

Abstract
Pseudomonas aeruginosa is a Gram-negative bacterium that causes opportunistic infections in immunocompromised individuals. P. aeruginosa employs a type III secretion system to inject effector molecules into the cytoplasm of the host cell. This interaction with the host cell leads to inflammatory responses that eventually result in cell death. We show that infection of macrophages with P. aeruginosa results in activation of caspase-1 in an IPAF-dependent, but flagellin-independent, manner. Macrophages deficient in IPAF or caspase-1 were markedly resistant to P. aeruginosa–induced cell death and release of the proinflammatory cytokine interleukin (IL)-1β. A subset of P. aeruginosa isolates express the effector molecule exoenzyme U (ExoU), which we demonstrate is capable of inhibiting caspase-1–driven proinflammatory cytokine production. This study shows a key role for IPAF and capase-1 in innate immune responses to the pathogen P. aeruginosa, and also demonstrates that virulent ExoU-expressing strains of P. aeruginosa can circumvent this innate immune response.
Pseudomonas aeruginosa causes a variety of acute infections, such as ventilator-associated pneumonias, burn superinfections, and medical device–related infections. In addition, P. aeruginosa chronically infects cystic fibrosis patients and causes significant morbidity and mortality in this population. Like many other pathogenic Gram-negative bacteria, P. aeruginosa utilizes a type III secretion system (TTSS) to inject effector molecules into the cytoplasmic compartment of the host cell. Four effector molecules, exoenzyme S (ExoS), ExoT, ExoU, and ExoY, are variably expressed by P. aeruginosa strains and are able to initiate inflammatory events that may lead to apoptotic or necrotic cell death (1, 2). The TTSS is a complex macromolecular structure that spans both bacterial membranes and includes a long, needlelike structure through which the effector molecules pass. Access of the effector molecules into the cytoplasm of the host cell requires disruption of the plasma membrane by a proteinaceous structure that forms a pore (3). This structure, called the translocon, is made up of two bacterial proteins, PopB and PopD, inserted into the host cell membrane (4, 5). P. aeruginosa mutants that have a defective TTSS have been shown to be less virulent than their wild-type counterparts (6–8). Although other Gram-negative bacteria, such as Salmonella typhimurium (9) and Shigella flexneri (10, 11), have been shown to mediate a caspase-1–dependent macrophage death that is dependent on a functional TTSS, the mechanisms by which P. aeruginosa mediates TTSS-dependent macrophage cell death is largely unknown (12).
The mammalian NLR family is composed of >20 members that contain a C-terminal leucine-rich repeat domain, a central nucleotide-binding NACHT domain, and an N-terminal protein–protein interaction domain composed of a caspase activation and recruitment domain (CARD) or Pyrin domain (13–16). These proteins promote the assembly of multiprotein complexes, termed inflammasomes, which are required for the activation of inflammatory caspases. Caspase-1 is a key component of the inflammasome, and is able to initiate a cell death program, as well as process the proinflammatory cytokines pro-IL-1β and pro-IL-18 into their mature forms, IL-1β and -18, respectively (17). IPAF (also known as NLRC4, CARD12, or CLAN), which is a member of the NLR family, has been shown to be responsible for caspase-1 activation in response to S. typhimurium and Legionella pneumophila (18, 19). It has been suggested that for Salmonella and Legionella, cytosolic flagellin is required for the activation of caspase-1 through an IPAF-dependent pathway (20–23).
In this study, we show that ExoU-deficient strains of P. aeruginosa kill macrophages in an IPAF/caspase-1–dependent manner that also results in the secretion of caspase-1–driven proinflammatory cytokines. In contrast, ExoU-expressing P. aeruginosa strains kill macrophages independently of caspase-1. In addition, ExoU can inhibit caspase-1 activation and thereby inhibit the secretion of IL-1β and -18. Unlike previous reports, which have suggested that flagellin activates the IPAF inflammasome, we show that P. aeruginosa activation of IPAF and caspase-1 is independent of flagellin, but does require a functional TTSS/translocon.
RESULTS
P. aeruginosa–induced macrophage cell death is caspase-1 dependent
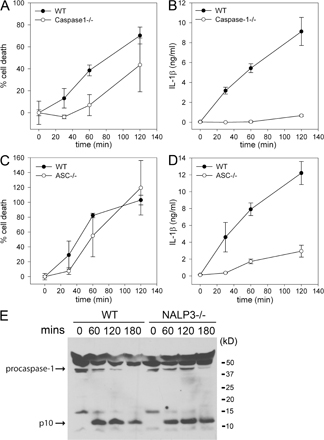
Caspase-1 is responsible for initiating cell death in response to several stimuli. We tested whether P. aeruginosa–induced macrophage cell death was dependent on caspase-1. WT macrophages infected with the P. aeruginosa strain PAK underwent rapid cell death, as measured by lactate dehydrogenase (LDH) release (Fig. 1 A and Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20071239/DC1). In contrast, caspase-1–deficient macrophages had a marked delay in cell death in response to infection with P. aeruginosa (Fig. 1 A and Fig. S1 A). WT macrophages infected with P. aeruginosa also secreted IL-1β in a caspase-1–dependent manner (Fig. 1 B and Fig. S1 B), demonstrating that caspase-1 plays a role in both P. aeruginosa–induced macrophage cell death and the secretion of IL-1β.
Figure 1.

P. aeruginosa–induced macrophage cell death and IL-1β secretion are dependent on IPAF and caspase-1. LPS-primed macrophages from WT, caspase-1–, ASC-, NALP3-, or IPAF-deficient mice were infected with P. aeruginosa strain PAK at a MOI of 20 bacteria per macrophage. Culture supernatants were collected 1 h after infection (A–D) or as indicated (E and F). Cytotoxicity was measured by LDH release and expressed as a percentage of LDH release by Triton X-100 detergent (A, C, and E). IL-1β release into culture supernatants was measured by ELISA (B, D, and F). Determinations were performed in triplicate and expressed as the mean ± the SD. *, P = 0.01. Results are representative of two (E and F) and three (A–D) separate experiments. (G and H) Lysates from LPS-primed WT, IPAF-, and ASC-deficient macrophages infected with P. aeruginosa strain PAK at a MOI of 20 bacteria per macrophage for the indicated times were immunoblotted with antibodies against the p10 subunit of caspase-1. Results are representative of two independent experiments.
NALP3 (also known as NLRP3, CIAS1, and cryopyrin), along with the adaptor molecule ASC, can activate caspase-1 in response to a variety of stimuli, including ATP, nigericin, maitotoxin, uric acid, and the pathogens Listeria monocytogenes and Staphylococcus aureus (24–27). ASC- and NALP3-deficient macrophages were as susceptible to P. aeruginosa–induced macrophage cell death as WT macrophages (Fig. 1 C and Fig. S1 C). However, IL-1β secretion in response to P. aeruginosa infection was partially dependent on ASC (Fig. 1 D and Fig. S1 D). NALP3-deficiency did not have any effect on IL-1β secretion in response to P. aeruginosa infection (Fig. 1 D).
P. aeruginosa activation of caspase-1 is dependent on IPAF
We used IPAF-deficient macrophages to test if IPAF was required for P. aeruginosa–induced caspase-1 activation. IPAF-deficient macrophages infected with the P. aeruginosa strain PAK displayed a marked delay in cell death compared with WT macrophages (Fig. 1 E). Macrophage cell death did eventually occur in IPAF-deficient macrophages, suggesting that P. aeruginosa possesses additional mechanisms by which to kill host cells. IPAF-deficient macrophages also secreted markedly less IL-1β in response to infection with PAK (Fig. 1 F). The defect in cell death and IL-1β secretion observed in IPAF-deficient macrophages in response to infection with P. aeruginosa was not caused by differences in binding of bacteria to the macrophages. We found an equal number of bacteria bound to both WT and IPAF-deficient macrophages 30 min after infection with P. aeruginosa strain PAK (unpublished data).
