

Success in crystallization of a functionally intact Hsp70 chaperone required genetic engineering to minimize polydispersity and modulate interdomain interactions, as well as high concentrations of the potent structure stabilizer TMAO. These approaches may be generally useful in crystallization of conformationally flexible proteins that exhibit interdomain motions.
Keywords: Hsp70, Hsc70, chaperones
Abstract
Hsp70s are essential chaperones with roles in a variety of cellular processes and representatives in all kingdoms of life. They are comprised of a nucleotide-binding domain (NBD) and a protein substrate-binding domain (SBD). Structures of isolated NBDs and SBDs have been reported but, until recently, a functionally intact Hsp70 containing both the NBD and SBD has resisted structure determination. Here, it is reported that preparation of diffraction-quality crystals of functionally intact bovine Hsc70 required (i) deletion of part of the protein to reduce oligomerization, (ii) point mutations in the interface between the SBD and NBD and (iii) use of high concentrations of the structure-stabilizing agents glycerol and trimethylamine oxide (TMAO). The introduction of point mutations in interdomain interfaces and the use of the potent structure stabilizer TMAO may be generally useful in crystallization of multidomain proteins that exhibit interdomain motions.
1. Introduction
Hsp70 and Hsc70 proteins play central roles in a large number of cellular processes such as protein folding and translocation and the remodeling of a variety of protein complexes (Young et al., 2003 ▶; Bukau & Horwich, 1998 ▶; Schmid & Rothman, 1985 ▶; James et al., 1997 ▶). They are comprised of a 44 kDa nucleotide-binding domain (NBD) and an 18 kDa substrate-binding domain (SBD), both of which are well conserved, and a variable 10 kDa C-terminal domain that is dispensable for chaperone function but is important for its oligomerization (Young et al., 2003 ▶). ATP binding to the NBD communicates a signal to the SBD which alters the latter’s conformation, apparently opening a helical lid that covers the substrate-binding pocket in the ADP state to allow substrate release (Hightower et al., 1994 ▶; Zhu et al., 1996 ▶). A global conformational change associated with ATP binding is also suggested by small-angle X-ray scattering studies (Wilbanks et al., 1995 ▶; Shi et al., 1996 ▶) and such a change may provide for a power stroke in processes such as the movement of proteins through translocation pores (Voisine et al., 1999 ▶).
Because of their critical roles in so many cellular processes and evidence that their mechanisms involve cycles of conformational change, there has been strong interest in determining the structures of Hsp70s. Crystal structures of NBDs from bovine and humans and of the 10 kDa rat Hsc70 oligomerization domain and NMR and crystal structures of SBDs from rat Hsc70 and Escherichia coli Hsp70 (DnaK) have been determined (Flaherty et al., 1990 ▶, 1994 ▶; O’Brien & McKay, 1993 ▶; Sriram et al., 1997 ▶; Wang et al., 1998 ▶; Morshauer et al., 1999 ▶; Zhu et al., 1996 ▶; Chou et al., 2003 ▶). However, the crystal structure of a functionally intact chaperone containing both NBD and SBD has, until recently, not been described.
The barriers to preparation of diffraction-quality crystals of intact Hsp70s can probably be ascribed to two of these proteins’ properties. The first is the propensity of the full-length proteins to heterogeneously oligomerize (Kim et al., 1992 ▶), a behavior recognized as inimical to crystallization (Veesler et al., 1994 ▶; Ferre-D’Amare & Doudna, 1997 ▶). The second is the likely existence of interdomain motion (Buchberger et al., 1995 ▶; Wilbanks et al., 1995 ▶; Shi et al., 1996 ▶; Palleros et al., 1992 ▶; Banecki et al., 1992 ▶; Dale et al., 2003 ▶). We report here that these barriers were overcome for bovine Hsc70 (bHsc70) by engineering the protein to limit oligomerization and to modulate interdomain interactions and by including high concentrations of protein structure-stabilizing agents during crystallization.
