

Abstract
Langerin is a C-type lectin receptor that recognizes glycosylated patterns on pathogens. Langerin is used to identify human and mouse epidermal Langerhans cells (LCs), as well as migratory LCs in the dermis and the skin draining lymph nodes (DLNs). Using a mouse model that allows conditional ablation of langerin+ cells in vivo, together with congenic bone marrow chimeras and parabiotic mice as tools to differentiate LC- and blood-derived dendritic cells (DCs), we have revisited the origin of langerin+ DCs in the skin DLNs. Our results show that in contrast to the current view, langerin+CD8− DCs in the skin DLNs do not derive exclusively from migratory LCs, but also include blood-borne langerin+ DCs that transit through the dermis before reaching the DLN. The recruitment of circulating langerin+ DCs to the skin is dependent on endothelial selectins and CCR2, whereas their recruitment to the skin DLNs requires CCR7 and is independent of CD62L. We also show that circulating langerin+ DCs patrol the dermis in the steady state and migrate to the skin DLNs charged with skin antigens. We propose that this is an important and previously unappreciated element of immunosurveillance that needs to be taken into account in the design of novel vaccine strategies.
C-type lectin receptors are predominantly expressed by DCs and function as pattern recognition receptors by interacting with glycosylated moieties on pathogens. This usually results in internalization of the pathogen, lysosomal degradation, and subsequent loading of pathogen-derived peptides into MHC molecules for antigen presentation (1). Langerin (CD207) is a type II lectin receptor containing only one carbohydrate recognition domain (2, 3). Although the ligands of langerin have not been clearly identified, langerin has recently been shown to bind HIV-1 gp120 through calcium-dependent mannose recognition (4). Langerin is also a potent inducer of Birbeck granules, a hallmark of epidermal DCs, also called Langerhans cells (LCs), which consist of zippered membranes that are visible by electron microscopy (2). Upon antigen capture, langerin associates to Birbeck granules and facilitates the routing of captured mannosylated ligands from the cell surface into a nonclassical antigen-processing pathway (2). Therefore, expression of the langerin receptor is likely to reflect a functionally distinct cell population with specific antigen uptake and processing capacity.
Langerin is expressed at high levels on mouse and human LCs (2, 3), but is absent on dermal interstitial DCs (5), providing a unique tool to study the biology of cutaneous DC populations. Cutaneous DCs include langerin+ MHC class II+ LCs in the epidermis (6) and langerin− MHC class II+ interstitial DCs in the dermis (7–10). In addition to interstitial DCs, the dermis contains migratory langerin+ MHC class II+ LCs en route to the skin draining LNs (DLNs) (11–14). In the LNs, DCs are categorized as blood-derived CD8+ and CD8− DCs and migratory DCs recruited from the periphery through the lymphatics (15). In the skin DLNs, migratory DCs include langerin+ LCs and dermal langerin− DCs (14, 16). However, it is now clear that in mice, langerin expression is not restricted to LCs, but is also expressed on CD8+ DCs. Indeed, CD8+langerinlo DCs are present in all lymphoid organs, including LN, spleen, and thymus (3, 17, 18). In contrast to migratory LCs, CD8+ DCs express much lower levels of langerin and lack Birbeck granules, suggesting a non-LC origin of these cells (19). Therefore, in skin DLNs, langerin+ cells are thought to include CD8+langerinlo blood-derived DCs and CD8−langerinhi LC-derived DCs. Langerin expression has also been recently identified on DCs in the lung mucosa and vascular wall (20) and in the gut serosa (21).
Although the mechanisms that regulate the trafficking of tissue DCs to the DLN during inflamed conditions have been well studied (22), much less is known about the trafficking of DC precursors from the blood to tissue and from tissue to DLN in the steady state. In this study, we have revisited the origin and homeostasis of langerin+ DCs in the skin DLNs in the absence of overt tissue injury. To examine the homeostasis of langerin+ DCs, we used a recently described mouse model that allows the specific depletion of langerin+ cells, while sparing other DC populations. This model consists of a knock-in mouse expressing enhanced GFP (EGFP) fused with a diphtheria toxin (DT) receptor (DTR), under the control of the langerin promoter (Langerin-DTR/EGFP) on the C57BL/6 background (18, 23). Administration of DT leads in 24 h to the elimination of all langerin+ cells, including LCs, without affecting the langerin− DC compartment and without skin or systemic toxicity (18, 23). After toxin treatment, LCs repopulate the epidermis in 6–8 wk, offering a window of time to investigate the relative contribution of DC precursors to the skin DLN DC compartments (18, 23). The original description of this model showed that the repopulation of langerin+ DCs in the skin DLNs precedes LC repopulation in the epidermis by weeks (18, 23). Intrigued by these results, we sought to further clarify the origin and the homeostasis of skin DLN langerin+ DCs.
We have previously used congenic parabiotic pairs and congenic BM chimeric mice as tools to differentiate epidermal LCs from circulating DC precursors. We have established that in mice that have been lethally irradiated and reconstituted with donor congenic BM, epidermal LCs remained of host origin throughout life, despite a full donor engraftment in the blood (24). We also demonstrated that in congenic parabiotic mice that share the same blood circulation but separate organs for long period of time, LCs do not mix in the skin and remain of host origin >18 mo after initiation of the parabiosis (24). Therefore, expression of the congenic allele in each parabiont and in recipients of congenic BM grafts can be used to trace the blood- versus LC-derived origin of skin DLN langerin+ DCs. We have excluded from our analysis LN langerinloCD8+ DCs, as they are thought to derive from blood circulating precursors. In contrast, skin DLN langerinhiCD8− DCs are thought to derive exclusively from epidermal LCs and were the focus of this study. Our results reveal for the first time the presence in mice of a population of langerin+ DCs, independent of LCs, in the dermis and the skin DLNs. Our study also reveals that LC-independent langerin+ DCs are recruited from the blood to the dermis in the steady state to capture tissue antigens before emigrating to the skin DLNs, where they present skin-derived peptide-MHC complexes.
RESULTS
Repopulation of langerin+ DCs in skin DLNs precedes the repopulation of LCs in the epidermis
Injection of DT into Langerin-DTR/EGFP mice leads to elimination of langerin+ DCs in the absence of overt skin or systemic toxicity, and therefore can be used to examine the repopulation of langerin+ DCs. As previously described, DT eliminated the entire LC pool in the epidermis, in addition to langerinloCD8+ DCs and langerinhiCD8− DCs in the skin DLNs (Fig. 1, A and B). LCs were totally eliminated 2 d after DT injection and remained absent >14 d after treatment in the epidermis (Fig. 1, A and B). Langerin+ DCs in the skin DLNs were also efficiently depleted after DT, but in contrast to LCs, their repopulation occurred much earlier (Fig. 1, A and B). Langerin+ DCs in the skin DLNs include CD8+ DCs and CD8− DCs. LangerinloCD8+ DCs recovered to normal levels in <5 d after DT administration. Their rapid repopulation after DT administration is consistent with the fact that langerinloCD8+ DCs are thought to derive from circulating precursors with a turnover time of a few days (25, 26). In contrast, langerin+CD8− DCs, a population that is thought to derive exclusively from epidermal LCs, started to repopulate the skin DLNs long before LCs reappeared in the epidermis (Fig. 1, A and B), accounting for 25–30% of total langerin+CD8− DCs in normal untreated skin DLNs. Langerin+CD8− DCs were not detected in nonskin DLNs, such as mesenteric LNs, after DT treatment (unpublished data). Based on these results, we conclude that a population of LC-independent langerin+ DCs, distinct from CD8+ DCs, exists in the skin DLNs. The level of langerin expression on repopulating DCs in the skin DLNs was measured by EGFP fluorescence and by langerin intracellular staining. LC-independent langerin+ DCs in the skin DLNs expressed higher levels of langerin compared with CD8+ DCs and similar levels compared with skin DLN langerin+ DCs in untreated mice (Fig. 1 C). Using immunostaining and confocal analysis of DT-treated skin DLNs, we were able to visualize in situ the presence of LC-independent langerin+ DCs in the T cell areas of the skin DLNs 2 wk after DT, a time when no LCs are present in the skin (Fig. 1 D).
Figure 1.

