

Abstract
Integrins are critical for hemostasis and thrombosis because they mediate both platelet adhesion and aggregation. Talin is an integrin-binding cytoplasmic adaptor that is a central organizer of focal adhesions, and loss of talin phenocopies integrin deletion in Drosophila. Here, we have examined the role of talin in mammalian integrin function in vivo by selectively disrupting the talin1 gene in mouse platelet precursor megakaryocytes. Talin null megakaryocytes produced circulating platelets that exhibited normal morphology yet manifested profoundly impaired hemostatic function. Specifically, platelet-specific deletion of talin1 led to spontaneous hemorrhage and pathological bleeding. Ex vivo and in vitro studies revealed that loss of talin1 resulted in dramatically impaired integrin αIIbβ3-mediated platelet aggregation and β1 integrin–mediated platelet adhesion. Furthermore, loss of talin1 strongly inhibited the activation of platelet β1 and β3 integrins in response to platelet agonists. These data establish that platelet talin plays a crucial role in hemostasis and provide the first proof that talin is required for the activation and function of mammalian α2β1 and αIIbβ3 integrins in vivo.
Thrombosis and hemostasis depend on platelet function. Upon disruption of vascular integrity, platelets adhere to sites of injury and aggregate, thereby preventing excessive bleeding (1). Stable platelet adhesion to the injured blood vessel and subsequent aggregation in turn depend on integrin adhesion receptors. This point is well illustrated in patients with genetic defects in integrin subunits αIIb or β3 that cause the bleeding disorder Glanzmann thrombasthenia due primarily to defective platelet aggregation or in animals lacking all (α2β1, α5β1, and α6β1) (2) or certain (α2β1) (3) platelet β1 integrins that manifest milder bleeding defects due to reduced platelet adhesion to vascular surfaces (3, 4).
The ability of platelets to increase integrin affinity (operationally defined as integrin activation) is critical for normal platelet function. Circulating platelets are usually in a resting state. Upon stimulation through agonist receptors, such as those for ADP, collagen, or thrombin, signaling events within the platelet lead to complex biological effects including activation of β1 and β3 integrins (5). Activated αIIbβ3 then binds plasma proteins such as fibrinogen, leading to platelet aggregation, whereas activation of β1 integrins leads to adhesion of platelets to vessel wall components such as collagen (1, 5). The molecular events that link agonist receptors to integrin activation are incompletely understood; however, experiments in cultured cells have indicated that this signaling results in increased association of the cytoplasmic protein talin with the integrin β subunit cytoplasmic domain, inducing an increase in integrin affinity via a long-range allosteric change in the integrin's conformation (6). The requirement of talin for integrin activation has been examined in vitro; however, its role in vivo remains to be determined.
Talin is a 270-kD protein composed of a 50-kD head domain and a 220-kD rod domain. It was identified in platelets where it comprises 3–8% of total platelet protein (7). The head domain contains binding sites for β1A, β1D, β2, β3, β5, and β7 (8) integrin subunits and for another membrane protein, layilin (9). The rod domain contains binding sites for vinculin and F-actin. Thus, talin serves as a critical link between integrins and the actin cytoskeleton (10). Furthermore, in invertebrates, talin is necessary for formation of the integrin-associated cytoplasmic protein complex that includes proteins such as paxillin, vinculin, integrin-linked kinase, PINCH, and parvin (11). Lack of talin phenocopies lack of integrins in Drosophila, probably due to disrupted linkage to the actin cytoskeleton (11, 12). There are two mammalian talin isoforms encoded by distinct genes: talin1 is expressed ubiquitously, and talin2 is highly expressed in brain and striated muscle (13). In mice, global deletion of talin1 results in embryonic lethality between embryonic days 8.5 and 9.5 due to defects in cell migration before and during gastrulation (14). Thus, based on studies in vitro and in invertebrates, talin is essential for the function of certain integrins and for integrin activation. To examine the role of talin in integrin function in vivo, we selectively deleted talin1 in platelets and megakaryocytes in mice and found that platelet talin1 is essential for hemostasis because it is required for the function and activation of platelet α2β1 and αIIbβ3 integrins.
RESULTS AND DISCUSSION
Platelet-specific deletion of talin1
Global genetic deletion of talin1 in mice is lethal by embryonic day 9 (14). To circumvent this early embryonic lethality, we deleted talin1 specifically in platelet precursor megakaryocytes by crossing mice harboring a floxed talin1 allele (Tln1) (Fig. 1 A) with platelet factor 4–Cre (PF4-Cre) mice that express Cre recombinase selectively in platelets and megakaryocytes (15). Mice homozygous for the floxed Tln1 allele and positive for the PF4-Cre transgene (Tln1 fl/fl Cre+) had slightly reduced platelet counts compared with Tln1 fl/fl Cre− littermates (Table S1, available at http://www.jem.org/cgi/content/full/jem.20071800/DC1) that were still in the normal range (16). SDS-PAGE analysis of platelet lysates revealed a selective loss in the band corresponding to talin in Tln1 fl/fl Cre+ platelets (Fig. 1 D). In addition, loss of talin was observed by immunofluorescence in CD41+ megakaryocytes from the bone marrow of Tln1 fl/fl Cre+ mice (Fig. 1 E). Deletion of talin in Tln1 fl/fl Cre+ mice was only detectable in platelets and megakaryocytes as CD41− bone marrow cells from Tln1 fl/fl Cre+ mice showed similar low levels of talin immunofluorescence as that from Tln1 fl/fl Cre− mice (not depicted), consistent with PF4-Cre–mediated recombination being selective for the megakaryocytic lineage as reported previously (15).
Figure 1.

