

Abstract
Transport and sorting of lipids must occur with specific mechanisms because the membranes of intracellular organelles differ in lipid composition even though most lipid biosynthesis begins in the ER. In yeast, ceramide is synthesized in the ER and transferred to the Golgi apparatus where inositolphosphorylceramide (IPC) is formed. These two facts imply that ceramide can be transported to the Golgi independent of vesicular traffic because IPC synthesis still continues when vesicular transport is blocked in sec mutants. Nonvesicular IPC synthesis in intact cells is not affected by ATP depletion. Using an in vitro assay that reconstitutes the nonvesicular pathway for transport of ceramide, we found that transport is temperature and cytosol dependent but energy independent. Preincubation of ER and Golgi fractions together at 4°C, where ceramide transport does not occur, rendered the transport reaction membrane concentration independent, providing biochemical evidence that ER-Golgi membrane contacts stimulate ceramide transport. A cytosolic protease-sensitive factor is required after establishment of ER-Golgi contacts.
Keywords: ceramide transport; sphingolipids; membrane contacts; secretion; Saccharomyces cerevisiae
Introduction
The membranes of various intracellular compartments of all eukaryotic cells have distinct lipid compositions (van Meer, 1998; Schneiter et al., 1999). The molecular mechanisms that establish and maintain these different lipid compositions are largely unresolved. These processes must be highly regulated to ensure correct membrane assembly for normal cellular function (Futerman et al., 1998). Lipid localization can be achieved by the dynamic interplay between local synthesis, modification, transport, and sorting of various lipids. The first step in the biosynthesis of most membrane lipids occurs in the ER, and then lipids travel to their final destination. This movement must occur selectively by specific mechanisms for each lipid (McIntyre and Sleight, 1994; Trotter and Voelker, 1994; Liscum and Munn, 1999; van Meer and Holthuis, 2000; Voelker, 2000). It is assumed that transport of lipids between intracellular compartments occurs via either vesicular or nonvesicular transport or both. Nonvesicular transport could occur by transport of lipid monomers through the cytosol or by transfer of lipids at regions of close apposition between membranes. Although several lipid transfer proteins and mutants defective in lipid transport have been identified and isolated (Nishijima et al., 1997; Emoto et al., 1999; Liscum and Munn, 1999; Raggers et al., 2000), the precise roles of proteins in net transport and sorting of lipids remain unclear.
In mammalian cells, ceramide is formed by decarboxylation and condensation of L-serine with palmitoyl-CoA, followed by reduction of 3-ketodihydrosphingosine to generate dihydrosphingosine (DHS),* N-acylation of DHS with a fatty acid, and finally desaturation of dihydroceramide (DHS-Cer) (Merrill and Jones, 1990). The biosynthesis of ceramide seems to occur at the cytosolic surface of the ER in mammalian cells (Mandon et al., 1992; Hirschberg et al., 1993), although the ceramide synthase has not been identified. Ceramide, a common precursor for all complex sphingolipids, is then transported to the Golgi apparatus for conversion to both sphingomyelin and glucosylceramide by yet-unknown mechanisms (van Meer and Holthuis, 2000). Recently, it has been shown by in vivo analysis of a mutant resistant to lysenin, an SM-directed cytolysin, that the transport of ceramide to the sites of sphingomyelin and glucosylceramide synthesis occurs via a mechanism that differs from the ER-to-Golgi vesicular transport of glycoproteins (Fukasawa et al., 1999).
In yeast cells, the steps in ceramide biosynthesis are similar to those in mammalian cells except that DHS-Cer is hydroxylated by Sur2p to give phytoceramide (Haak et al., 1997; Grilley et al., 1998), but the exact localization and topology of the reactions of ceramide biosynthesis have not been studied. In contrast to the situation in mammalian cells, it was assumed that ceramide is converted to inositolphosphorylceramide (IPC), one of three major classes of yeast sphingolipids, by addition of inositolphosphate from phosphatidylinositol (PI) in the ER. Subsequently, IPC was thought to be delivered to the Golgi apparatus where IPC is mannosylated to form mannosyl-IPC followed by mannosyl-diinositolphosphorylceramide (Dickson and Lester, 1999; Schneiter, 1999). This idea was based on the fact that IPC synthesis continued at nonpermissive temperature in sec mutants that block ER-to-Golgi vesicular transport of proteins (Puoti et al., 1991). Recently, Aur1p, a protein required for IPC synthesis, has been localized to the Golgi apparatus (Levine et al., 2000), implying that ceramide is delivered to the Golgi apparatus. This suggests that a nonvesicular transport mechanism exists for ceramide transport to the Golgi apparatus.
In vitro systems that reproduce transport events occurring in intact cells are powerful tools to understand molecular mechanisms (Wuestehube and Schekman, 1992; Moreau et al., 1993; Barlowe et al., 1994; Funakoshi et al., 2000). Therefore, we have developed a cell-free system with isolated donor and acceptor membranes that allows us to study the mechanisms of ceramide transport and ceramide synthesis. Here, we show that ceramide is transported from the ER to the Golgi apparatus via two distinct pathways, one vesicle dependent and the other vesicle and ATP independent. We provide biochemical evidence that ER-Golgi membrane contacts play an important role in nonvesicular ceramide transport. A cytosolic factor is required for transport after formation of ER-Golgi contacts.
Results
ER to Golgi sec mutants do not completely block sphingolipid synthesis
In yeast, IPC, the simplest sphingolipid, is made by transferring inositolphosphate from PI onto ceramide by Aur1p, a Golgi enzyme (Dickson and Lester, 1999; Schneiter, 1999; Levine et al., 2000). Previous studies showed that IPC can be synthesized from radiolabeled DHS or myo-inositol, even when ER-to-Golgi vesicular transport is blocked in temperature-sensitive secretion mutants (sec12, -16, -17, -18, and -23) at restrictive temperature (Puoti et al., 1991; Reggiori and Conzelmann, 1998). To rule out the possibility that the lack of complete inhibition was due to newly synthesized IPC synthase remaining blocked in the ER, the experiment was repeated in the presence of the protein synthesis inhibitor cycloheximide (Chx). Wild-type or sec18 mutant cells were grown at 24°C and then metabolically labeled with [3H]DHS for 2 h at 24 or 37°C after preincubations with Chx (Fig. 1 A). Lipids were extracted and deacylated by mild alkaline hydrolysis to identify radiolabeled lipids and separated by TLC. Phosphatidylethanolamine, phosphatidylcholine (PC), and PI were completely sensitive to mild alkaline hydrolysis, whereas sphingolipids were resistant. [3H]myo-inositol–radiolabeled lipid mixtures and [3H]DHS-1-phosphate (DHS-1-P) prepared in vitro from [3H]DHS with yeast cytosol, and ATP regenerating system were used as standards to identify PI, IPC/B, IPC/C, and DHS-1-P. At 37°C, wild-type cells showed a decrease in IPC/C synthesis compared with 24°C. This decrease is due to the presence of Chx because no reduction was found in absence of Chx (unpublished data). As expected, sec18 made only 30–45% of the amount of IPC/C synthesized in the wild-type at 37°C. This was also seen when the labeling was performed for 30 min, although the reduction in IPC/C synthesis was increased slightly (Fig. 1 B). Similar results can be observed in other mutants such as sec17, sec12, and sec23, although the amount of remaining transport varies. The results with the sec12 and sec23 mutants, which are more specific for ER to Golgi transport than sec18, suggest that ∼40–50% of ER to Golgi ceramide transport is nonvesicular under our conditions. Our results confirm previous studies reporting that IPC/C can be synthesized independently from ER-to-Golgi vesicular transport and show the importance of the nonvesicular pathway.
Figure 1.