Kinetics of langerin+ repopulation in the epidermis and in skin DLNs. 6–8-wk-old Langerin-DTR/EGFP mice received a single intraperitoneal injection of DT (1 μg) and the kinetics of reappearance of langerin+ was followed by flow cytometry in the epidermis and the skin DLNs at different time points after depletion (Fig. S1). (A, top) Dot plots show I-Ab and langerin expression (EGFP) among living (DAPI−) CD45+ cells in the epidermis at different times after DT treatment. The remaining CD45+I-Ab−langerin-EGFP− cells represent dendritic epidermal T cells (DETC). (A, bottom) The percentage of cells expressing CD8 and langerin among DAPI−CD11c+I-Ab+ DCs is shown for skin DLNs. (B) Graphs in log scale present the kinetics of reappearance of LCs in the epidermis and CD8−langerin+ (▪) versus CD8+langerinlo (•) in the skin DLNs. Mean values (± the SD) are shown. (right) Histogram shows the percentage of CD8−langerin+ (mean values ± the SD) among CD11c+I-Ab+ cells before and after DT treatment (DAY14). (C) Overlaid histograms show the langerin levels of expression measured either by EGFP fluorescence (top) or langerin intracellular staining (bottom) in skin DLN CD11c+I-Ab+ CD8− langerin+ cells before and after DT treatment (DAY14). C57BL/6 control and isotype control are shown in black for Langerin-EGFP or langerin intracellular staining, respectively. Representative data from three independent experiments are shown. Each experiment included at least three separately analyzed mice per time point; error bars represent the SD between the results obtained from each of the three mice. (D) Double immunostaining (B220 in blue and langerin in red) of skin DLNs from Langerin-DTR/EGFP (Langerin-EGFP in green) before and after DT treatment (DAY14). Fig. S1 is available at http://www.jem.org/cgi/content/full/jem.20071733/DC1. Bars: (top) 200 mm; (bottom) 50 mm.
The repopulation of langerin+CD8− DCs in skin DLNs correlates with the recruitment of circulating langerin+ DCs to the dermis
Langerin+CD8− DCs in the skin DLNs can derive either from the skin or from blood circulating precursors that enter directly into the LN from the blood. Because LCs are absent when langerin+CD8− DCs reappear in the skin DLNs, we looked for langerin+ DCs in the dermis. We detected langerin+ DCs in the dermis long before LCs reappeared in the skin (Fig. 2, A and B). Dermal langerin+ DCs appeared around day 5 and increased up to day 10 after DT injection, after which they remained stable in the dermis, reaching 20% of total dermal langerin+ cells, 10% of total dermal DCs, and 2% of total dermal leukocytes.
Figure 2.

Kinetics of langerin+ repopulation in the dermis. Kinetics of reappearance of langerin+ DCs was followed by flow cytometry in the dermis. (A) Dots plots show langerin expression (EGFP) among live (DAPI−) CD45+I-Ab+ cells in the dermis at different times after DT treatment. (B) Graph in log scale presents the percentage of I-Ab+ langerin+ DCs among total dermal DAPI−CD45+ cells (•) versus epidermal LCs (▪) at different times after DT treatment. (right) Histogram shows the percentage of I-Ab+ langerin+ DCs among DAPI−CD45+ cells before and after DT treatment (DAY14). Mean values (± the SD) are shown. Representative data from three independent experiments are shown. Each experiment included at least three separately analyzed mice per time point; error bars represent the SD between the results obtained from each of the three mice.
Dual origin of langerin+CD8− DCs in parabiotic animals
The aforementioned results suggest that a population of langerin+ DCs, independent of LCs, resides in the dermis and in the skin DLNs. To directly examine the origin of skin DLN langerin+ DCs, we used parabiotic mice in which C57BL/6 CD45.2+ mice were paired with congenic CD45.1+ mice so that they shared the same blood circulation, but separate organs, for prolonged periods of time. As previously described, epidermal LCs remained of recipient origin >6 mo after parabiosis, which is consistent with their origin from skin-resident precursors (Fig. 3, A and B). Therefore, the presence of CD45.1+langerin+ DCs in the dermis of the CD45.2+ parabiont should establish their independence from LCs and their recruitment from the blood to the dermis in the steady state. As shown in Fig. 3, we were able to detect blood-derived langerin+CD8− DCs in the skin (Fig. 3, A and B) and the skin DLNs (Fig. 3, C and D) of each parabiont. Total dermal DC chimerism was low, but significant, compared with LC chimerism as previously described (27), reflecting either prolonged or partial self-replenishment (27) or, as recently suggested, an uneven distribution of DC precursors (26). Nevertheless, the presence of donor-derived dermal langerin+ DCs suggests that, in contrast to LC-derived dermal DCs, dermal langerin+ DCs can originate from blood-derived precursors in the steady state. Interestingly, the chimerism of langerin+ DCs was lower than langerin− DCs. Because host langerin+ DCs consist of a mixture of dermal langerin+ DCs and migrating LCs, it is likely that the lower chimerism of langerin+ compared with langerin− DCs reflects the contribution of migrating LCs. Therefore, to better reveal the recruitment of blood-derived langerin+ cells to the skin and to the skin DLNs, we parabiosed Langerin-DTR/EGFP CD45.2+ C57BL/6 animals with CD45.1+ C57BL/6 and followed the repopulation of langerin+ DCs in the CD45.2+ parabionts at different times after DT treatment. 2 d after DT injection, langerin+ DCs were eliminated from the skin and the skin DLNs of the CD45.2+ Langerin-DTR/EGFP parabionts (unpublished data). 18 d after DT administration, a time point where LCs are absent (Fig. 3 E, top), the percentage of donor CD45.1+langerin+ DCs increased to the level of langerin− DCs in the dermis (Fig. 3, E and F, bottom) and in the skin DLNs (Fig. 3, G and H) of the CD45.2+ Langerin-DTR/EGFP parabionts. These results establish that in the steady state, circulating langerin+ DCs are recruited to the dermis and to the skin DLNs independently of LCs.
Figure 3.

Homeostasis of langerin+ DCs in parabiotic mice. Each parabiotic pair consisted of one CD45.1+ C57BL/6 mouse with either a CD45.2+ C57BL/6 mouse (A–D) or CD45.2+ Langerin-DTR/EGFP C57BL/6 mouse for DT depletion experiments (E–H). (A–D) Skin and skin DLN chimerism was examined by flow cytometry in untreated CD45.2+ parabiont of a CD45.1/CD45.2 parabiotic pair at 2 mo after the initiation of parabiosis. (A) Dot plots show the chimerism of gated LCs (CD45+I-Ab+ langerin+) in the epidermis (top) and the chimerism of gated langerin− and langerin+ DCs (CD45+I-Ab+) in the dermis (bottom). (B) Bar graphs show the percentages of donor (CD45.1+)-derived cells among gated LCs (top) and langerin− or langerin+ dermal DCs (bottom). (C) Dot plots show the percentage of host and donor cells among gated CD11c+ I-Ab+ DCs, langerin−CD8− DCs and langerin+CD8− DCs. (D) Mean values of langerin− versus langerin+ DCs chimerism in CD45.2+ parabionts. Similar data are found in the skin and skin DLNs of untreated CD45.2 Langerin-DTR/EGFP parabionts (unpublished data). Representative data from 5 separate experiments are shown. Each experiment included two to four mice. Skin was separately analyzed for each mouse, whereas skin DLNs were pooled (pool of two mice); error bars represent the SD between the results obtained from each mouse (skin) or from pooled DLNs (skin DLN). (E–H) Similar data to that found in A–D was obtained from DT-treated CD45.2+ Langerin-DTR/EGFP parabiont of a CD45.1/CD45.2 Langerin-DTR/EGFP parabiotic pair. Data presented are from 18 d after DT treatment, a time when LCs are still depleted (top), whereas langerin+ dermal DCs are recovered. Representative data from two separate experiments are shown. Each experiment included two separately analyzed mice, except for the skin DLNs that were pooled. For the skin, error bars represent the SD between the results obtained from each of the two mice.