Deletion of talin1 in platelets and megakaryocytes. (A) Scheme of the targeting strategy. Homologous recombination of the Tln1 conditional targeting vector into the Tln1 gene of embryonic stem cells introduced a loxP site (triangle) downstream of exon 4 and a floxed Neo cassette upstream of exon 1 to generate the targeted Neo allele (flN). Partial Cre-mediated recombination in vivo was used to delete only the floxed Neo cassette leaving floxed exons 1–4 (conditional allele, fl). In cells expressing both the Tln1 conditional allele and PF4-Cre, Cre recombinase–mediated recombination will result in deletion of coding exons 1–4, generating the Tln1-deleted allele (2). E, EcoRI; probe, external probe for Southern analysis. Primers used for genotyping mice are indicated. (B) Demonstration of homologous recombination in embryonic stem cells by Southern blotting. Genomic DNA from embryonic stem clones was digested with EcoRI and probed with an external probe shown in A. The wild-type allele gives rise to a 13.8-kb band (wt/wt), whereas the targeted allele gives rise to a 7.6-kb band due to introduction of an internal EcoRI site (flN/wt). (C) PCR genotyping of mice possessing the conditional allele. Genomic DNA isolated from ear biopsies of Tln1 fl/fl, Tln1 fl/+, and Tln1 fl/+ mice was analyzed by PCR using the primer pair shown in A. (D) Coomassie blue–stained SDS-PAGE gel of platelet lysates from Tln1 fl/fl Cre+ and Tln1 fl/fl Cre2 mice shows reduction of talin expression in Tln1 fl/fl Cre+ samples, whereas other protein bands are expressed at similar levels. (E) Immunostaining of freshly isolated bone marrow samples showing reduced talin expression in CD41+ megakaryocytes from Tln1 fl/fl Cre+ mice. Bar, 20 mm.
These results show that terminal megakaryocyte development and platelet formation do not require talin. Large megakaryocytes from Tln1 fl/fl Cre+ mice were devoid of talin as judged by staining of bone marrow cells with the 8d4 monoclonal antibody. Importantly, we targeted the talin1 allele and not talin2, an isoform that is very similar to talin1 (13). Nevertheless, hematopoietic cells express little talin2 (13). Furthermore, the 8d4 antibody reacts with both talin isoforms, and thus the absence of 8d4 staining of megakaryocytes from Tln1 fl/fl Cre+ mice confirms the elimination of talin expression in mature megakaryocytes. Previous work establishes that the PF4-cre mice we used express Cre specifically in megakaryocytes, including large megakaryocytes (15), and we observed that talin was still present in other cells of the hematopoietic lineage in the Tln1 fl/fl Cre+ mice. In spite of elimination of most of the talin, we observed abundant megakaryocytes in the bone marrow of the platelet talin1–deficient mice. Talin-deficient platelets were present at normal abundance, indicating that talin is not required for the formation of platelets from megakaryocytes. It is noteworthy that there was a slight reduction in platelet count in Tln1 fl/fl Cre+ mice relative to Tln1 fl/fl Cre− littermates. This reduction was not due to the presence of the PF4-Cre transgene in the absence of the floxed Tln1 allele because Tln1 fl/+ Cre+ mice had platelet counts similar to that of Tln1 fl/fl Cre− mice (755 ± 24 vs. 777 ± 74, × 109 platelets/ml ± SEM, Tln1 fl/fl Cre− vs. Tln1 fl/+ Cre+). Hence, it will be of interest in future work to examine the response of these animals to challenges to megakaryocytopoiesis.
Bleeding diathesis in platelet talin–deficient mice
Despite normal platelet counts in adult Tln1 fl/fl Cre+ mice (Table S1), these mice showed dramatically impaired hemostasis. In a tail bleeding assay, Tln1 fl/fl Cre+ mice bled continuously for the 10-min duration of the assay, whereas Tln1 fl/fl Cre− mice stopped bleeding an average of 4.6 min after tail resection (Fig. 2 A). Platelet talin deficiency was also associated with spontaneous bleeding. Lethal spontaneous internal hemorrhage, most often localized to the abdominal cavity, was observed in 8% of 1–2-d-old Tln1 fl/fl Cre+ mice (Fig. 2 B). By 3 wk of age, 45% fewer Tln1 fl/fl Cre+ than Tln1 fl/fl Cre− mice were alive. In addition, 12 out of 76 Tln1 fl/fl Cre+ mice were found dead between 3 and 9 wk of age compared with 3 out of 139 Tln1 fl/fl Cre− mice (Fig. 2 B). Tln1 fl/fl Cre+ mice that survived to 8 wk of age had a 95% incidence of gastrointestinal bleeding compared with 7% of Tln1 fl/fl Cre− littermates as judged by an assay for fecal blood (Fig. 2 C). Gastrointestinal bleeding in Tln1 fl/fl Cre+ mice was associated with profound anemia as manifested by significantly reduced red blood cell counts and hemoglobin concentration (Fig. 2 D and Table S1).
Figure 2.

Reduced survival and perinatal hemorrhage in Tln1fl/fl Cre+ mice. (A) Tln1 fl/fl Cre+ mice have prolonged bleeding times in a tail bleeding assay. Time to cessation of bleeding after tail resection was recorded for up to 10 min, at which time bleeding was stopped by cauterization. (B) Example of 1-d-old Tln1 fl/fl Cre+ pup found dead with visible internal hemorrhage (arrows). Table showing reduced survival of Tln1 fl/fl Cre+ mice. The number of animals obtained from Tln1 fl/fl Cre2 × Tln1 fl/fl Cre+ breeding at 3 wk of age is shown. (C) Incidence of fecal blood in Tln1 fl/fl Cre+ and Tln1 fl/fl Cre2 mice at 8–10 wk of age was determined by a guaiac-based hemoccult assay. (D) Peripheral red blood cell counts from 10-wk-old Tln1 fl/fl Cre+ and Tln1 fl/fl Cre2 littermates.