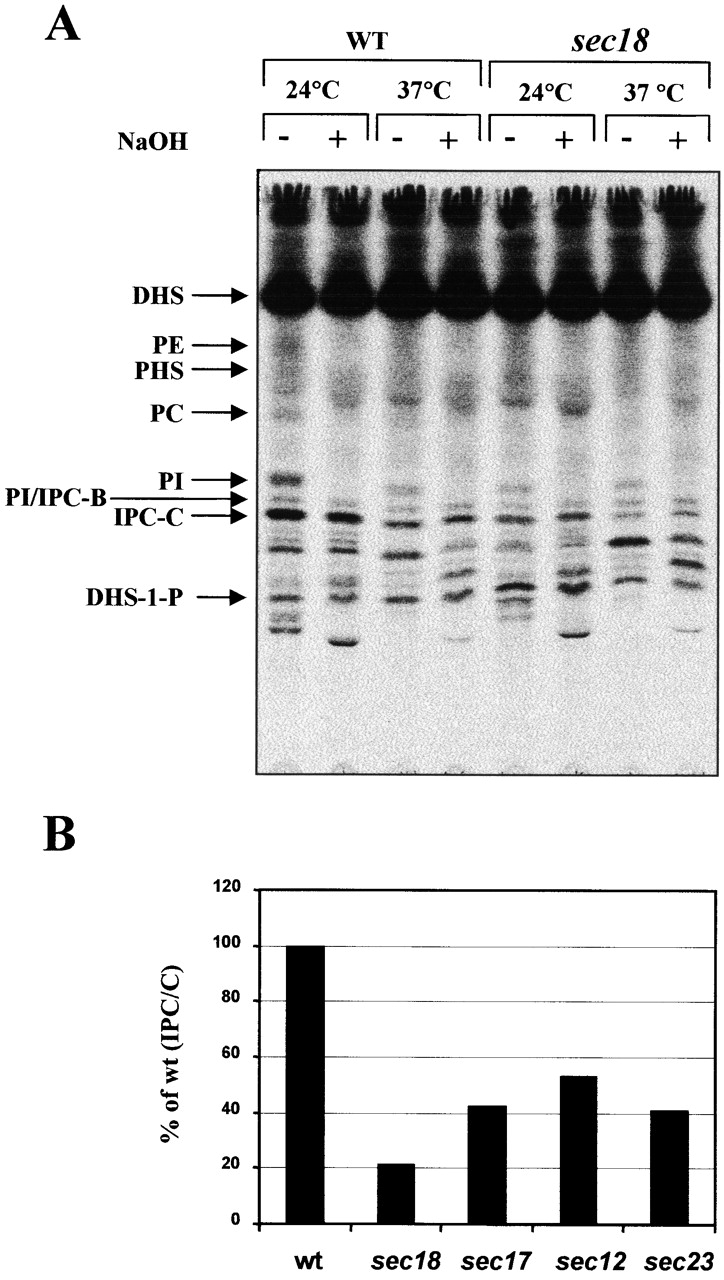
Incorporation of [ 3 H]DHS into sphingolipids in sec mutants. (A) Wild-type (RH448) and sec18 mutant (RH1737) cells were preincubated with Chx for 20 min at 24°C, then incubated for 20 min at 24 or 37°C, and labeled with [3H]DHS for 2 h at the same temperature. The labeled lipids were subjected (+) or not (−) to mild alkaline hydrolysis with NaOH and applied to TLC plates using solvent I. DHS, phosphatidylethanolamine, PHS, PC, PI, the different hydroxylation forms of IPC (Fig. 2 A, IPC-A, -B, and -C), and DHS-1-P are marked. (B) Wild-type and sec mutants (RH1737, RH1592, RH1491, and RH1436) were preincubated with Chx for 20 min at 24°C, then incubated for 20 min at 37°C, and labeled with [3H]DHS for 30 min at 37°C. The labeled lipids treated with NaOH were applied to TLC plates as above. Incorporation of radioactivity into IPC/C was quantified, and the relative amount of IPC synthesis was determined as the percentage of the amount in wild-type cells.
Ceramide and IPC synthesis in vitro
Our goal is to study the mechanisms underlying transport of newly synthesized ceramide to its site of conversion to IPC. Because an in vitro system using isolated organelles would be a powerful tool, we first examined the synthesis of IPC by microsomal membranes from [3H]DHS. When microsomal membranes were incubated at 24°C with cytosol, ATP-regenerating system, GDP-mannose, coenzyme A, and liposomes containing hexacosanoic acid (C26) and PI, two subclasses of IPC (IPC/B and IPC/C), were synthesized. The IPC/B and IPC/C comigrated with [3H]myo-inositol–labeled standards were base resistant, PI-PLC sensitive (Fig. 2 A), and their synthesis was blocked by aureobasidin A (AbA), an inhibitor of IPC synthesis (Fig. 3 A) (Zhong et al., 1999).
Figure 2.

Analysis of lipids labeled with [ 3 H]DHS in vitro. (A) In vitro [3H]DHS labeling with microsomal membranes and cytosol from RH981 was performed at 10°C for 15 min and then at 4 or 24°C for 2 h as described in Materials and methods. Aliquots of the labeled lipids were treated or not with NaOH or PI-PLC. TLC was performed using solvent I. (B and C) In vitro [3H]DHS labeling and NaOH treatment of the labeled lipids were performed as above. IPCs (lanes 1–4) and presumed ceramides (lanes 5–8) were purified from the lipids treated with NaOH. Aliquots of the purified lipids were treated or not with PI-PLC or HCl. TLC was performed using solvent I (B) or III (C). (D) Lipids *I and *II visible in C, lane 6, were purified and treated or not with HCl. TLC was performed using solvent I. X, Y, and Z are unidentified lipids.
Figure 3.
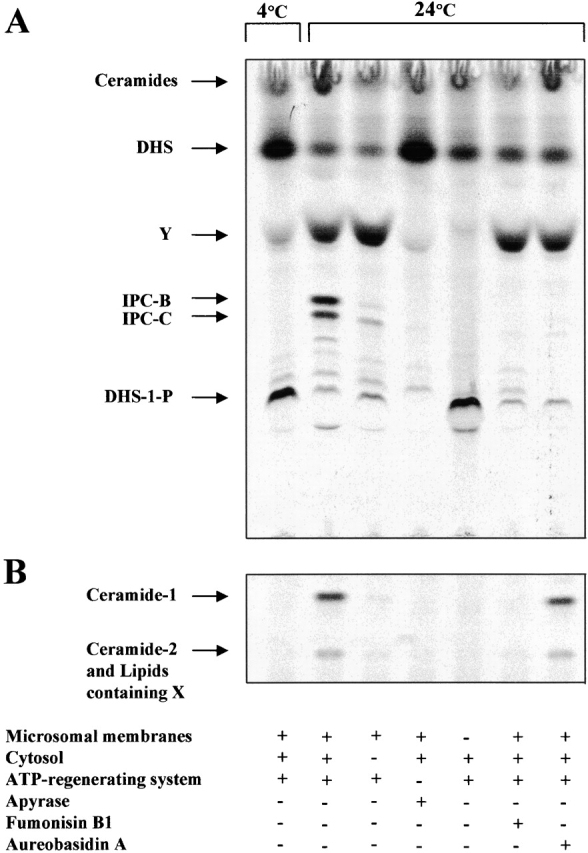
Requirements for ceramide and IPC synthesis in vitro. (A and B) In vitro [3H]DHS labeling under different conditions as indicated and NaOH treatment of the labeled lipids were performed as described in the legend to Fig. 2 A. To the samples containing no ATP-regenerating system, apyrase (0.5 U, 10 U/ml) was added. FuB and AbA were used at 100 μM and 50 nM, respectively. TLC was performed using solvent I (A) or III (B).
The identity of ceramide was established by its chemical characteristics and by using specific inhibitors of ceramide synthesis. Base-resistant IPCs and presumed ceramides synthesized in vitro were purified from TLC plate by scraping followed by PI-PLC and/or HCl treatments (Fig. 2, B–D). The presumed ceramides (Fig. 2 B, lane 5) comigrated with ceramides derived from base-resistant IPCs treated with PI-PLC (Fig. 2 B, lane 2) and yielded the sphingoid bases DHS and PHS and an unidentified lipid X on strong HCl hydrolysis (Fig. 2 B, lanes 3, 4, 7, and 8) in solvent system I. When solvent system III was used, two different ceramides could be resolved from PI-PLC–treated IPCs (Fig. 2 C, lane 2). From the presumed ceramide fractions, we observed one extra lipid Z, which migrates further than the two ceramides and was hydrolyzed by HCl (Fig. 2 C, lanes 5 and 6). We have not further investigated the identity of this lipid. Two ceramides, *I and *II (Fig. 2 C, lane 6) were purified and hydrolyzed individually with HCl. This treatment produced DHS from ceramide *I. PHS and X were produced from ceramide *II (Fig. 2 D). These results show that ceramide-1 (*I) is DHS-Cer and ceramide-2 (*II) is a complex of phytoceramide and a ceramide having X. Ceramide-1 and -2 formation was blocked by specific inhibitors of ceramide synthesis, fumonisin B1 (FuB) (Fig. 3 B) (Merrill et al., 1993) and australifungin (unpublished data) (Mandala et al., 1995). The inhibition by FuB reveals that ceramide synthesis is based on the fatty acyl-CoA–dependent reaction, not on the reversal of ceramidases (Mao et al., 2000a,b).
This in vitro system using microsomal membranes showed that ceramide synthesis is temperature dependent and requires ATP, cytosol, and a membrane fraction (Fig. 3 B). When the liposomes containing hexacosanoic acid (C26) and PI were omitted from the reaction mixture, a small reduction (18%) in the synthesis of IPCs was found. We also found that N-ethylmaleimide (NEM) completely inhibited ceramide synthesis, suggesting the requirement of an NEM-sensitive protein or compound (unpublished data).
Ceramide and IPC synthase activities are localized in the ER and Golgi apparatus, respectively
In yeast, no evidence concerning the site of ceramide synthesis has been published. However, IPC synthase activity and Aur1p have been localized to the Golgi apparatus (Levine et al., 2000). Therefore, to investigate the transport of ceramide it was important to define where ceramide synthesis activity is located. Subcellular fractionation by sucrose density gradient of [3H]DHS-labeled membranes expressing Aur1p-hemagglutinin (HA) showed that the majority of synthesized ceramides and DHS and DHS-1-P behaved as the ER marker Wbp1p, whereas IPCs cosedimented with Aur1p-HA and Sed5, a marker for Golgi membranes (unpublished data; Banfield et al., 1994; Schröder et al., 1995; Levine et al., 2000). This result suggests the ER and Golgi localizations of ceramide and IPC synthesis, respectively.