Caspase-1 activation involves autocatalytic processing of the 45-kD pro-caspase-1 to generate two subunits, p20 and p10. Caspase-1 activation in PAK-infected WT macrophages was detected by Western blot by the appearance of the p10 cleavage product as early as 30 min after infection (Fig. 1, G and H). We did not observe caspase-1 activation in response to P. aeruginosa infection in either IPAF- or ASC-deficient macrophages (Fig. 1, G and H). NALP3-deficient macrophages did not display any defect in caspase-1 activation in response to P. aeruginosa infection (Fig. S1 E). These data identify IPAF as the critical inflammasome component required for caspase-1–mediated cell death and IL-1β secretion in response to infection with P. aeruginosa.
ExoU induces macrophage cell death independently of IPAF
The TTSS is capable of delivering up to four different effector proteins, ExoS, ExoT, ExoU, and ExoY. Two of these effector molecules, ExoS and ExoU, have direct cytotoxic effects on the host cell (1, 2); however, ExoU, which possesses phospholipase A2 activity (2, 28, 29), is markedly more cytotoxic than the ADP-ribosyltransferase/Rho GTPase-activating protein ExoS (30). ExoU is expressed by ∼20% of clinical isolates, but is not produced by the P. aeruginosa strain PAK, which expresses the effectors ExoS, ExoT, and ExoY (30). To examine the role of ExoU in P. aeruginosa–induced macrophage cell death, we used the P. aeruginosa strain PA103, which expresses ExoU and ExoT, but not ExoS and ExoY. WT macrophages infected with PA103 rapidly underwent cell death, as measured by LDH release (Fig. 2, A and B). IPAF-deficient macrophages infected with PA103 also underwent rapid cell death (Fig. 2, A and C) in contrast to the protection observed in IPAF-deficient macrophages infected with the P. aeruginosa strain PAK. We also observed cell death in WT macrophages infected with the ExoU-deficient strain PA103ΔU, which is consistent with the findings of others (12); however, IPAF-deficient macrophages infected with PA103ΔU were resistant to cell death (Fig. 2, A–C). These data suggest that P. aeruginosa has at least two distinct methods by which it can induce cell death in macrophages. The first is by the previously described ExoU pathway, whereas the second is a caspase-1–dependent cell death, which is observable when ExoU is not present. IPAF-deficient macrophages infected with the ExoT-deficient strain PA103ΔT remained susceptible to cell death, and infection with PA103ΔUT, which is deficient in both ExoU and ExoT, had a similar cell death phenotype to that observed with PA103ΔU (Fig. 2 A). Thus, PA103ΔUT, which produces none of the known effector molecules, still kills macrophages in an IPAF-dependent manner (Fig. 2 A), suggesting that the IPAF inflammasome is not activated by any of the four exotoxins.
Figure 2.

ExoU can mediate macrophage cell death in an IPAF-independent manner. (A) LPS-stimulated macrophages from WT or IPAF-deficient mice were infected with the P. aeruginosa strain PA103, PA103ΔU, PA103ΔT, or PA103ΔUT at a MOI of 20 bacteria per macrophage. 1 h after infection, culture supernatants were collected and cytotoxicity was measured by LDH release and expressed as a percentage of LDH release by Triton X-100 detergent. (B and C) Nonstimulated WT (B) or IPAF−/− (C) macrophages were infected with the P. aeruginosa strain PA103 or PA103ΔU at a MOI of 20 bacteria per macrophage. At the indicated times after infection, culture supernatants were collected and cytotoxicity was measured by LDH release into the culture supernatant. Determinations were performed in triplicate and expressed as the mean ± the SD. Results are representative of two to three separate experiments.
ExoU inhibits caspase-1 activation and secretion of IL-1β
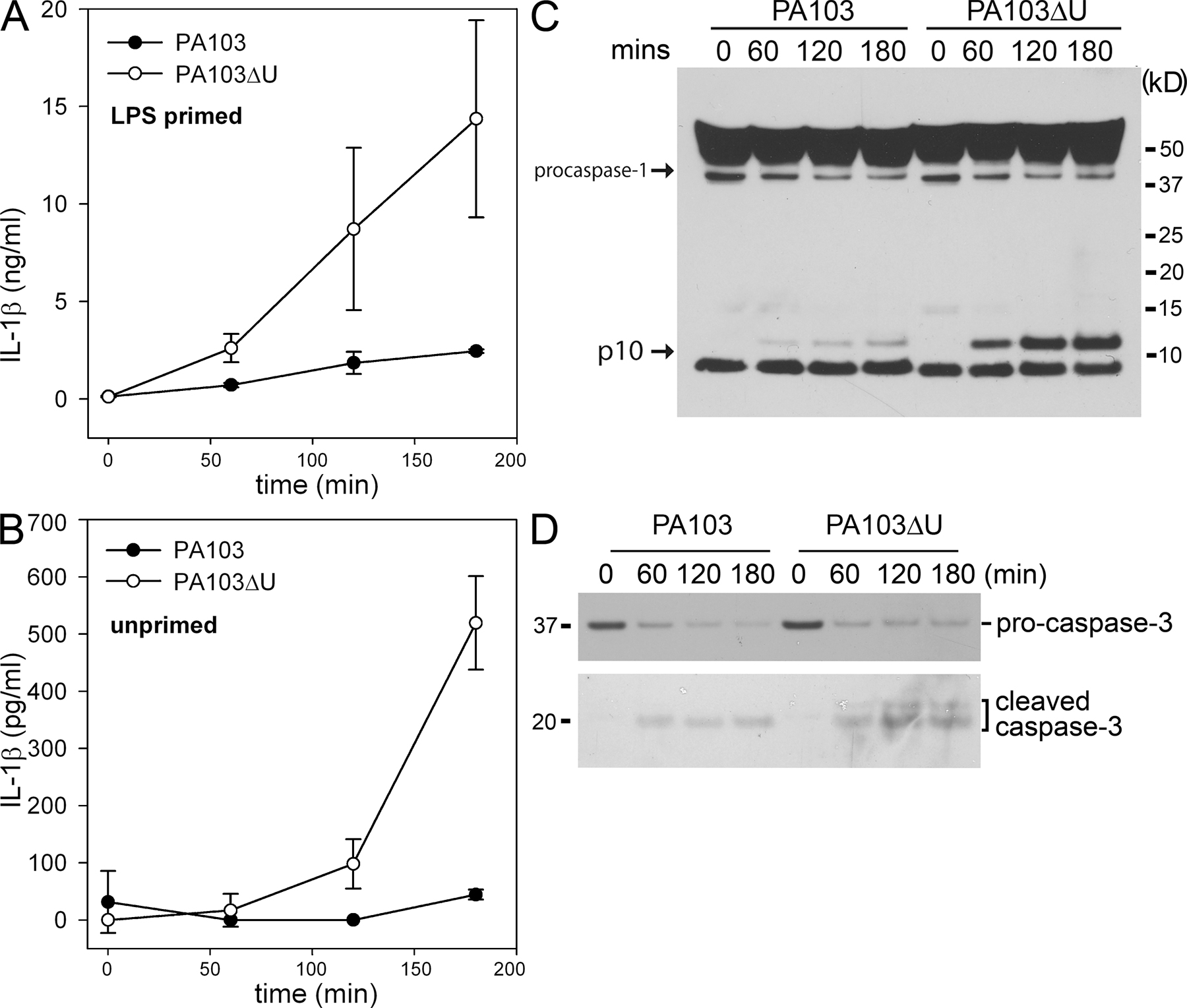
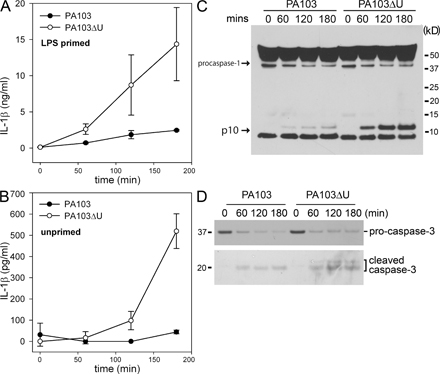
We observed that WT macrophages infected with either PA103 or PA103ΔU both underwent cell death with similar kinetics as observed by LDH release (Fig. 2 B). However, PA103ΔU-infected macrophages secreted markedly more IL-1β (Fig. 3 A) compared with macrophages infected with PA103, suggesting that ExoU actively inhibits the secretion of IL-1β from macrophages. TNFα (Fig. 3 B) levels were also elevated in PA103ΔU-infected macrophages compared with macrophages infected with PA103. However, IL-6 (Fig. 3 C) secretion induced by infection with PA103ΔU or PA103 was not significantly different. A similar difference in IL-1β secretion was also observed in LPS-primed macrophages infected with PA103ΔU versus PA103 (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20071239/DC1). As seen in Fig. S2 (A and B) LPS-primed macrophages produced substantially more IL-1β in response to infection with either PA103 or PA103ΔU compared with unprimed macrophages caused by the large intracellular pool of pro-IL-1β generated after its induction by LPS treatment. However, we were unable to measure P. aeruginosa–induced TNFα in LPS-primed macrophages because LPS treatment alone induced robust TNFα production. Both PA103 and PA103ΔU induced equal amounts of pro-IL-1β production in unprimed macrophages, but PA103 failed to activate caspase-1, as observed during Western blotting by the absence of the caspase-1 p10 subunit in PA103-infected macrophages (Fig. 3 D). PA103 was able to activate caspase-1 in LPS-primed macrophages, but this activation was markedly reduced compared with infection with PA103ΔU (Fig. S2 C). To determine if ExoU was specifically inhibiting caspase-1 activation, we examined the