. Blachly-Dyson, and M. Forte. 1996. VDAC, a channel in the outer mitochondrial membrane. Ion Channels, Vol. 4. Plenum, New York. 169–202. [DOI] [PubMed]
- Crompton, M. 1999. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341:233–249. [PMC free article] [PubMed] [Google Scholar]
- de Pinto, V., G. Prezioso, and F. Palmieri. 1987. A simple and rapid method for the purification of the mitochondrial porin from mammalian tissues. Biochim. Biophys. Acta. 905:499–502. [DOI] [PubMed] [Google Scholar]
- Davies, K.J. 1999. The broad spectrum of responses to oxidants in proliferating cells: a new paradigm for oxidative stress. IUBMB Life. 48:41–47. [DOI] [PubMed] [Google Scholar]
- Desagher, S., and J.C. Martinou. 2000. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 10:369–377. [DOI] [PubMed] [Google Scholar]
- Du, C., M. Fang, Y. Li, L. Li, and X. Wang. 2000. Smac, a mitochondrial protein that promotes cyto c-dependent caspase activation by eliminating IAP inhibition. Cell. 102:33–42. [DOI] [PubMed] [Google Scholar]
- Duchen, M.R. 2000. Mitochondria and Ca(2+) in cell physiology and pathophysiology. Cell Calcium. 28:339–348. [DOI] [PubMed] [Google Scholar]
- Eskes, R., B. Antonsson, A. Osen-Sand, S. Montessuit, C. Richter, R. Sadoul, G. Mazzei, A. Nichols, and J.C. Martinou. 1998. Bax-induced cyto c release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol. 143:217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum, G. 2000. Mitochondrial participation in ischemic and traumatic neural cell death. J. Neurotrauma. 17:843–855. [DOI] [PubMed] [Google Scholar]
- Ghafourifar, P., U. Schenk, S.D. Klein, and C. Richter. 1999. Mitochondrial nitric-oxide synthase stimulation causes cyto c release from isolated mitochondria. Evidence for intramitochondrial peroxynitrite formation. J. Biol. Chem. 274:31185–31188. [DOI] [PubMed] [Google Scholar]
- Gottlieb, E., M.G. Vander Heiden, and C.B. Thompson. 2000. Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor α-induced apoptosis. Mol. Cell. Biol. 20:5680–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger, D.N., M.E. Hollwarth, and D.A. Parks. 1986. Ischemia-reperfusion injury: role of oxygen-derived free radicals. Acta. Physiol. Scand. 548:47–63. [PubMed] [Google Scholar]
- Green, D.R., and J.C. Reed. 1998. Mitochondria and apoptosis. Science. 281:1309–1312. [DOI] [PubMed] [Google Scholar]
- Green, D.R. 2000. Apoptotic pathways: paper wraps stone blunts scissors. Cell. 102:1–4. [DOI] [PubMed] [Google Scholar]
- Gross, A., J. Jockel, M.C. Wei, and S.J. Korsmeyer. 1998. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 17:3878–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, M.H., and C.B. Thompson. 2000. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 7:1182–1191. [DOI] [PubMed] [Google Scholar]
- Hajnóczky, G., G. Csordás, M. Madesh, and P. Pacher. 2000. Control of apoptosis by IP3 and ryanodine receptor driven calcium signals. Cell Calcium. 28:349–363. [DOI] [PubMed] [Google Scholar]
- Heiskanen, K.M., M.B. Bhat, H.W. Wang, J. Ma, and A.L. Nieminen. 1999. Mitochondrial depolarization accompanies cyto c release during apoptosis in PC6 cells. J. Biol. Chem. 274:5654–5658. [DOI] [PubMed] [Google Scholar]
- Hennet, T., C. Richter, and E. Peterhans. 1993. Tumour necrosis factor-alpha induces superoxide anion generation in mitochondria of L929 cells. Biochem. J. 289:587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi, M., T. Honda, R.J. Proske, and E.T. Yeh. 1998. Regulation of reactive oxygen species-induced apoptosis and necrosis by caspase 3-like proteases. Oncogene. 17:2753–2760. [DOI] [PubMed] [Google Scholar]
- Hildeman, D.A., T. Mitchell, T.K. Teague, P. Henson, B.J. Day, J. Kappler, and P.C. Marrack. 1999. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 10:735–744. [DOI] [PubMed] [Google Scholar]
- Hockenbery, D.M., Z.N. Oltvai, X.M. Yin, C.L. Milliman, and S.J. Korsmeyer. 1993. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 75:241–251. [DOI] [PubMed] [Google Scholar]
- Jacobson, M.D. 1996. Reactive oxygen species and programmed cell death. Trends Biochem. Sci. 21:83–86. [PubMed] [Google Scholar]