Next, we investigated whether ceramide and IPC synthesis could be reconstituted using purified ER- and Golgi-enriched membranes. ER-enriched membranes were prepared as described previously (Wuestehube and Schekman, 1992), and Golgi-enriched membranes were isolated by differential centrifugation (see Materials and methods). Western blotting analysis showed that Wbp1p was found exclusively in P27 fraction and P45 fraction, resulting from further fractionation of S27, whereas Emp47p, a Golgi marker (Schröder et al., 1995) and Aur1p-HA were found mainly in the final supernatant (S45) (Fig. 4 A). Therefore, we used the S45 fraction as source of Golgi-enriched membranes even though a small amount of ER membranes remained in this fraction. Fractionation of the P27 fraction on a sucrose gradient resulted in further purification of ER membranes (at the 40/50% sucrose interface) as shown in Fig. 4 B. Fig. 4 C shows an experiment in which ceramide and IPC synthase activities were examined with the purified ER- and Golgi-enriched membranes. As expected, efficient synthesis and subsequent conversion of ceramide to IPC required the addition of both ER- and Golgi-enriched membranes. When only ER membranes were incubated, the amount of ceramide synthesized remained high (87% of complete incubation), but the level of IPC synthesis was low. This confirms that IPC synthase activity is localized in Golgi membranes. In contrast, addition of only Golgi membranes resulted in significant reductions of ceramide and IPC synthesis by 41 and 27%, respectively. Since a detectable amount of Wbp1p was found in the Golgi membrane fraction, the lack of complete loss of ceramide synthase activity is most likely due to ER contamination in the Golgi fraction. The above results are consistent with the localization of ceramide synthesis to the ER. Ceramide would then be transported to the Golgi apparatus for conversion to IPC.
Figure 4.
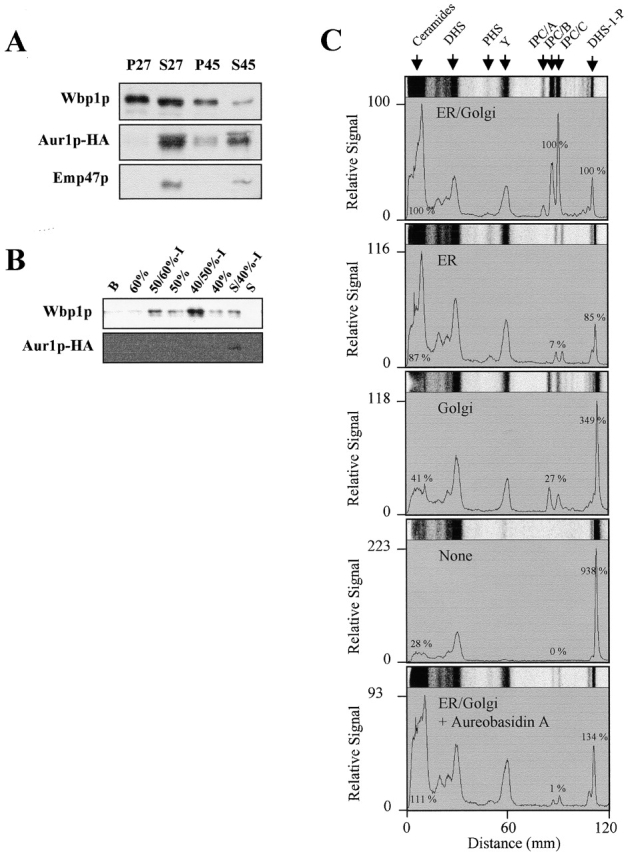
Reconstitution of ceramide and subsequent IPC synthesis with ER- and Golgi-enriched membranes. (A and B) Purification of ER- and Golgi-enriched membranes was performed as described in Materials and methods. (A) Microsomal membranes from RH5256 were subjected to sequential centrifugation to obtain pellet (P27), supernatant (S27) fraction, and from the S27 fraction, pellet (P45), supernatant (S45) fraction. (B) The P27 fraction was subjected to sucrose density step gradient, and the resulting gradient fractions were collected from the top. Equal fractions were analyzed for the distribution of Aur1p-HA, Emp47p, and Wbp1p by Western blotting. (C) In vitro labeling using [3H]DHS with either ER- (40/50%-I fraction, 5–10 μg) or Golgi- (S45 fraction, 25–50 μg) enriched membranes or both (40/50%-I fraction, 5–10 μg and S45 fraction, 25–50 μg), and NaOH treatment of the labeled lipids were performed as described in the legend to Fig. 2 A. Control experiments were done without membranes or with both membranes in the presence of AbA (50 nM). The lipids were applied to TLC plates using solvent I. Images of portions of individual lanes were shown, and the maximal signal of [3H]ceramide in the experiment, which was done with both membranes, was set to 100 as the relative signal. In these diagrams, the total amount of incorporation of [3H]DHS into each lipid was expressed as the percentage of that of [3H]DHS with both membranes.
Properties of ceramide transport in vitro
To study the transport of ceramide from ER to the Golgi compartment, we next developed a two stage in vitro assay that is based on the conversion of ceramide to IPC. In the first stage, radiolabeled ceramides are synthesized by incubating ER-enriched membranes in the presence of cytosol, ATP-regenerating system, substrates, and AbA for 2 h at 24°C. Ceramide synthesis from [3H]DHS with the ER-enriched membranes also requires ATP and cytosol and is temperature and time dependent (unpublished data). AbA was used to block any IPC synthase activity coming from contamination of Golgi membranes in the ER fraction. After ceramide synthesis, the ER membranes were washed by centrifugation (27,000 g, 10 min, 4°C). The recovery of radiolabeled ceramides was >85%. In the second stage, an acceptor membrane and cytosol, ATP-regenerating system, substrates, AbA, and FuB (to block further ceramide synthesis) were added to the ER membrane fraction containing [3H]ceramide. Microsomal membranes prepared from an AbA-resistant strain were used as acceptor membranes because the AbA-resistant but not wild-type cells can make IPCs in the presence of AbA in vivo (Fig. 5 A). Microsomal membranes prepared from the AbA-resistant cells are resistant to AbA but are sensitive to FuB (unpublished data). Fig. 5 B shows the basic features of the in vitro ceramide transport assay. When acceptor membranes from the AbA-resistant strain were used in a complete reaction mixture, ceramides were converted to IPC, indirectly assaying transport of ceramide from ER to the Golgi membranes. The amount of ceramide remaining in the reaction mixture was reduced after incubation at 24°C compared with at 4°C (unpublished data). As a negative control, no conversion was detected with acceptor membranes from a wild-type strain. The conversion of ceramide to IPC had a strong requirement for cytosol. The reaction continued for 120 min with a lag of 15 min (Fig. 5 C), which varied depending upon the experiment and preparations (see Fig. 7 , A–C). The reaction was temperature dependent because no conversion of ceramide to IPC occurs at 4°C (unpublished data). Depletion of ATP did not affect the conversion of ceramide to IPC, suggesting that ceramide transport occurs in our system in an ATP-independent manner. The reaction was dependent on the amount of acceptor membranes, being linear up to ∼50 μg (protein concentration) of acceptor membranes. (Fig. 5 D). To test for specificity of transfer of ceramide from ER to Golgi, we took 25 μg of acceptor membranes and added increasing concentrations of competitor membranes. If transfer is specific, then microsomal membranes from wild-type cells (AbA sensitive) should inhibit IPC synthesis. This was the case (Fig. 5 E). If the transfer of ceramide occurs to any membrane, then mitochondria should also be able to compete, which they did not (Fig. 5 E).
Figure 5.
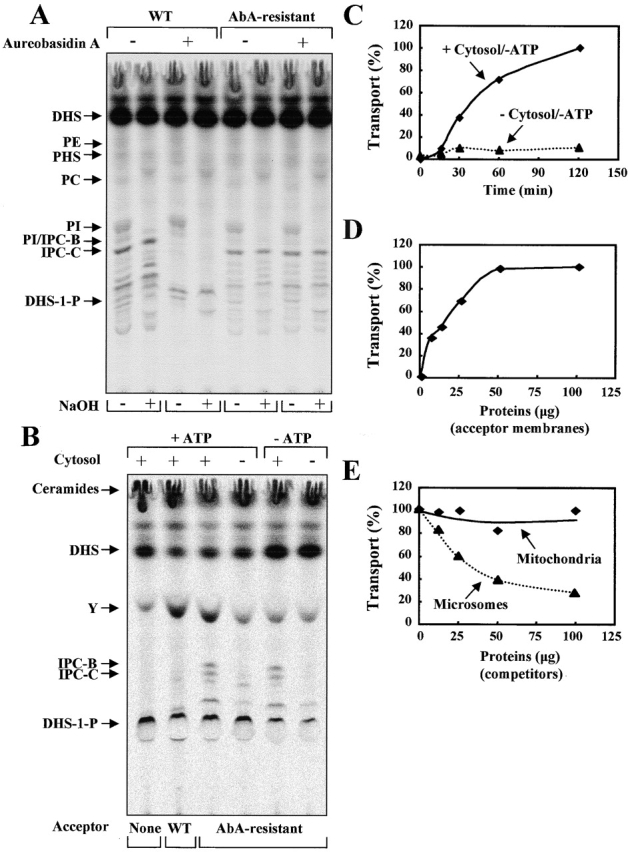
Cytosol- but not ATP-dependent transport of ceramide in vitro. (A) Wild-type (WT, RH5247) and AbA-resistant (RH5255) cells were preincubated with ethanol or AbA (20 μg/ml) for 20 min at 24°C and then labeled with [3H]DHS for 30 min at the same temperature. Lipid analysis was performed as described in the legend to Fig. 1 A. (B) In vitro ceramide transport assay was performed in the presence (+) or absence (−) of cytosol or ATP as described in Materials and methods. To the samples containing no ATP, apyrase was added, and lipid analysis was performed as described in the legend to Fig. 3 A. Control experiments were done without or with acceptor membranes from wild-type (RH5247). (C and D) In vitro ceramide transport assay was performed in the presence or absence of cytosol for indicated times (C) and in the presence of cytosol for 60 min with indicated amounts of the acceptor membranes (D) under ATP-depletion conditions as described above. (E) In vitro ceramide transport assay was performed with 25 μg of acceptor membranes as described in D. The indicated amounts of microsomal membranes (RH5247) or purified mitochondria (D273-10B) from wild-type strains were added as competitors before the second stage reaction. Ceramide transport (%) was determined by quantification of the total amount of the incorporation of [3H]ceramide into IPC and was expressed as the percentage of values of the experiment without competitor membranes.