effects of PA103 and PA103ΔU infection on the NF-κB and MAPK signaling cascades. Phosphorylation (not depicted) and degradation of IκBα in macrophages infected with either PA103 or PA103ΔU occurred at similar rates (Fig. 3 E). Phosphorylation of p38 and ERK1/2 was not inhibited in macrophages infected with PA103 as compared with PA103ΔU (Fig. 3 E); in fact, ERK1/2 phosphorylation occurred more quickly in PA103-infected macrophages. However, caspase-1 activation was not observed in the presence of ExoU (Fig. 3 E), suggesting that ExoU inhibition of caspase-1 activation leads to markedly reduced secretion of IL-1β. Activation of caspase-3 in macrophages infected with either PA103 or PA103ΔU occurred at similar rates (Fig. S2 D), further suggesting that the effects of ExoU were specific to caspase-1. TNFα production is independent of caspase-1 activation; however, the increased TNFα production by macrophages infected with PA103ΔU as compared with PA103 (Fig. 3 B) may be caused by the indirect effects of increased IL-1β production.
Figure 3.

P. aeruginosa ExoU inhibits caspase-1 activation. (A–C) Nonstimulated WT macrophages were infected with P. aeruginosa strain PA103 or PA103ΔU at a MOI of 20 bacteria per macrophage. At the indicated times after infection, culture supernatants were collected and IL-1β, TNF-α, and IL-6 release into culture supernatants was measured by ELISA. Results are expressed as the mean ± SEM of four separate experiments, each performed in triplicate. (D and E) Lysates from nonstimulated WT macrophages infected with P. aeruginosa strain PA103 or PA103ΔU at a MOI of 20 bacteria per macrophage for the indicated times were immunoblotted with antibodies against IL-β the p10 subunit of caspase-1, IκBα, phosphorylated-p38, p38, phosphorylated-ERK1/2, or ERK1/2. Results are representative of two independent experiments.
To determine whether the phospholipase activity of ExoU is required for inhibition of caspase-1–dependent cytokine secretion, we used a reversible inhibitor of iPLA2 and cPLA2, AACOCF3. Pretreatment of LPS-primed macrophages with AACOCF3 had no effect on cell death or IL-1β secretion induced by infection with PA103ΔU (Fig. 4, A and B). AACOCF3 did, however, significantly decrease the amount of cell death induced by the ExoU containing strain PA103 (Fig. 4 A), confirming the findings of others (28, 29). AACOCF3 pretreatment of LPS-primed macrophages also resulted in an increased amount of secreted IL-1β (Fig. 4 B). In addition, AACOCF3 pretreatment of unprimed macrophages infected with PA103 resulted in the activation of caspase-1 in contrast to untreated macrophages in which PA103 infection failed to activate caspase-1 (Fig. 4 C). These results demonstrate that the phospholipase activity of ExoU not only affects macrophage cell death, but also has dramatic effects on the ability of the macrophage to effectively activate caspase-1 and secrete proinflammatory cytokines. It was interesting to find that the inhibition of ExoU phospholipase A2 activity with AACOCF3 resulted in an increase of IPAF-driven IL-1β secretion, whereas AACOCF3 completely inhibited the NALP3-driven production of IL-1β by treatment with ATP (Fig. 4 B).
Figure 4.

P. aeruginosa ExoU phospholipase activity is required for inhibition of caspase-1 activation. (A and B) LPS-stimulated WT macrophages were either left untreated or pretreated with 100 μM AACOCF3 for 30 min, and then infected with the P. aeruginosa strain PA103 or PA103ΔU at a MOI of 20 bacteria per macrophage. 1 h after infection, culture supernatants were collected. Parallel wells of untreated or AACOCF3-pretreated LPS-stimulated WT macrophages were stimulated with 5 mM ATP for 30 min, and then culture supernatants were collected. Cytotoxicity was measured by LDH release into culture supernatants, and IL-1β release was measured by ELISA. Determinations were performed in triplicate and expressed as the mean ± the SD. *, P = 0.03; **, P = 0.0001. Results are representative of two separate experiments. (C) Nonstimulated WT macrophages were either left untreated or pretreated with 100 μM AACOCF3 for 30 min, and then infected with the P. aeruginosa strain PA103 or PA103ΔU at a MOI of 20 bacteria per macrophage. 90 min after infection, the combined lysate of cells and supernatant were immunoblotted with antibodies against the p10 subunit of caspase-1. (D) Nonstimulated WT macrophages were infected with the P. aeruginosa strain PA103, PA103ΔUT, PA103ΔU, PA103ΔT, PA103ΔUT/LS608, or PA103ΔUT/S142A at a MOI of 20 bacteria per macrophage. 3 h after infection, culture supernatants were collected and IL-1β release was measured by ELISA. Determinations were performed in triplicate and expressed as the mean ± the SD. Results are representative of three separate experiments.
The role of ExoU PLA2 activity in inhibiting caspase-1 activation was also tested using ExoU point mutants. ExoU bearing an alanine substitution for the putative catalytic serine residue (referred to as ExoU-S142A) has previously been shown to lack PLA2 activity and to be noncytotoxic (31). The ExoU-LS608 variant of ExoU contains a 5 amino acid insertion in the localization domain (immediately before N608) and is not appropriately targeted to the plasma membrane; this protein also lacks PLA2 activity and is noncytotoxic (31, 32). PA103ΔUT expressing either ExoU-S142A or -LS608 were unable to inhibit IL-1β secretion from WT macrophages (Fig. 4 D), again suggesting that the phospholipase activity of ExoU is required for the inhibition of caspase-1 activation.
