

This article reports the production, crystallization and X-ray structure determination of perdeuterated human carbonic anhydrase (HCA II). The refined structure is shown to be highly isomorphous with hydrogenated HCA II, especially with regard to the active site architecture and solvent network.
Keywords: human carbonic anhydrase II, perdeuteration, neutron diffraction
Abstract
Human carbonic anhydrase II (HCA II) is a zinc metalloenzyme that catalyzes the reversible hydration and dehydration of carbon dioxide and bicarbonate, respectively. The rate-limiting step in catalysis is the intramolecular transfer of a proton between the zinc-bound solvent (H2O/OH−) and the proton-shuttling residue His64. This distance (∼7.5 Å) is spanned by a well defined active-site solvent network stabilized by amino-acid side chains (Tyr7, Asn62, Asn67, Thr199 and Thr200). Despite the availability of high-resolution (∼1.0 Å) X-ray crystal structures of HCA II, there is currently no definitive information available on the positions and orientations of the H atoms of the solvent network or active-site amino acids and their ionization states. In preparation for neutron diffraction studies to elucidate this hydrogen-bonding network, perdeuterated HCA II has been expressed, purified, crystallized and its X-ray structure determined to 1.5 Å resolution. The refined structure is highly isomorphous with hydrogenated HCA II, especially with regard to the active-site architecture and solvent network. This work demonstrates the suitability of these crystals for neutron macromolecular crystallography.
1. Introduction
The mammalian α-class of carbonic anhydrases (α-CAs) are monomeric zinc metalloenzymes with a molecular weight of ∼29 kDa. Humans have 14 α-CAs (HCA I–XIV) that differ with regard to expression patterns, expression levels, subcellular location, kinetic properties and sensitivity to inhibitors (Parkkila, 2000 ▶; Tufts et al., 2003 ▶). Carbonic anhydrases catalyze the reversible hydration/dehydration reaction of CO2/HCO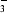 (equations 1 and 2; Silverman & Lindskog, 1988 ▶; Christianson & Fierke, 1996 ▶; Lindskog, 1997 ▶). Upon binding of CO2 in a hydrophobic cleft of the active site, the hydration reaction is initiated by nucleophilic attack on the substrate carbon by the zinc-bound OH− to form HCO. The HCO is displaced from the zinc by water,
(equations 1 and 2; Silverman & Lindskog, 1988 ▶; Christianson & Fierke, 1996 ▶; Lindskog, 1997 ▶). Upon binding of CO2 in a hydrophobic cleft of the active site, the hydration reaction is initiated by nucleophilic attack on the substrate carbon by the zinc-bound OH− to form HCO. The HCO is displaced from the zinc by water,
The zinc-bound OH− is regenerated for the next round of catalysis by the transport of a proton from the zinc-bound water to the bulk-solvent environment (B),
This proton transfer has been shown to be the rate-limiting step. It is now well established that in HCA II, one of the most efficient of the α-CA isozymes, His64 shuttles a proton between the zinc-bound solvent and the bulk-solvent environment (2). Mutation of His64 to an alanine in HCA II reduces the enzymatic activity by tenfold to 50-fold (Tu et al., 1989 ▶). His64 sits on the rim of the active-site cleft and is ∼7.5 Å away from the zinc-bound solvent. It has been postulated that the proton transfer occurs through a chain of water molecules, W1, W2, W3a and W3b (Fig. 1 ▶ a; Christianson & Fierke, 1996 ▶; Lindskog, 1997 ▶; Lindskog & Silverman, 2000 ▶; Fisher et al., 2005 ▶). Crystal structures of HCA II at various pH values have shown that His64 can occupy dual conformations, the so-called ‘in’ and ‘out’ positions, and it has been suggested that this flexibility is required for efficient proton transfer (Nair & Christianson, 1991 ▶; Fisher et al., 2005 ▶). In HCA II, several amino acids (Tyr7, Asn62, Asn67, Thr199 and Thr200) seem to be critical in the stabilization of the active-site solvent network (Zn-H2O/OH−, W1, W2, W3a and W3b; Fig. 1 ▶ a). The hydroxyl group of Thr199 stabilizes the zinc-bound solvent and is thought to be important for catalysis, positioning the Zn-bound OH− lone electron pair for the nucleophilic attack of the incoming CO2 substrate (Merz, 1990 ▶). Solvent molecule W1 is oriented by the zinc-bound solvent, the side chain of Thr200 and W2; solvent molecule W2 is further stabilized by W3a, which is stabilized by the hydroxyl group of Tyr7, and W3b, which is held in place by the side chains of Asn62 and Asn67 (Fisher et al., 2005 ▶). This arrangement orients the three solvent molecules W2, W3a and W3b within close proximity of the ‘in’ conformation of His64. This solvent network is conserved in HCA II in the pH range 5.0–10.0 (Fisher et al., 2005 ▶).