Figure 7.
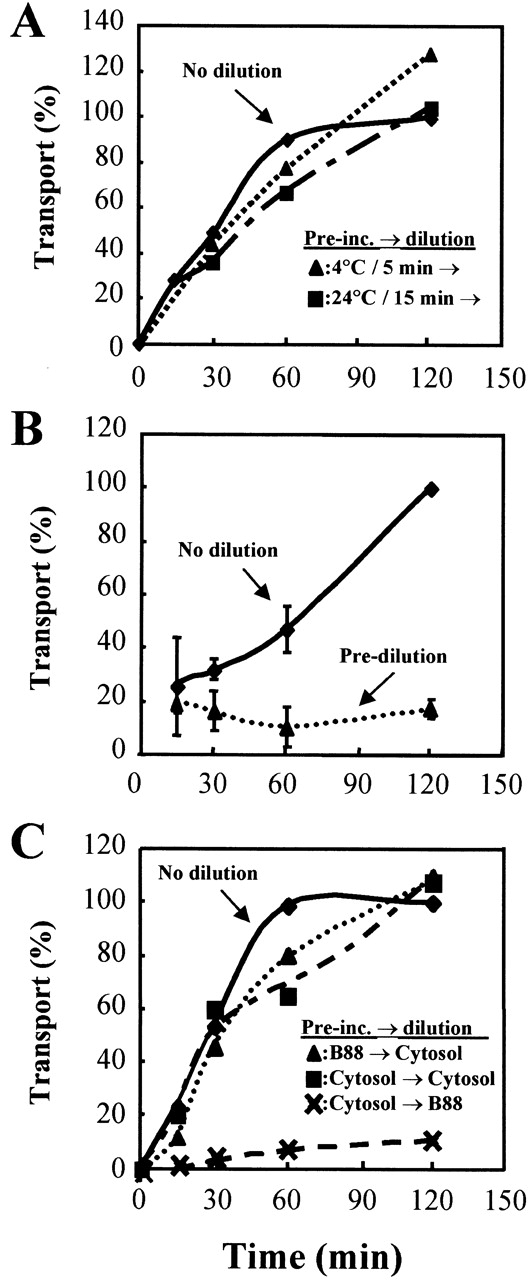
Ceramide transport occurs through ER-Golgi membrane contacts. In vitro ceramide transport assay was performed under ATP depletion conditions as described in the legend to Fig. 5 C. (A) For dilution experiments, the reaction mixtures for transport assay prepared on ice were preincubated at 4°C for 5 min or at 24°C for 15 min and then diluted 10-fold with B88 containing cytosol (2 mg/ml), GDP-mannose (50 μM), liposomes composed of PI (250 μM), and Apyrase (10 U/ml). (B) The reaction mixtures containing donor and acceptor membranes were prediluted independently fivefold with dilution buffer before the mixing. (C) The reaction mixtures prepared in the presence or absence of cytosol were preincubated at 4°C for 5 min and then diluted 10-fold with dilution buffer with or without cytosol. At the indicated times for 24°C incubation, 50-μl aliquots from the undiluted or 500-μl aliquots from the diluted reaction mixtures were withdrawn. Lipid analysis of the mixtures was performed, and ceramide transport (transport in undiluted reaction set to 100%) was determined as described in the legend to Fig. 5 C.
Since cytosol was required for conversion of ceramide to IPC, we next tested whether the cytosolic component was specific and proteinaceous (Table I). The cytosol requirement was specific because BSA could not substitute for cytosol. Furthermore, addition of Triton X-100 resulted in an almost complete loss of the cytosol requirement, suggesting that cytosol is required for ceramide transport but not IPC synthase activity. Cytosol treated with trypsin or treated at 95°C for 15 min did not support the efficient conversion to IPC. These results indicate that a heat-labile and trypsin-sensitive cytosol protein(s) is required for ceramide transport. In contrast, treatment of donor, acceptor, or both membranes with trypsin had little inhibitory effect on conversion of ceramide to IPC, although under the same conditions trypsin treatment resulted in complete removal of the HA epitope tag from the COOH terminus of Aur1p (unpublished data).
Table I. Cytosolic protein is required for ATP-independent transport of ceramide.
| Transport
|
|||
|---|---|---|---|
| Addition | +ATP | −ATP | |
| (%)
|
|||
| Untreated cytosol | 100 | 100 | |
| No cytosol (B88) | 8 | 0 | |
| No cytosol (B88 containing BSA) | 0 | 0 | |
| Treated cytosol | |||
| + Trypsin, 30 min, 4°C, + trypsin inhibitor | 9 | 16 | |
| + Trypsin + trypsin inhibitor, 30 min, 4°C | 87 | 117 | |
| 15 min, 95°C | 0 | 0 | |
| Untreated cytosol + 1% Triton X-100 | 100 | ND | |
| No cytosol (B88) + 1% Triton X-100 | 99 | ND | |
Cytosolic protein is required for ATP-independent transport of ceramide. In vitro ceramide transport assay was performed with untreated or treated cytosol in the presence or absence of ATP as described in the legend to Fig. 5 B. For trypsin treatment, cytosol was treated with trypsin (1.25 mg/ml) for 30 min at 4°C and quenched with soybean trypsin inhibitor (5 mg/ml). In control treatments, soybean trypsin inhibitor was added before addition of trypsin. For heat treatment, cytosol was incubated for 15 min at 95°C and centrifuged for 1 min at 12,000 g, and the resulting supernatant was used for the assay. If present, Triton X-100 (1%) was added to reaction mixtures for 20 min at 4°C before the reaction. Lipid analysis was performed and ceramide transport (%) was determined as described in the legend to Fig. 5 C. ND, not determined.
The next set of experiments was performed to determine whether ceramide transport in our assay requires ER-to-Golgi vesicular transport. Several studies using in vitro cell-free assays have shown that NEM and nonhydrolyzable analogs of GTP such as GTPγS inhibit ER-to-Golgi-vesicular transport (Baker et al., 1990; Barlowe et al., 1994). As seen in Fig. 6 A, neither GTPγS nor NEM had a strong inhibitory effect on ceramide transport. The nonvesicular nature of the transport reaction was confirmed by using cytosol and microsomal membranes from sec18 mutant cells, which are defective for delivery of ER-derived vesicles to the Golgi at restrictive temperature in vitro (Muñiz et al., 2001). Formation of IPC was monitored by a one step assay at permissive (24°C) and restrictive (30 and 37°C) temperatures. Although high temperature reduced the extent of IPC synthesis in both wild-type and sec18 extracts, no specific defect inthe sec18 mutant was detected (Fig. 6 B). Therefore, our results demonstrate that transport of ceramide from ER to the Golgi compartment in our assay occurs by nonvesicular transport.
Figure 6.

Ceramide is transported via a nonvesicular pathway. (A) In vitro ceramide transport assay were performed in the presence of cytosol and ATP as described in the legend to Fig. 5 B. GTPγS (0.4 mM), GTP (0.4 mM), GTP (0.4 mM) plus Sar1p (90 μg/ml), or NEM (10 mM) was added just before the second stage reaction. (B) In vitro [3H]DHS labeling with cytosol and microsomal membranes from wild-type (RH981) or sec18 mutant (RH2043) cells at different temperatures and lipid analysis were performed as described in the legend to Fig. 2 A.