The hemostatic defects observed in Tln1 fl/fl Cre+ mice are at least as severe as those observed in β3 integrin null mice (17). In our hands, 22.9% fewer β3−/− than β3+/+ offspring from β3+/− by β3+/− matings survived to 3 wk of age, a smaller reduction in survival compared with Tln1 fl/fl Cre+ mice (45% fewer Tln1 fl/fl Cre+ than Tln1 fl/fl Cre−). However, because the Tln1 fl/fl and β3 null mice were both on mixed genetic backgrounds (C57BL/6-Sv129), it is not possible to make definitive statements regarding the relative hemostatic impairment in these mice.
Platelet talin is required for thrombus formation
Thrombus formation in mice with talin-deficient platelets was examined in vivo by ferric chloride–induced injury of the common carotid artery. Complete occlusion of the carotid arteries of Tln1 fl/fl Cre− mice occurred 7.0 ± 0.9 min after injury, whereas none of the Tln1 fl/fl Cre+ mice tested showed reduced flow during the 20-min assay (Fig. 3). Histological examination of the carotid arteries of these animals after the thrombosis experiment indicated a similar extent of ferric chloride–induced vessel injury in Tln1 fl/fl Cre+ and Tln1 fl/fl Cre− mice (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20071800/DC1). Collectively, our results show that deletion of talin1 in platelets leads to markedly impaired hemostasis and thrombus formation in vivo.
Figure 3.

Impaired thrombus formation in Tln1fl/fl Cre+ mice in vivo and ex vivo. (A) Time to occlusion of the carotid artery was determined with a Doppler flow probe after a 3-min application of 10% ferric chloride. The experiment was stopped 20 min after injury in all animals. (B) Adhesion of Tln1 fl/fl Cre+, Tln1 fl/fl Cre2, and b3(L476A) platelets to collagen and subsequent thrombus formation in flowing blood were analyzed by epifluorescence and confocal microscopy at a shear rate of 1,500 s21. Heparinized whole blood containing 10 mM mepacrine to render platelets fluorescent was perfused over glass coated with fibrillar type I collagen for 2 min. Tln1 fl/fl Cre2 platelets adhere to the collagen-coated surface and form thrombi (larger aggregates of bright fluorescence). In contrast, Tln1 fl/fl Cre+ platelets form only transient contacts resulting in sparse coverage of platelets on the surface. b3(L746A) platelets form a monolayer that cover much of the surface but do not form thrombi, as seen by the lack of highly fluorescent aggregates that form with Tln1 fl/fl Cre2 platelets. Images shown are single frames from a real-time recording (Video S1). Bar, 20 mm. (C) Percent of the collagen-coated surface covered with platelets was calculated as the number of fluorescent pixels (due to adhesion of fluorescently labeled platelets) divided by the total number of pixels (representing the total surface). *, P < 0.0005; NS, not significant. (D) Quantification of the volume of the thrombi formed on the collagen-coated surface after perfusion for 2 min with blood from Tln1 fl/fl Cre+, Tln1 fl/fl Cre2, and b3(L746A) mice. Confocal serial Z-section reconstructions of the platelet thrombi were used to calculate the thrombi volume as described previously (reference 32). *, P < 0.0005.
α2β1 integrin–mediated adhesion of platelets to exposed subendothelial collagen after vascular trauma is thought to be a key step in hemostasis. To examine the ability of Tln1 fl/fl Cre+ platelets to adhere to collagen under physiological conditions, we measured platelet adhesion and thrombus formation to collagen in flowing blood. Platelets from Tln1 fl/fl Cre− mice stably adhered to the collagen-coated surface and subsequently formed platelet-rich thrombi visible as highly fluorescent aggregates (Fig. 3 B, middle). In contrast, Tln1 fl/fl Cre+ platelets formed only transient contacts with the collagen-coated surface and did not form thrombi (Fig. 3 and Video S1, which is available at http://www.jem.org/cgi/content/full/jem.20071800/DC1). Interestingly, platelets from β3(L746A) mice, in which β3 integrin–talin interactions are selectively disrupted (18), formed stable adhesions to collagen indicated by the platelet monolayer shown in Fig. 3 B and quantified in Fig. 3 C. Nevertheless, the β3(L746A) platelets failed to undergo the integrin αIIbβ3–mediated platelet–platelet interactions required for thrombus formation (Fig. 3, B and D). Thus, lack of platelet talin impairs α2β1 integrin–dependent adhesion to collagen in flow and integrin αIIbβ3–dependent platelet thrombus formation.
Platelet talin is required for integrin-mediated platelet adhesion to collagen and platelet aggregation
We examined platelet adhesion and aggregation in vitro to directly assess the effects of talin deficiency on these integrin-dependent processes. In static adhesion assays, the talin-deficient platelets showed a marked defect in adhesion to soluble type I collagen (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20071800/DC1). Furthermore, like β3(L746A) platelets (18), talin-deficient platelets manifested profoundly impaired aggregation in response to stimulation with ADP or PAR4 peptide (Fig. S1 B). Thus, the talin-deficient platelets manifest virtual absence of platelet functions mediated by both α2β1 and αIIbβ3 integrins, thus accounting for their profound hemostatic defect.