In vivo production of IL-1β and -18 in response to P. aeruginosa peritonitis is dependent on IPAF
To assess whether ExoU inhibits caspase-1–driven cytokine production in vivo, we used a P. aeruginosa peritonitis model. WT mice were infected intraperitoneally with 1 × 107 CFU of either P. aeruginosa strain PA103 or PA103ΔU, and serum cytokines were analyzed 4 h after infection. The caspase-1–driven cytokines IL-1β (P = 0.036) and IL-18 (P < 0.0001) were both significantly higher in serum from mice infected with PA103ΔU compared with PA103 (Fig. 5, A and B), whereas serum TNFα (P = 0.313) levels were not statistically different between PA103- and PA103ΔU-infected mice (Fig. 5 C). Collectively, these data recapitulate in vivo our in vitro finding that ExoU is capable of inhibiting caspase-1 activation.
Figure 5.

In vivo cytokine production induced by P. aeruginosa peritonitis. (A–C) WT mice were injected intraperitoneally with 1 × 107 CFU of either PA103 (n = 15 mice) or PA103ΔU (n = 15 mice); 4 h after infection, mice were killed and blood was collected by cardiac puncture. Serum IL-1β, IL-18, and TNFα concentrations were measured by ELISA. Data represent the mean ± the SEM. *, P = 0.036; **, P < 0.0001. (D–F) WT or IPAF−/− mice were injected intraperitoneally with 1 × 107 CFU of PA103ΔU; 4 h after infection, mice were killed and blood was collected by cardiac puncture. Serum IL-1β (n = 10 mice per group), IL-18 (n = 10 mice per group), and TNFα (n = 10 WT mice, n = 9 IPAF−/− mice) concentrations were measured by ELISA. Data represent the mean ± the SEM, ***, P = 0.023; ****, P = 0.014. (G) WT (n = 8), IPAF+/− (n = 6), or IPAF−/− (n = 10) mice were injected intraperitoneally with 5 × 106 CFU of PA103ΔU; 12 h after infection, mice were killed and peritoneal lavage with 10 ml of PBS was collected, spleens were removed and homogenized, and dilutions were plated on Vogel-Bonner minimal media for enumeration of CFUs. Bacterial counts from peritoneal lavage and spleen of IPAF−/− mice were significantly higher compared with WT mice (P < 0.0001 and P = 0.0009, respectively) and with IPAF+/− mice (P = 0.0005 and P = 0.0017, respectively).
We next asked whether IPAF was responsible for the secretion of caspase-1–driven cytokines in vivo in response to infection with the ExoU-deficient P. aeruginosa strain PA103ΔU. Serum cytokines were analyzed from WT and IPAF−/− mice 4 h after intraperitoneal infection with 1 × 107 CFU of PA103ΔU. Serum IL-1β (P = 0.023) and IL-18 (P = 0.014) were both significantly decreased in PA103ΔU-infected IPAF−/− mice compared with WT (Fig. 5, D and E). Serum TNFα (P = 0.357) levels were not statistically different between PA103ΔU-infected WT and IPAF−/− mice (Fig. 5 F). These data support a significant role for IPAF in the production of caspase-1–driven cytokines in vivo in response to infection with P. aeruginosa. Bacterial burdens from peritoneal lavage and spleen of WT, IPAF+/−, and IPAF−/− mice 12 h after intraperitoneal infection with 5 × 106 CFU of PA103ΔU were determined (Fig. 5 G). Bacterial burdens in the peritoneal lavage fluid and spleen were significantly higher in IPAF-deficient mice compared with WT (P < 0.0001 and P = 0.0009, respectively) or IPAF+/− mice (P = 0.0005 and P = 0.0017, respectively). Hence, IPAF is important in innate immune defenses required to control the replication of PA103ΔU in vivo.
An intact type III secretion system is required for P. aeruginosa–induced activation of caspase-1
Because no P. aeruginosa effector molecule appeared to be responsible for activation of caspase-1, we examined if the TTSS itself was required for P. aeruginosa-mediated caspase-1 activation. We used two TTSS mutants, PA103pscJ∷Tn5 (MutN), which fails to assemble a TTSS apparatus, and PA103pcrV∷Tn5 (Mut1), which assembles a TTSS apparatus lacking translocon components. Neither PA103MutN nor PA103Mut1 were able to induce macrophage cell death, as has been previously described (12, 33), or to induce IL-1β secretion upon infection (Fig. 6, A and B). We also failed to detect caspase-1 activation by Western blot after infection of macrophages with either PA103MutN or PA103Mut1 (Fig. 6 C), suggesting that an intact TTSS is required for P. aeruginosa–induced caspase-1 activation. Pseudomonas type IV pili are not required for effector molecule delivery by the TTSS, though they appear to render the process more efficient (34). Infection of macrophages with the type IV pilus mutant, PA103ΔpilA, still resulted in caspase-1 activation, macrophage cell-death, and IL-1β secretion, although to a lesser extent than PA103ΔUT (Fig. 6, A and B). IPAF-deficient macrophages failed to secrete IL-1β when infected with PA103ΔpilA, demonstrating that IL-1β secretion triggered by PA103ΔpilA was still IPAF dependent (unpublished data).
Figure 6.

An intact TTSS is required for P. aeruginosa-mediated caspase-1 activation. LPS-stimulated WT macrophages were infected with the P. aeruginosa strain PA103ΔUT, PA103MutN, PA103Mut1, PA103ΔpilA, PA103ΔpopB, or PA103ΔpopD at a MOI of 20 bacteria per macrophage. At the indicated time (A and B) or 3 h (D and E) after infection, culture supernatants were collected and cytotoxicity was measured by LDH release and expressed as a percentage of LDH release by Triton X-100 detergent. IL-1β release into the culture supernatant was measured by ELISA. Determinations were performed in triplicate and expressed as the mean ± the SD. Results are representative of two (A and B) or three (D and E) separate experiments. (C) Lysates from LPS-stimulated WT macrophages that were infected with the P. aeruginosa strain PA103ΔUT, PA103MutN, PA103Mut1, or PA103ΔpilA for 3 h were immunoblotted with antibodies against the p10 subunit of caspase-1.
PA103Mut1, which was not able to activate caspase-1, does not produce PopB or PopD, the two proteins that form the translocon pore within the host cell membrane. We examined whether both proteins were required for caspase-1 activation by infecting macrophages with PA103ΔpopB and PA103ΔpopD, which are deficient in PopB and PopD, respectively. Infection with either PA103ΔpopB or PA103ΔpopD did not induce macrophage cell death or induce IL-1β secretion (Fig. 6, D and E), suggesting that membrane disruption by the translocon is a required step for P. aeruginosa–induced caspase-1 activation.
IPAF-dependent activation of caspase-1 by P. aeruginosa is not dependent on bacterial flagellin
Recent studies have suggested that IPAF recognizes cytosolic flagellin (20, 21, 23). As PA103ΔU does not assemble flagella on its surface, but nonetheless activates caspase-1 in an IPAF-dependent manner, we decided to test the requirement for flagellin explicitly. We used PAK and an isogenic mutant deficient in flagellin (PAKΔfliC) to determine if the IPAF-dependent caspase-1 activation we observed in response to P. aeruginosa infection was caused by flagellin. We first confirmed that the flagellin-deficient P. aeruginosa mutant, PAKΔfliC, did not express flagellin (35). Polyclonal antibody against PAK FliC protein was used to probe whole cell extract of the PAK strain and the PAKΔfliC mutant by Western blot. Cell extracts from the PAK strain immunoblotted with anti-FliC polyclonal antibody revealed a 50-kD product, which was not observed in cell extracts from the PAKΔfliC mutant (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20071239/DC1). WT macrophages infected with PAKΔfliC still secreted a large amount of IL-1β, although less than compared with that induced by PAK (Fig. 7 A). Importantly, the IL-1β secreted in response to infection with both PAK and PAKΔfliC was IPAF dependent, as IPAF-deficient macrophages secreted greatly diminished IL-1β in response to infection with PAK and PAKΔfliC than did WT macrophages (Fig. 7, A and B). Caspase-1 activation in PAKΔfliC-infected WT macrophages was detected by Western blot by the appearance of the p10 cleavage product (Fig. 7 C). PAKΔfliC failed to activate caspase-1 in IPAF-deficient macrophages (Fig. 7 C). These data demonstrate that flagellin is not required for P. aeruginosa–induced caspase-1 activation.
