

Introduction
The activity state of mitochondrial branched-chain α-keto acid dehydrogenase (BCKDH) is highly regulated according to the physiological requirements for protein synthesis or degradation of excess branched-chain amino acids.1 This regulation is primarily exerted through reversible phosphorylation of the catalytic subunit of BCKDH by a specific protein kinase and a specific phosphoprotein phosphatase. Two phosphorylation sites exist on the E1α subunit although regulation of BCKDH activity results exclusively from phosphorylation of site 1, Ser-293 in rat E1α. Phosphorylation of site 2, Ser-303 in rat E1α, occurs at a significantly slower rate2 and is silent with respect to regulation of BCKDH activity.3 Thiamine pyrophosphate (TPP) and branched-chain α-keto acids (cofactor and substrates for the BCKDH-catalyzed reaction) inhibit the phosphorylation by BCKDH kinase, and it has been suggested that BCKDH kinase inactivates BCKDH E1 by placing a covalently linked phosphate directly into the active site of the dehydrogenase.4 Studies of the mechanism of inactivation of BCKDH by phosphorylation require a ready source of purified subunits of this multienzyme complex that can be reconstituted into an active BCKDH complex under various conditions. To this end we have expressed the BCKDH E1 component as a recombinant enzyme5 in a form that can be easily purified and reconstituted with the purified transacylase core (E2) of the BCKDH complex. The purified recombinant E1 subunit is enzymatically active when combined with E2, can be inactivated by treatment with the purified recombinant BCKDH kinase and ATP, and can be manipulated by site-directed mutagenesis.
Materials
R408 and VCS M13 are purchased from Stratagene (La Jolla, CA). Vectors pET15 and pET21 are purchased from Novagen (Madison, WI). Nickel-NTA-agarose is purchased from Qiagen (Valencia, CA). Restriction enzymes, restriction enzyme buffers, and other DNA-modifying enzymes are purchased from BRL (Gaithersburg, MD). All chemical reagents for enzyme assay are purchased from Sigma (St. Louis, MO). The site-directed mutagenesis system is purchased from Amersham (Arlington Heights, IL). Native rat liver BCKDH E2 subunit is prepared by a modification of the method of Cook et al.6 according to Shimomura et al.7 The pGroESL plasmid was a kind gift of A. Gatenby (Central Research and Development, Du Pont, Wilmington, DE).
Construction of Prokaryotic Expression Vector for Branched-Chain α-Keto Acid Dehydrogenase E1
A prokaryotic expression vector5 containing rat BCKDH E1α and E1β cDNAs is constructed as follows. A BamHI site is introduced into the 5′ end of rat E1β cDNA by site-directed mutagenesis. The 1220-base pair (bp) BamHI fragment, containing nucleotides 62–1281 of rat E1β, is ligated to the BamHI site of pET-21a (Novagen) to produce plasmid pET-E1β. This plasmid carries the E1α-coding sequence in frame for expression from the T7 promoter. A 2474-bp MaeI fragment, containing nucleotides 87–1683 of rat E1α cDNA and nucleotides 450–1301 of pUC19, is isolated from the rat E1α cDNA and ligated to the NdeI site of pET-15b (Novagen) to produce the plasmid pET-E1α. This plasmid carries the E1α cDNA with an in-frame fusion of a hexahistidine tag and a T7 promoter. A fragment carrying the T7 promoter, the E1α-coding region, and the T7 terminator is isolated from the pET-E1α plasmid by ClaI digestion, followed by “blunting” with T4 DNA polymerase, and SphI digestion. The pET-E1β plasmid is then digested with BglII followed by “blunting” with T4 DNA polymerase and digestion with SphI. The E1α blunt-ended SphI fragment is then ligated to the blunt-ended SphI-cut pET E1β plasmid. This ligation produced an expression vector with two separate “cassettes” with a T7 promoter and T7 terminator for the E1α and E1β cDNAs. The final expression vector (Fig. 1) was prepared by performing site-directed mutagenesis to remove 57 extra bases (encoding vector sequence and a portion of the mitochondrial leader sequence) from the 5′ end of the E1β cDNA. E1α in this construct has 11 extra amino acids at the N terminus corresponding to the leader sequence, and 19 extra amino acids from the vector that include the histidine tag and a thrombin cleavage site. This vector is designated pET-E1.
Fig. 1.
