

Low iodide concentrations were sufficient to allow SAD and SIRAS phasing of cubic crystals of a novel fatty acid isomerase using Cu Kα radiation.
Keywords: fatty-acid isomerases, Propionibacterium acnes
Abstract
The polyenoic fatty-acid isomerase from Propionibacterium acnes (PAI) catalyzes the double-bond isomerization of linoleic acid to conjugated linoleic acid, which is a dairy- or meat-derived fatty acid in the human diet. PAI was overproduced in Escherichia coli and purified to homogeneity as a yellow-coloured protein. The nature of the bound cofactor was analyzed by absorption and fluorescence spectroscopy. Single crystals of PAI were obtained in two crystal forms. Cubic shaped crystals belong to space group I213, with a unit-cell parameter of 160.4 Å, and plate-like crystals belong to the monoclinic space group C2, with unit-cell parameters a = 133.7, b = 60.8, c = 72.2 Å, β = 115.8°. Both crystal forms contain one molecule per asymmetric unit and diffract to a resolution of better than 2.0 Å. Initial phases were obtained by SIRAS from in-house data from a cubic crystal that was soaked with an unusually low KI concentration of 0.25 M.
1. Introduction
Conjugated linoleic acid (CLA) is a collective term for positional and geometric isomers of linoleic acid (9-cis,12-cis octadecadienoic acid). Two isomers, 9-cis,11-trans and 10-trans,12-cis CLA, have been shown to exert anti-carcinogenic, anti-atherogenic, anti-obesity, anti-inflammatory and antidiabetic effects in mammals (Choi et al., 2001 ▶). The main source of CLA in the human diet is meat and milk from ruminants and these essential fatty acids can be limiting. Commercial food enrichment with CLA relies on the chemical isomerization of plant oils rich in linoleic acid (LA), which yields several isomers in an ill-defined mixture. As it has been shown that individual CLA isomers have different effects in vivo (Wahle et al., 2004 ▶), an alternative and more specific route for CLA production would be via enzymatic isomerization of easily accessible precursors.
To this end, the gene coding for the Propionibacterium acnes polyenoic fatty-acid isomerase (PAI; EMBL accession code q6a8x5) has been cloned, the protein recombinantly produced in Escherichia coli and its biochemical properties assessed (Hornung et al., 2006 ▶). PAI catalyzes the isomerization of LA to 10-trans,12-cis CLA, but also accepts other C18 fatty acids such as linolenic (6-cis,9-cis,12-cis octatrienoic) acid as substrates. Two other polyenoic fatty-acid isomerases from Butyrivibrio fibrisolvens (Kepler et al., 1970 ▶, 1971 ▶) and the algae Ptilota filicina (Sanders et al., 2001 ▶; Zheng et al., 2002 ▶) have been described. The algal enzyme does not share significant sequence homology with PAI and there is no sequence information for the Butyrivibrio enzyme. In addition, no structure of any fatty-acid isomerase is available. With the aim of obtaining a structural description of the polyenoic fatty-acid isomerization reaction, we have crystallized PAI in two forms and employed halide-based phasing to generate initial electron-density maps. Furthermore, we initially characterized by absorption and fluorescence spectroscopy a yellow-coloured cofactor, probably a flavin, which is likely to be involved in catalysis.
2. Methods
2.1. Protein purification and spectroscopic procedures
PAI was cloned from genomic DNA of P. acnes and overproduced in E. coli as described by Hornung et al. (2006 ▶). Purification of PAI will be described in detail elsewhere (Liavonchanka et al., 2006 ▶). Absorption spectra of 40 µM solutions of FAD and PAI were recorded at room temperature in 50 mM Tris–HCl pH 7.5 on a single-beam Xe-flashlamp absorption spectrometer (Fig. 1 ▶ a). Fluorescence emission spectra in the range 460–650 nm (10 nm bandwidth) were measured at 298 K on a FluoroMax3 spectrofluorimeter at 2 µM (FAD) and 6 µM (PAI) concentrations in 40 mM sodium phosphate pH 7.5 after excitation at 450 nm (1 nm bandwidth) and an integration time of 1 s (Fig. 1 ▶ b).
Figure 1.