Figure 7.

IPAF-dependent activation of caspase-1 by P. aeruginosa does not require flagellin. (A) LPS-stimulated macrophages from WT or IPAF-deficient mice were infected with P. aeruginosa strain PAK or the flagellin-deficient PAKΔfliC at a MOI of 20 bacteria per macrophage. 1 h after infection, culture supernatants were collected, and IL-1β release into culture supernatants was measured by ELISA. Determinations were performed in triplicate and expressed as the mean ± the SD. Results are representative of three separate experiments. (B and C) LPS-stimulated macrophages from WT or IPAF-deficient mice were infected with P. aeruginosa strain PAKΔfliC at a MOI of 20 bacteria per macrophage. (B) At the indicated times after infection, culture supernatants were collected and IL-1β release into culture supernatants was measured by ELISA. (C) Lysates were immunoblotted with antibodies against the p10 subunit of caspase-1. Determinations were performed in triplicate and expressed as the mean ± the SD. Results are representative of two separate experiments.
DISCUSSION
Caspase-1 and IL-1β have been postulated to have deleterious effects for the host in acute P. aeruginosa-infections. Schultz et al. found that IL-1R deficiency had a protective effect in response to pulmonary infection with P. aeruginosa (36). In another study, neutralization of IL-1β with antibodies protected acid sphingomyelinase-deficient mice from lethal P. aeruginosa pneumonia (37). In addition, Thakur et al. found in a P. aeruginosa keratitis model, that caspase-1 deficiency results in less corneal damage (38). However, it remains unclear from these studies if the adverse effects mediated by IL-1β and caspase-1 are caused by inflammatory damage or lack of control of bacterial replication. In contrast to these studies, IL-1R–deficient mice had greater colonization with P. aeruginosa compared with WT mice after oral exposure to the pathogen in drinking water (39). We show that the IPAF-inflammasome plays an important role in host defenses against P. aeruginosa. IPAF-deficiency resulted in increased bacterial burdens in the peritoneum and spleen of mice infected intraperitoneally with PA103ΔU, which also correlated with a decrease in serum IL-1β and IL-18.
Our results demonstrate that P. aeruginosa–induced macrophage cell death and secretion of IL-1β occur in a manner that is dependent on activation of the IPAF inflammasome. After macrophage infection with P. aeruginosa, caspase-1 is activated through an IPAF-dependent mechanism. The resulting activation of caspase-1 leads to the processing and secretion of IL-1β and the initiation of macrophage cell death. The adaptor molecule ASC contains a Pyrin and a CARD domain and can form a multiprotein complex along with NALP3 and caspase-1 that results in caspase-1 activation and processing of pro-IL-1β (15). As IPAF can interact directly with pro-caspase-1 through a CARD–CARD interaction, the role of ASC in IPAF-mediated caspase-1 activation remains unclear. We found that ASC-deficient macrophages were partially defective in IL-1β secretion, but still underwent P. aeruginosa–induced cell death with similar kinetics compared with WT macrophages (Fig. S1, C and D). This was surprising, as caspase-1 activation is not observed in ASC-deficient macrophages after P. aeruginosa infection (Fig. 1 H). However, this is consistent with the finding that S. typhimurium–induced caspase-1–dependent cell death, although IPAF dependent, was only partially dependent on ASC (18). These observations suggest that ASC is, in fact, crucial for IPAF-mediated caspase-1 activation, and that in the absence of ASC, IPAF may partner with another caspase, such as caspase-11, to mediate cell death.
Recent studies have suggested that cytosolic flagellin can activate the IPAF inflammasome (20–23). The intracellular pathogen, L. pneumophila can activate caspase-1 upon infection of macrophages, a process that requires IPAF and a second NLR protein called Naip5 (Birc1e) (19). L. pneumophila mutants deficient in the gene encoding flagellin fail to activate caspase-1 (22, 23). S. typhimurium also activates caspase-1 in an IPAF-dependent manner, but does not require Naip5 (18). S. typhimurium mutants deficient in flagellin were found to be defective in their ability to activate caspase-1 (20, 21). In contrast, we found that infection with either the nonflagellated strain PA103ΔU or the flagellin-deficient mutant strain PAKΔfliC, resulted in the robust activation of caspase-1 and secretion of IL-1β (Fig. 7, A–C). At higher multiplicity of infection (MOI), flagellin-deficient S. typhimurium strains were also capable of inducing macrophage secretion of IL-1β (21). Collectively, these data suggest that IPAF-dependent activation of caspase-1 occurs independently of bacterial flagellin. P. aeruginosa flagellin may act to enhance TTSS-dependent caspase-1 activation by aiding bacterial adhesion to the host cell in a manner similar to type IV pili.
A functional TTSS that contains the translocon proteins PopB and PopD was required for P. aeruginosa–induced caspase-1 activation and IL-1β secretion. Both S. typhimurium and S. flexneri also require their TTSS for activation of caspase-1. Likewise, L. pneumophila–induced caspase-1 activation is dependent on the Dot-Icm type IV secretion system, which is structurally unrelated but functionally analogous to the TTSS (19). Thus, the type III and IV secretion systems are a common feature of Gram-negative pathogens shown to activate the IPAF inflammasome. We have demonstrated that flagellin is not required for IPAF-dependent caspase-1 activation by P. aeruginosa. We favor the hypothesis that in the case of P. aeruginosa, IPAF detects membrane damage induced by the type III secretion system translocon rather than any specific pathogen-derived molecule.
In addition to the IPAF/caspase-1–dependent macrophage cell death that we have described, P. aeruginosa also possesses multiple additional methods to induce host cell death. Upon infection of epithelial cells, P. aeruginosa induces the up-regulation of CD95/CD95 ligand on the cell surface. Ligation of CD95 by the CD95 ligand results in the activation of caspases 8 and 3 and the initiation of apoptosis of the epithelial cell (40). Additional studies are needed to determine if the cell death pathways initiated by P. aeruginosa in macrophages are the same as those in epithelial cells (12, 41). The P. aeruginosa TTSS is capable of delivering a combination of four different effector proteins, ExoS, ExoT, ExoU, and ExoY, into the host cell. Of these, ExoS and ExoU have been shown to be cytotoxic. Using the P. aeruginosa mutant strain PA103ΔUT, which has an intact TTSS but lacks all known effector molecules, we were able to demonstrate that caspase-1–dependent macrophage cell death and IL-1β secretion do not require the action of any described TTSS effectors. In addition, ExoU-expressing P. aeruginosa were still capable of killing macrophages, even in the absence of IPAF. These data identify at least two pathways that P. aeruginosa can exploit to kill macrophages, i.e., IPAF-dependent caspase-1 activation and IPAF-independent ExoU-mediated cytotoxicity/necrosis.