Figure 1.

Active site of (a) hydrogenated and (b) perdeuterated HCA II. Important catalytic side chains in the active sites are shown in yellow ball-and-stick representation and are as labeled. The Zn atom is shown as a black sphere, while solvent molecules are in red. Inferred hydrogen bonds are shown as orange dashed lines. (a) Hydrogenated HCA II (PDB code 1tbt; Fisher et al., 2005 ▶) and (b) perdeuterated HCA II; the blue electron density shown is a 2F o − F c map contoured at 1.5σ. The figure was generated and rendered using MOLSCRIPT and RASTER3D (Esnouf, 1999 ▶; Merritt & Bacon, 1997 ▶).
However, from the X-ray crystal structures of HCA II we still have to infer the positions of H atoms, amino-acid protonation states and plausible hydrogen-bond patterns in the active site (Duda et al., 2003 ▶). In this regard, there are still many unanswered questions about the precise structural organization of the hydrogen-bonded solvent network and its contribution to the catalytic mechanism of this highly efficient carbonic anhydrase. At what pH is the zinc-bound solvent a hydroxide/water? How is the hydrogen of the hydroxyl group of Thr199 oriented? Is His64 doubly or singularly protonated in the ‘in’ and ‘out’ conformations? Do one or all of the solvent molecules W2, W3a and W3b form hydrogen bonds with His64? Is W2 a kinetically trapped hydronium ion? And if W2 is a hydronium ion, does it form an Eigen cation with W1, W3a and W3b?
Assigning the positions of H atoms is only possible in X-ray crystallography if ultrahigh-resolution (≤1.0 Å) data can be collected. Even then the data can be ambiguous, especially when assigning the positions of H atoms of water molecules with high (<20 Å2) thermal parameters (Gutberlet et al., 2001 ▶). However, neutron crystallography at medium (∼2.0 Å) resolution can complement medium-to-high-resolution (∼2.0–1.0 Å) X-ray crystallography and allow the placement and analysis of key H atoms (Habash et al., 2000 ▶; Coates et al., 2001 ▶). This is because hydrogen has a neutron scattering length (−3.7 × 10−15 m or −3.7 fm) similar in magnitude but opposite in sign to other atoms found in proteins (O, 5.8 fm; N, 9.4 fm; C, 6.6 fm; S, 2.8 fm). One disadvantage is that the negative scattering length of hydrogen can result in partial cancellation of the positive signal from the O/N/C/S atoms (Habash et al., 2000 ▶). Moreover, hydrogen has a large incoherent neutron scattering cross-section (∼80 barns) that contributes to a large and limiting experimental background. In contrast, deuterium has a positive scattering length (6.7 fm) and a significantly smaller incoherent neutron-scattering cross-section (∼2.0 barns). The use of fully deuterated protein thus reduces the scattering background, providing a near order of magnitude improvement in the signal-to-noise ratio of the data, and makes it easier to locate D atoms in the resulting neutron density maps (Shu et al., 2000 ▶). In the case of D2O water molecules, neutron density maps often allow location of the two D atoms, revealing the orientation of the D2O molecule (Habash et al., 2000 ▶; Coates et al., 2001 ▶). In order to address the question of how the active-site structure of HCA II promotes proton shuttling, we will use both X-ray and neutron diffraction methods to study crystals of perdeuterated HCA II. These approaches should facilitate the direct determination of H/D positions. Here, we present the production, crystallization and X-ray structure of perdeuterated HCA II and compare it with a previously determined X-ray structure of hydrogenated HCA II (Fisher et al., 2005 ▶).