2. Materials and methods
2.1. Mutant construction and protein purification
Mutations were introduced into the bHsc70ΔCterm vector using a two-stage PCR protocol (Wang & Malcolm, 1999 ▶). Proteins were expressed in E. coli BL21/DE3/pLysS (Novagen) at 303 K. Cell pellets from a 6 l culture were resuspended in 90 ml 10 mM Tris pH 8.0, 1 mM EDTA, 1 mM DTT, 5% glycerol (buffer A) and lysed by sonication. 5 ml of 10% polymin P pH 8.0 were added and incubated with stirring for 10 min at 277 K, followed by centrifugation at 15 000 rev min−1 for 15 min. The pellet was discarded and 4 g ammonium sulfate was added for every 10 ml of supernatant; the solution was stirred for 15 min at 277 K and centrifuged at 15 000 rev min−1 for 30 min. The supernatant was discarded and the pellet was resuspended in 60 ml of buffer A and dialyzed overnight against buffer A at 277 K. The sample was loaded onto a 60 ml Q-Sepharose anion-exchange column (Pharmacia) and eluted with a 500 ml linear gradient of buffer A and buffer A plus 0.5 M NaCl. Fractions containing bHsc70ΔCterm eluted between 0.25 and 0.35 M NaCl and were pooled and dialyzed against 10 mM Tris pH 6.5, 1 mM EDTA, 1 mM DTT, 5% glycerol (buffer B). The sample was loaded onto a 50 ml SP-Sepharose cation-exchange column (Pharmacia) and eluted with a 500 ml linear gradient of buffer B and buffer B plus 0.5 M NaCl. Fractions containing bHsc70ΔCterm eluted between 0.15 and 0.25 M NaCl and were pooled and concentrated to 15 ml using an Amicon stir cell with a 50 kDa moelcular-weight cutoff membrane. The concentrated sample was loaded onto a 600 ml Sephacryl S-200 (Pharmacia) gel-exclusion column and eluted with buffer A. Fractions containing monomeric bHsc70ΔCterm were identified by SDS–PAGE and native PAGE, concentrated to 5–15 mg ml−1, dialyzed for 48 h against buffer A plus 50% glycerol and stored at 253 K.
2.2. Crystallization
Screening was performed by mixing 1 µl of 13–40 mg ml−1 protein in buffer A plus 50% glycerol with 1 µl of commercial crystal screen solutions (Hampton, Wizard) in clear conical 96-well polycarbonate microtiter dishes. Drops were overlaid with 30 µl paraffin oil and incubated at 289 K. If after 4 d a drop remained clear, another microlitre of crystallization solution was added. This was repeated every 4 d until crystals or precipitates appeared. For optimization, pH, precipitate and protein concentration and additives were systematically screened by the microbatch method. Once optimal conditions were identified, crystals suitable for data collection were grown by the macrobatch method with drops of 20–50 µl overlaid with 0.5 ml paraffin oil. Typically, 5 µl of 13 mg ml−1 Hsc70ΔCtermE213A/D214A was mixed with 5 µl of 4 M TMAO and 10 µl of 18% PEG 8000, overlaid with oil and incubated at 289 K for 2–3 weeks.
2.3. Data collection, processing and structure determination
Crystals were mounted on nylon loops directly from the mother liquor, which provided sufficient cryoprotection. Data were collected on a Rigaku Cu rotating-anode X-ray source with imaging-plate detector. Data were processed with DENZO (Otwinowski & Minor, 1997 ▶). Molecular replacement and refinement were performed with MOLREP (Vagin & Teplyakov, 1997 ▶) and the CNS suite of programs (Brünger et al., 1998 ▶) and model building was performed with O (Jones et al., 1991 ▶).
3. Results
3.1. Deletion of the Hsc70 10 kDa C-terminal domain reduces oligomerization and allows crystallization
Deletion of the 10 kDa C-terminal domain of bHsc70 yields a functional chaperone which does not oligomerize (Wilbanks et al., 1995 ▶; Ungewickell et al., 1997 ▶). This is illustrated in Fig. 1 ▶, where lanes 1 and 2 show purified bovine bHsc70ΔCterm (amino acids 1–554) and full-length bHsc70 (amino acids 1–623), respectively, resolved by denaturing (SDS) PAGE. Both samples exhibit apparent single-band purity. Lanes 3 and 4 show the same samples run on a native gel. The Hsc70ΔCterm protein (lane 3) runs as a single band, while the full-length protein exhibits several bands against a background smear. Efforts to crystallize full-length bHsc70 yielded precipitates but no crystals, while crystals of bHsc70ΔCterm were obtained in the first screens.