The origin of langerin+CD8− DCs in congenic BM chimeras
Congenic BM chimeric mice provide another model in which the CD45 congenic allele can be used to distinguish LCs from blood-derived DCs in the skin DLNs. The advantage of this model is that the majority of circulating leukocytes express the congenic allele, whereas in parabiotic mice, no more than 50% of leukocytes express the congenic allele (26). To generate congenic BM chimeras, C57BL/6 CD45.2+ mice were lethally irradiated and reconstituted with BM cells isolated from C57BL/6 CD45.1+ mice. As previously described, a few weeks after transplant, most blood leukocytes originate from donor BM–derived precursors and express CD45.1, whereas the majority of epidermal LCs are maintained by radioresistant precursors, and remain of host (CD45.2+) origin throughout life, in the absence of skin injury (24) (Fig. 4 B). Using flow cytometry analysis of skin samples isolated at different times after transplant, we found that a substantial population of langerin+ DCs in the dermis of BM chimeras expressed donor CD45.1 antigens, suggesting that they derived from donor BM-derived circulating precursors and not from the host remaining CD45.2+ LCs (Fig. 4, A and B). Consistent with our results (Fig. 2), this population corresponded to ∼10% of dermal MHC class II+ cells. Blood-derived CD45.1+langerin+ DCs in the dermis were also MHC class II+, CD11cint, CD11b+, CX3CR1−, CD8−, a phenotype similar to that expressed by classical LCs with the exception of the langerin levels, but different from that of dermal langerin− DCs (Fig. 4 C). We also discovered that, in the skin DLNs, an increased proportion of langerin+CD8− DCs express CD45.1 (60–70%), and therefore are derived from blood-precursors and not from epidermal LCs (Fig. 4, D and E). Blood- and epidermal-derived langerin+CD8− DCs were indistinguishable phenotypically and expressed higher levels of MHC class II and langerin and lower levels of CD11c compared with langerinloCD8+ DCs (Fig. 4 F). Interestingly, langerin expression formed a smear on dermal blood–derived langerin+ DCs, which may suggest that the langerin receptor was being acquired upon entry in the dermis and up-regulated during the migration to the skin DLNs. These results are consistent with the data from a companion paper showing that blood-derived langerin+ DCs that emigrate from skin explants have similar langerin levels compared with LCs (28). Donor-derived langerin+ DCs were visualized in situ by confocal microscopy analysis of skin sections isolated from (CD45.2+Langerin-EGFP BM→CD45.1+C57BL/6) or the reverse (CD45.1+C57BL/6 BM→CD45.2+Langerin-EGFP) mice chimeras (Fig. 4 G). Donor-derived langerin+ DCs appear scattered in the dermis, without any conspicuous localization near the hair follicle. Donor-derived CD45.2+ MHC class II+ Langerin-EGFP+ DCs were also purified by FACS and stained for langerin on cytospins (Fig. 4 H). Altogether, our results suggest that, although undistinguishable phenotypically, blood- and epidermal-derived langerin+CD8− DCs coexist in the skin DLNs of BM chimeric mice.
Figure 4.

Homeostasis of langerin+ DCs after congenic BM transplantation. Lethally irradiated host CD45.2+C57BL/6 were transplanted with congenic donor CD45.1+C57BL/6 BM (CD45.1 into CD45.2 chimeric mice). 2 mo after reconstitution, host versus donor chimerism was measured in the langerin+ DC population in the skin (A and B) and in skin DLNs (D and E). (A) Dot plots show I-Ab and langerin expression among dermal CD45+ cells, and the host versus donor chimerism of langerin+ dermal DCs (CD45+I-Ab+). (B) Mean values of langerin+ DCs chimerism in the dermis. LC (CD45+I-Ab+ langerin+) chimerism in the epidermis is shown as control. (C) Expression of different markers was compared by flow cytometry between host remaining (CD45.2+I-Ab+ langerin+) LCs and donor-derived langerin− or langerin+ dermal DCs. (D) Dot plots show CD8 versus langerin expression among CD11c+I-Ab+ cells in the skin DLNs and host versus donor chimerism of CD8− langerin+ DCs. (E) Mean values of langerin+ DC chimerism. (F) Phenotype of donor-derived CD8− DC langerin+, CD8+ DC langerin−, and host remaining LCs. (G) Back skin cross-sections isolated from CD45.2 Langerin-EGFP into CD45.1 chimeric mice (left) or the reverse CD45.1 into CD45.2 Langerin-EGFP chimeric mice (right) were stained with anti-langerin antibody. Nuclei were counterstained with DAPI. Bar, 50 μm. (H) Dermal cell suspensions were prepared from CD45.2 Langerin-EGFP into CD45.1 chimeric mice. Donor langerin+ dermal DCs were sorted on the criteria of CD45.2, I-Ab, and EGFP expression and harvested onto cytospin slides for langerin staining. Inset shows langerin staining control. Bar, 10 μm. Representative data from four independent experiments are shown. Each experiment included at least three separately analyzed mice; error bars represent the SD between the results obtained from each of the three mice.
Mechanisms that regulate the trafficking of langerin+CD8− DCs to the skin DLNs
The presence of blood-derived langerin+CD8− DCs in the skin DLNs always paralleled their presence in the skin. However, the percentage of blood (CD45.1+)-derived langerin+ DCs among total langerinhi DCs was always higher in the skin DLNs compared with the dermis (compare Fig. 4 B with Fig. 4 E), suggesting that these cells either transit more rapidly than migratory LCs, but do not accumulate in the skin, or that they can also be recruited to the skin DLNs directly from the blood. To address these questions, we reconstituted lethally irradiated CD45.1+C57BL/6 recipient mice with BM cells isolated from congenic CD45.2+ mice that lack L-selectin (CD62L KO) or the chemokine receptor CCR7 (CCR7 KO). L-selectin is constitutively expressed on most leukocytes and plays an important role in leukocyte homing into lymphoid tissue from the blood (29). CCR7 is a chemokine receptor that plays a critical role in the migration of DCs from the skin to the DLNs (30); therefore, absence of CCR7 should interfere with the migration of DCs from the skin to the skin DLNs. 2 mo after reconstitution, the percentage of CCR7 KO and CD62L KO circulating langerin+ DCs in the dermis was similar to their wild-type counterpart (Fig. 5, A and B). In contrast, the percentage of CCR7 KO langerin+CD8− DCs was markedly reduced compared with CCR7+/+ langerin+CD8− DCs in the skin DLNs (Fig. 5, C and D). No differences were observed between the percentages of CD62LKO versus CD62L+/+ langerin+CD8− DCs. These results suggest that circulating langerin+CD8− DCs reach the skin DLNs through the skin and not directly from the blood.
Figure 5.

Mechanisms involved in the recruitment to the dermis and the migration to the skin DLNs of dermal langerin+ DCs. (A–D) Host congenic CD45.1+C57BL/6 were lethally irradiated and transplanted with donor CD45.2+ C57BL/6 (Control), CCR7-deficient (CCR7 KO), or CD62L-deficient (CD62L KO) BM. After complete donor engraftment, the chimerism of total langerin+ cells in the dermis and in the skin DLNs was measured by flow cytometry. (A) Dot plots show CD45.1 (host) versus CD45.2 (donor) expression in langerin+ dermal DCs from control, CCR7KO, and CD62LKO chimeric mice. (B) Mean values (± the SD) of langerin+ dermal DCs chimerism. (C and D) Same as in A and B, but for CD8−langerin+ DCs in the skin DLNs of control, CCR7 KO, or CD62L KO groups. (E) Lethally irradiated host wild-type CD45.2+C57BL/6 (Control) or CD45.2+ P/E-selectin deficient (P/E-selectin KO) were lethally irradiated and transplanted with congenic CD45.1+C57BL/6 BM. After complete donor engraftment, the chimerism of total CD45+I-Ab+ langerin+ DCs in the dermis was measured by flow cytometry. Bar graph shows the chimerism of langerin+ dermal DCs in control group (black bars) versus P/E-selectin KO group (white bars) 2 mo after transplantation. Mean values (± the SD) are shown. (F) CD45.1+ C57BL/6 were lethally irradiated and reconstituted with a mixture of CD45.1+ WT BM and CD45.2+ BM isolated from WT, CCR2 KO, or CCR6 KO BM, as described in the Materials and methods. 2 mo after transplant, chimerism of total langerin+ DCs in the dermis was measured. Bar graph shows the mean value of the percentage of CD45.2+ cells among langerin+ and langerin− dermal DCs in control group (black bars), CCR2 KO group (white bars), and CCR6 KO group (gray bars). Mean values (± the SD) are shown. Representative data from two independent experiments are shown. Each experiment included at least three separately analyzed mice; error bars represent the SD between the results obtained from each of the three mice.