2. Experimental procedures
2.1. Expression and purification of HCA II from high-cell density cultures
Perdeuterated HCA II was produced in Escherichia coli BL21(DE3) adapted for growth in fully deuterated medium in a three-step process (LB to minimal media in H2O with glycerol and then to minimal media in D2O with deuterated glycerol) that involved growing the cells on plated media at 310 K. When the cells were fully adapted, they were transferred to fully deuterated liquid media (Table 1 ▶). Deuterated glycerol was added at various concentrations depending on the growth step. Deuterium-adapted cells were used to directly inoculate 0.5 l medium into a 1.5 l fermenter under high cell-density culture growth conditions at 303 K in the presence of 30 mg l−1 kanamycin. Cells were harvested 16 h after induction with 0.2 mM IPTG at a final OD600 of 15, with a yield of ∼1 g wet cell paste per millilitre of perdeuterated glycerol provided; from 5 g of these cells, ∼60 mg of perdeuterated HCA II could be obtained. The HCA II was purified using pAMBS affinity chromatography as has been described elsewhere (Khalifah et al., 1977 ▶). All buffers used during purification were made with H2O. After purification, the exchangeable H atoms were back-exchanged to deuteriums against deuterated crystallization buffer (100 mM Tris–DCl pH 7.8). Protein purity and solubility were assessed by SDS–PAGE and the absorbance ratio (392/280 nm), respectively. The deuteration levels of HCA II were determined by mass spectrometry.
Table 1. Minimal medium composition.
| (NH4)2SO4 | 6.86 g l−1 |
| KH2PO4 | 1.56 g l−1 |
| Na2HPO4 | 5.16 g l−1 |
| Ammonium citrate | 0.49 g l−1 |
| MgSO4 | 0.3 g l−1 |
| Metal salts solution† | 1 ml l−1 |
0.5 g l−1 CaCl2, 0.18 g l−1 FeCl3, 0.18 g l−1 ZnSO4, 0.16 g l−1 CuSO4, 0.15 g l−1 MnSO4, 0.18 g l−1 CoCl2, 20.1 g l−1 EDTA.
2.2. Crystallization and X-ray data collection
Initially, crystals of perdeuterated HCA II were grown using the hanging-drop vapor-diffusion technique (McPherson, 1982 ▶) using 5 µl perdeuterated protein (20–30 mg ml−1) mixed with 5 µl precipitant solution (100 mM Tris–DCl pH 7.8, 1.15 M sodium citrate) at 277 K. Crystal size and quality were further improved using a temperature-control device developed at the EMBL, Grenoble (work to be published). Crystals were cryoprotected prior to data collection by quick-dipping them in 20% d8-glycerol mother liquor. Synchrotron diffraction data were collected at the ESRF (beamline ID29). Several crystals were used for data collection with 1° oscillation angle and an exposure time of 1 s at 105, 125 and 185 mm crystal-to-detector distances. X-ray data processing was performed using DENZO and data were scaled and reduced with SCALEPACK (Otwinowski & Minor, 1997 ▶).