Figure 1.
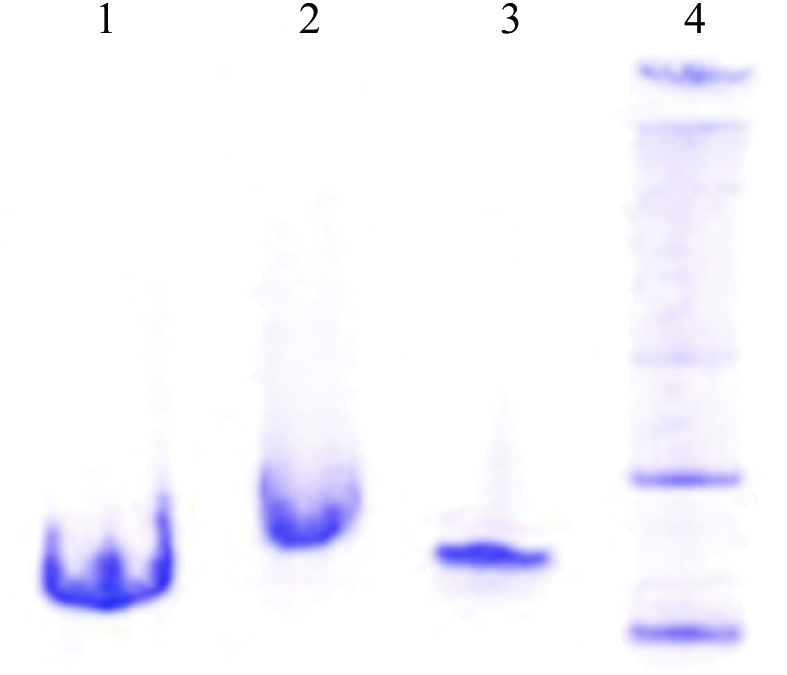
Deletion of the 10 kDa C-terminal domain of Hsc70 reduces oligomerization. Lanes 1 and 2: SDS–PAGE of Hsc70ΔCterm and full-length Hsc70, respectively. Lanes 3 and 4: native PAGE of Hsc70ΔCterm and full-length Hsc70, respectively.
bHsc70ΔCterm crystals grew as aggregates of thin plates or dendrites (Fig. 2 ▶ a) with 3K–8K PEGs as precipitants and 50–250 mM acetate, citrate or nitrate as buffers between pH 6.5 and 9.0 and with glycerol at 12–25%(v/v). Through optimization, we were able to obtain single thin plates that diffracted poorly (Fig. 2 ▶ b). To improve these crystals, we generated an N-terminally His-tagged protein to facilitate the preparation of purer protein. The tag changed the behavior of the protein during expression: the untagged protein formed inclusion bodies at 310 K and had to be expressed at 303 K to be soluble, while the tagged protein remained soluble when expressed at 310 K. The tag also altered the crystallizability of the protein: although all but three amino acids (Gly-Ser-His) were removed by thrombin digestion, these three amino acids prevented crystallization under conditions that gave crystals of the untagged protein. Extensive screening of the tagged protein (following thrombin digestion) did not identify any conditions under which the tagged protein would crystallize. We therefore abandoned use of the tagged protein and used anion-exchange, cation-exchange and gel-exclusion chromatography to further purify the untagged protein. With the purer protein, crystals with a better shape that diffracted beyond 3 Å with a rotating-anode source were grown (Fig. 2 ▶ c). However, crystal decay prevented the collection of complete data sets and merging R factors were high (9.9% for data to 6 Å and >30% for higher resolution data). More complete data sets were collected at the SBC-19BM beamline at the Brookhaven Synchrotron, but statistics remained poor (R merge of 20.4% for a 3.8 Å data set). Quantitative methylation of accessible lysines (Rayment et al., 1993 ▶) blocked crystallization.
Figure 2.