Analysis of the cofactor and crystallization of PAI. (a) UV spectra of 40 µM solutions of free FAD (black curve) and PAI (red curve) show the two characteristic absorption maxima for an oxidized flavin (FAD or FMN) at 360–390 nm and 440–470 nm. (b) The fluorescence emission spectra of free FAD (2 µM, black curve) and PAI (6 µM, red curve) show the characteristic flavin emission at 525 nm. Cofactor fluorescence is strongly quenched when bound to the protein (compare second ordinate). (c) Cubic PAI crystals from 0.1 M Tris–HCl pH 8.5, 2 M Li2SO4, 2%(v/v) PEG 400. (d) Monoclinic PAI crystals from 0.1 M MES–NaOH pH 6.0, 2 M (NH4)2SO4, 5%(v/v) PEG 400. The longest dimension in both crystal forms is 0.2 mm. (e) and (f) show diffraction patterns of the cubic and monoclinic crystals, respectively, collected in-house on a MAR345 imaging-plate detector at a distance of 150 mm after 10 min exposure. The outer resolution is 1.86 Å.
2.2. Crystallization and crystal characterization
Initial PAI crystals of irregular shape were obtained from condition No. 39 of Crystal Screen I (Hampton Research), i.e. 0.1 M HEPES–NaOH pH 7.5, 2 M (NH4)2SO4, 2%(v/v) PEG 400 at 283 K using a protein concentration of 5 mg ml−1 by sitting-drop vapour diffusion. Based on this lead condition, cubic shaped crystals of PAI were grown by mixing equal volumes of 10 mg ml−1 PAI with 0.1 M Tris–HCl pH 8.5, 2 M Li2SO4 and either 2%(v/v) PEG 400 or 2%(v/v) 1,4-butanediol (Fig. 1 ▶ c). Plate-like crystals grew from 0.1 M MES–NaOH pH 6.0, 2 M (NH4)2SO4, 5%(v/v) PEG 400 (Fig. 1 ▶ d). The cubic crystals were directly cryocooled in liquid nitrogen without additional cryoprotectant, while plates were cryoprotected in mother liquor supplemented with 12.5%(v/v) PEG 400. Crystals were mounted in an arbitrary orientation and data were collected in-house on a MAR345dtb image-plate detector mounted on a MicroMax 007 generator operating with a copper target. The exposure time was 10 min, the oscillation angle was 0.5° and the crystal-to-detector was distance 150 mm. Data reduction was performed with the HKL suite of programs (Otwinowski & Minor, 1997 ▶) (Table 1 ▶). Indexing of the diffraction pattern of the cubic crystals (Fig. 1 ▶ e) was consistent with space groups I23 or I213, which cannot be distinguished based on systematic absences. The diffraction pattern of the plate-shaped crystals (Fig. 1 ▶ f) indexed in monoclinic space group C2. There is one molecule per asymmetric unit in both crystal forms.
Table 1. X-ray data-collection statistics.
Values in parentheses are for data in the highest resolution shell (2.00–1.95 Å for the native data sets and 2.29–2.21 Å for the KI soak).
| Data set | Native I | KI soak | Native II |
|---|---|---|---|
| Space group | I213 | I213 | C2 |
| Unit-cell parameters (Å, °) | a = 160.4 | a = 160.3 | a = 133.7, b = 60.8, c = 72.2, β = 115.8 |
| Matthews coefficient (Å3 Da−1) | 3.7 | 3.7 | 2.8 |
| Solvent content (%) | 65 | 65 | 55 |
| Wavelength (Å) | 1.54 | 1.54 | 1.54 |
| Resolution range (Å) | 50–1.95 | 50–2.21 | 50–1.95 |
| Unique reflections | 49143 | 58536 | 37527 |
| Completeness (%) | 98.4 (78.2) | 87.9 (19.7) | 100 (98.1) |
| Mean I/σ(I) | 14.0 (1.3) | 12.1 (1.0) | 11.4 (2.5) |
| Rmerge† | 9.9 (75.6) | 12.9 (55.5) | 12.3 (61.2) |
R
merge = 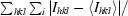
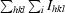 .
.