Talin is required for activation of platelet α2β1 and αIIbβ3 integrins
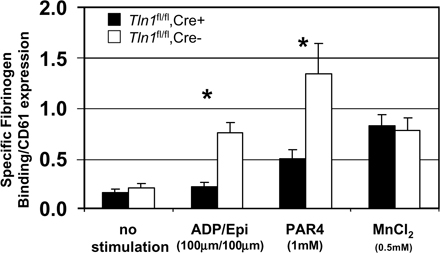
Agonist-induced increase in integrin αIIbβ3 affinity (activation) is required for platelet aggregation (19). Indeed, β3(L746A) mice, in which αIIbβ3–talin interactions are disrupted, have impaired αIIbβ3 integrin activation and platelet aggregation (18). To test the requirement of talin for the activation of αIIbβ3, we measured binding of FITC-labeled fibrinogen to the surface of washed Tln1 fl/fl Cre+ and Tln1 fl/fl Cre− platelets by flow cytometry. Stimulation of Tln1 fl/fl Cre− platelets with a combination of ADP/epinephrine (100 μM each) or PAR4 peptide (1 mM) led to an increase in the amount of bound fibrinogen. In contrast, the amount of agonist-induced fibrinogen binding was greatly reduced in Tln1 fl/fl Cre+ platelets (Fig. 4 A and Fig. S3, which is available at http://www.jem.org/cgi/content/full/jem.20071800/DC1). In the presence of 0.5 mM MnCl2, however, Tln1 fl/fl Cre+ and Tln1 fl/fl Cre− platelets bound similar amounts of fibrinogen, indicating that the αIIbβ3 present on Tln1 fl/fl Cre+ platelets is capable of binding fibrinogen if activated exogenously (Fig. 4 B and Fig. S3). Thus, with regards to αIIbβ3 activation, platelet talin deficiency phenocopies the β3(L746A) mutation in which the β3 integrin–talin interaction is disrupted. Collectively, these data firmly establish talin as a critical regulator of αIIbβ3 integrin activation in vivo.
Figure 4.

Impaired agonist-induced activation of b3 and b1 integrins in Tln1fl/fl Cre+ platelets. (A) The amount of FITC-labeled fibrinogen bound to platelets from Tln1 fl/fl Cre+ or Tln1 fl/fl Cre2 mice was measured by flow cytometry and expressed as the amount of fibrinogen bound to platelets in each group relative to the amount of fibrinogen bound to platelets in the presence of 0.5 mM MnCl2. *, P < 0.001. (B) Fibrinogen binding to Tln1 fl/fl Cre+ and Tln1 fl/fl Cre2 platelets was similar in the presence of 0.5 mM MnCl2. (C) Activation of b1 integrin in Tln1 fl/fl Cre+, Tln1 fl/fl Cre2, and b3(L746A) platelets after stimulation with 1 mM PAR4, 100 mm ADP, and 100 mM epinephrine was measured by binding of the conformation-sensitive antibody 9EG7 and expressed relative to total b1 integrin surface expression measured by the conformation-insensitive antibody HMb1-1. *, P < 0.05; **, P < 0.0005.
The impaired adhesion of Tln1 fl/fl Cre+ platelets to collagen noted above suggests that α2β1 integrin activation may also be impaired in Tln1 fl/fl Cre+ platelets. To examine the activation of β1 integrins in Tln1 fl/fl Cre+ platelets, we measured the binding of the conformation-sensitive β1 integrin antibody 9EG7 to agonist-stimulated platelets. Tln1 fl/fl Cre− platelets bound significantly more 9EG7 upon stimulation. This response was largely ablated in platelets from Tln1 fl/fl Cre+ mice (Fig. 4 C). These results show that talin expression is required for agonist-induced activation of both αIIbβ3 and α2β1 integrins in platelets. In addition, these data suggest that impaired activation of α2β1 integrins contributes to the spontaneous bleeding observed in Tln1 fl/fl Cre+ mice.
Thus, the principle that talin is required for activation of β1 and β3 integrins, which was suggested by in vitro studies (20), applies in vivo. Furthermore, the central role of talin in integrin function in invertebrates (11, 12, 21) extends to vertebrates. A β3(L746A) mutation that selectively disrupts the ability of β3 integrin to bind talin leads to impaired agonist-induced activation of platelet αIIbβ3 (18). Together with the present finding of impaired agonist-induced activation of αIIbβ3 in talin-deficient platelets, these data show that talin binding to integrin β cytoplasmic domains is a final common step in integrin activation in vivo, and that disruption of this interaction has a profound impact on integrin-dependent adhesive functions in mammals.
Talin-deficient platelets are nearly completely deficient in hemostatic function. It is noteworthy that the pathological bleeding observed in Tln1 fl/fl Cre+ mice is absent in β3(L746A) mice despite having comparable impairments in αIIbβ3 activation and platelet aggregation (18). One obvious explanation for this more severe phenotype in the platelet talin-deficient animals is the impairment in the activation and function of platelet β1 integrins. Furthermore, in Drosophila, talin deletion results in marked weakening of the connections of integrins with the actin cytoskeleton; hence, a defect in the connection of β1 and β3 integrins to the actin cytoskeleton may also contribute to the severe phenotype observed. Deletion of platelet β1 integrins or point mutations that would disrupt β1–talin interactions can result in defects in platelet function and in hemostasis (4, 22). Given the hemostatic defects that result from lack of platelet β3 or β1 integrins, and our finding that platelet integrin function is virtually completely dependent on talin, it is likely that the hemostatic defect in Tln1 fl/fl Cre+ mice is ascribable largely to the lack of integrin function.