The effector molecule ExoU also had a dramatic and unexpected effect on cytokine secretion. In vivo infection of mice with the P. aeruginosa mutant strain PA103ΔU led to substantially more IL-1β and -18 production than infection with ExoU-expressing PA103. This effect was mediated by the inhibition of caspase-1 activation by ExoU. As either pharmacologic inhibition of PLA2 activity or infection with strains expressing ExoU mutants lacking PLA2 activity resulted in enhanced caspase-1 activation and IL-1β secretion, ExoU phospholipase activity appears central to its ability to suppress caspase-1 activation. It is unclear if ExoU-mediated caspase-1 inhibition occurs directly or through the effects of ExoU on another intermediate. Although less than one-third of clinical isolates express ExoU, strains that secrete ExoU cause more severe disease than strains that do not (7). Disruption of the exoU gene resulted in decreased virulence of P. aeruginosa in mouse models of acute pneumonia (42). In addition, patients with nosocomial P. aeruginosa pneumonias who were infected with ExoU-secreting isolates had worse outcomes than patients infected with isolates that did not secrete type III effectors (43). The ability of ExoU to inhibit caspase-1 production may serve as an additional virulence mechanism that ExoU-expressing strains of P. aeruginosa use to evade the immune system.
It has recently been demonstrated that membrane damage induced by pore-forming toxins, such as listeriolysin O and aerolysin, can activate caspase-1 through a NALP3-dependent pathway (25, 44). Disruption of the phagosome by Francisella tularensis has also been demonstrated to activate caspase-1 in an ASC-dependent manner (45). It is possible that IPAF and NALP3 recognize an endogenous molecule that is induced or modified by pathogen-induced membrane disruption rather than direct recognition of the pathogen itself. This has been described for the Arabidopsis R protein RPS5, which, like the NLRs, contains a nucleotide-binding site and leucine-rich repeats. In Arabidopsis, resistance to P. syringae strains expressing AvrPphB requires RPS5 and the protein kinase PBS1 (46). Proteolytic cleavage of PBS1 by AvrPphB is required for RPS5-mediated resistance. This suggests that RPS5 does not directly detect AvrPphB, but rather recognizes this protein indirectly through its cleavage of endogenous PBS1. Further studies to identify the downstream events after membrane disruption by the TTSS that result in IPAF inflammasome activation will be critical to understanding how IPAF participates in pathogen sensing.
MATERIALS AND METHODS
Mice.
The generation of NALP3-, ASC-, caspase-1–, and IPAF-deficient mice has been previously described (27, 47). ASC and NALP3-deficient mice were backcrossed onto the C57BL/6 genetic background for at least 8 generations. IPAF- and caspase-1–deficient mice were backcrossed onto the C57BL/6 genetic background for at least 5 generations. Age- and sex-matched C57BL/6 mice purchased from The Jackson Laboratory were used as WT controls for most studies. IPAF+/− mice were intercrossed to generate mutant and control mice for experiments using IPAF-deficient mice. All protocols used in this study were approved by the Yale Institutional Animal Care and Use Committee.
Bacterial strains and growth conditions.
The P. aeruginosa strain PAK and the isogenic PAKΔfliC mutant, in which fliC is disrupted by a tetracycline resistance cassette, were obtained from J. Mattick (University of Queensland, Brisbane, Australia). All other P. aeruginosa mutants used in this study are isogenic with PA103 (48), which is a nonflagellated clinical isolate that produces two Type III secreted exotoxins, ExoU and ExoT. The type III secretion mutant PA103 pscJ∷Tn5 (mutN) (33), the type III translocation-deficient mutant PA103 pcrV∷Tn5 (mut1) (49), the pilin-deficient strain PA103pilA∷aacC1 (50), and the strains lacking ExoU (PA103ΔU), ExoT (PA103ΔT), or both exotoxins (PA103ΔUT) have been previously described (51). The ExoU variants PA103ΔUT/S142A and PA103ΔUT/LS608 were expressed in P. aeruginosa by integrating a single copy of the mutated exoU allele with its endogenous promoter into the chromosomal aatB site of strain PA103ΔUT, as previously described (31).
Unmarked in-frame deletions of popB (Δaa 11–371) and popD (Δaa 7–274) were constructed by allelic exchange (52). 5′ and 3′ sequences flanking each gene were amplified by PCR and cloned in tandem into pEX18Gm (53) to create the vectors pEX18-ΔpopB and pEX18-ΔpopD. The plasmids were mobilized into PA103 by conjugation; exconjugants were initially selected on Vogel-Bonner media plus gentamicin (100 μg/ml), and then subcultured to VBM plus 5% sucrose to select for vector backbone excision. Candidate ΔpopB and ΔpopD isolates were screened by PCR and confirmed by Southern blotting (unpublished data).
P. aeruginosa strains were cultured in Luria–Bertani broth overnight, diluted 1:50 (volume/volume), and grown for 2 h. Bacteria were washed and resuspended in PBS.
Macrophages.
Thioglycollate-elicited peritoneal macrophages were generated by injecting 1 ml of 3% thioglycollate solution into the peritoneal cavity of mice. 3–4 d later, macrophages were collected by peritoneal lavage and plated in DME supplemented with 10% FCS, 2 mM l-glutamine, 100 U/ml penicillin G, and 100 μg/ml streptomycin. Unless otherwise indicated, macrophages were activated by stimulating with 50 ng/ml LPS from E. coli serotype 0111:B4 (Invivogen) for 12–16 h before infection with P. aeruginosa.
In vitro bacterial infection.
Macrophages that were either unstimulated or had been activated with 50 ng/ml LPS for 12–16 h were washed, and the medium was replaced with antibiotic-free DME supplemented with 10% FCS and 2 mM l-glutamine. Macrophages were then infected with P. aeruginosa at a MOI of 20 bacteria per macrophage unless otherwise indicated; plates were then centrifuged for 3 min at 700 rpm in a swinging-bucket centrifuge. At the indicated time after infection, supernatants were collected. Culture supernatants were then assayed for IL-1β and TNFα. Antibody pairs for the IL-1β and TNFα ELISAs were purchased from R&D Systems and eBioscience, respectively. Antibody pairs for the IL-6 ELISA were obtained from BD Biosciences. Macrophage cell death was determined at the indicated time points by LDH release using a cytotoxicity (LDH) detection kit (Promega).
Bacterial peritonitis.
Mice (8–12 wk old) were injected intraperitoneally with 1 × 107 CFU of either P. aeruginosa strain PA103 or PA103ΔU in 250 μl of DME. 4 h after infection, mice were killed and blood was collected by cardiac puncture. Serum was diluted two- to threefold in PBS and assayed for IL-1β, TNFα, and IL-18 levels by ELISA. Antibody pairs for the IL-18 ELISA were purchased from MBL. To determine bacterial loads in the peritoneum and spleen, mice (8–12 wk old) were injected intraperitoneally with 5 × 106 CFU of P. aeruginosa strain PA103ΔU in 250 μl of DME. 12 h after infection, mice were killed, peritoneal lavage with 10 ml of PBS was collected, and the spleen was homogenized in PBS. Dilutions were plated on Vogel-Bonner minimal medium to determine CFUs.
Western blotting.
Electrophoresis of proteins was performed using the NuPAGE system (Invitrogen) according to manufacturer's protocol. In brief, after infection with P. aeruginosa, macrophages and supernatants were lysed in lysis buffer (50 mM Tris-HCL, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, and a protease inhibitor cocktail [Roche]) and stored at −80oC until analyzed. Proteins were separated on a NuPAGE gel, and transferred to PVDF membrane by electroblotting. To detect caspase-1 a rabbit polyclonal anti–mouse casapse-1 p10 antibody (Santa Cruz Biotechnology, Inc.) was used. Antibodies to phosphorylated-IκBα, IκBα, phosphorylated-p38, p38, phosphorylated-ERK1/2, ERK1/2, and caspase-3 were purchased from Cell Signaling Technology. The anti–IL-1β antibody used for Western blotting was purchased from Chemicon International.
Statistical analysis.
We performed statistical analysis by using an unpaired Student's t test for all studies, except bacterial burdens, which were compared by the nonparametric Mann-Whitney test. We considered values of P < 0.05 to be statistically significant.
Online supplemental material.