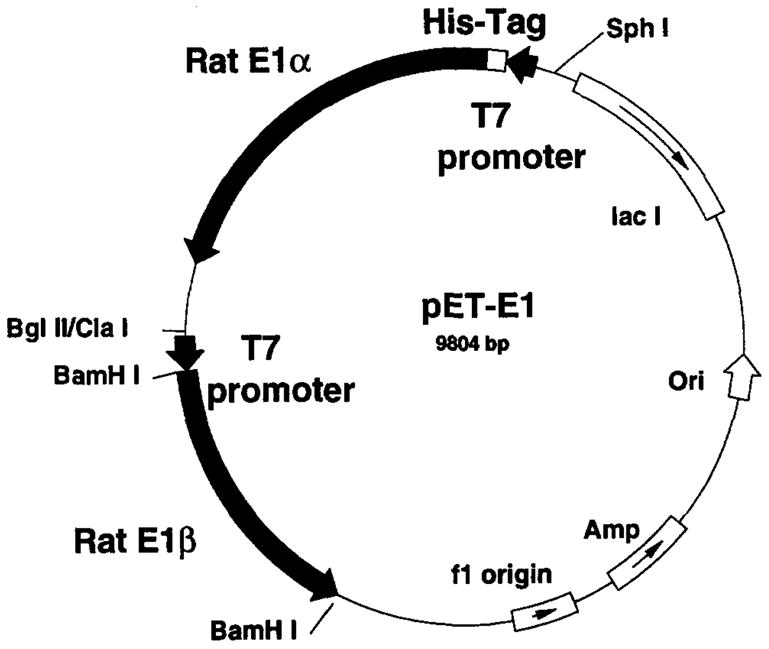
Prokaryotic vector construct for expression of rat BCKDH E1.
Site-Directed Mutagenesis of Branched-Chain α-Keto Acid Dehydrogenase E1α
Single-stranded pET-E1 DNA is isolated by single-strand rescue, using either R408 or VCS M13 helper phage. Single-strand rescue and purification of the single-stranded DNA are performed exactly according to the manufacturer protocols (Stratagene). Site-directed mutagenesis5,8 is performed with the Sculptor system (Amersham), following the manufacturer protocol. Mutants are identified by dideoxynucleotide sequencing methods, using the double-stranded plasmid DNA as template.
Expression and Purification of Recombinant Branched-Chain α-Keto Acid Dehydrogenase E1
In this procedure 5,8 BL21(DE3) Escherichia coli ceils are double transfected with the pET-E1 plasmid (ampicillin resistant) and the pGroESL plasmid (chloramphenicol resistant). Double transfectants are selected by growth at 37° in TY medium containing both kanamycin and chloramphenicol (70 μg/ml each). When the cell cultures obtain an optical density at 600 nm (OD600 nm) of approximately 0.8, isopropyl-β-D-thiogalactopyranoside (IPTG) is added to a final concentration of 0.5 mM and growth is continued for 18–20 hr at 37°. The induced bacterial cultures are then pelleted and resuspended in 10 volumes of buffer A [20 mM Tris-HCl (pH 8.0), 0.5 M NaCl, 10 mM 2-mercaptoethanol, 5 mM imidazole] containing 0.5% (v/v) Triton X-100, and a 100-μg/ml concentration of both benzamidine and phenylmethylsulfonyl fluoride (PMSF). Cells are lysed by sonication and the resulting homogenates are centrifuged at 20,000g for 30 min at 4°. The soluble fraction is applied to a 10-ml nickel-NTA (Qiagen) column previously equilibrated in the same buffer. The column is washed with 5 volumes of buffer A containing 20 mM imidazole, 5 volumes of buffer A containing 40 mM imidazole, and 5 volumes of buffer A containing 60 mM imidazole. The histidine-tagged protein is eluted with buffer A containing 200 mM imidazole. Elution fractions containing E1 protein are pooled, brought to 20% (v/v) glycerol, and stored at −70°. Purified enzyme is analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS--PAGE) and appears as two bands (E1α and E1β proteins) in equal relative quantities. The coexpression of GroEL and GroES increases the yield of soluble E1 by approximately threefold in this system.
Assay of Branched-Chain α-Keto Acid Dehydrogenase Activity
Enzyme activity is measured spectrophotometrically at 340 nm by monitoring the generation of NADH produced at 30° by the combined action of the BCKDH E1, E2, and E3 subunits. Each assay consists of 1 ml of cocktail containing 30 mM potassium phosphate (pH 7.5), 0.1% (v/v) Triton X-100, 1 mM NAD+, 2 mM dithiothreitol (DTT), 0.4 mM coenzyme A (CoA), 0.4 mM TPP, 2 mM MgCl2, 7.5 units of porcine heart dihydrolipoamide reductase (E3), 1 μg of E1, and 0.75 μg of E2. A baseline absorbance is measured for several minutes and the reaction is then initiated by the addition of 0.5 mM α-ketoisovalerate (KIV). In this assay, a nonlinear phase, or “lag phase,” is observed for the activity of the reconstituted complex. This nonlinear phase extends for approximately 5 min, followed by a linear rate of NADH production. The cause of this initial nonlinear phase has not been determined. For kinetic studies, the KIV solutions are standardized by an end-point assay using purified BCKDH complex.9 The concentrations of TPP solutions are measured by absorbance at 267 nm (E = 8520 M−1 cm−1).
Reconstitution of Branched-Chain α-Keto Acid Dehydrogenase E1/E2 Complex
BCKDH E1/E2 subcomplex is reconstituted 5,8 with purified recombinant E1s and purified native E2. Increasing the ratio of E1 to E2 protein in the reconstituted complex increases BCKDH activity until the E1/E2 ratio is approximately 1.10 Activity reaches a plateau at E1/E2 ratios above 1, suggesting that E1 is rate limiting below a ratio of 1 and that E2 is rate-limiting above a ratio of 1 (Fig. 2).