Morphology and surface receptor expression is normal in talin-deficient platelets
To examine the effect of deleting talin on platelet structure, we examined Tln1 fl/fl Cre+ and Tln1 fl/fl Cre− platelet morphology by electron microscopy. Platelet shape, α granules, mitochondria, open canalicular system, and microtubule coils appeared similar in Tln1 fl/fl Cre+ and Tln1 fl/fl Cre− platelets (Fig. 5 A).Tln1 fl/fl Cre+ mice had slightly larger platelets than Tln1 fl/fl Cre− mice as judged by flow cytometry (forward scatter, 13.9 ± 0.2 vs. 15.3 ± 0.4 arbitrary units, Tln1 fl/fl Cre− vs. Tln1 fl/fl Cre+, P < 0.005) and by measurement of the area of at least 140 randomly selected electron microscopic platelet profiles (0.67 ± 0.03 μm2 vs. 0.97 ± 0.4 μm2, Tln1 fl/fl Cre− vs. Tln1 fl/fl Cre+ , P < 0.005). As noted above, the Tln1 fl/fl Cre+ mice have active gastrointestinal bleeding, suggesting that the slightly increased platelet size could be due to an increased proportion of circulating young platelets. In contrast to β3 integrin null platelets (17), talin-deficient platelets had normal fibrinogen content, suggesting that talin-dependent activation of αIIbβ3 in mature megakaryocytes is not required for fibrinogen uptake (Fig. 5 B). In addition, the surface expression of several adhesion receptors, as measured by flow cytometry, was not significantly different in Tln1 fl/fl Cre+ and Tln1 fl/fl Cre− platelets (Fig. 5 C). Of particular importance, the quantity of surface P-selectin was similar on both resting and stimulated Tln1 fl/fl Cre+ and Tln1 fl/fl Cre− platelets, confirming the presence of platelet α granules in the absence of talin. Furthermore, both platelet genotypes exhibited a similar fourfold increase in P-selectin surface expression in response to ADP/epinephrine/PAR4 peptide stimulation, confirming that the mutant platelets were capable of responding to platelet agonists. Collectively, these data demonstrate that talin is dispensable for the formation of platelets that can respond to platelet agonists and manifest a normal complement of granule constituents and adhesion receptors. Thus, the observed adhesion defects in Tln1 fl/fl Cre+ mice are ascribable to loss of talin-mediated integrin functions and not a general disruption of platelet structure and function.
Figure 5.

Platelets from Tln1fl/fl Cre+ mice are structurally normal and express normal levels of surface receptors. (A) Electron micrographs showing normal structural features of Tln1 fl/fl Cre+ platelets. Platelets from Tln1 fl/fl Cre+ (top left) and Tln1 fl/fl Cre2 (bottom left) mice both display a discoid shape characteristic of resting platelets and similar granular contents. Insets show microtubule coils in platelets from both Tln1 fl/fl Cre+ and Tln1 fl/fl Cre2 mice. Equatorial section of a Tln1 fl/fl Cre+ platelet (top right) shows circumferential microtubule coil (arrow heads). Bars, 1 mm. (B) Platelet fibrinogen content was determined by Coomassie blue staining of platelet lysates separated by SDS-PAGE. 5 mg of purified human fibrinogen served as a marker for the prominent protein band corresponding to fibrinogen in the platelet lysate samples. Consistent with previous reports (reference 17), fibrinogen content is reduced in platelets from b3 integrin null mice. (C) Surface expression of integrin aIIb, a2, b1, b3, and P-selectin was measured by flow cytometry. For P-selectin expression, platelets were incubated with or without PAR4/ADP/epinephrine (1 mm/100 mM/100 mM) for 5 min before the addition of an FITC-conjugated P-selectin antibody.
Platelet shape has been proposed to depend on the cortical actin cytoskeleton. Platelet filamin and spectrin play important roles in this cortical cytoskeleton (23), whereas talin is cytoplasmic in resting platelets and only recruited to the cortical cytoskeleton after platelet activation (24, 25). The normal shape of the talin-deficient platelets provides direct proof that platelet talin makes little if any contribution to the integrity of the cortical cytoskeleton in resting platelets. Furthermore, the talin-deficient platelets contained normal-appearing α granules and comparable contents of fibrinogen and P-selectin to littermate control mice. P-selectin is synthesized by megakaryocytes, whereas the bulk of platelet fibrinogen is taken up from the plasma; that uptake depends on integrin αIIbβ3 (26). The ability of talin-deficient megakaryocytes to package fibrinogen into α granules is noteworthy in light of the defect in activation of integrin αIIbβ3 in these platelets. It is possible that residual talin in early megakaryocytes may permit normal fibrinogen uptake; however, we have also observed normal platelet fibrinogen in the β3(L746A) platelets that have a similar defect in αIIbβ3 integrin activation (unpublished data), and human platelets with a β3(S752P) mutation also contain normal quantities of fibrinogen in spite of manifesting defective αIIbβ3 activation (27). Thus, even though integrin αIIbβ3 ligand binding function is required for normal fibrinogen uptake in megakaryocytes (26), activation of the integrin is not needed. In addition, talin-deficient platelets increased their surface expression of P-selectin in response to platelet agonists, indicating that the platelets could respond to the agonists, a conclusion supported by the normal shape change in the platelet aggregation tracings. Similarly, the increased surface P-selectin suggests that α granule secretion does not depend on talin; notably, this process is PIP2 dependent (28), and talin can recruit and regulate one isoform of PI5 kinase (29), a rate-limiting step in PIP2 synthesis. In summary, the talin-deficient platelets exhibit normal morphology and respond to platelet agonists.
Here, we have shown that platelet talin is essential for platelet-dependent hemostasis because it is required for the function and activation of β1 and αIIbβ3 integrins. Despite Tln1 fl/fl Cre+ mice having normal platelet counts, these animals exhibited both lethal spontaneous bleeding and resistance to induced thrombosis. These in vivo findings were ascribable to profound defects in the function of multiple platelet integrins, as platelets from Tln1 fl/fl Cre+ mice failed to adhere to collagen or to form platelet-rich thrombi ex vivo. In vitro studies documented the profound impairment of integrin-mediated adhesion and platelet aggregation in talin1-deficient platelets and showed that these platelets were deficient in the agonist-induced activation of both α2β1 and αIIbβ3 integrins, despite maintaining the capacity to respond to the agonists as indicated by surface display of P-selectin. Thus, talin is necessary for the activation of α2β1 and αIIbβ3 integrins in vivo, and platelet talin is absolutely required for hemostasis because it is necessary for the adhesive functions of these integrins.