(a) Hsc70ΔCterm crystals obtained in initial screens (microbatch under oil: 1 µl of 20 mg ml−1 protein in 10 mM Tris pH 8.0, 1 mM EDTA, 1 mM DTT, 50% glycerol plus 1 µl 0.2 M sodium acetate, 0.1 M Tris 8.5, 30% PEG 4000). (b) Optimized crystals [inset; grown as in (a) but with 0.5 M KI added] and diffraction pattern. (c) Further optimization of crystals [inset; grown as in (b) but the protein was further purified by cation-exchange and gel-exclusion chromatography] and a representative diffraction pattern (P212121; a = 78.3, b = 95.2, c = 178.5 Å).
3.2. Identification of a surface mutant of Hsc70ΔCterm with improved crystallization properties
Derewenda (2004 ▶) has reported that mutation of exposed charged groups to alanines can improve a protein’s crystallization properties. We selected eight clusters of charged side chains located on exposed loops of the bHsc70 NBD and mutated these to alanines (Fig. 3 ▶; K108A/E110A/K112A, D186A/K187A/K188A, K250A/K251A/D252A, E283A/D285A/E289A, K325A/D327A/K328A, K357A/E358A/K361A, D383A/K384A/E386A and E213A/D214A). These mutations were constructed in both His-tagged and untagged backgrounds. Native gels of the purified proteins revealed that they were monodisperse and showed shifts in migration consistent with the charge changes introduced by the mutation (Fig. 4 ▶). All 16 proteins (eight mutants in tagged and untagged versions) were exhaustively screened by microbatch under oil using commercially available screens.
Figure 3.

Crystal structure of bovine Hsc70ΔCterm with NBD in yellow and SBD in green and residues mutated to alanines highlighted. I, K108/E110/K112; II, D186/K187/K188; III, K250/K251/D252; IV, E283/D285/E289; V, K325/D327/K328; VI, K357/E358/K361; VII, D383/K384/E386; VIII, E213/D214.
Figure 4.

Native PAGE of wild-type (WT) and mutant bHsc70ΔCterms. Above each lane the type of residues mutated and the net effect on the protein’s charge are given (i.e. in lane 1 DKK to AAA changes one negative and two positive side chains to neutral, leading to a net change of −1). Changes in electrophoretic migration are consistent with the expected net changes in charge.
One mutant (E283A/D285A/E289A), which ran as a smear on native gels (Fig. 4 ▶, lane 6), crystallized more poorly than the wild-type protein. The other six triple mutants crystallized similarly to the wild type. Small aggregated crystals of the E213A/D14A mutant were obtained with 1 µl of 13 mg ml−1 protein in 10 mM Tris pH 8.0, 1 mM DTT, 1 mM EDTA mixed with 1 µl 0.2 M TMAO, 0.1 M Tris pH 8.5, 20% PEG monomethyl ether 2000. These crystals appeared to be chunkier than the thin plates that were obtained with the wild type. Optimization yielded thick plates of up to 50 µm in thickness and 300 µm on an edge (Fig. 5 ▶), which diffracted to beyond 2.6 Å on a rotating-anode source. The optimized conditions [1 M TMAO, 12.5%(v/v) glycerol, 2.5 mM Tris pH 8.0, 1 mM DTT, 3 mg ml−1 protein, 9–11% PEG 8000] were notable for the low protein concentration (required to minimize nucleation and growth of aggregates) and the high concentrations of glycerol and TMAO, two potent structure-stabilizing agents. A 2.6 Å data set was collected from a single crystal of Hsc70ΔCterm E213A/D214A and molecular replacement with structures of isolated bHsc70 NBD and DnaK SBD was used to solve the structure (Table 1 ▶), which has been described elsewhere (Jiang et al., 2005 ▶).
Figure 5.

Crystal of Hsc70ΔCtermE213A/D214A mutant (macrobatch under oil at 289 K: 5 µl of 13 mg ml−1 protein in 10 mM Tris pH 8.0, 50% glycerol, 1 mM EDTA, 1 mM DTT plus 5 µl of 4 M TMAO plus 10 µl of 18% PEG 8000).