3. Results and discussion
3.1. Spectroscopic characterization of the yellow cofactor
Upon concentration, it was noticed that PAI is yellow in colour. Protein aggregation can lead to yellow colouring owing to light scattering, but no such scattering was observed in absorption measurements and PAI eluted in a monodisperse form from a gel-filtration column (data not shown). Thus, a cofactor of the flavin type or an iron cluster could be present in PAI. The absorption spectrum of PAI in the 300–500 nm range shows two bands at 370 and 460 nm (Fig. 1 ▶ a) indicative of the isoalloxazine ring of an oxidized flavin. A control measurement with FAD seconded this hypothesis. Flavins also fluoresce at 525 nm after excitation at 450 nm (Chapman & Reid, 1999 ▶). Typical flavin fluorescence was found when the emission spectra of FAD and PAI were compared (Fig. 1 ▶ b). Upon binding of flavins to proteins, their fluorescence is strongly quenched (Chapman & Reid, 1999 ▶), which is also the case for PAI. The maximal fluorescence emission intensity of PAI is >80-fold lower than that of free FAD. Taken together, these spectroscopic data indicate the presence of a flavin, FAD or FMN, in PAI, which is likely to function as a cofactor in fatty-acid isomerization.
3.2. In-house SIRAS phasing after halide soaking
Halide soaking is a potentially rapid method for either in-house (using iodide) or synchrotron (using bromide) phasing by SAD or SIRAS (Dauter et al., 2000 ▶). The high symmetry (I23 or I213) and solvent content (65%) of the cubic crystal form are beneficial factors for density modification once a halide substructure has been found. Crystals were briefly (<1 min) soaked in mother liquor supplemented with 1.0 M KI, but tended to crack and failed to diffract X-rays. As halides weakly and non-covalently bind to both ionic and hydrophobic areas of the protein surface, high concentrations of >0.5 M of the halide salt are usually used (Uson et al., 2003 ▶). Exceptions are lysozyme and xylanase, where 0.35 and 0.5 M KI were used, respectively (Dauter et al., 2000 ▶). In the case of PAI, the highest KI concentration that still retained crystal morphology and diffraction (Table 1) was 0.25 M, which might result in reduced occupancy of the derivative sites. An anomalous difference Patterson map at Harker section u = 0.5 calculated to 3 Å resolution with the origin removed showed a number of significant peaks >10σ, indicating successful derivatization (Fig. 2 ▶ a). Analysis for anomalous signal content with SHELXC by comparing the anomalous signal-to-noise ratio based on the variances of F + and F − revealed ΔF/σ(ΔF) ratios of >1.16 for reflections of <3.5 Å resolution, where a value of 0.8 indicates no anomalous signal (Zwart, 2005 ▶). Substructure-determination calculations were performed with SHELXD (Schneider & Sheldrick, 2002 ▶) using data truncated at a resolution of 3.5 Å in space groups I23 and I213. A SAD experiment in space group I213 resulted in a 14-atom substructure with occupancies >0.2 (correlation coefficient CC of 0.47/0.26 for all/weak reflections). This substructure yielded interpretable electron-density maps after a quite extensive density-modification and phase-extension scheme of 300 cycles in SHELXE (estimated map CC of 0.85 to 2.5 Å resolution; data not shown) and also established the space group as I213.
Figure 2.

SIRAS phasing of cubic PAI. (a) Anomalous difference Patterson map at section u = 0.5 calculated to a resolution of 3 Å showing several distinct peaks for bound iodide. Contours are in steps of 1σ and are coloured differently for each level. (b) Graphical representation of Table 2 ▶ showing a drop in occupancy to ∼0.2 at site No. 14. Iodide 2 is located on a crystallographic twofold. The top 13 sites were used as input for density modification and enantiomer selection in SHELXE. (c) Progress of density modification and phase extension monitored by the protein/solvent contrast versus modification cycle. While the correct hand of the substructure is immediately apparent, the quality of the electron density requires at least 60 cycles for convergence. (d) Estimated map correlation coefficient as a function of resolution calculated with density-modified phases. (b), (c) and (d) were generated with HKL2MAP (Pape & Schneider, 2004 ▶). (e) Experimental electron-density map contoured at 2.0σ after density modification and phase extension in SHELXE. The 30 Å section encompasses three molecules and shows a clear solvent boundary.