2.3. Structure determination and model refinement
All refinement and model modifications were performed using CNS v.1.1 (Brünger et al., 1998 ▶) and Coot (Emsley & Cowtan, 2004 ▶), respectively. The perdeuterated HCA II X-ray diffraction data were isomorphous to the previously reported hydrogenated HCA II structure (PDB code 1tbt; Fisher et al., 2005 ▶). Hence, the hydrogenated HCA II coordinates, with the Zn atom and solvent molecules removed (to prevent model bias), were used to calculate the initial phases for the perdeuterated HCA II data. After one cycle of rigid-body refinement, annealing by heating to 3000 K with gradual cooling, geometry-restrained position and individual temperature-factor refinement, F o − F c and 2F o − F c electron-density maps were generated. These maps clearly showed the position of the Zn atom, which was then included in the model and subsequent refinement cycles. After several further cycles of refinement, solvent molecules were incorporated into the model using the automated solvent-picking program implemented in CNS until no more solvent molecules were found in F o − F c maps contoured at the 2.0σ level and refinement continued until the R cryst and R free converged (Table 2 ▶).
Table 2. Data-collection and model refinement statistics.
Data for the highest resolution shell are given in parentheses.
| Data-collection statistics | |
| Molecular weight (Da) | 30664 |
| Temperature (K) | 100 |
| Wavelength (Å) | 1.005 |
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 42.1, b = 41.0, c = 72.0, β = 104.4 |
| Resolution (Å) | 20.0–1.50 (1.55–1.50) |
| No. of unique reflections | 37372 (3689) |
| Completeness (%) | 97.0 (96.8) |
| Redundancy | 3.0 (2.3) |
| I/σ(I) | 28.0 (6.4) |
| Rmerge† | 0.09 (0.17) |
| Refinement statistics | |
| No. of protein atoms/solvent atoms | 2058/222 |
| Rcryst/Rfree‡ | 0.195/0.206 |
| R.m.s.d. bond lengths (Å) | 0.005 |
| R.m.s.d. bond angles (°) | 1.396 |
| Average B factors (Å2) | |
| Main chain | 10.5 |
| Side chain | 12.9 |
| Solvent | 23.3 |
| Zn atom | 4.9 |
| Ramachandran statistics | |
| Most favoured (%) | 87.6 |
| Additionally and generously allowed (%) | 12.4 |
R
merge = 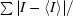
 , where I is the intensity of a reflection and 〈I〉 is the average intensity.
, where I is the intensity of a reflection and 〈I〉 is the average intensity.
R
cryst = 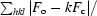
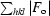 ; R
free is calculated from 5.0% of data for cross-validation.
; R
free is calculated from 5.0% of data for cross-validation.
3. Results
3.1. Deuteration level
The deuteration level of HCA II prior to crystallization was determined by time-of-flight electrospray mass spectrometry. The software Isotopic Pattern Calculator v.1.6.5 for Macintosh (http://www.shef.ac.uk/chemistry/chemputer/isotopes.html) was used to calculate the theoretical isotopic distribution and the pattern was then matched to the experimental spectrum. The analysis corresponded to a deuteration level of ∼99%.
3.2. X-ray analysis
The perdeuterated HCA II crystals grew in approximately 15 d to dimensions of approximately 0.4 × 0.4 × 0.1 mm (Fig. 2 ▶). Several data sets were collected and three were processed and scaled together from 20.0–1.5 Å resolution for a total of 220° of data. The perdeuterated HCA II crystals were shown to belong to the monoclinic space group P21, with unit-cell parameters a = 42.1, b = 41.0, c = 72.5 Å, β = 104.4°, with a scaling R merge of 0.090 and were isomorphous with hydrogenated HCA II crystals (Fisher et al., 2005 ▶). Data-collection statistics are shown in Table 2 ▶.
Figure 2.

Optical photograph of a perdeuterated HCA II crystal of approximate dimensions 0.4 × 0.4 × 0.1 mm.