Mechanisms that regulate the recruitment of circulating langerin+ DCs to the dermis
Previous results suggest that circulating DC precursors roll continuously along noninflamed mouse dermal endothelium in vivo, an interaction shown to be strictly dependent on endothelial selectins (31). To examine if endothelial selectins play a role in the homeostasis of dermal langerin+ DCs, we reconstituted lethally irradiated C57BL/6 mice that lack both P- and E-selectins (P/E-selectin KO), or wild-type C57BL/6 controls with BM cells isolated from congenic CD45.1+C57BL/6 mice. 2 mo after reconstitution, we found that P/E-selectin KO have similar numbers of CD45.1+ circulating B cells, monocytes, and neutrophils compared with wild-type controls, suggesting that absence of P- or E-selectins did not affect BM engraftment (unpublished data). In contrast, the percentage of dermal langerin+ DCs in P/E-selectin KO mice was 30–50% lower than the percentage of dermal langerin+ DCs in wild-type mice (Fig. 5 E). Similar results were observed for dermal langerin− DCs (unpublished data).
We previously established that CCR2 chemokine ligands are required for the recruitment of circulating dermal DC precursors to the skin (27). We also established that CCR2 and CCR6 chemokine ligands are required for the recruitment of circulating LC precursors and the repopulation of LCs in inflamed skin (32, 33). To examine the role of these molecules in the recruitment of circulating donor langerin+ DC precursors to the dermis, we reconstituted lethally irradiated CD45.1+C57BL/6 mice with a 1:1 mixture of hematopoietic progenitors cells that consisted of CD45.2+ BM cells that either lacked CCR2 (CCR2 KO) or CCR6 (CCR6 KO) and wild-type CD45.1+ BM. 2 mo after reconstitution, we found that CCR2−/−, CCR6−/−, and wild-type CD45.1+ BM gave rise to similar numbers of circulating B cells and neutrophils, suggesting that the absence of CCR2 and CCR6 did not affect BM engraftment (unpublished data). In contrast, CCR2−/−CD45.2+CD115+ circulating monocytes represented 30–40% of circulating CD45.2+CCR6−/− or CD45.1+ wild-type monocytes (unpublished data), confirming that absence of CCR2 expression on hematopoietic progenitors affects monocyte repopulation in the blood (27, 34). We found that CCR2−/− dermal langerin+ DCs were reduced in the skin compared with their wild-type counterpart (Fig. 5 F), whereas CCR6 KO langerin+ dermal DCs were not affected (Fig. 5 F). Similar results were observed for dermal langerin− DCs (Fig. 5 F). These differences underline the essential role of CCR2 in the recruitment of dermal langerin+ DCs to the skin, whereas the role of CCR6 seems to be restricted to the repopulation of epidermal LCs.
Role of langerin+CD8− DCs in skin immunity
Our results reveal for the first time that circulating langerin+ DC precursors are recruited to the dermis in the steady state or after minor injuries (i.e., DT or radiation injuries). Our results also suggest that circulating langerin+ precursors that are recruited to the dermis do not establish in the epidermis, but emigrate to the skin DLNs. To examine if blood-derived dermal langerin+ DCs play a role in skin immunity, we sought to analyze whether they can capture, process, and present skin-derived antigen in the skin DLNs under steady-state conditions. To assess antigen presentation, we took advantage of the YAe monoclonal antibody that recognizes an MHC class II I-E–derived peptide presented in the context of MHC class II I-Ab molecules. We reconstituted lethally irradiated F1 (C57BL/6 I-Ab–/– x BALB/c I-Ed+) recipient mice with BM cells isolated from CD45.1+ C57BL/6 I-Ab+/+ Langerin-EGFP BM. In transplanted mice, only I-Ab+/+ donor cells, and not host residual LC I-Ab−/−, can present I-Ed–derived peptide recognized by YAe. In the skin of F1 (C57BL/6xBALB/c) animals, I-Ed is expressed by host radioresistant LCs (24) and some activated keratinocytes (35) (unpublished data). Therefore, donor I-Ab+/+ DCs should be able to present I-Ed–derived peptides and stain positive for YAe only if they can capture, process, and present skin-derived antigens. We found that, in the dermis, donor-derived circulating dermal langerin+ DCs stained positive for YAe, as did langerin− dermal DCs, establishing their capacity to capture and present exogenous skin-derived antigens (Fig. 6, A and B, top). Our results also show that blood-derived dermal langerin+ DCs that migrate to the skin DLNs also stain positive for YAe (Fig. 6, A and B, bottom). A higher percentage of blood-derived dermal langerin+ DCs were positive for YAe in the skin DLNs compared with the dermis (Fig. 6, A and B, bottom), suggesting that dermal-derived langerin+ DCs process and present skin-derived antigens en route to the skin DLNs. Similar results were obtained when corresponding purified dermal DC populations were examined on cytospins for langerin and YAe expression (Fig. 6 C).
Figure 6.

Phagocytic processing and migration capacities of blood-derived langerin+ DCs. Lethally irradiated F1 (C57BL/6 I-Ab−/−/BALB/c I-Ed+) mice were reconstituted with BM cells isolated from donor Langerin-EGFP CD45.1+ C57BL/6 I-Ab+ mice. (A, top) Overlaid histograms show YAe staining on gated host LCs (CD45.2+CD11c+Langerin-EGFP−), donor langerin+ dermal DCs (CD45.1CD11c+Langerin-EGFP+), and donor langerin− dermal DCs (CD45.2+CD11c+Langerin-EGFP−). LCs isolated from F1 (C57BL/6 I-Ab+/+/BALB/c I-Ed+) mice were used as controls. (A, bottom) YAe staining is shown for host LC-derived DCs (CD45.2+CD11c+Langerin-EGFP−), donor langerin+ DCs (CD45.1+CD11c+CD8a−Langerin-EGFP+), and donor langerin− DCs (CD45.2+CD11c+CD8−langerin−) in the skin DLNs. Corresponding YAe isotype control (blue) and WT F1-positive control (green) are shown. (B) Mean values (± the SD) of the percentage of YAe+ cells among donor langerin+ or langerin− DCs in the dermis and skin DLNs. Representative data from two independent experiments are shown. Each experiment included at least three separately analyzed mice; error bars represent the SD between the results obtained from each of the three mice. (C) Host LCs in the epidermis, donor langerin+, or langerin− DCs in the dermis, LC-derived DCs, or donor langerin+ or langerin− DCs in the skin DLNs were purified by cell sorter and stained on cytospin for YAe and langerin as described in Materials and methods. Insets show YAe isotype control for each tested DC population. Dashed white lines indicate where cells were spliced from different fields to present more cells within a panel. Bar, 10 μm.
DISCUSSION
Using three separate models to trace the turnover of circulating leukocytes, our study reveals the presence of a population of langerin+ DCs, independent of LCs, in the dermis and skin DLNs of mice in the steady state.
We used parabiotic mice to establish the constitutive recruitment of blood-derived langerin+ DCs to the dermis and their subsequent emigration to the skin DLNs. Parabiotic mice provide a useful model to study the physiological turnover of leukocytes (36). Although recent data suggest that circulating DC precursors do not reach complete equilibrium in parabionts (26), this model continues to be very useful to compare trafficking patterns among DC subsets. Parabiotic mice are particularly useful to distinguish LC-derived DCs from blood-derived dermal langerin+ DCs in the steady state because LCs fail to mix in the epidermis >18 mo after the parabiosis is established (24). Using the congenic CD45.1 marker to trace circulating leukocytes in the CD45.2 parabiont, our results reveal that blood-borne dermal langerin+ DCs are constitutively recruited to the dermis and the skin DLNs in the absence of overt injuries. Similar results are reported in a companion paper (28). In this study, the authors show that in congenic BM chimeric mice, langerin+ DCs express higher levels of CD45 compared with LCs and used the levels of CD45 expression to identify langerin+ DCs, independent of LCs, in the dermis of naive nontransplanted animals. Altogether, these results strongly support the recruitment of circulating langerin+ DCs, independent of LCs, to the dermis in the steady state.