A higher quality substructure solution was obtained in a SIRAS experiment against data set native I (Table 1 ▶). The derived substructure of 13 sites with relative occupancy >0.2 had a CC of 0.51 and 0.39 for all and for weak reflections, respectively (Fig. 2 ▶ b and Table 2 ▶). Density modification and phase extension using this substructure immediately established the correct hand of the substructure, but required at least 60 cycles to converge at the maximum protein/solvent contrast (Fig. 2 ▶ c). Electron density calculated with these phases had an overall estimated map CC of 0.85 to 2.2 Å resolution (Fig. 2 ▶ d) and showed a clear solvent boundary (Fig. 2 ▶ e).
Table 2. Fractional coordinates of the ten most occupied iodide sites as determined by SHELXD .
| Site | x | y | z | Occupancy |
|---|---|---|---|---|
| 1 | 0.232 | 0.101 | 0.250 | 1.00 |
| 2 | 0.250 | 0.112 | 0.500 | 0.47 |
| 3 | 0.356 | 0.153 | 0.448 | 0.93 |
| 4 | 0.167 | 0.022 | 0.128 | 0.67 |
| 5 | 0.163 | 0.074 | 0.050 | 0.62 |
| 6 | 0.370 | 0.209 | 0.456 | 0.55 |
| 7 | 0.124 | 0.039 | −0.046 | 0.41 |
| 8 | 0.213 | 0.006 | 0.220 | 0.41 |
| 9 | 0.381 | 0.067 | 0.478 | 0.40 |
| 10 | 0.158 | 0.021 | 0.211 | 0.37 |
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 523; TP A16 to MGR). AL is supported by IMPRS Molecular Biology, Göttingen, Germany.
References
- Chapman, S. K. & Reid, G. A. (1999). Methods Mol. Biol.131, 27–28.
- Choi, Y., Park, Y., Pariza, M. W. & Ntambi, J. M. (2001). Biochem. Biophys. Res. Commun.284, 689–693. [DOI] [PubMed] [Google Scholar]
- Dauter, Z., Dauter, M. & Rajashankar, K. R. (2000). Acta Cryst. D56, 232–237. [DOI] [PubMed] [Google Scholar]
- Hornung, E., Krueger, C., Pernstich, C., Gipmans, M., Porzel, A. & Feussner, I. (2006). In the press. [DOI] [PubMed]
- Kepler, C. R., Tucker, W. P. & Tove, S. B. (1970). J. Biol. Chem.245, 3612–3620. [PubMed] [Google Scholar]
- Kepler, C. R., Tucker, W. P. & Tove, S. B. (1971). J. Biol. Chem.246, 2765–2771. [PubMed] [Google Scholar]
- Liavonchanka, A., Hornung, E., Feussner, I. & Rudolph, M. G. (2006). In the press.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pape, T. & Schneider, T. R. (2004). J. Appl. Cryst.37, 843–844. [Google Scholar]
- Sanders, D. A., Staines, A. G., McMahon, S. A., McNeil, M. R., Whitfield, C. & Naismith, J. H. (2001). Nature Struct. Biol.8, 858–863. [DOI] [PubMed] [Google Scholar]
- Schneider, T. R. & Sheldrick, G. M. (2002). Acta Cryst. D58, 1772–1779. [DOI] [PubMed] [Google Scholar]
- Usón, I., Schmidt, B., von Bulow, R., Grimme, S., von Figura, K., Dauter, M., Rajashankar, K. R., Dauter, Z. & Sheldrick, G. M. (2003). Acta Cryst. D59, 57–66. [DOI] [PubMed] [Google Scholar]
- Wahle, K. W., Heys, S. D. & Rotondo, D. (2004). Prog. Lipid Res.43, 553–587. [DOI] [PubMed] [Google Scholar]
- Zheng, W., Wise, M. L., Wyrick, A., Metz, J. G., Yuan, L. & Gerwick, W. H. (2002). Arch. Biochem. Biophys.401, 11–20. [DOI] [PubMed] [Google Scholar]
- Zwart, P. H. (2005). Acta Cryst. D61, 1437–1448. [DOI] [PubMed] [Google Scholar]