- Jurgensmeier, J.M., Z. Xie, Q. Deveraux, L. Ellerby, D. Bredesen, and J.C. Reed. 1998. Bax directly induces release of cyto c from isolated mitochondria. Proc. Natl. Acad. Sci. USA. 95:4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane, D.J., T.A. Sarafian, R. Anton, H. Hahn, E.B. Gralla, J.S. Valentine, T. Ord, and D.E. Bredesen. 1993. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 262:1274–1277. [DOI] [PubMed] [Google Scholar]
- Korsmeyer, S.J., M.C. Wei, M. Saito, S. Weiler, K.J. Oh, and P.H. Schlesinger. 2000. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cyto c. Cell Death Differ. 7:1166–1173. [DOI] [PubMed] [Google Scholar]
- Krajewski, S., M. Krajewska, L.M. Ellerby, K. Welsh, Z. Xie, Q.L. Deveraux, G.S. Salvesen, D.E. Bredesen, R.E. Rosenthal, G. Fiskum, and J.C. Reed. 1999. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA. 96:5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer, G., B. Dallaporta, and M. Resche-Rigon. 1998. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60:619–642. [DOI] [PubMed] [Google Scholar]
- Kroemer, G., and J.C. Reed. 2000. Mitochondria control of cell death. Nat. Med. 6:513–519. [DOI] [PubMed] [Google Scholar]
- Lewis, W., E.S. Levine, B. Griniuviene, K.O. Tankersley, J.M. Colacino, J.P. Sommadossi, K.A. Watanabe, and F.W. Perrino. 1996. Fialuridine and its metabolites inhibit DNA polymerase gamma at sites of multiple adjacent analog incorporation, decrease mtDNA abundance, and cause mitochondrial structural defects in cultured hepatoblasts. Proc. Natl. Acad. Sci. USA. 93:3592–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P., D.Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri, and X. Wang. 1997. Cyto c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 91:479–489. [DOI] [PubMed] [Google Scholar]
- Lin, K.T., J.Y. Xue, M. Nomen, B. Spur, and P.Y. Wong. 1995. Peroxynitrite-induced apoptosis in HL-60 cells. J. Biol. Chem. 270:16487–16490. [DOI] [PubMed] [Google Scholar]
- Liu, X., C.N. Kim, J. Yang, R. Jemmerson, and X. Wang. 1996. Induction of apoptotic program in cell-free extracts: requirement for dATP and cyto c. Cell. 86:147–157. [DOI] [PubMed] [Google Scholar]
- Madesh, M., O. Benard, and K.A. Balasubramanian. 1999. Apoptotic process in the monkey small intestinal epithelium: 2. Possible role of oxidative stress. Free Radic. Biol. Med. 26:431–438. [DOI] [PubMed] [Google Scholar]
- Marchetti, P., M. Castedo, S.A. Susin, N. Zamzami, T. Hirsch, A. Macho, A. Haeffner, F. Hirsch, M. Geuskens, and G. Kroemer. 1996. Mitochondrial permeability transition is a central coordinating event of apoptosis. J. Exp. Med. 184:1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo, I., C. Brenner, J.M. Zamzami, S.A. Jurgensmeier, H.L. Susin, M.C. Vieira, M.C. Prevost, Z. Xie, S. Matsuyama, J.C. Reed, and G. Kroemer. 1998. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 281:2027–2031. [DOI] [PubMed] [Google Scholar]
- Narita, M., S. Shimizu, T. Ito, T. Chittenden, R.J. Lutz, H. Matsuda, and Y. Tsujimoto. 1998. Bax interacts with the permeability transition pore to induce permeability transition and cyto c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA. 95:14681–14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher, P., P. Csordás, T. Schneider, and G. Hajnóczky. 2000. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J. Physiol. 529:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino, J.G., S.T. Chen, M. Tafani, J.W. Snyder, and J.L. Farber. 1998. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J. Biol. Chem. 273:7770–7775. [DOI] [PubMed] [Google Scholar]
- Patel, M., B.J. Day, J.D. Crapo, I. Fridovich, and J.O. McNamara. 1996. Requirement for superoxide in excitotoxic cell death. Neuron. 16:345–355. [DOI] [PubMed] [Google Scholar]
- Petit, P.X., H. Lecoeur, E. Zorn, C. Dauguet, B. Mignotte, and M.L. Gougeon. 1995. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J. Cell Biol. 130:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein, J.D., L.A. Bristol, B. Hosler, R.H. Brown, Jr., and R.W. Kuncl. 1994. Chronic inhibition of superoxide dismutase produces apoptotic death of spinal neurons. Proc. Natl. Acad. Sci. USA. 91:4155–4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, M., S.J. Korsmeyer, and P.H. Schlesinger. 2000. BAX-dependent transport of cyto c reconstituted in pure liposomes. Nat. Cell Biol. 2:553–555. [DOI] [PubMed] [Google Scholar]