Congenic BM chimeric mice provide another tool to distinguish LCs from blood-derived DCs, as LCs remain of host origin months after transplant despite complete donor-derived chimerism in the blood. In this model, blood-derived langerin+ DCs were also present in the dermis and accounted for 30% of total langerin+ dermal DCs. Surprisingly, the percentage of blood-derived langerin+ DCs is increased in the skin DLNs, reaching up to 60% of langerin+ DCs. The higher percentage of donor-derived langerin+ DCs in the skin DLNs, compared with dermal langerin+ DCs in congenic BM chimeras, is intriguing, and could be partially caused by a minor contribution of donor-derived LCs, which represent 5–10% of total LCs in areas of skin exposed to fight- or grooming-related injuries, such as the neck and the back skin. However, the constant percentage of donor-derived langerin+ in skin DLNs draining different parts of the skin, including those containing host LCs exclusively, argues against this hypothesis (unpublished data). Alternatively, the radiation-induced injuries that might occur in transplanted BM chimera mice may also affect the homeostasis of dermal langerin+ DCs and increase their recruitment to the skin DLNs. Another possibility is that blood-derived langerin+ DCs are recruited directly from the blood to the skin DLNs. Several lines of evidence argue against this possibility. First, the presence of langerin+CD8− DCs in the skin DLNs always paralleled their presence in the skin, with the latter reconstituting the dermis before the skin DLNs in depletion experiments. Second, their recruitment to the skin DLNs was independent of the L-selectin (CD62L), but required the chemokine receptor CCR7. CD62L is constitutively expressed on most leukocytes, including DC precursors, and plays a critical role in the first steps of leukocyte extravasation from the blood to the LN, allowing the rolling of blood leukocytes on high endothelial venules (HEVs) (29). CCR7 is the receptor for two structurally related chemokines, CCL19 and CCL21 (37). CCL19 and CCL21 are expressed by stromal cells within the lymphoid T cell zones, and CCL21 is expressed by HEVs and at lower levels by the lymphatic endothelium (38, 39). In mice lacking CCR7 (30) or CCR7 ligands (40), DCs fail to migrate from the skin to the T cell areas of the skin DLNs, providing the most compelling evidence for an essential role of CCR7 in this process. Altogether, these studies led to the current concept that migrating DCs express CCR7 and become chemotactically attracted to CCL21-expressing lymphatic vessels. The migrating DCs reach the subcapsular sinus of the DLNs and follow a gradient of CCL21 and CCL19 to move into the lymphoid T zone (38). Unlike all other chemokine receptors, CCR7 is resistant to ligand-induced down-regulation (41), which may explain how DCs can use CCR7 to perform all the critical steps of migration. The failure of blood-derived langerin+ DCs to enter the skin DLNs in the absence of CCR7, but not in the absence of CD62L, strongly suggests that these cells reach the skin DLNs through the skin lymphatics and not directly from the HEVs. However, CD8+ DCs, a population thought to be recruited to the LN through the HEVs, were also able to reach the skin DLNs in the absence of CD62L; however, in contrast to CD8− langerin+ DCs, the lack of CCR7 did not affect their recruitment to the skin DLNs (unpublished data). These results were surprising, as we expected that, similar to naive T cells (39), expression of CD62L and CCR7 would both be required for efficient homing to the HEVs. However, these data support previous findings, showing that CD62L is not required for the migration of spleen DCs on resting or activated endothelial cells in vitro (42), and suggest that other molecules might orchestrate the recruitment of blood DCs through the HEVs. A potential candidate might be “chemerin,” a proteolytically regulated chemoattractant protein expressed at high levels by HEVs and shown to chemoattract in vitro ChemR23+ DCs, which include human blood DCs, but not LCs (43, 44).
We also examined the mechanisms that regulate the recruitment of blood-derived langerin+ DCs to the dermis. Our data suggest that blood-derived langerin+ DCs use E- and P-selectin to enter the skin in the absence of skin injuries. These data are consistent with a previous study showing that human CD34+-derived DCs injected into immunocompromised mice roll continuously along noninflamed mouse dermal endothelium in an E- and P-selectin–dependent manner (31). Our data also show that CCR2 controls the recruitment of blood-derived langerin+ DCs to the quiescent dermis. Expression of CCR2 (45) on circulating leukocytes was shown to be required for the repopulation of LCs (32, 46) and dermal langerin− DCs in inflamed skin (27). Our results represent the first indication for a role of CCR2 in cutaneous DC homeostasis in the absence of overt skin inflammation. However, it remains possible that the transplant model used to reveal the role of CCR2 might lead to x ray–induced skin injuries, triggering the release of CCR2 ligands and the recruitment of circulating DCs that will not be encountered in the steady state. CCR2 also plays a critical role in the release of BM monocytes into the bloodstream (34), and mice reconstituted with CCR2−/− BM have a decreased number of circulating monocytes (27). Therefore, it is possible that the decrease of CCR2−/− blood-derived langerin+ DCs in the dermis reflects a reduced capacity for CCR2−/− monocytes to reach the peripheral blood and differentiate into dermal DCs in the quiescent skin. These results support the recent findings that monocytes participate in DC homeostasis in peripheral tissues, but not in lymphoid organs (47), and the role of CCR2 in the development of nonlymphoid DC populations other than skin is currently being analyzed in the laboratory.
Consistent with previous studies, our results show that after systemic depletion of langerin+ cells in vivo, the repopulation of dermal and skin DLN langerin+ DCs precedes the repopulation of epidermal LCs (18, 23). We also demonstrate that dermal langerin+ DCs, independent of LCs, participate in the repopulation of the langerin+ DC pool in the skin DLNs, whereas the epidermis remains empty of LCs. It is intriguing that langerin+ dermal DCs migrate to the skin DLNs before seeding an epidermis depleted of LCs, suggesting that blood-derived langerin+ DCs cannot differentiate into LCs in vivo. Another possibility is that the toxin used to deplete langerin+ cells leads to inflammatory signals that trigger the migration of blood-derived dermal langerin+ DCs to the skin DLNs before they can reach the epidermis. This is unlikely because we have shown that circulating LC precursors can seed an inflamed epidermis and differentiate into LCs (33). In addition, we and others have not detected signs of skin inflammation or injuries after DT injection ([18] and unpublished data). A more likely explanation is that in vivo conditional LC ablation with DT frees available LC niches, but fails to induce epidermal injury signals, precluding the recruitment of blood-derived precursors to the epidermis. It will be interesting to examine if induction of inflammatory cytokines and chemokines in the epidermis after DT treatment accelerates the recruitment of dermal langerin+ DCs to the epidermis. If this is true, these data, together with our previous data showing that monocytes are the precursors of LCs in inflamed skin, will support the hypothesis that monocytes are constitutively recruited to the dermis where they differentiate into langerin+ DCs dedicated to survey the epidermis or the dermal–epidermal junction.
The acquisition of the langerin receptor on skin-infiltrating monocytes or a committed DC precursor might represent the signature of a “LC-like” differentiation program induced by environmental factors such as TGFβ, for example (48). Supporting this hypothesis is our finding that dermal langerin+ DCs seem to progressively acquire the LC marker langerin while losing the monocyte marker CX3CR1, a marker that is also absent on LCs, but partially expressed by dermal langerin− DCs (Fig. 4 C). Whether dermal langerin+ and dermal langerin− DCs represent the progeny of a precursor that follows a separate differentiation pathway, with the dermal langerin+ DCs being an intermediate precursor to LCs, is plausible, but difficult to test because of the limitation of an experimental system dealing with a low number of cells. More information will be needed to compare the nature of the antigen cargo displayed by these different sources of antigen-presenting cells in the skin DLNs to assess the immune relevance of each of these DC populations.