Table 1. Data-collection and refinement statistics.
| Space group | P21 |
| Unit-cell parameters (Å, °) | a = 65.7, b = 50.1, c = 87.2, β = 99.9 |
| Solvent content | 47% (1 molecule per ASU) |
| Resolution (Å) | 50–2.6 (2.69–2.60) |
| Wavelength (Å) | 1.54 |
| Rmerge (%) | 7.9 (45.9) |
| I/σ(I) | 11.6 (2.4) |
| Unique reflections | 16521 |
| Completeness (%) | 98.7 |
| R factor | 22.0 |
| Rfree | 29.7 |
| R.m.s.d. bonds (Å) | 0.011 |
| R.m.s.d. angles (°) | 1.57 |
| Non-Gly Ramachandran allowed (%) | 99.2 |
4. Discussion and conclusions
The observation that the removal of a subdomain that promotes heterogeneous oligomerization allowed the crystallization of Hsc70 is consistent with the view that polydispersity interferes with crystallization (Veesler et al., 1994 ▶; Dale et al., 2003 ▶). It is more difficult to understand why a six-His tag on the N-terminus should alter Hsc70 behavior during expression or why the tag alters crystallizability even after all but three amino acids from the tag are removed. In the E213A/D214A crystal the protein N-terminus is involved in packing interactions, but this would not explain the effects of the tag on solubility during expression, nor why exhaustive screening failed to yield any crystals with any of the proteins that contained the three extra amino acids at the N-terminus. It has been reported that tags added to the N-terminus of DnaK interfere with allosteric communication between the NBD and SBD (Boice & Hightower, 1997 ▶). The structure of Hsc70ΔCterm E213A/D214A reveals that the N-terminus of the NBD is close to the SBD and to the interdomain linker that has been proposed to play a critical role in interdomain communication (Jiang et al., 2005 ▶). It seems possible that changes at the N-terminus affect the interaction between the NBD and SBD so as to globally alter the conformational properties of the protein and in this way change its solubility and crystallization properties.
The role of interdomain interactions in obtaining suitable crystals is consistent with the result that of the eight mutants made, only the E213A/D214A mutation is near the interdomain interface (Figs. 3 ▶ and 6 ▶). If the crystal structure of the bHsc70 NBD (Flaherty et al., 1990 ▶) is superimposed on the structure of Hsc70ΔCterm E213A/D214A, the Glu213 side chain of the NBD clashes with Asn417 of the SBD (Fig. 6 ▶). Removal of this clash could stabilize the interface, but it seems likely that the flexible Glu or Asp side chains could move to relieve this clash in the wild-type protein. Moreover, the introduced mutations also eliminate a favorable interaction with Arg418 of the SBD (Fig. 6 ▶) and this would tend to destabilize the interface. The precise mechanism by which the mutation affects the interface to allow crystallization is therefore unclear. Interface stabilization, destabilization or shifting of the orientation of the two domains to facilitate packing are all possible explanations, but we conclude that the Derewenda (2004 ▶) approach of mutating mobile charged residues on a protein’s surface to improve its crystallizability may also be useful when applied to the interdomain interface of proteins showing interdomain movement. However, in such instances it is essential to demonstrate that the introduced mutations do not compromise the protein’s function. Critically, we found that in a clathrin cage disassembly assay, which recapitulates the in vivo function of the chaperone in removing clathrin triskelia from coated vesicles, both the stochiometry and kinetics of the E213A/D214A mutant were indistinguishable from the wild type (Jiang et al., 2005 ▶).
Figure 6.

Location of E213A/D214A mutations at the interdomain interface. The interface between the SBD (green) and NBD (yellow) from the Hsc70ΔCtermE213A/D214A structure is shown with the A213/A214 mutant side chains in black and Asn417 and Arg418 in yellow and blue, respectively. Superimposed on this structure is the Cα trace for the isolated bovine Hsc70 ATPase domain (in blue) with the E213/D214 side chains in red.