Two mechanisms for nonvesicular transport between membranes have been postulated. One is transfer of lipid through the cytosol and the other is transfer of lipid through direct membrane contacts (McIntyre and Sleight, 1994; Trotter and Voelker, 1994). The former should be strongly dependent on ER and Golgi membrane concentrations throughout the reaction. However, preliminary experiments suggested that the reaction was dilution independent at several time points of the reaction at 24°C. Therefore, we examined the effect of dilution on ceramide transport after a preincubation of donor and acceptor membranes together at 4°C where transport does not occur. Fig. 7 A shows the kinetics of ceramide transport with different preincubation conditions. IPC synthesis was not affected by dilution after premixing of membranes at 4°C for 5 min. Control experiments showed that when membranes were diluted independently before the mixing (0 min preincubation), no time-dependent conversion of ceramide to IPC was observed (Fig. 7 B). Therefore, once the two membranes have been preincubated at 4°C, synthesis of IPC is a zero order reaction with respect to membranes. This strongly suggests that ceramide transport occurs through ER-Golgi membrane contacts. In addition, we found that when donor and acceptor membranes were preincubated at 4°C with cytosol and then diluted without cytosol, no efficient conversion of ceramide to IPC was observed (Fig. 7 C), demonstrating that cytosol is required after ER-Golgi membrane contacts. In contrast, preincubation at 4°C without cytosol and subsequent dilution with cytosol did not affect the conversion of ceramide to IPC. Therefore, cytosol is not required under our conditions to establish ER-Golgi membrane contacts.
Depletion of ATP does not affect SEC18-independent ceramide transport in vivo
IPC synthesis still continues even though inhibited when ER-to-Golgi vesicular transport is blocked in sec mutant cells (Fig. 1, A and B). Our in vitro results suggest that SEC-independent IPC synthesis is mediated by ATP-independent transport of ceramide (Fig. 5, B and C, and Fig. 6 A). Since incorporation of [3H]myo-inositol into PI occurs at a lower temperature than the incorporation into IPC (Puoti et al., 1991), this difference allowed us to examine the effect of ATP depletion on SEC18-independent IPC synthesis in intact cells in an experiment that measures the synthesis of sphingolipids from endogenously synthesized ceramides. Mutant sec18 cells were labeled with [3H]myo-inositol at 1–3°C for 45 min and further incubated with NaF and NaN3 at 1–3°C for 10 min to allow depletion of ATP. Subsequently, the cells were chased at 37°C for 30 min. During the pulse at 1–3°C, very little IPC/C was synthesized. When sec18 mutant cells were chased at 37°C, approximately three times more IPC/C was produced. This increase in IPC/C synthesis was not affected by depletion of ATP (Fig. 8) . We conclude that transport of ceramide from the ER to the Golgi apparatus by the nonvesicular (SEC18-independent) pathway in intact cells does not require ATP, and therefore this process reflects the ceramide transport we reconstituted in vitro.
Figure 8.
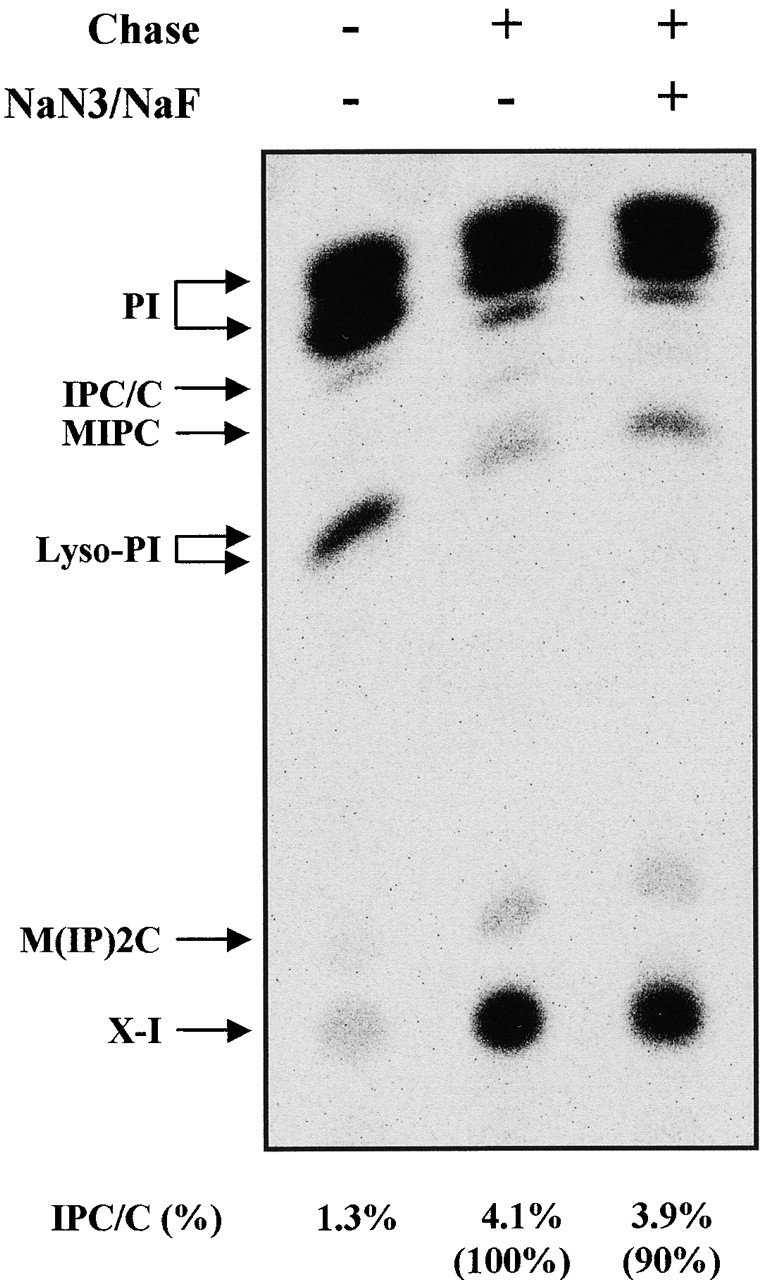
SEC-independent IPC biosynthesis does not require ATP in intact cells. Mutant sec18 (RH1737) cells labeled with [3H]myo-inositol were subjected to ATP depletion procedure and then chased at restrictive temperature as described in Materials and methods. The labeled lipids were applied to TLC plates using solvent II. PI, IPC/C, lysophosphatidylinositol (Lyso-PI), mannosyl-IPC (MIPC), mannosyl-diinositolphosphorylceramide (M[IP]2C), and X-I (unidentified lipid) are marked.
Discussion
Transport of lipids between subcellular compartments must be achieved by one or more of the following possible mechanisms: vesicular transport with budding of vesicles from donor membranes and targeting/fusion with the acceptor membranes, transfer of lipid monomers through the cytosol, or transfer of lipid through membrane contacts (McIntyre and Sleight, 1994; Trotter and Voelker, 1994; Liscum and Munn, 1999; van Meer and Holthuis, 2000; Voelker, 2000).
Our study provides evidence that ceramide is transported from the ER to the Golgi apparatus by both vesicular and nonvesicular means in yeast. From in vivo experiments, IPC synthesis from radiolabeled DHS was reduced but still observed when ER-to-Golgi vesicular transport was blocked in temperature-sensitive secretion mutants even under the conditions where protein synthesis was also blocked in sec18 mutant cells. This SEC18-independent IPC synthesis was also not affected by ATP depletion. Consistent with this, IPC synthesis was inhibited partially by ATP depletion in wild-type cells (unpublished data). These results suggest that ceramide is transported from the ER to the Golgi apparatus via two distinct pathways in intact cells, one vesicle dependent and the other nonvesicular. Analysis using an in vitro assay demonstrated that transport of ceramide from the ER to the Golgi compartment was not sensitive to ATP depletion and was SEC18 independent, indicating a nonvesicular transport mechanism. The correlation between in vivo and in vitro experiments provides evidence that the in vitro assay for ceramide transport measures a physiologically relevant transport process.
Some previous studies have suggested that nonvesicular transport of ceramide in vitro does not require cytosol (Moreau et al., 1993; Kok et al., 1998). Recently a cytosolic protein requirement has been demonstrated using semiintact cells to measure ceramide transport from the ER to the site of synthesis of sphingomyelin in the Golgi compartment (Funakoshi et al., 2000). In vitro analysis with the LY-A mutant, displaying a specific defect in ER-to-Golgi trafficking of ceramide to the site of sphingomyelin synthesis, showed that LY-A mutant cells are defective in a cytosolic protein involved in the ceramide transport. Since ER-to-Golgi transport of GPI-anchored or transmembrane proteins in LY-A cells appeared to be normal, it was suggested that ceramide is delivered to the Golgi apparatus by a mechanism that differs from ER-to-Golgi vesicular transport of proteins. Although the delivery of ceramide from the ER to the Golgi apparatus is mediated by two pathways, ATP-dependent and ATP-independent (nonvesicular), the defect in LY-A cells is specific to ATP-dependent trafficking of ceramide (Fukasawa et al., 1999). It is not clear whether the ATP-dependent delivery of ceramide is via one of multiple vesicular pathways that is specific for ceramide or nonvesicular pathways. In this respect, evidence for the existence of two distinct vesicle populations upon budding from the ER was provided recently (Muñiz et al., 2001).