Fig. S1 (A–D) shows kinetics of macrophage cell death and IL-1β secretion in WT, ASC-, and caspase-1–deficient macrophages in response to infection with P. aeruginosa strain PAK. Fig. S1 E shows that NALP3-deficient macrophages are not defective in caspase-1 activation in response to infection with PAK. Fig. S2 (A–C) shows that, similar to unprimed macrophages, caspase-1 activation and IL-1β secretion in LPS-primed macrophages is inhibited by ExoU. Fig. S2 D shows that caspase-3 activation occurs with similar kinetics in unprimed macrophages infected with either PA103 or PA103ΔU. Fig. S3 shows the absence of flagellin expression in the P. aeruginosa strain PAKΔfliC. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20071239/DC1.
Supplemental Material
Acknowledgments
We thank Jorge Galán and Maria Lara-Tejero for helpful discussion and Suzanne Cassel and Lauren Zenewicz for critical review of the manuscript. We thank Anthony Coyle, Ethan Grant, and John Bertin for providing mutant mice and Alan Hauser for providing the P. aeruginosa ExoU variants PA103ΔUT/S142A and PA103ΔUT/LS608.
This work was supported by grants from the Ellison Foundation (R.A.F), the Patrick and Catherine Weldon Donaghue Medical Research Foundation (B.I. Kazmierczak), National Institutes of Health (NIH) K08 AI065517 (F.S. Sutterwala), NIH F31 AI061882 (L.A. Mijares), and NIH R01 AI054920 (B.I. Kazmierczak). F.S. Sutterwala was supported by a Pfizer Fellowship in Infectious Diseases. B.I. Kazmierczak is a Donaghue Investigator. R.A. Flavell is an investigator of the Howard Hughes Medical Institute.
The authors have no conflicting financial interests.
Abbreviations used: CARD, caspase activation and recruitment domain; LDH, lactate dehydrogenase; MOI, multiplicity of infection; TTSS, type III secretion system.
B.I. Kazmierczak and R.A. Flavell contributed equally to this paper.
F.S. Sutterwala's present address is Inflammation Program, Dept. of Medicine, University of Iowa, Coralville, IA 52241.
References
- 1.Barbieri, J.T., and J. Sun. 2004. Pseudomonas aeruginosa ExoS and ExoT. Rev. Physiol. Biochem. Pharmacol. 152:79–92. [DOI] [PubMed] [Google Scholar]
- 2.Sato, H., and D.W. Frank. 2004. ExoU is a potent intracellular phospholipase. Mol. Microbiol. 53:1279–1290. [DOI] [PubMed] [Google Scholar]
- 3.He, S.Y., K. Nomura, and T.S. Whittam. 2004. Type III protein secretion mechanism in mammalian and plant pathogens. Biochim. Biophys. Acta. 1694:181–206. [DOI] [PubMed] [Google Scholar]
- 4.Dacheux, D., J. Goure, J. Chabert, Y. Usson, and I. Attree. 2001. Pore-forming activity of type III system-secreted proteins leads to oncosis of Pseudomonas aeruginosa-infected macrophages. Mol. Microbiol. 40:76–85. [DOI] [PubMed] [Google Scholar]
- 5.Frithz-Lindsten, E., A. Holmström, L. Jacobsson, M. Soltani, J. Olsson, R. Rosqvist, and A. Forsberg. 1998. Functional conservation of the effector protein translocators PopB/YopB and PopD/YopD of Pseudomonas aeruginosa and Yersinia pseudotuberculosis.Mol. Microbiol. 29:1155–1165. [DOI] [PubMed] [Google Scholar]
- 6.Roy-Burman, A., R.H. Savel, S. Racine, B.L. Swanson, N.S. Revadigar, J. Fujimoto, T. Sawa, D.W. Frank, and J.P. Wiener-Kronish. 2001. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J. Infect. Dis. 183:1767–1774. [DOI] [PubMed] [Google Scholar]
- 7.Schulert, G.S., H. Feltman, S.D. Rabin, C.G. Martin, S.E. Battle, J. Rello, and A.R. Hauser. 2003. Secretion of the toxin ExoU is a marker for highly virulent Pseudomonas aeruginosa isolates obtained from patients with hospital-acquired pneumonia. J. Infect. Dis. 188:1695–1706. [DOI] [PubMed] [Google Scholar]
- 8.Vance, R.E., A. Rietsch, and J.J. Mekalanos. 2005. Role of the type III secreted exoenzymes S, T, and Y in systemic spread of Pseudomonas aeruginosa PAO1 in vivo. Infect. Immun. 73:1706–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hersh, D., D.M. Monack, M.R. Smith, N. Ghori, S. Falkow, and A. Zychlinsky. 1999. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA. 96:2396–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, Y., M.R. Smith, K. Thirumalai, and A. Zychlinsky. 1996. A bacterial invasin induces macrophage apoptosis by binding directly to ICE. EMBO J. 15:3853–3860. [PMC free article] [PubMed] [Google Scholar]
- 11.Hilbi, H., J.E. Moss, D. Hersh, Y. Chen, J. Arondel, S. Banerjee, R.A. Flavell, J. Yuan, P.J. Sansonetti, and A. Zychlinsky. 1998. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 273:32895–32900. [DOI] [PubMed] [Google Scholar]
- 12.Hauser, A.R., and J.N. Engel. 1999. Pseudomonas aeruginosa induces type-III-secretion-mediated apoptosis of macrophages and epithelial cells. Infect. Immun. 67:5530–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fritz, J.H., R.L. Ferrero, D.J. Philpott, and S.E. Girardin. 2006. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 7:1250–1257. [DOI] [PubMed] [Google Scholar]
- 14.Inohara, N., M. Chamaillard, C. McDonald, and G. Núñez. 2005. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu. Rev. Biochem. 74:355–383. [DOI] [PubMed] [Google Scholar]
- 15.Mariathasan, S., and D.M. Monack. 2007. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 7:31–40. [DOI] [PubMed] [Google Scholar]
- 16.Ting, J.P., and B.K. Davis. 2005. CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu. Rev. Immunol. 23:387–414. [DOI] [PubMed] [Google Scholar]
- 17.Dinarello, C.A. 1998. Interleukin-1β, interleukin-18, and the interleukin-1β converting enzyme. Ann. N. Y. Acad. Sci. 856:1–11. [DOI] [PubMed] [Google Scholar]
- 18.Mariathasan, S., K. Newton, D.M. Monack, D. Vucic, D.M. French, W.P. Lee, M. Roose-Girma, S. Erickson, and V.M. Dixit. 2004. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 430:213–218. [DOI] [PubMed] [Google Scholar]
- 19.Zamboni, D.S., K.S. Kobayashi, T. Kohlsdorf, Y. Ogura, E.M. Long, R.E. Vance, K. Kuida, S. Mariathasan, V.M. Dixit, R.A. Flavell, et al. 2006. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7:318–325. [DOI] [PubMed] [Google Scholar]
- 20.Franchi, L., A. Amer, M. Body-Malapel, T.D. Kanneganti, N. Ozoren, R. Jagirdar, N. Inohara, P. Vandenabeele, J. Bertin, A. Coyle, et al. 2006. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin-1β in salmonella-infected macrophages. Nat. Immunol. 7:576–582. [DOI] [PubMed] [Google Scholar]
- 21.Miao, E.A., C.M. Alpuche-Aranda, M. Dors, A.E. Clark, M.W. Bader, S.I. Miller, and A. Aderem. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin-1β via Ipaf. Nat. Immunol. 7:569–575. [DOI] [PubMed] [Google Scholar]
- 22.Molofsky, A.B., B.G. Byrne, N.N. Whitfield, C.A. Madigan, E.T. Fuse, K. Tateda, and M.S. Swanson. 2006. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203:1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren, T., D.S. Zamboni, C.R. Roy, W.F. Dietrich, and R.E. Vance. 2006. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanneganti, T.D., N. Ozoren, M. Body-Malapel, A. Amer, J.H. Park, L. Franchi, J. Whitfield, W. Barchet, M. Colonna, P. Vandenabeele, et al. 2006. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 440:233–236. [DOI] [PubMed] [Google Scholar]