Growth of diffraction-quality crystals also required the structure-stabilizing agents TMAO and glycerol, with the best crystals obtained at 12.5%(v/v) glycerol and 1 M TMAO. TMAO has been shown to be an exceptionally potent protein structure-stabilizing agent (Auton & Bolen, 2005 ▶) and is even capable of forcing otherwise unstructured proteins to assume native-like folds (Baskakov et al., 1999 ▶; Baskakov & Bolen, 1998 ▶). These observations have led to the suggestion that TMAO might facilitate structural studies of proteins that are otherwise too conformationally flexible to crystallize (Baskakov & Bolen, 1998 ▶; Bolen, 2004 ▶). We identified an initial crystal ‘hit’ with a commercial screen (Hampton Index HT; condition F2) that contained 0.2 M TMAO and found in subsequent optimization that the single most important factor in improving the crystal was to increase the TMAO concentration from 0.15 to 1.0 M. TMAO may help stabilize more compact and crystallizable protein conformations and its effects are likely analogous to the effects of polyols such as glycerol, which has been frequently used to crystallize conformationally flexible or unstable proteins (Sousa, 1995 ▶). There are as yet only a few reports of the use of TMAO in protein crystallization (Doolittle, 2003 ▶; Hill et al., 2002 ▶). However, TMAO has the potential to be more effective than glycerol (or any other polyol) in this respect because its structure-stabilizing effects are quantitatively larger (Auton & Bolen, 2005 ▶; Bolen, 2004 ▶). The ability of TMAO to overcome the barrier to crystallization posed by excessive conformational flexibility needs to be more generally explored.
Supplementary Material
PDB reference: Hsc70, 1yuw, r1yuwsf
Acknowledgments
We thank Dr D. McKay for an expression plasmid for bHsc70ΔCterm. This work was supported by GM-52522 and AQ-1486 from the NIH and Welch Foundation (to RS) and MDA3473 and NS29051 from the MDA and NINDS (to EML). Support for the X-ray Crystallography Core and the UTHSCSA Center for Macromolecular Interactions from the UTHSCSA Executive Research Committee and San Antonio Cancer Institute is acknowledged.
References
- Auton, M. & Bolen, D. W. (2005). Proc. Natl Acad. Sci. USA, 102, 15065–15068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banecki, B., Zylicz, M., Bertoli, E. & Tanfani, F. (1992). J. Biol. Chem.267, 25051–25058. [PubMed] [Google Scholar]
- Baskakov, I. V. & Bolen, D. W. (1998). J. Biol. Chem.273, 4831–4834. [DOI] [PubMed] [Google Scholar]
- Baskakov, I. V., Kumar, R., Srinivasan, G., Ji, Y.-S., Bolen, D. W. & Thompson, E. B. (1999). J. Biol. Chem.274, 10693–10696. [DOI] [PubMed] [Google Scholar]
- Boice, J. A. & Hightower, L. E. (1997). J. Biol Chem.272, 24825–24831. [DOI] [PubMed] [Google Scholar]
- Bolen, D. W. (2004). Methods, 34, 312–322. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Buchberger, A., Theyssen, H., Schroder, H., McCart, J. S., Giuseppe, V., Milkereit, P., Reinstein, J. & Bukau, B. (1995). J. Biol. Chem.270, 16903–16910. [DOI] [PubMed] [Google Scholar]
- Bukau, B. & Horwich, A. L. (1998). Cell, 92, 351–366. [DOI] [PubMed] [Google Scholar]
- Chou, C. C., Forouhar, F., Yeh, Y. H., Shr, H. L., Wang, C. & Hsiao, C. D. (2003). J. Biol. Chem.278, 30311–30317. [DOI] [PubMed] [Google Scholar]
- Dale, G. E., Oefner, C. & D’Arcy, A. (2003). J. Struct. Biol.142, 88–97. [DOI] [PubMed] [Google Scholar]
- Derewenda, Z. S. (2004). Structure, 12, 529–535. [DOI] [PubMed] [Google Scholar]
- Doolittle, R. F. (2003). Biophys. Chem.100, 307–313. [DOI] [PubMed] [Google Scholar]
- Ferre-d’Amare, A. R. & Doudna, J. A. (1997). Methods Mol. Biol.74, 371–377. [DOI] [PubMed] [Google Scholar]
- Flaherty, K. M., De Luca-Flaherty, C. & McKay, D. B. (1990). Nature (London), 346, 623–628. [Google Scholar]
- Flaherty, K. M., Wilbanks, S. M., De Luca-Flaherty, C. & McKay, D. B. (1994). J. Biol. Chem.269, 12899–12907. [PubMed] [Google Scholar]
- Hightower, L. E., Sadis, S. E. & Takenaka, I. M. (1994). The Biology of Heat Shock Proteins and Molecular Chaperones, edited by R. I. Morimoto, A. Tissieres & C. Georgepoulos, pp. 179–207. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press.