3.3. Structure determination and refinement
The final R cryst and R free of the perdeuterated HCA II structure to 1.5 Å resolution were 19.5 and 20.6%, respectively. The model is of good quality, with root-mean-square deviations (r.m.s.d.s) for bond lengths and angles of 0.005 Å and 1.4°, respectively. A Ramachandran plot also shows that 87.6% of the residues are in the most favored region, while the other 12.4% are in the additional and generously allowed regions (Laskowski et al., 1993 ▶). Refinement and final model statistics are given in Table 2 ▶. The r.m.s.d. between the hydrogenated HCA II (PDB code 1tbt; Fisher et al., 2005 ▶) and perdeuterated structures was 0.2 Å for all Cα atoms. However, when only the active-site residues [Tyr7, Asn62, His64 (‘in’ and ‘out’), Asn67, His94, His96, His119, Thr199 and Thr200] and solvent molecules (Zn-H2O/OH−, W1, W2, W3 and W3b) are superimposed, this value decreased to <0.1 Å (compare Figs. 1 ▶ a and 1 ▶ b). The relative positions and distances between the solvent molecules and the side chains that coordinate them are very similar and are shown in Fig. 1 ▶ and Table 3 ▶.
Table 3. Distances (Å) between solvent molecules and active-site residues for hydrogenated and perdeuterated HCA II.
| Residues and solvent molecules | Hydrogenated | Perdeuterated |
|---|---|---|
| Zn2+—ZnH2O/OH− (D2O/OD−) | 2.03 | 1.91 |
| ZnH2O/OH−(D2O/OD−)—W1† | 2.56 | 2.80 |
| W1—W2 | 2.79 | 2.81 |
| W2—W3a | 2.74 | 2.86 |
| W2—W3b | 2.92 | 2.83 |
| ZnH2O/OH− (D2O/OD−)—Thr199 OG1 | 2.69 | 2.64 |
| W1—Thr200 OG1 | 2.73 | 2.71 |
| W3a—Tyr7 OH | 2.72 | 2.74 |
| W3b—Asn62 ND1 | 2.67 | 2.60 |
| W3b—Asn67 OD1 | 3.17 | 3.03 |
W represents either H2O or D2O in the hydrogenated or perdeuterated structures, respectively.
4. Discussion and conclusions
We report the successful large-scale expression, crystallization and X-ray structure determination of perdeuterated HCA II. These results show that it is possible to (i) overexpress perdeuterated HCA II in E. coli, once the optimal conditions for growth of the bacteria have been established, as has also been shown for cytochrome P450cam (Meilleur et al., 2005 ▶), and (ii) crystallization conditions for perdeuterated HCA II can be readily obtained in order to grow crystals of sufficient size and quality for neutron diffraction studies, as has also been shown for human aldose reductase (Hazemann et al., 2005 ▶).
It is well known and described in the literature that perdeuteration has subtle effects on the physical, chemical and functional properties of proteins (Brockwell et al., 2001 ▶; Meilleur et al., 2004 ▶). Our results demonstrate that the perdeuterated HCA II structure is highly isomorphous with its hydrogenated counterpart (0.2 Å r.m.s.d. for all Cα atoms) at 1.5 Å resolution (Fig. 1 ▶). Not only are the overall HCA II structures very similar, but the key residues in the active site (Tyr7, Asn62, His64, Asn67, Thr199 and Thr200) and the solvent molecules (zinc-bound solvent, W1, W2, W3a and W3b), which are known to be critical in mediating proton transfer, are identical (<0.1 Å r.m.s.d.) within experimental error (Fig. 1 ▶; Table 3 ▶).
Our data represent proof-of-principle that the perdeuterated and hydrogenated HCA II structures are extremely similar. This result validates the use of perdeuterated HCA II for neutron crystallography in order to obtain detailed information on the active site that can be directly correlated with X-ray structures for functional annotation and direct visualization of H/D atoms.
Supplementary Material
PDB reference: perdeuterated human carbonic anhydrase II, 2ax2, r2ax2sf
Acknowledgments
The authors thank William Shepard for help and support during X-ray data collection at the ESRF (beamline ID29). This work was supported by grants from the National Institutes of Health GM25154 (DNS and RM) and the Maren Foundation (RM), and has benefited from the activities of the EU-funded DLAB project under contracts HPRI-CT-2001-50035 and RII3-CT-2003-505925.