- Shafir, I., W. Feng, and V. Shoshan-Barmataz. 1998. Voltage-dependent anion channel proteins in synaptosomes of the torpedo electric organ: immunolocalization, purification, and characterization. J. Bioenerg. Biomembr. 30:499–510. [DOI] [PubMed] [Google Scholar]
- Shimizu, S., M. Narita, and Y. Tsujimoto. 1999. Bcl-2 family proteins regulate the release of apoptogenic cyto c by the mitochondrial channel VDAC. Nature. 399:483–487. [DOI] [PubMed] [Google Scholar]
- Shimizu, S., Y. Matsuoka, Y. Shinohara, Y. Yoneda, and Y. Tsujinoto. 2001. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J. Cell Biol. 152:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin, S.A., H.K. Lorenzo, N. Zamzami, I. Marzo, B.E. Snow, G.M. Brothers, J. Mangion, E. Jacotot, P. Costantini, M. Loeffler, et al. 1999. a. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 397:441–446. [DOI] [PubMed] [Google Scholar]
- Susin, S.A., H.K. Lorenzo, N. Zamzami, I. Marzo, C. Brenner, N. Larochette, M.C. Prevost, P.M. Alzari, and G. Kroemer. 1999. b. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med. 189:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai, G., R. Krishnamurthy, and G. Hajnóczky. 1999. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 18:6349–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen, A.M., P.G. Ekert, M. Pakusch, J. Silke, L.M. Connolly, G.E. Reid, R.L. Moritz, R.J. Simpson, and D.L. Vaux. 2000. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 102:43–53. [DOI] [PubMed] [Google Scholar]
- Vander Heiden, M.G., N.S. Chandel, P.T. Schumacker, and C.B. Thompson. 1999. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol. Cell. 3:159–167. [DOI] [PubMed] [Google Scholar]
- von Ahsen, O., C. Renken, G. Perkins, R.M. Kluck, E. Bossy-Wetzel, and D.D. Newmeyer. 2000. Preservation of mitochondrial structure and function after Bid- or Bax-mediated cyto c release. J. Cell Biol. 150:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse, N.J., J.C. Goldstein, O. von Ahsen, M. Schuler, D.D. Newmeyer, and D.R. Green. 2001. Cyto c maintains mitochondrial transmembrane potential and atp generation after outer mitochondrial membrane permeabilization during the apoptotic process. J. Cell Biol. 153:319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, M.C., T. Lindsten, V.K. Mootha, S. Weiler, A. Gross, M. Ashiya, C.B. Thompson, and S.J. Korsmeyer. 2000. tBid, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14:2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Wei, M.C., W.X. Zong, E.H. Cheng, T. Lindsten, V. Panoutsakopoulou, A.J. Ross, K.A. Roth, G.R. MacGregor, C.B. Thompson, and S.J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 292:727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J.C., and G.A. Cortopassi. 1998. Induction of the mitochondrial permeability transition causes release of the apoptogenic factor cytochrome c. Free Radic. Biol. Med. 24:624–631. [DOI] [PubMed] [Google Scholar]
- Yuan, J., and B.A. Yankner. 2000. Apoptosis in the nervous system. Nature. 407:802–809. [DOI] [PubMed] [Google Scholar]
- Zamzami, N., P. Marchetti, M. Castedo, D. Decaudin, A. Macho, T. Hirsch, S.A. Susin, P.X. Petit, B. Mignotte, and G. Kroemer. 1995. a. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 182:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami, N., P. Marchetti, M. Castedo, C. Zanin, J.L. Vayssiere, P.X. Petit, and G. Kroemer. 1995. b. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med. 181:1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami, N., S.A. Susin, P. Marchetti, T. Hirsch, I. Gomez-Monterrey, M. Castedo, and G. Kroemer. 1996. Mitochondrial control of nuclear apoptosis. J. Exp. Med. 183:1533–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti, M., and I. Szabo. 1995. The mitochondrial permeability transition. Biochim. Biophys. Acta. 1241:139–176. [DOI] [PubMed] [Google Scholar]
- Zou, H., W.J. Henzel, X. Liu, A. Lutschg, and X. Wang. 1997. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cyto c-dependent activation of caspase-3. Cell. 90:405–413. [DOI] [PubMed] [Google Scholar]