Our study also reports a striking difference between the homeostasis and trafficking pattern of LCs and dermal langerin+ DCs. After conditional ablation of langerin+ DCs in vivo, we were surprised to discover that dermal langerin+ DCs reappear in 5 d and start to emigrate to the skin DLNs and repopulate the langerin+ DC pool immediately after, whereas LCs remained absent from the epidermis for >2 wk after their elimination. The constitutive recruitment of blood-derived langerin+ DCs to the dermis, together with their rapid transit through the skin before reaching the skin DLNs, strongly support their role in skin immunosurveillance. This role is further supported by our findings showing that blood-derived dermal langerin+ DCs capture and process skin-derived antigens before emigrating to the T cell areas of the skin DLNs, where they present skin-derived peptides in the context of MHC class II molecules. These findings establish that, in addition to LCs and dermal langerin− DCs, an additional population of antigen-presenting cells may play a role in skin immunity. The expression of the langerin receptor on the surface of dermal DCs should allow the sampling of antigen patterns by LCs, but not by dermal langerin− DCs.
Our results reveal a novel pathway of cutaneous DC differentiation and homeostasis that add to the complexity of the DC network system developed to support skin immune responses (Fig. 7). We identified a population of circulating langerin+ DCs, independent of LCs, that constitutively patrols the dermis to capture and process skin antigens before emigrating to the skin DLNs, where they present skin-derived peptides–MHC complexes (Fig. 7). This study uncovers a previously unappreciated element of skin immunosurveillance that is likely to impact the design of vaccine strategies.
Figure 7.

Origin of langerin+ DC in mice. This diagram illustrates a hypothetical view on cutaneous DC differentiation pathways in mice in the steady state. Epidermal LCs are maintained by radioresistant hematopoietic progenitors that have taken residence in the skin before birth, whereas the majority of dermal DCs derive from circulating DC precursors. Our results now suggest that circulating blood-derived CCR2+ DCs are constitutively recruited to the dermis. E- and/or P-selectin direct this recruitment in transplanted animals, and studies are conducted in the laboratory to examine if this is also true in nontransplanted mice. In response to cutaneous factors, CCR2+ DCs differentiate into dermal langerin+ DCs, but might also be able to differentiate into dermal langerin−CX3CR1+/− DCs. Dermal langerin+ DCs transit, but do not accumulate in the dermis, where they capture skin antigens, before emigrating to the skin DLNs to present skin-derived peptides in the context of MHC molecules. Both LCs and dermal DCs require CCR7 to migrate to the T cells area of skin DLNs. In the skin DLNs, dermal langerin+ DCs are undistinguishable from LC-derived DCs, and they are characterized as CD8−langerin+CD11clo, but differ from blood-derived CD8+ langerinloCD11hi DCs. Whether CCR2+ blood precursors that give rise to dermal langerin+ DCs and contribute to CD8−langerin+ CD11clo in the skin DLNs represent a committed DC precursor or a circulating monocyte is currently under study in the laboratory.
MATERIALS AND METHODS
Mice.
BALB/c, C57BL/6 (CD45.2+), and congenic C57BL/6 CD45.1+ mice 5–8 wk of age were purchased from The Jackson Laboratory. Langerin-EGFP, Langerin-DTR/EGFP mice were generated as previously described (18). CCR2-deficient C57BL/6 (45) and CCR6-deficient C57BL/6 (49), CD62L−/− (50) and CCR7−/− (30) (a gift from J. Lowe, Cleveland Clinic, Cleveland, OH), and P/E-selectin–deficient (P/E−/−) mice (51) (a gift from P. Frenette, Mount Sinai School of Medicine, New York, NY) were bred and maintained at our animal facility (Mount Sinai School of Medicine, New York, NY). CD45.1+ Langerin-EGFP mice were generated by crossing of congenic C57BL/6 CD45.1+ with CD45.2+ Langerin-EGFP. C57BL/6 BALB/c F1 and I-Ab−/−C57BL/6 BALB/c F1 were generated by breeding BALB/c with C57BL/6 or I-Ab−/−C57BL/6 (The Jackson Laboratory), respectively. All mice used for experiments were between 8 and 12 wk of age. Parabiotic mice were generated as previously described (36). Pairs of parabiotic mice consisted of congenic C57BL/6 CD45.1+ mice linked to either C57BL/6 CD45.2+ or Langerin-DTR/EGFP C57BL/6 CD45.2+ mice. Parabiotic mice were killed ∼2 mo after initiation of parabiosis and subjected to tissue analysis. To confirm efficient blood mixing in parabiotic mice, the percentage of CD45.1+ and CD45.2+ cells among blood leukocytes was analyzed in each animal. All animal protocols were approved by the Institutional Committee on Animal Welfare of the Mount Sinai Medical School.
Flow cytometry and cell sorting.
Multiparameter analyses of stained cell suspensions were made on an LSR II flow cytometer (Becton Dickinson) and were analyzed with FlowJo software (Tree Star, Inc.). Fluorochrome- or biotin-conjugated monoclonal antibodies specific to mouse B220 (RA3-6B2), CD8a (53–6.7), I-Ab (AF6-120.1), IA/IE (M5/114.15.2), CD11b (M1/70), CD11c (N418), CD45 (30F11), CD45.1 (A20), CD45.2 (104), CD115 (AFS98), Gr-1Ly6C (1A8), Gr-1Ly6C/G (RB6-8C5), CD3 (17A2), CD4 (clone L3T4), anti–mouse Eα52-68 peptide bound to the I-Ab/MHC Class II (YAe), the corresponding isotype controls, and the secondary reagents (allophycocyanin, peridinin chlorophyll protein, and phycoerythrin–indotricarbocyanine–conjugated streptavidin) were purchased either from BD Biosciences or eBioscience. For YAe staining, naive untreated cells from C57BL/6 mice that lack I-E molecules were used as negative controls, and cells from C57BL/6 BALB/c F1 mice were used as positive controls. Anti–mouse CD16-32 (BD Biosciences) and biotinylated mouse IgG2b isotype control (BD Biosciences) were used to block Fc III/II receptors and as a YAe isotype control antibody, respectively. F4/80 (A3-1) was purchased from Serotec. Polyclonal antibody to langerin was purchased from Santa Cruz Biotechnology. Intracellular staining against langerin was performed with the BD Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's protocol. For cytospin immunostaining, stained cell suspensions were sorted using an inFlux cell sorter (Cytopeia, Inc.) and achieve 97–99% purity.
Transplantation of BM cells.
8-wk-old recipient CD45.2+ or CD45.1+ C57BL/6 mice were lethally irradiated with 1,200 rads, delivered in 2 doses of 600 rads each, 3 h apart, and were injected i.v. with 106 BM cells obtained from congenic CD45.1+ or CD45.2+ C57BL/6 adult mice, respectively. To address the role of CCR2 and CCR6 in dermal DC langerin+/− homeostasis, lethally irradiated CD45.1+ C57BL/6 mice were reconstituted with a 1:1 mixture of WT CD45.1+ BM cells and CD45.2+ BM isolated from WT, CCR2 KO, or CCR6 KO mutant mice. In all transplanted mice, levels of blood donor chimerism were analyzed by measuring the percentage of donor cells among total B220+ B cells, Ly6C/G+CD115− granulocytes, and CD115+ monocytes in the blood 3 wk after transplantation.
Preparation of epidermal and dermal cell suspension.
Skin cell suspensions were isolated as previously described (24) and were analyzed by flow cytometry. In brief, mouse ears were split in two parts (dorsal and ventral) and incubated for 60 min in PBS containing 0.5% trypsin with 5 mM EDTA (Invitrogen) to allow for separation of dermal and epidermal sheets. Epidermal and dermal sheets were then cut into small pieces and incubated for 2.5 h in collagenase D (1.6 mg/ml, working activity of 226 U/mg; Worthington) to obtain homogeneous cell suspension. The analysis of hematopoietic cell populations present in skin cell suspensions was assessed by flow cytometry by gating on DAPI−CD45+ cells.
Immunofluorescence analysis of mouse skin.