The dilution independence of ceramide transport after ER and Golgi membranes have been incubated together at 4°C, where transport does not occur, strongly suggests that stable membrane contacts are formed and that these are relevant to the nonvesicular transport process. The ER-to-Golgi membrane contacts are likely to be saturable and specific for ceramide transport, since microsomes from wild-type cells inhibited ceramide transport in vitro, whereas addition of purified mitochondria did not. Our data provide important biochemical evidence for a role of membrane–membrane contacts in intracellular lipid transport and provide a system to begin to characterize this process.
Experimental evidence for an important function of membrane contacts has been found for ER-to-mitochondria trafficking of phosphatidylserine (Voelker, 1993; Achleitner et al., 1999). In addition, a close apposition of trans-ER with trans-Golgi cisternae has been reported by a high voltage electron microscope tomographic study (Ladinsky et al., 1999). Since the trans-ER lacks buds, it is most likely that this close apposition is associated with transport of lipids via nonvesicular rather than with vesicular transport of proteins.
ER-to-Golgi transport of ceramide for glucosylceramide synthesis was almost independent of ATP and cytosol (Funakoshi et al., 2000). In contrast, our experiments have shown that ATP-independent nonvesicular transport of ceramide requires a heat-labile and trypsin-sensitive cytosol protein(s). To our knowledge, this is the first clear evidence for a cytosolic protein requirement for nonvesicular transport of ceramide. What could be the function of this cytosolic factor?
Our dilution experiments have shown that the cytosol requirement is involved in the late step of ceramide transport after establishment of ER-Golgi membrane contacts. Thus, the cytosolic protein(s) seems to be required for transfer of ceramide from ER to Golgi membranes. It is unlikely that a cytosolic factor would be required for the transbilayer movement of ceramide after it has reached the Golgi compartment. Therefore, the late cytosolic requirement also makes it unlikely that ceramide is transferred from ER to Golgi during our short incubation at 4°C. The processes of membrane–membrane contact and transfer could be obligatorily coupled because cytosol could not efficiently extract ceramide out of the ER without acceptor membranes (unpublished data).
Analysis using our in vitro transport assay also revealed that ER-to-Golgi transport of ceramide continues for >60 min with a lag period from 5 to 15 min. The rather long time requirement could be due to ceramide transport to the Golgi complex and/or reflect the need to translocate ceramide from the cytosolic to the lumenal side of Golgi apparatus. Based on the predicted topology of Aur1p and localization of its putative active site in the lumen of the Golgi compartment, this translocation step would be required in our assay (Levine et al., 2000). A lumenal active site is consistent with our findings that trypsin treatment of membranes did not destroy conversion of ceramide to IPC. The half times for spontaneous translocation of short acyl chain ceramide (C5-DMB-ceramide) across lipid bilayers has been reported to be 22 min (Bai and Pagano, 1997), but measurements for natural ceramides are not available. Since short acyl chain lipids appear to undergo rapid transbilayer movement, on the basis of the correlation between the hydrophobicity of acyl chain and the rate of transbilayer movement (Bai and Pagano, 1997), it would follow that the spontaneous translocation of natural ceramide would occur in the order of hours and that specific proteins must be involved in the movement of ceramide. This translocation should be ATP independent because nonvesicular transport of ceramide does not require ATP. Such specific lipid translocators or flippases exist in the ER, Golgi apparatus, and plasma membrane (Raggers et al., 2000). Most of them require energy (e.g., ATP), but some proteins such as scramblase are involved in ATP-independent transbilayer lipid movement. In addition, ATP-independent protein-mediated lipid movement has been postulated in the translocation of glucosylceramide from the cytosolic to the lumenal leaflet of the Golgi membranes.
Our in vitro transport assay seems to reconstitute only the nonvesicular ceramide transport pathway. The reasons for the lack of reconstitution of the vesicular pathway are not clear, but in yeast the Golgi apparatus is made up of apparently unconnected compartments. Aur1p is located primarily in the medial-Golgi but not cis-Golgi compartments (Levine et al., 2000). Therefore, it seems plausible that ceramide transported to the cis-Golgi compartment via vesicular mechanism could be not delivered to the IPC synthase compartment efficiently in our assay because it involves too many transport steps. This may help explain why GTP plus Sar1p inhibits ceramide transport in our assay. GTP and Sar1p stimulate vesicle budding (Barlowe et al., 1994), and ceramide could be sequestered in these vesicles. In any event, it seems likely that nonvesicular pathway is used directly to generate IPC. To show that the direct fusion of ER with Golgi membranes did not occur under our assay conditions, we assayed for the acquisition of α-1,6 mannose modifications onto the ER form of the GPI-anchored protein, Gas1p (Muñiz et al., 2001). Less than 5% (background) of the Gas1p received this modification. Therefore, this cannot account for the >20% transport of ceramide that we see in our assay (unpublished data).
We postulate that the two different pathways of ceramide transport may have different functions. Vesicular-dependent transport would function to establish and maintain the lipid composition of ER and Golgi membranes during ER to early Golgi trafficking associated with possible functions of ceramide in regulation of cellular processes (Futerman et al., 1998; Hannun and Luberto, 2000) such as protein transport and signal transduction. For example, the sorting of GPI-anchored proteins from the ER is regulated by sphingoid base or ceramide synthesis (Horvath et al., 1994; Sütterlin et al., 1997). Nonvesicular transport would function to provide ceramide directly for de novo biosynthesis of sphingolipids for their important roles. These hypotheses will be addressed by further studies of the molecular mechanisms underlying the nonvesicular transport of ceramide.
Materials and methods
Strains
The secretion mutant strains used in this work were RH1737 (MATa sec18–20 his4 leu2 ura3 bar1), RH2043 (MATa sec18–20, his4 leu2 ura3 bar1 pep4::URA3), RH1592 (MATa sec17–1 his4 leu2 ura3 lys2 bar1), RH1491 (MATa sec12–4 his4 leu2 ura3 lys2 bar1), RH1436 (MATa sec23–1 his4 leu2 ura3 bar1), and the corresponding wild-type strains RH448 (MATa his4 leu2 ura3 lys2 bar1) and RH981 (MATa his4 leu2, ura3 lys2 bar1 pep4::URA3). Three copies of the HA epitope were introduced at the COOH terminus of Aur1p as described previously (Levine et al., 2000) by transformation of strain RH5247 (MATa his3 leu2 ura3 lys2 barl pep4–3) to yield RH5256 (MATa aur1::AUR1 3xHA HIS5Sp his3 leu2 ura3 lys2 barl pep4–3). Epitope tagging was confirmed by Western blotting. An AbA-resistant strain, RH5255 (MATa aur1::AUR1-C his3 leu2 ura3 lys2 barl pep4–3), was generated by transformation of RH5247 with the StuI/SmaI DNA fragment from pAUR101 integrating vector, which has mutant AUR1-C (Takara Shuzo Co., Ltd.) (Hashida-Okado et al., 1996, 1998) and isolated on YPD plates containing 2 μg/ml of AbA. Strain (D273–10B, MATα ATCC25657) was used for isolation and purification of mitochondria as described (Glick and Pon, 1995).
In vivo labeling with [3H]DHS and [3H]myo-inositol
Cell cultures and labeling of lipids with [3H]DHS was performed as described previously (Zanolari et al., 2000). If present, Chx (200 μg/ml; Fig. 1, A and B) and AbA (20 μg/ml; Fig. 5 A) were added, and cells were incubated for 20 min at 24°C before preincubation and at the start of the preincubation, respectively.
For the energy deprivation experiments, sec18 cells were preincubated for 20 min at 1–3°C and labeled with [3H]myo-inositol at 1–3°C. After 45 min, myo-inositol (100 μg/ml) and NaF/NaN3 (each 10 mM) were added and incubated for 20 min at 1–3°C. The cells were then chased for 30 min at 37°C after the addition of 4 vol of SD medium containing myo-inositol.
After the incubations, cells were placed on ice and washed, and cell pellets were subjected to lipid extraction (Zanolari et al., 2000). If necessary, the extracted lipids were submitted to mild alkaline hydrolysis with NaOH. After neutralization with acetic acid, the lipids were desalted by partitioning between N-butanol and water. The pooled organic phases were dried under nitrogen. The lipids were dissolved in chloroform/methanol/water (CMW; 10/10/3 [vol/vol/vol]) for TLC on Kieselgel 60 plates (20 × 20; Merck) and developed in solvent I (chloroform/methanol/4.2 N NH4OH, 9/7/2 [vol/vol/vol]) or solvent II (chloroform/methanol/0.25% KCl, 55/45/10 [vol/vol/vol]). Radiolabeled lipids were visualized and quantified on a Cyclone Storage Phosphor System using a tritium-sensitive screen (Packard).