- 25.Mariathasan, S., D.S. Weiss, K. Newton, J. McBride, K. O'Rourke, M. Roose-Girma, W.P. Lee, Y. Weinrauch, D.M. Monack, and V.M. Dixit. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 440:228–232. [DOI] [PubMed] [Google Scholar]
- 26.Martinon, F., V. Petrilli, A. Mayor, A. Tardivel, and J. Tschopp. 2006. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 440:237–241. [DOI] [PubMed] [Google Scholar]
- 27.Sutterwala, F.S., Y. Ogura, M. Szczepanik, M. Lara-Tejero, G.S. Lichtenberger, E.P. Grant, J. Bertin, A.J. Coyle, J.E. Galán, P.W. Askenase, and R.A. Flavell. 2006. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 24:317–327. [DOI] [PubMed] [Google Scholar]
- 28.Phillips, R.M., D.A. Six, E.A. Dennis, and P. Ghosh. 2003. In vivo phospholipase activity of the Pseudomonas aeruginosa cytotoxin ExoU and protection of mammalian cells with phospholipase A2 inhibitors. J. Biol. Chem. 278:41326–41332. [DOI] [PubMed] [Google Scholar]
- 29.Sato, H., D.W. Frank, C.J. Hillard, J.B. Feix, R.R. Pankhaniya, K. Moriyama, V. Finck-Barbançon, A. Buchaklian, M. Lei, R.M. Long, et al. 2003. The mechanism of action of the Pseudomonas aeruginosa-encoded type III cytotoxin, ExoU. EMBO J. 22:2959–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, V.T., R.S. Smith, B. Tummler, and S. Lory. 2005. Activities of Pseudomonas aeruginosa effectors secreted by the Type III secretion system in vitro and during infection. Infect. Immun. 73:1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rabin, S.D., and A.R. Hauser. 2005. Functional regions of the Pseudomonas aeruginosa cytotoxin ExoU. Infect. Immun. 73:573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabin, S.D., J.L. Veesenmeyer, K.T. Bieging, and A.R. Hauser. 2006. A C-terminal domain targets the Pseudomonas aeruginosa cytotoxin ExoU to the plasma membrane of host cells. Infect. Immun. 74:2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang, P.J., A.R. Hauser, G. Apodaca, S.M. Fleiszig, J. Wiener-Kronish, K. Mostov, and J.N. Engel. 1997. Identification of Pseudomonas aeruginosa genes required for epithelial cell injury. Mol. Microbiol. 24:1249–1262. [DOI] [PubMed] [Google Scholar]
- 34.Jendrossek, V., S. Fillon, C. Belka, I. Muller, B. Puttkammer, and F. Lang. 2003. Apoptotic response of Chang cells to infection with Pseudomonas aeruginosa strains PAK and PAO-I: molecular ordering of the apoptosis signaling cascade and role of type IV pili. Infect. Immun. 71:2665–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray, T.S., and B.I. Kazmierczak. 2006. FlhF is required for swimming and swarming in Pseudomonas aeruginosa.J. Bacteriol. 188:6995–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schultz, M.J., A.W. Rijneveld, S. Florquin, C.K. Edwards, C.A. Dinarello, and T. van der Poll. 2002. Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L285–L290. [DOI] [PubMed] [Google Scholar]
- 37.Grassmé, H., V. Jendrossek, A. Riehle, G. von Kurthy, J. Berger, H. Schwarz, M. Weller, R. Kolesnick, and E. Gulbins. 2003. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat. Med. 9:322–330. [DOI] [PubMed] [Google Scholar]
- 38.Thakur, A., R.P. Barrett, S. McClellan, and L.D. Hazlett. 2004. Regulation of Pseudomonas aeruginosa corneal infection in IL-1β converting enzyme (ICE, caspase-1) deficient mice. Curr. Eye Res. 29:225–233. [DOI] [PubMed] [Google Scholar]
- 39.Reiniger, N., M.M. Lee, F.T. Coleman, C. Ray, D.E. Golan, and G.B. Pier. 2007. Resistance to Pseudomonas aeruginosa chronic lung infection requires cystic fibrosis transmembrane conductance regulator-modulated interleukin-1 (IL-1) release and signaling through the IL-1 receptor. Infect. Immun. 75:1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grassmé, H., S. Kirschnek, J. Riethmueller, A. Riehle, G. von Kurthy, F. Lang, M. Weller, and E. Gulbins. 2000. CD95/CD95 ligand interactions on epithelial cells in host defense to Pseudomonas aeruginosa.Science. 290:527–530. [DOI] [PubMed] [Google Scholar]
- 41.Coburn, J., and D. Frank. 1999. Macrophages and epithelial cells respond differently to the Pseudomonas aeruginosa type III secretion system. Infect. Immun. 67:3151–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finck-Barbançon, V., J. Goranson, L. Zhu, T. Sawa, J.P. Wiener-Kronish, S.M. Fleiszig, C. Wu, L. Mende-Mueller, and D.W. Frank. 1997. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol. Microbiol. 25:547–557. [DOI] [PubMed] [Google Scholar]
- 43.Hauser, A.R., E. Cobb, M. Bodi, D. Mariscal, J. Valles, J.N. Engel, and J. Rello. 2002. Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa.Crit. Care Med. 30:521–552. [DOI] [PubMed] [Google Scholar]
- 44.Gurcel, L., L. Abrami, S. Girardin, J. Tschopp, and F.G. van der Goot. 2006. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 126:1135–1145. [DOI] [PubMed] [Google Scholar]
- 45.Mariathasan, S., D.S. Weiss, V.M. Dixit, and D.M. Monack. 2005. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202:1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shao, F., C. Golstein, J. Ade, M. Stoutemyer, J.E. Dixon, and R.W. Innes. 2003. Cleavage of Arabidopsis PBS1 by a bacterial type III effector. Science. 301:1230–1233. [DOI] [PubMed] [Google Scholar]
- 47.Lara-Tejero, M., F.S. Sutterwala, Y. Ogura, E.P. Grant, J. Bertin, A.J. Coyle, R.A. Flavell, and J.E. Galan. 2006. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J. Exp. Med. 203:1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu, P.V. 1966. The roles of various fractions of Pseudomonas aeruginosa in its pathogenesis: Identity of the lethal toxins produced in vitro and in vivo. J. Infect. Dis. 116:481–489. [DOI] [PubMed] [Google Scholar]
- 49.Hauser, A.R., P.J. Kang, S.J.M. Fleiszig, K. Mostov, and J. Engel. 1998. Defects in type III secretion correlate with internalization of Pseudomonas aeruginosa by epithelial cells. Infect. Immun. 66:1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Comolli, J.C., L.L. Waite, K.E. Mostov, and J.N. Engel. 1999. Pili binding to asialo-GM1 on epithelial cells can mediate cytotoxicity or bacterial internalization by Pseudomonas aeruginosa.Infect. Immun. 67:3207–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garrity-Ryan, L., B. Kazmierczak, R. Kowal, J. Comolli, A. Hauser, and J.N. Engel. 2000. The arginine finger domain of ExoT is required for actin cytoskeleton disruption and inhibition of internalization of Pseudomonas aeruginosa by epithelial cells and macrophages. Infect. Immun. 68:7100–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schweizer, H.P., and T.T. Hoang. 1995. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa.Gene. 158:15–22. [DOI] [PubMed] [Google Scholar]
- 53.Hoang, T.T., R.R. Karkhoff-Schweizer, A.J. Kutchma, and H.P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 212:77–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}