References
- Brockwell, D., Yu, L., Cooper, S., McCleland, S., Cooper, A., Attwood, D., Gaskell, S. J. & Barber, J. (2001). Protein Sci.10, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Christianson, D. W. & Fierke, C. A. (1996). Acc. Chem. Res.29, 331–339.
- Coates, L., Erskine, P. T., Wood, S. P., Myles, D. A. A. & Cooper, J. B. (2001). Biochemistry, 40, 13149–13157. [DOI] [PubMed] [Google Scholar]
- Duda, D., Govindasamy, L., Agbandje-McKenna, M., Tu, C. K., Silverman, D. N. & McKenna, R. (2003). Acta Cryst. D59, 93–104. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Esnouf, R. M. (1999). Acta Cryst. D55, 938–940. [DOI] [PubMed] [Google Scholar]
- Fisher, S. Z., Hernandez Prada, J., Tu, C. K., Duda, D., Yoshioka, C., An, H., Govindasamy, L., Silverman, D. N. & McKenna, R. (2005). Biochemistry, 44, 1097–1105. [DOI] [PubMed] [Google Scholar]
- Gutberlet, T., Heinemann, U. & Steiner, M. (2001). Acta Cryst. D57, 349–354. [DOI] [PubMed] [Google Scholar]
- Habash, J., Raftery, J., Nuttall, R., Price, H. J., Wilkinson, C., Kalb (Gilboa), A. J. & Helliwell, J. R. (2000). Acta Cryst. D56, 541–550. [DOI] [PubMed] [Google Scholar]
- Hazemann, I., Dauvergne, M. T., Blakeley, M. P., Meilleur, F., Haertlein, M., Van Dorsselaer, A., Mitschler, A., Myles, D. A. & Podjarny, A. (2005). Acta Cryst. D61, 1413–1417. [DOI] [PubMed] [Google Scholar]
- Khalifah, R. G., Strader, D. J., Bryant, S. H. & Gibson, S. M. (1977). Biochemistry, 16, 2241–2247. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Lindskog, S. (1997). Pharmacol. Ther.74, 1–20. [DOI] [PubMed] [Google Scholar]
- Lindskog, S. & Silverman, D. N. (2000). The Carbonic Anhydrases: New Horizons, edited by W. R. Chegwidden, N. D. Carter & Y. H. Edwards, pp. 175–195. Basel, Switzerland: Birkhäuser Verlag.
- McPherson, A. (1982). Preparation and Analysis of Protein Crystals. New York: Wiley.
- Meilleur, F., Contzen, J., Myles, D. A. A. & Jung, C. (2004). Biochemistry, 43, 8744–8753. [DOI] [PubMed] [Google Scholar]
- Meilleur, F., Dauvergne, M. T., Schlichting, I. & Myles, D. A. A. (2005). Acta Cryst. D61, 539–544. [DOI] [PubMed] [Google Scholar]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol.277, 505–524. [DOI] [PubMed]
- Merz, K. M. (1990). J. Mol. Biol.214, 799–802. [DOI] [PubMed] [Google Scholar]
- Nair, S. K. & Christianson, D. W. (1991). J. Am. Chem. Soc.113, 9455–9458.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Parkkila, S. (2000). The Carbonic Anhydrases: New Horizons, edited by W. R. Chegwidden, N. D. Carter & Y. H. Edwards, pp. 79–93. Basel, Switzerland: Birkhäuser Verlag.
- Shu, F., Ramakrishnan, V. & Schoenborn, B. P. (2000). Proc. Natl Acad. Sci. USA, 97, 3872–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman, D. N. & Lindskog, S. (1988). Acc. Chem. Res.21, 30–36.
- Tu, C. K., Silverman, D. N., Forsman, C., Jonsson, B.-H. & Lindskog, S. (1989). Biochemistry, 28, 7913–7918. [DOI] [PubMed] [Google Scholar]
- Tufts, B. L., Esbaugh, A. & Lund, S. G. (2003). Comput. Biochem. Physiol. A, 136, 259–269. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: perdeuterated human carbonic anhydrase II, 2ax2, r2ax2sf