MATERIALS AND METHODS
Generation of mice.
Conditional talin1 knockout mice were generated by introducing loxP sites flanking coding exons 1–4 of the Tln1 gene by gene targeting. Targeting of the Tln1 locus was confirmed by Southern blot of EcoR1-digested genomic DNA hybridized with a 5′ cDNA probe. Mice were genotyped by PCR using the following primers indicated in Fig. 1 A: primer a: 5′-aagcaggaacaaaagtaggtctcc-3′ and primer b: 5′-gcatcgtcttcaccacattcc-3′. Mice homozygous for the Tln1 floxed allele (Tln1 fl/fl) on a mixed C57BL/6-Sv129 genetic background were crossed with PF4-Cre (Cre+) mice on a C57BL/6 background (15). To obtain mice with talin1-deficient platelets, Tln1 fl/fl Cre+ males were bred with Tln1 fl/fl Cre− females. In all experiments, Tln1 fl/fl Cre+ mice were compared with Tln1 fl/fl Cre− sex-matched littermates. The generation of β3(L746A) mice has been recently described (18). Mice were housed in the University of California, San Diego, animal facility, and experiments were approved by the university's Institutional Animal Care and Use Committee.
SDS-PAGE.
For examination of platelet talin protein content, washed platelets were lysed by adding 1 vol of 2X modified RIPA buffer (300 mM NaCl, 100 mM Tris, pH 7.4, 0.2% SDS, 2% Triton X-100, 2% sodium deoxycholate, 2 mM PMSF, 2 mM NaVO4, 2 mM NaF, 2 mM EDTA, and complete protease inhibitor; Roche), and samples were clarified by centrifugation at 13,000 g for 10 min at 4°C. Laemmli buffer containing 10 mM DTT was added to 10 μg of protein lysates, and samples were boiled for 5 min before being separated on 6% Tris-glycine gels (Invitrogen) and stained with Coomassie blue. For analysis of platelet fibrinogen content, platelets were lysed in Laemmli buffer in the absence of any reducing agent and separated on a 6% Tris-glycine gel. Fibrinogen was identified as a Coomassie blue–stained band migrating with an apparent molecular mass of 340,000.
Immunofluorescence.
After lysing red blood cells with RBC lysis buffer (155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) bone marrow cells from the femurs of 6–9-wk-old mice were fixed in 3.7% formaldehyde/PBS and applied to fibrinogen-coated (100 μg/ml) glass slides by Cytospin preparation (Thermo Fisher Scientific). Cells were permeabilized with 0.1% Triton X-100/PBS containing 5% BSA for 1 h at room temperature and incubated with talin antibody 8d4 (1:50 dilution; Sigma-Aldrich) overnight at 4°C. After washing with PBS, cells were incubated with 2.5 μg/ml FITC-conjugated anti-CD41 (BD Biosciences) and 5 μg/ml Alexa-568 goat anti–mouse IgG for 2 h at room temperature. After washing, slides were mounted with coverslips using Vectashield anti-fade media (Vector Laboratories) and observed on a Leica DM LS fluorescence microscope. Images were captured with a spot color digital camera (National Instruments) using manual exposure settings that were identical for Tln1 fl/fl Cre+ and Tln1 fl/fl Cre− samples.
Hemostasis assays.
The presence of fecal blood was detected with a guaiac-based hemoccult detection assay (Helena Laboratories) on freshly obtained stool samples.
Tail bleeding assays were performed by resecting 1 mm of the tail, followed by immersion in 37°C isotonic saline as described previously (17). All experiments were terminated at 10 min by cauterizing the tail.
Platelet isolation and functional assays.
Washed platelets were obtained as described previously (30). Soluble fibrinogen binding was measured by incubating platelets for 20 min with 150 μg/ml FITC-labeled fibrinogen, followed by fixation with 1% formaldehyde for 10 min at room temperature. Specific fibrinogen binding was determined by subtracting the amount of fibrinogen bound in the presence of 5 mM EDTA. Bound fibrinogen was detected with a FACScan flow cytometer (Becton Dickinson). β1 integrin activation was measured by the binding of an FITC-labeled confirmation-sensitive β1 integrin antibody 9EG7 (BD Biosciences), or the confirmation-insensitive PE-conjugated β1 antibody HMβ1-1 (BD Biosciences). Washed platelets were incubated with or without agonists for 10 min at room temperature, followed by the addition of 3 μg/ml of either 9EG7 or HMβ1-1 for 20 min and detected by flow cytometry. Similarly, surface expression of P-selectin was measured by the binding of FITC–anti–P-selectin (BD Biosciences) following the same protocol as described above for measuring β1 integrin activation. Surface expression of β3, αIIb, and α2 integrins were measured by flow cytometry with the following antibodies: FITC–anti-CD61, FITC–anti-CD41 (BD Biosciences), and PE–anti-α2 integrin (eBioscience).
For analysis of static adhesion of platelets to collagen, 96-well plates (Immulon HB2; Dynex Technologies) were coated with 2 μg of acid-soluble type I collagen from rat tail (Sigma-Aldrich) in 100 μl PBS overnight at 4°C. After two washes with PBS and blocking with 5% BSA/PBS for 2 h at room temperature, 5 × 106 washed platelets suspended in platelet incubation buffer were added to each well and allowed to adhere for 1 h at room temperature. Wells were then washed three times with platelet incubation buffer, and adherent platelets were quantified by acid-phosphatase assay (18). Percent platelet adhesion was calculated as the number of adherent platelets relative to the number of platelets in wells that were not washed (total platelets per well). Platelet aggregation was performed as described previously (18) using platelet-rich plasma (PRP) diluted to a platelet concentration of 3 × 108 platelets/ml with platelet-poor plasma.
Ex vivo adhesion to collagen.
Adhesion and thrombus formation in flowing blood was performed and analyzed as described previously (31, 32).