Cytosol and membrane preparations
Cytosols were prepared from RH981 and RH2043 strains as described (Salama et al., 1993). The preparation of microsomal membranes and ER-enriched membranes were performed as described (Baker et al., 1990; Wuestehube and Schekman, 1992) with a few modifications. In brief, spheroplasts (from 0.5 OD600 U of cells in log phase, total 4 × 1010 cells) were lysed in lysis buffer (0.1 M sorbitol, 20 mM Hepes, pH 7.4, 150 mM potassium acetate, 2 mM EDTA, 1 mM DTT, 1 mM PMSF, and 1 μg/ml protease inhibitor mixture [pepstatin, leupeptin, antipain]). Unbroken cells and cell debris were removed by centrifugation (3000 g, 10 min, 4°C), and the resulting supernatants were centrifuged (100,000 g, 1 h, 4°C) to collect the microsomal membrane fraction. The pellet was washed twice and resuspended in 1 ml (25–50 mg protein) of B88 (20 mM Hepes-KOH, pH 6.8, 150 mM potassium acetate, 5 mM magnesium acetate, 250 mM sorbitol). Aliquots were frozen in liquid nitrogen and stored at −80°C. For ER- and Golgi-enriched membranes, supernatants containing microsomal membranes were subjected to two successive centrifugation steps (27,000 g, 10 min, 4°C) giving rise to pellet (P27) and supernatant (S27) fractions, and S27 was centrifuged (45,000 g, 15 min, 4°C), resulting in P45 and S45. S45 containing Golgi-enriched membranes was spun (100,000 g, 1 h, 4°C), washed twice, and resuspended in 1 ml (10–20 mg protein) of B88. P27 was resuspended in lysis buffer and loaded onto a 7.5 ml sucrose density step gradient (2.5 ml each of 40, 50, and 60% sucrose in 10 mM Hepes, pH 7.4, 1 mM MgCl2). The gradient was centrifuged at 200,000 g for 2 h and 20 min at 4°C in TST41.14 rotor (Kontron Instruments). The ER-enriched membrane fraction at the 40/50% sucrose interface was collected, washed twice by centrifugation (27,000 g, 10 min, 4°C), and resuspended in 1 ml (2–4 mg protein) of B88. Aliquots of each membrane fraction were frozen in liquid nitrogen and stored at −80°C. Protein concentrations were determined using the protein assay kit from Bio-Rad Laboratories.
In vitro labeling with [3H]DHS
To assay for ceramide and IPC synthase activities, microsomal membranes (100–200 μg) or membrane fractions (ER-enriched membranes, 5–10 μg; Golgi-enriched membranes, 25–50 μg), cytosol (100 μg), ATP regenerating system (1 mM ATP, 40 mM phosphocreatine, 0.2 mg/ml creatine phosphokinase), GDP-mannose (50 μM), and a mix of [3H]DHS and unlabeled DHS (10 and 40 pmoles, respectively, 0.5 μCi) were first incubated (15 min, 10°C) for incorporation of DHS into membranes. Coenzyme A (50 μM) and liposomes containing hexacosanoic acid (C26) and PI (50 and 250 μM, respectively) prepared as described (Funato et al., 1992) were added, and the mixture was incubated (2 h, 24°C) in a final volume of 50 μl of B88. The reaction was stopped by adding 333 μl of chloroform/methanol (CM; 1/1 [vol/vol]). The organic phase was collected after centrifuging at 13,000 g for 5 min, and the pellet was reextracted with 250 μl of CMW. The extracted lipids were submitted to a mild alkaline treatment, N-butanol extraction, and analyzed by TLC with solvent I as described above. Radiolabeled ceramide was analyzed with solvent III (chloroform/acetic acid, 9/1 [vol/vol]) (Morell and Radin, 1970).
In vitro ceramide transport assay
Radiolabeled ceramide was synthesized in vitro from [3H]DHS using ER-enriched membranes (10–20 μg) as described above except that GDP-mannose was omitted, AbA (0.2 μM) was included in the assay mixture, and liposomes containing hexacosanoic acid (C26) and PC were used instead of liposomes containing hexacosanoic acid (C26) and PI. The membranes were then recovered by centrifugation at 27,000 g for 10 min at 4°C, washed twice, resuspended in B88, and used as the donor membrane in the in vitro transport assay. The recovery of radiolabeled lipids was 50–75% percent of the total. Subsequently, the donor membranes were incubated for 2 h at 24°C in the total reaction mixture (50 μl) containing cytosol (2 mg/ml), ATP regenerating system, GDP-mannose (50 μM), liposomes composed of PI (250 μM), FuB (200 μM), AbA (0.4 μM), and microsomal membranes (50–100 μg) prepared from AbA-resistant strain (RH5255) as the acceptor membrane to allow transport to occur. The reaction was stopped by adding 333 μl of CM. The preparation of the lipid extracts and analysis were done as described above.
Lipid analysis
Radiolabeled lipids were collected from TLC plates by scraping and eluting with CMW. The isolated lipid extract was dried under nitrogen and partitioned between N-butanol and water. Lipid mixtures (Fig. 2 A) or isolated lipids (Fig. 2, B–D) were subjected to phosphoinositide-specific PLC digestion as described previously (Hamburger et al., 1995). Strong HCl hydrolysis of lipids was performed as described (Puoti et al., 1991). After enzymatic and/or chemical treatments, lipids were dried under nitrogen and partitioned between N-butanol and water. After drying, the lipids were dissolved in CMW and analyzed by TLC as described above.
Acknowledgments
We thank R. Schekman for strains and antibodies, S. Munro for plasmids, J. Holenstein and T. Aust for technical assistance, and members of the Riezman lab for comments on this article.
This work was supported by a grant from the Swiss National Science Foundation and a European Community grant (HPRN-2000-00077) funded through the Bundesamt für Bildung und Wåssenschatt (to H. Riezman).
Footnotes
Abbreviations used in this paper: AbA, aureobasidin A; Chx, cycloheximide; DHS, dihydrosphingosine; DHS-Cer, dihydroceramide; DHS-1-P, [3H]DHS-1-phosphate; FuB, fumonisin B1; HA, hemagglutinin; IPC, inositolphosphorylceramide; NEM, N-ethylmaleimide; PC, phosphatidylcholine; PHS, phytosphingosine; PI, phosphatidylinositol.
References
- Achleitner, G., B. Gaigg, A. Krasser, E. Kainersdorfer, S.D. Kohlwein, A. Perktold, G. Zellnig, and G. Daum. 1999. Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact. Eur. J. Biochem. 264:545–553. [DOI] [PubMed] [Google Scholar]
- Bai, J., and R.E. Pagano. 1997. Measurement of spontaneous transfer and transbilayer movement of BODIPY-labeled lipids in lipid vesicles. Biochemistry. 23:4977–4983. [DOI] [PubMed] [Google Scholar]
- Baker, D., L. Wuestehube, R. Schekman, D. Botstein, and N. Segev. 1990. GTP-binding Ypt1 protein and Ca2+ function independently in a cell-free protein transport reaction. Proc. Natl. Acad. Sci. USA. 87:355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield, D.K., M.J. Lewis, C. Rabouille, G. Warren, and H.R. Pelham. 1994. Localization of Sed5, a putative vesicle targeting molecule, to the cis-Golgi network involves both its transmembrane and cytoplasmic domains. J. Cell Biol. 127:357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe, C., L. Orci, T. Yeung, M. Hosobuchi, S. Hamamoto, N. Salama, M.F. Rexach, M. Ravazzola, M. Amherdt, and R. Schekman. 1994. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 77:895–907. [DOI] [PubMed] [Google Scholar]
- Dickson, R.C., and R.L. Lester. 1999. Yeast sphingolipids. Biochim. Biophys. Acta. 1426:347–357. [DOI] [PubMed] [Google Scholar]
- Emoto, K., O. Kuge, M. Nishijima, and M. Umeda. 1999. Isolation of a Chinese hamster ovary cell mutant defective in intramitochondrial transport of phosphatidylserine. Proc. Natl. Acad. Sci. USA. 96:12400–12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa, M., M. Nishijima, and K. Hanada. 1999. Genetic evidence for ATP-dependent endoplasmic reticulum-to-Golgi apparatus trafficking of ceramide for sphingomyelin synthesis in Chinese hamster ovary cells. J. Cell Biol. 144:673–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi, T., S. Yasuda, M. Fukasawa, M. Nishijima, and K. Hanada. 2000. Reconstitution of ATP- and cytosol-dependent transport of de novo synthesized ceramide to the site of sphingomyelin synthesis in semi-intact cells. J. Biol. Chem. 275:29938–29945. [DOI] [PubMed] [Google Scholar]
- Funato, K., R. Yoda, and H. Kiwada. 1992. Contribution of complement system on destabilization of liposomes composed of hydrogenated egg phosphatidylcholine in rat fresh plasma. Biochim. Biophys. Acta. 1103:198–204. [DOI] [PubMed] [Google Scholar]
- Futerman, A.H., R. Ghidoni, and G. van Meer. 1998. Lipids: regulatory functions in membrane traffic and cell development. EMBO J. 17:6772–6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grilley, M.M., S.D. Stock, R.C. Dickson, R.L. Lester, and J.Y. Takemoto. 1998. Syringomycin action gene SYR2 is essential for sphingolipid 4-hydroxylation in Saccharomyces cerevisiae. J. Biol. Chem. 273:11062–11068. [DOI] [PubMed] [Google Scholar]