For staining of frozen sections, skin samples or skin DLNs were first fixed for 2 h in PBS 4% paraformaldehyde containing 20% sucrose to preserve EGFP fluorescence and were rinsed in PBS before freezing in optimum cutting temperature compound. 8-μm-thick skin sections were prepared and stained with anti-langerin (polyclonal goat IgG; Santa Cruz Biotechnology) for 1 h, washed in PBS, and incubated with secondary reagent donkey anti–goat Cy3 (Jackson Immunoresearch Laboratories). Skin DLN sections were first stained with anti-langerin (polyclonal goat IgG), followed by donkey anti–goat Cy3, and were stained after with biotinylated anti-B220 (RA3-6B2; BD Biosciences) revealed with streptavidin-Cy5 (Vector Laboratories). Slides were mounted with Vectashield containing DAPI (4,6-diamidino-2-phenylindole; Vector Laboratories). Images were acquired with a laser scanning confocal microscope (TCS-SP; Leica) and analyzed with Image J software (National Institutes of Health).
Immunofluorescence analysis of cytospins.
Cytospins were prepared from 5–10,000 sorted cells, air-dried, and fixed in 3% paraformaldehyde. Slides were incubated with 0.8 μg/ml anti-langerin goat polyclonal antibody or corresponding control (goat IgG; Sigma-Aldrich). For YAe staining, after blocking with 5 μg/ml mouse IgG2b (eBioscience) and 2.5 μg/ml anti-CD16/32 antibodies (BD Biosciences), slides were incubated with 2.5 μg/ml anti-YAe biotinylated antibody (eBioscience) and 0.8 μg/ml anti-langerin goat polyclonal antibody or isotype controls. Secondary detection of YAe was achieved using biotinyl-tyramide amplification (kit NEL700A; PerkinElmer) combined with 1 μg/ml Cy5-conjugated streptavidin (Jackson Immunoresearch Laboratories) and 1 μg/ml Cy3-conjugated donkey anti–goat (Jackson Immunoresearch Laboratories). GFP fluorescence was usually not detectable in these preparations. Images were acquired using an Axiophot microscope at 40× magnification (Carl Zeiss, Inc.).
Online supplemental material.
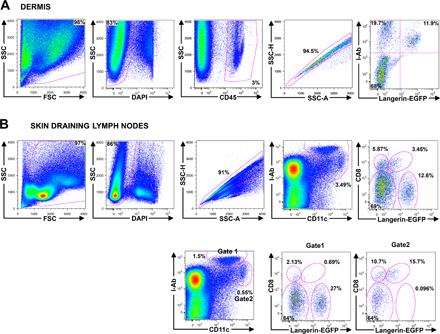
Fig. S1 depicts the gating strategy used in the dermis and in the skin DLNs to analyze all DC subsets by flow cytometry. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20071733/DC1.
Supplemental Material
Acknowledgments
The authors would like to thank Drs. J. Donovan and Y. Kirkorian for their review of the manuscript.
This work was supported by grants from the National Institutes of Health (RO1-CA112100), the Leukemia and Lymphoma Society (FG, 3220-08), the Leukaemia Research Fund Bennett Senior Fellowship (M.P. Collin), and the Amy Strelzer Manasevit Research program (M. Bogunovic).
The authors have no conflicting financial interests.
Abbreviations used: EGFP, enhanced GFP; DLN, draining LN; DT, diphtheria toxin; DTR, DT receptor; HEV, high endothelial venule; LC, Langerhans cell.
A. Kissenpfennig's present address is Infection and Immunity Group, Center for Cancer Research and Cell Biology, School of Biomedical Sciences, Queen's University, Belfast, Northern Ireland.
References
- 1.Figdor, C.G., Y. van Kooyk, and G.J. Adema. 2002. C-type lectin receptors on dendritic cells and Langerhans cells. Nat. Rev. Immunol. 2:77–84. [DOI] [PubMed] [Google Scholar]
- 2.Valladeau, J., O. Ravel, C. Dezutter-Dambuyant, K. Moore, M. Kleijmeer, Y. Liu, V. Duvert-Frances, C. Vincent, D. Schmitt, J. Davoust, et al. 2000. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 12:71–81. [DOI] [PubMed] [Google Scholar]
- 3.Valladeau, J., V. Clair-Moninot, C. Dezutter-Dambuyant, J.J. Pin, A. Kissenpfennig, M.G. Mattei, S. Ait-Yahia, E.E. Bates, B. Malissen, F. Koch, et al. 2002. Identification of mouse langerin/CD207 in Langerhans cells and some dendritic cells of lymphoid tissues. J. Immunol. 168:782–792. [DOI] [PubMed] [Google Scholar]
- 4.de Witte, L., A. Nabatov, M. Pion, D. Fluitsma, M.A. de Jong, T. de Gruijl, V. Piguet, Y. van Kooyk, and T.B. Geijtenbeek. 2007. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat. Med. 13:367–371. [DOI] [PubMed] [Google Scholar]
- 5.Turville, S.G., P.U. Cameron, A. Handley, G. Lin, S. Pohlmann, R.W. Doms, and A.L. Cunningham. 2002. Diversity of receptors binding HIV on dendritic cell subsets. Nat. Immunol. 3:975–983. [DOI] [PubMed] [Google Scholar]
- 6.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 7.Cerio, R., C.E. Griffiths, K.D. Cooper, B.J. Nickoloff, and J.T. Headington. 1989. Characterization of factor XIIIa positive dermal dendritic cells in normal and inflamed skin. Br. J. Dermatol. 121:421–431. [DOI] [PubMed] [Google Scholar]
- 8.Nestle, F.O., L.A. Turka, and B.J. Nickoloff. 1994. Characterization of dermal dendritic cells in psoriasis. Autostimulation of T lymphocytes and induction of Th1 type cytokines. J. Clin. Invest. 94:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenz, A., M. Heine, G. Schuler, and N. Romani. 1993. Human and murine dermis contain dendritic cells. Isolation by means of a novel method and phenotypical and functional characterization. J. Clin. Invest. 92:2587–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dupasquier, M., P. Stoitzner, A. van Oudenaren, N. Romani, and P.J. Leenen. 2004. Macrophages and dendritic cells constitute a major subpopulation of cells in the mouse dermis. J. Invest. Dermatol. 123:876–879. [DOI] [PubMed] [Google Scholar]
- 11.Larsen, C.P., R.M. Steinman, M. Witmer-Pack, D.F. Hankins, P.J. Morris, and J.M. Austyn. 1990. Migration and maturation of Langerhans cells in skin transplants and explants. J. Exp. Med. 172:1483–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lukas, M., H. Stossel, L. Hefel, S. Imamura, P. Fritsch, N.T. Sepp, G. Schuler, and N. Romani. 1996. Human cutaneous dendritic cells migrate through dermal lymphatic vessels in a skin organ culture model. J. Invest. Dermatol. 106:1293–1299. [DOI] [PubMed] [Google Scholar]
- 13.Weinlich, G., M. Heine, H. Stossel, M. Zanella, P. Stoitzner, U. Ortner, J. Smolle, F. Koch, N.T. Sepp, G. Schuler, and N. Romani. 1998. Entry into afferent lymphatics and maturation in situ of migrating murine cutaneous dendritic cells. J. Invest. Dermatol. 110:441–448. [DOI] [PubMed] [Google Scholar]
- 14.Stoitzner, P., S. Holzmann, A.D. McLellan, L. Ivarsson, H. Stossel, M. Kapp, U. Kammerer, P. Douillard, E. Kampgen, F. Koch, et al. 2003. Visualization and characterization of migratory Langerhans cells in murine skin and lymph nodes by antibodies against Langerin/CD207. J. Invest. Dermatol. 120:266–274. [DOI] [PubMed] [Google Scholar]
- 15.Shortman, K., and Y.J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 16.Henri, S., D. Vremec, A. Kamath, J. Waithman, S. Williams, C. Benoist, K. Burnham, S. Saeland, E. Handman, and K. Shortman. 2001. The dendritic cell populations of mouse lymph nodes. J. Immunol. 167:741–748. [DOI] [PubMed] [Google Scholar]
- 17.Takahara, K., Y. Omatsu, Y. Yashima, Y. Maeda, S. Tanaka, T. Iyoda, B.E. Clausen, K. Matsubara, J. Letterio, R.M. Steinman, et al. 2002. Identification and expression of mouse Langerin (CD207) in dendritic cells. Int. Immunol. 14:433–444. [DOI] [PubMed] [Google Scholar]
- 18.Kissenpfennig, A., S. Henri, B. Dubois, C. Laplace-Builhe, P. Perrin, N. Romani, C.H. Tripp, P. Douillard, L. Leserman, D. Kaiserlian, et al. 2005. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 22:643–654. [DOI] [PubMed] [Google Scholar]