Transmission electron microscopy.
Blood was drawn by cardiac puncture into 0.1 vol of 0.13 M sodium citrate. After adding 1 vol modified Tyrode's buffer (140 mM NaCl, 2.7 mM KCl, 0.4 mM NaH2PO4, 10 mM NaHCO3, 5 mM dextrose, and 10 mM Hepes) samples were centrifuged for 5 min at 200 g at room temperature to obtain PRP. The PRP was incubated for 30 min at 37°C before fixing by the addition of 1 vol of 2X fixative (3% gluteraldehyde and 6% paraformaldehyde in 0.2 M cacodylate buffer plus 10% sucrose, pH 7.4) and incubated for 15 min at room temperature. Platelets were centrifuged at 700 g for 5 min and resuspended and stored overnight in 1X fixative. Samples were processed as described previously (33), and images were obtained with a JEOL 1200 EX II electron microscope.
Ferric chloride–induced thrombosis.
Ferric chloride–induced thrombosis was performed as described previously (18) by applying a 1.2 X 1.2–mm piece of filter paper soaked in 10% ferric chloride to each side of the common carotid artery of a mouse under isoflurane anesthesia.
Statistics.
Statistical analyses of mouse survival and spontaneous death were performed with χ2 and Fisher's exact tests, respectively. The statistical significance of all other data were determined using Student's t test. A p-value of <0.05 was considered statistically significant. All error bars represent standard error of the mean.
Online supplemental material.
Video S1 shows adhesion of fluorescently labeled platelets to fibrillar type I collagen in flowing blood. Time shown is from the beginning of flow over the collagen-coated surface. Video S1 is available at http://www.jem.org/cgi/content/full/jem.20071800/DC1.
Supplemental Material
Acknowledgments
We gratefully acknowledge Timo Meerloo and Dr. Marilyn Farquhar for help with electron microscopy. We are grateful to Dr. Catrin Pritchard for advice in generating mice.
This work was supported by grants from the National Institutes of Health (HL31950 and HL078784), the Cell Migration Consortium, NIH (U54 GM064346), and the Wellcome Trust (077532). B.G. Petrich is a fellow of the American Heart Association.
The authors have no conflicting financial interests.
References
- 1.Ruggeri, Z.M. 2002. Platelets in atherothrombosis. Nat. Med. 8:1227–1234. [DOI] [PubMed] [Google Scholar]
- 2.Nieswandt, B., C. Brakebusch, W. Bergmeier, V. Schulte, D. Bouvard, R. Mokhtari-Nejad, T. Lindhout, J.W. Heemskerk, H. Zirngibl, and R. Fassler. 2001. Glycoprotein VI but not alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J. 20:2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen, J., T.G. Diacovo, D.G. Grenache, S.A. Santoro, and M.M. Zutter. 2002. The alpha(2) integrin subunit-deficient mouse: a multifaceted phenotype including defects of branching morphogenesis and hemostasis. Am. J. Pathol. 161:337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarratt, K.L., H. Chen, M.M. Zutter, S.A. Santoro, D.A. Hammer, and M.L. Kahn. 2005. GPVI and alpha2beta1 play independent critical roles during platelet adhesion and aggregate formation to collagen under flow. Blood. 106:1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shattil, S.J., and P.J. Newman. 2004. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 104:1606–1615. [DOI] [PubMed] [Google Scholar]
- 6.Ratnikov, B.I., A.W. Partridge, and M.H. Ginsberg. 2005. Integrin activation by talin. J. Thromb. Haemost. 3:1783–1790. [DOI] [PubMed] [Google Scholar]
- 7.O'Halloran, T., M.C. Beckerle, and K. Burridge. 1985. Identification of talin as a major cytoplasmic protein implicated in platelet activation. Nature. 317:449–451. [DOI] [PubMed] [Google Scholar]
- 8.Calderwood, D.A., R. Zent, R. Grant, D.J. Rees, R.O. Hynes, and M.H. Ginsberg. 1999. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 274:28071–28074. [DOI] [PubMed] [Google Scholar]
- 9.Borowsky, M.L., and R.O. Hynes. 1998. Layilin, a novel talin-binding transmembrane protein homologous with C-type lectins, is localized in membrane ruffles. J. Cell Biol. 143:429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Critchley, D.R. 2004. Cytoskeletal proteins talin and vinculin in integrin-mediated adhesion. Biochem. Soc. Trans. 32:831–836. [DOI] [PubMed] [Google Scholar]
- 11.Brown, N.H., S.L. Gregory, W.L. Rickoll, L.I. Fessler, M. Prout, R.A. White, and J.W. Fristrom. 2002. Talin is essential for integrin function in Drosophila. Dev. Cell. 3:569–579. [DOI] [PubMed] [Google Scholar]
- 12.Tanentzapf, G., and N.H. Brown. 2006. An interaction between integrin and the talin FERM domain mediates integrin activation but not linkage to the cytoskeleton. Nat. Cell Biol. 8:601–606. [DOI] [PubMed] [Google Scholar]
- 13.Monkley, S.J., C.A. Pritchard, and D.R. Critchley. 2001. Analysis of the mammalian talin2 gene TLN2. Biochem. Biophys. Res. Commun. 286:880–885. [DOI] [PubMed] [Google Scholar]
- 14.Monkley, S.J., X.H. Zhou, S.J. Kinston, S.M. Giblett, L. Hemmings, H. Priddle, J.E. Brown, C.A. Pritchard, D.R. Critchley, and R. Fassler. 2000. Disruption of the talin gene arrests mouse development at the gastrulation stage. Dev. Dyn. 219:560–574. [DOI] [PubMed] [Google Scholar]