- Hill, M. C., Bates, I. R., White, G. F., Hallett, F. R. & Harauz, G. (2002). J. Struct. Biol.139, 13–26. [DOI] [PubMed] [Google Scholar]
- James, P., Pfund, C. & Craig, E. A. (1997). Science, 275, 387–389. [DOI] [PubMed] [Google Scholar]
- Jiang, J., Prasad, K., Lafer, E. M. & Sousa, R. (2005). Mol. Cell, 20, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kim, D., Lee, Y. J. & Corry, P. M. (1992). J. Cell. Physiol.153, 353–361. [DOI] [PubMed] [Google Scholar]
- Morshauer, R. C., Hu, W., Wang, H., Pang, Y., Flynn, G. C. & Zuiderweg, E. R. P. (1999). J. Mol. Biol.289, 1387–1403. [DOI] [PubMed] [Google Scholar]
- O’Brien, M. C. & McKay, D. B. (1993). J. Biol. Chem.268, 24323–24329. [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Palleros, D. R., Reid, K. L., McCarty, J. S., Walker, G. C. & Fink, A. L. (1992). J. Biol. Chem.267, 5279–5285. [PubMed] [Google Scholar]
- Rayment, I., Rypniewski, W. R., Schmidt-Base, K., Smith, R., Tomchick, D. R., Benning, M. M., Winkelmann, D. A., Wesenberg, G. & Holden, H. M. (1993). Science, 261, 50–58. [DOI] [PubMed] [Google Scholar]
- Schmid, S. L. & Rothman, J. E. (1985). J. Biol. Chem.260 10044. [PubMed] [Google Scholar]
- Shi, L., Kataoka, M. & Fink, A. (1996). Biochemistry, 35, 3297–3308. [DOI] [PubMed] [Google Scholar]
- Sousa, R. (1995). Acta Cryst. D51, 271–277. [DOI] [PubMed] [Google Scholar]
- Sriram, M., Osipiuk, J., Freeman, C., Morimoto, R. I. & Joachimiak, A. (1997). Structure, 5, 403–414. [DOI] [PubMed] [Google Scholar]
- Ungewickell, E., Ungewickell, H. & Holstein, S. E. H. (1997). J. Biol. Chem.272, 19594–19600. [DOI] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]
- Veesler, S., Marcq, S., Lafont, S., Astier, J. P. & Boistelle, R. (1994). Acta Cryst. D50, 355–360. [DOI] [PubMed] [Google Scholar]
- Voisine, C., Craig, E. A., Zufall, N., von Ahsen, O., Pfanner, N. & Voos, W. (1999). Cell, 97, 565–574. [DOI] [PubMed] [Google Scholar]
- Wang, H., Kurochkin, V., Pang, Y., Hu, W., Flynn, G. C. & Zuiderweg, E. R. P. (1998). Biochemistry, 37, 7929–7940. [DOI] [PubMed] [Google Scholar]
- Wang, W. & Malcolm, B. A. (1999). Biotechniques, 26, 680–682. [DOI] [PubMed] [Google Scholar]
- Wilbanks, S. M., Chen, L., Tsuruta, H., Hodgson, K. O. & McKay, D. B. (1995). Biochemistry, 34, 12095–12105. [DOI] [PubMed] [Google Scholar]
- Young, J. C., Barral, J. M. & Hartl, F. U. (2003). Trends Biochem. Sci.28, 541–547. [DOI] [PubMed] [Google Scholar]
- Zhu, X., Zhao, X., Burkholder, W. F., Gragerov, A., Ogata, C. M., Gottesman, M. E. & Hendrickson, W. A. (1996). Science, 272, 1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Hsc70, 1yuw, r1yuwsf