- 19.Douillard, P., P. Stoitzner, C.H. Tripp, V. Clair-Moninot, S. Ait-Yahia, A.D. McLellan, A. Eggert, N. Romani, and S. Saeland. 2005. Mouse lymphoid tissue contains distinct subsets of langerin/CD207 dendritic cells, only one of which represents epidermal-derived Langerhans cells. J. Invest. Dermatol. 125:983–994. [DOI] [PubMed] [Google Scholar]
- 20.Sung, S.S., S.M. Fu, C.E. Rose Jr., F. Gaskin, S.T. Ju, and S.R. Beaty. 2006. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 176:2161–2172. [DOI] [PubMed] [Google Scholar]
- 21.Flores-Langarica, A., S. Meza-Perez, J. Calderon-Amador, T. Estrada-Garcia, G. Macpherson, S. Lebecque, S. Saeland, R.M. Steinman, and L. Flores-Romo. 2005. Network of dendritic cells within the muscular layer of the mouse intestine. Proc. Natl. Acad. Sci. USA. 102:19039–19044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Randolph, G.J., V. Angeli, and M.A. Swartz. 2005. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 5:617–628. [DOI] [PubMed] [Google Scholar]
- 23.Bennett, C.L., E. van Rijn, S. Jung, K. Inaba, R.M. Steinman, M.L. Kapsenberg, and B.E. Clausen. 2005. Inducible ablation of mouse Langerhans cells diminishes but fails to abrogate contact hypersensitivity. J. Cell Biol. 169:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merad, M., M.G. Manz, H. Karsunky, A. Wagers, W. Peters, I. Charo, I.L. Weissman, J.G. Cyster, and E.G. Engleman. 2002. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat. Immunol. 3:1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamath, A.T., S. Henri, F. Battye, D.F. Tough, and K. Shortman. 2002. Developmental kinetics and lifespan of dendritic cells in mouse lymphoid organs. Blood. 100:1734–1741. [PubMed] [Google Scholar]
- 26.Liu, K., C. Waskow, X. Liu, K. Yao, J. Hoh, and M. Nussenzweig. 2007. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat. Immunol. 8:578–583. [DOI] [PubMed] [Google Scholar]
- 27.Bogunovic, M., F. Ginhoux, A. Wagers, M. Loubeau, L.M. Isola, L. Lubrano, V. Najfeld, R.G. Phelps, C. Grosskreutz, E. Scigliano, et al. 2006. Identification of a radio-resistant and cycling dermal dendritic cell population in mice and men. J. Exp. Med. 203:2627–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poulin, L.F., S. Henri, B. de Bovis, E. Devilard, A. Kissenpfennig, and B. Malissen. 2007. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J. Exp. Med. 204:3119 –3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butcher, E.C. 1991. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 67:1033–1036. [DOI] [PubMed] [Google Scholar]
- 30.Forster, R., A. Schubel, D. Breitfeld, E. Kremmer, I. Renner-Muller, E. Wolf, and M. Lipp. 1999. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 99:23–33. [DOI] [PubMed] [Google Scholar]
- 31.Robert, C., R.C. Fuhlbrigge, J.D. Kieffer, S. Ayehunie, R.O. Hynes, G. Cheng, S. Grabbe, U.H. von Andrian, and T.S. Kupper. 1999. Interaction of dendritic cells with skin endothelium: a new perspective on immunosurveillance. J. Exp. Med. 189:627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merad, M., P. Hoffmann, E. Ranheim, S. Slaymaker, M.G. Manz, S.A. Lira, I. Charo, D.N. Cook, I.L. Weissman, S. Strober, and E.G. Engleman. 2004. Depletion of host Langerhans cells before transplantation of donor alloreactive T cells prevents skin graft-versus-host disease. Nat. Med. 10:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ginhoux, F., F. Tacke, V. Angeli, M. Bogunovic, M. Loubeau, X.M. Dai, E.R. Stanley, G.J. Randolph, and M. Merad. 2006. Langerhans cells arise from monocytes in vivo. Nat. Immunol. 7:265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serbina, N.V., and E.G.P. Am. 2006. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 7:311–317. [DOI] [PubMed] [Google Scholar]
- 35.Breathnach, S.M., and S.I. Katz. 1983. Keratinocytes synthesize Ia antigen in acute cutaneous graft-vs-host disease. J. Immunol. 131:2741–2745. [PubMed] [Google Scholar]
- 36.Wright, D.E., A.J. Wagers, A.P. Gulati, F.L. Johnson, and I.L. Weissman. 2001. Physiological migration of hematopoietic stem and progenitor cells. Science. 294:1933–1936. [DOI] [PubMed] [Google Scholar]
- 37.Zlotnik, A., and O. Yoshie. 2000. Chemokines: a new classification system and their role in immunity. Immunity. 12:121–127. [DOI] [PubMed] [Google Scholar]
- 38.Cyster, J.G. 2000. Leukocyte migration: scent of the T zone. Curr. Biol. 10:R30–R33. [DOI] [PubMed] [Google Scholar]
- 39.Sallusto, F., and A. Lanzavecchia. 2000. Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol. Rev. 177:134–140. [DOI] [PubMed] [Google Scholar]
- 40.Gunn, M.D., S. Kyuwa, C. Tam, T. Kakiuchi, A. Matsuzawa, L.T. Williams, and H. Nakano. 1999. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 189:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sallusto, F., B. Palermo, D. Lenig, M. Miettinen, S. Matikainen, I. Julkunen, R. Forster, R. Burgstahler, M. Lipp, and A. Lanzavecchia. 1999. Distinct patterns and kinetics of chemokine production regulate dendritic cell function. Eur. J. Immunol. 29:1617–1625. [DOI] [PubMed] [Google Scholar]
- 42.Colvin, B.L., A.H. Lau, A.M. Schell, and A.W. Thomson. 2004. Disparate ability of murine CD8alpha- and CD8alpha+ dendritic cell subsets to traverse endothelium is not determined by differential CD11b expression. Immunology. 113:328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vermi, W., E. Riboldi, V. Wittamer, F. Gentili, W. Luini, S. Marrelli, A. Vecchi, J.D. Franssen, D. Communi, L. Massardi, et al. 2005. Role of ChemR23 in directing the migration of myeloid and plasmacytoid dendritic cells to lymphoid organs and inflamed skin. J. Exp. Med. 201:509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wittamer, V., J.D. Franssen, M. Vulcano, J.F. Mirjolet, E. Le Poul, I. Migeotte, S. Brezillon, R. Tyldesley, C. Blanpain, M. Detheux, et al. 2003. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 198:977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boring, L., J. Gosling, S.W. Chensue, S.L. Kunkel, R.V. Farese Jr., H.E. Broxmeyer, and I.F. Charo. 1997. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest. 100:2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merad, M., T. Sugie, E.G. Engleman, and L. Fong. 2002. In vivo manipulation of dendritic cells to induce therapeutic immunity. Blood. 99:1676–1682. [DOI] [PubMed] [Google Scholar]
- 47.Varol, C., L. Landsman, D.K. Fogg, L. Greenshtein, B. Gildor, R. Margalit, V. Kalchenko, F. Geissmann, and S. Jung. 2007. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J. Exp. Med. 204:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strobl, H., and W. Knapp. 1999. TGF-beta1 regulation of dendritic cells. Microbes Infect. 1:1283–1290. [DOI] [PubMed] [Google Scholar]
- 49.Cook, D.N., D.M. Prosser, R. Forster, J. Zhang, N.A. Kuklin, S.J. Abbondanzo, X.D. Niu, S.C. Chen, D.J. Manfra, M.T. Wiekowski, et al. 2000. CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity. 12:495–503. [DOI] [PubMed] [Google Scholar]
- 50.Xu, J., I.S. Grewal, G.P. Geba, and R.A. Flavell. 1996. Impaired primary T cell responses in L-selectin–deficient mice. J. Exp. Med. 183:589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frenette, P.S., T.N. Mayadas, H. Rayburn, R.O. Hynes, and D.D. Wagner. 1996. Susceptibility to infection and altered hematopoiesis in mice deficient in both P- and E-selectins. Cell. 84:563–574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}