- 15.Tiedt, R., T. Schomber, H. Hao-Shen, and R.C. Skoda. 2007. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood. 109:1503–1506. [DOI] [PubMed] [Google Scholar]
- 16.Tsakiris, D.A., L. Scudder, K. Hodivala-Dilke, R.O. Hynes, and B.S. Coller. 1999. Hemostasis in the mouse (Mus musculus): a review. Thromb. Haemost. 81:177–188. [PubMed] [Google Scholar]
- 17.Hodivala-Dilke, K.M., K.P. McHugh, D.A. Tsakiris, H. Rayburn, D. Crowley, M. Ullman-Cullere, F.P. Ross, B.S. Coller, S. Teitelbaum, and R.O. Hynes. 1999. Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J. Clin. Invest. 103:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petrich, B.G., P. Fogelstrand, A.W. Partridge, N. Yousefi, A.J. Ablooglu, S.J. Shattil, and M.H. Ginsberg. 2007. The antithrombotic potential of selective blockade of talin-dependent integrin alpha IIb beta 3 (platelet GPIIb-IIIa) activation. J. Clin. Invest. 117:2250–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett, J.S., and G. Vilaire. 1979. Exposure of platelet fibrinogen receptors by ADP and epinephrine. J. Clin. Invest. 64:1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tadokoro, S., S.J. Shattil, K. Eto, V. Tai, R.C. Liddington, J.M. de Pereda, M.H. Ginsberg, and D.A. Calderwood. 2003. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 302:103–106. [DOI] [PubMed] [Google Scholar]
- 21.Cram, E.J., S.G. Clark, and J.E. Schwarzbauer. 2003. Talin loss-of-function uncovers roles in cell contractility and migration in C. elegans. J. Cell Sci. 116:3871–3878. [DOI] [PubMed] [Google Scholar]
- 22.Chen, H., Z. Zou, K.L. Sarratt, D. Zhou, M. Zhang, E. Sebzda, D.A. Hammer, and M.L. Kahn. 2006. In vivo beta1 integrin function requires phosphorylation-independent regulation by cytoplasmic tyrosines. Genes Dev. 20:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hartwig, J.H., and M. DeSisto. 1991. The cytoskeleton of the resting human blood platelet: structure of the membrane skeleton and its attachment to actin filaments. J. Cell Biol. 112:407–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beckerle, M.C., D.E. Miller, M.E. Bertagnolli, and S.J. Locke. 1989. Activation-dependent redistribution of the adhesion plaque protein, talin, in intact human platelets. J. Cell Biol. 109:3333–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fox, J.E., L. Lipfert, E.A. Clark, C.C. Reynolds, C.D. Austin, and J.S. Brugge. 1993. On the role of the platelet membrane skeleton in mediating signal transduction. Association of GP IIb-IIIa, pp60c-src, pp62c-yes, and the p21ras GTPase-activating protein with the membrane skeleton. J. Biol. Chem. 268:25973–25984. [PubMed] [Google Scholar]
- 26.Handagama, P., R.M. Scarborough, M.A. Shuman, and D.F. Bainton. 1993. Endocytosis of fibrinogen into megakaryocyte and platelet alpha-granules is mediated by alpha IIb beta 3 (glycoprotein IIb-IIIa). Blood. 82:135–138. [PubMed] [Google Scholar]
-
27.Nurden, P., C. Poujol, J. Winckler, R. Combrie, J.P. Caen, and A.T. Nurden. 2002. A Ser752
 —Pro substitution in the cytoplasmic domain of beta3 in a Glanzmann thrombasthenia variant fails to prevent interactions between the alphaIIbbeta3 integrin and the platelet granule pool of fibrinogen. Br. J. Haematol.
118:1143–1151.
[DOI] [PubMed] [Google Scholar]
—Pro substitution in the cytoplasmic domain of beta3 in a Glanzmann thrombasthenia variant fails to prevent interactions between the alphaIIbbeta3 integrin and the platelet granule pool of fibrinogen. Br. J. Haematol.
118:1143–1151.
[DOI] [PubMed] [Google Scholar] - 28.Rozenvayn, N., and R. Flaumenhaft. 2001. Phosphatidylinositol 4,5-bisphosphate mediates Ca2+-induced platelet alpha-granule secretion: evidence for type II phosphatidylinositol 5-phosphate 4-kinase function. J. Biol. Chem. 276:22410–22419. [DOI] [PubMed] [Google Scholar]
- 29.Di Paolo, G., L. Pellegrini, K. Letinic, G. Cestra, R. Zoncu, S. Voronov, S. Chang, J. Guo, M.R. Wenk, and P. De Camilli. 2002. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 420:85–89. [DOI] [PubMed] [Google Scholar]
- 30.Law, D.A., L. Nannizzi-Alaimo, K. Ministri, P.E. Hughes, J. Forsyth, M. Turner, S.J. Shattil, M.H. Ginsberg, V.L. Tybulewicz, and D.R. Phillips. 1999. Genetic and pharmacological analyses of Syk function in alphaIIbbeta3 signaling in platelets. Blood. 93:2645–2652. [PubMed] [Google Scholar]
- 31.Konstantinides, S., J. Ware, P. Marchese, F. Almus-Jacobs, D.J. Loskutoff, and Z.M. Ruggeri. 2006. Distinct antithrombotic consequences of platelet glycoprotein Ibalpha and VI deficiency in a mouse model of arterial thrombosis. J. Thromb. Haemost. 4:2014–2021. [DOI] [PubMed] [Google Scholar]
- 32.Savage, B., F. Almus-Jacobs, and Z.M. Ruggeri. 1998. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 94:657–666. [DOI] [PubMed] [Google Scholar]
- 33.Head, B.P., H.H. Patel, D.M. Roth, F. Murray, J.S. Swaney, I.R. Niesman, M.G. Farquhar, and P.A. Insel. 2006. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J. Biol. Chem. 281:26391–26399. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}