- Glick, B.S., and L.A. Pon. 1995. Isolation of highly purified mitochondria from Saccharomyces cerevisiae. Methods Enzymol. 260:213–223. [DOI] [PubMed] [Google Scholar]
- Haak, D., K. Gable, T. Beeler, and T. Dunn. 1997. Hydroxylation of Saccharomyces cerevisiae ceramides requires Sur2p and Scs7p. J. Biol. Chem. 272:29704–29710. [DOI] [PubMed] [Google Scholar]
- Hamburger, D., M. Egerton, and H. Riezman. 1995. Yeast Gaa1p is required for attachment of a completed GPI anchor onto proteins. J. Cell Biol. 129:629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun, Y.A., and C. Luberto. 2000. Ceramide in the eukaryotic stress response. Trends Cell Biol. 10:73–80. [DOI] [PubMed] [Google Scholar]
- Hashida-Okado, T., A. Ogawa, M. Endo, R. Yasumoto, K. Takesako, and I. Kato. 1996. AUR1, a novel gene conferring aureobasidin resistance on Saccharomyces cerevisiae: a study of defective morphologies in Aur1p-depleted cells. Mol. Gen. Genet. 251:236–244. [DOI] [PubMed] [Google Scholar]
- Hashida-Okado, T., A. Ogawa, I. Kato, and K. Takesako. 1998. Transformation system for prototrophic industrial yeasts using the AUR1 gene as a dominant selection marker. FEBS Lett. 425:117–122. [DOI] [PubMed] [Google Scholar]
- Hirschberg, K., J. Rodger, and A.H. Futerman. 1993. The long-chain sphingoid base of sphingolipids is acylated at the cytosolic surface of the endoplasmic reticulum in rat liver. Biochem. J. 290:751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, A., C. Sütterlin, U. Manning-Krieg, N.R. Movva, and H. Riezman. 1994. Ceramide synthesis enhances transport of GPI-anchored proteins to the Golgi apparatus in yeast. EMBO J. 13:3684–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok, J.W., T. Babia, K. Klappe, G. Egea, and D. Hoekstra. 1998. Ceramide transport from endoplasmic reticulum to Golgi apparatus is not vesicle-mediated. Biochem. J. 333:779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladinsky, M.S., D.N. Mastronarde, J.R. McIntosh, K.E. Howell, and L.A. Staehelin. 1999. Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J. Cell Biol. 144:1135–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, T.P., C.A. Wiggins, and S. Munro. 2000. Inositol phosphorylceramide synthase is located in the Golgi apparatus of Saccharomyces cerevisiae. Mol. Biol. Cell. 11:2267–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscum, L., and N.J. Munn. 1999. Intracellular cholesterol transport. Biochim. Biophys. Acta. 1438:19–37. [DOI] [PubMed] [Google Scholar]
- Mandala, S.M., R.A. Thornton, B.R. Frommer, J.E. Curotto, W. Rozdilsky, M.B. Kurtz, R.A. Giacobbe, G.F. Bills, M.A. Cabello, I. Martin, et al. 1995. The discovery of australifungin, a novel inhibitor of sphinganine N-acyltransferase from Sporormiella australis. Producing organism, fermentation, isolation, and biological activity. J. Antibiot. 48:349–356. [DOI] [PubMed] [Google Scholar]
- Mandon, E.C., I. Ehses, J. Rother, G. van Echten, and K. Sandhoff. 1992. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J. Biol. Chem. 267:11144–11148. [PubMed] [Google Scholar]
- Mao, C., R. Xu, A. Bielawska, and L.M. Obeid. 2000. a. Cloning of an alkaline ceramidase from Saccharomyces cerevisiae. An enzyme with reverse (CoA-independent) ceramide synthase activity. J. Biol. Chem. 275:6876–6884. [DOI] [PubMed] [Google Scholar]
- Mao, C., R. Xu, A. Bielawska, Z.M. Szulc, and L.M. Obeid. 2000. b. Cloning and characterization of a Saccharomyces cerevisiae alkaline ceramidase with specificity for dihydroceramide. J. Biol. Chem. 275:31369–31378. [DOI] [PubMed] [Google Scholar]
- McIntyre, J.C., and R.G. Sleight. 1994. Mechanisms of intracellular membrane lipid transport. Curr. Topic Membranes. 40:453–474. [Google Scholar]
- Merrill, A.H., and D.D. Jones. 1990. An update of the enzymology and regulation of sphingomyelin metabolism. Biochim. Biophys. Acta. 1044:1–12. [DOI] [PubMed] [Google Scholar]
- Merrill, A.H., G. van Echten, E. Wang, and K. Sandhoff. 1993. Fumonisin B1 inhibits sphingosine (sphinganine) N-acyltransferase and de novo sphingolipid biosynthesis in cultured neurons in situ. J. Biol. Chem. 268:27299–27306. [PubMed] [Google Scholar]
- Moreau, P., C. Cassagne, T.W. Keenan, and D.J. Morre. 1993. Ceramide excluded from cell-free vesicular lipid transfer from endoplasmic reticulum to Golgi apparatus. Evidence for lipid sorting. Biochim. Biophys Acta. 1146:9–16. [DOI] [PubMed] [Google Scholar]
- Morell, P., and N.S. Radin. 1970. Specificity in ceramide biosynthesis from long chain bases and various fatty acyl coenzyme A's by brain microsomes. J. Biol. Chem. 245:342–350. [PubMed] [Google Scholar]
- Muñiz, M., P. Morsomme, and H. Riezman. 2001. Protein sorting upon exit from the endoplasmic reticulum. Cell. 104:313–320. [DOI] [PubMed] [Google Scholar]
- Nishijima, M., O. Kuge, and K. Hanada. 1997. Mammalian cell mutants of membrane phospholipid biogenesis. Trends Cell Biol. 7:324–329. [DOI] [PubMed] [Google Scholar]
- Puoti, A., C. Desponds, and A. Conzelmann. 1991. Biosynthesis of mannosylinositolphosphoceramide in Saccharomyces cerevisiae is dependent on genes controlling the flow of secretory vesicles from the endoplasmic reticulum to the Golgi. J. Cell Biol. 113:515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raggers, R.J., T. Pomorski, J.C.M. Holthuis, N. Kalin, and G. van Meer. 2000. Lipid traffic: the ABC of transbilayer movement. Traffic. 1:226–234. [DOI] [PubMed] [Google Scholar]
- Reggiori, F., and A. Conzelmann. 1998. Biosynthesis of inositol phosphoceramides and remodeling of glycosylphosphatidylinositol anchors in Saccharomyces cerevisiae are mediated by different enzymes. J. Biol. Chem. 273:30550–30559. [DOI] [PubMed] [Google Scholar]
- Salama, N.R., T. Yeung, and R.W. Schekman. 1993. The Sec13p complex and reconstitution of vesicle budding from the ER with purified cytosolic proteins. EMBO J. 12:4073–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiter, R. 1999. Brave little yeast, please guide us to thebes: sphingolipid function in S. cerevisiae. Bioessays. 21:1004–1010. [DOI] [PubMed] [Google Scholar]
- Schneiter, R., B. Brugger, R. Sandhoff, G. Zellnig, A. Leber, M. Lampl, K. Athenstaedt, C. Hrastnik, S. Eder, G. Daum, et al. 1999. Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J. Cell Biol. 146:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, S., F. Schimmöller, B. Singer-Kruger, and H. Riezman. 1995. The Golgi-localization of yeast Emp47p depends on its di-lysine motif but is not affected by the ret1-1 mutation in alpha-COP. J. Cell Biol. 131:895–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sütterlin, C., T.L. Doering, F. Schimmöller, S. Schröder, and H. Riezman. 1997. Specific requirements for the ER to Golgi transport of GPI-anchored proteins in yeast. J. Cell Sci. 110:2703–2714. [DOI] [PubMed] [Google Scholar]
- Trotter, P.J., and D.R. Voelker. 1994. Lipid transport processes in eukaryotic cells. Biochim. Biophys. Acta. 1213:241–262. [DOI] [PubMed] [Google Scholar]
- van Meer, G. 1998. Lipids of the Golgi membrane. Trends Cell Biol. 8:29–33. [DOI] [PubMed] [Google Scholar]
- van Meer, G., and J.C. Holthuis. 2000. Sphingolipid transport in eukaryotic cells. Biochim. Biophys. Acta. 1486:145–170. [DOI] [PubMed] [Google Scholar]
- Voelker, D.R. 1993. The ATP-dependent translocation of phosphatidylserine to the mitochondria is a process that is restricted to the autologous organelle. J. Biol. Chem. 268:7069–7074. [PubMed] [Google Scholar]
- Voelker, D.R. 2000. Interorganelle transport of aminoglycerophospholipids. Biochim. Biophys. Acta. 1486:97–107. [DOI] [PubMed] [Google Scholar]
- Wuestehube, L.J., and R.W. Schekman. 1992. Reconstitution of transport from endoplasmic reticulum to Golgi complex using endoplasmic reticulum-enriched membrane fraction from yeast. Methods Enzymol. 219:124–136. [DOI] [PubMed] [Google Scholar]
- Zanolari, B., S. Friant, K. Funato, C. Sütterlin, B.J. Stevenson, and H. Riezman. 2000. Sphingoid base synthesis requirement for endocytosis in Saccharomyces cerevisiae. EMBO J. 19:2824–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, W., D.J. Murphy, and N.H. Georgopapadakou. 1999. Inhibition of yeast inositol phosphorylceramide synthase by aureobasidin A measured by a fluorometric assay. FEBS Lett. 463:241–244. [DOI] [PubMed] [Google Scholar]