Fig. 2.

BCKDH activity of reconstituted complexes with different E1/E2 ratios. A constant amount of purified native E2 subunit (25 pmol) was reconstituted with different amounts of purified recombinant BCKDH E1 (0–30 pmol). Excess bovine E3 subunit was present in the assay mixutres. Inset: Western analysis with E2- and E1α-specific antisera. [Reproduced with permission from Y. Zhao, K. M, Popov, Y. Shimomura, N. Y. Kedishvili, J. Jaskiewicz, M. J. Kuntz, J. Kain, B. Zhang, and R. A. Harris, Arch. Biochem. Biophys. 308, 446 (1994).]
Assay of Branched-Chain α-Keto Acid Dehydrogenase E2 Binding by Mutant Branched-Chain α-Keto Acid Dehydrogenase E1 Enzymes
A number of site-directed mutants have been produced with this system. Mutations of the phosphorylation site 1, Ser-293, affect dehydrogenase activity and E1 kinetic parameters, as do other mutations surrounding this site. It is important to determine if such mutations block E1 activity by affecting the E1 active site, or by affecting the interaction of E1 and E2 during reconstitution of the complex. Interaction of inactive mutant E1 proteins with the BCKDH E2 subunit is measured by a method similar to that of Cook et al.6 This assay is based on the observation that inactive mutant E1 proteins can inhibit the reconstitution of enzymatically active complex with wild-type E1 and E2. A constant amount of E2 protein is combined with a constant amount of E1 protein, using various ratios of wild-type and mutant E1s. The amount of E1 is in excess of the amount of E2 to assure that E2 is rate limiting for BCKDH activity and that E1-binding sites on E2 are saturated. Reconstituted complexes with different ratios of mutant and wild-type E1 are assayed for BCKDH activity as described above. Mutant E1 proteins that appear to reconstitute with E2 produce approximately 50% inhibition when present at an equimolar ratio with wild-type E1 (Fig. 3).8
Fig. 3.
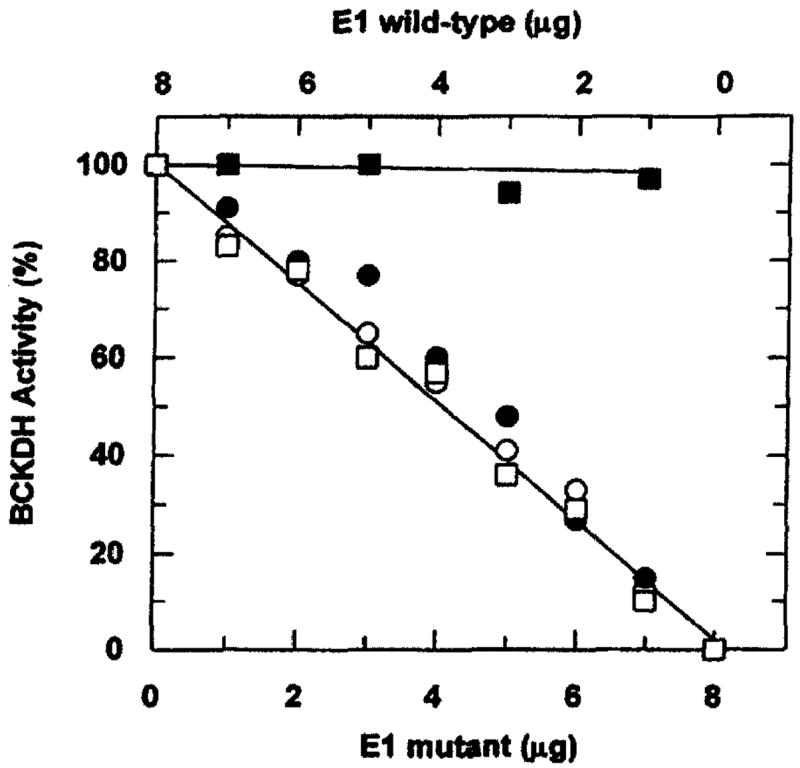
BCKDH activity of reconstituted complexes with different ratios of wild-type and mutant E1. A constant amount of purified native E2 (0.75 μg) was used for reconstitution with a constant total amount of E1 (8 μg) but with various ratios of mutant and wild-type E1 (■) Amount of wild-type E1 varied as indicated without addition of mutant E1; (○) amount of total E1 kept constant (8 μg) by varying R288A and wild-type E1; (●) by varying H292A and wild-type E1; (□) by varying D296A and wild-type E1. [Reproduced with permission from J. W. Hawes, R. J. Schnepf, A. E. Jenkins, Y. Shimomura, K. M. Popov, and R. A. Harris. J. Biol. Chem. 270, 31071 (1995).]
Characteristics of Wild-Type and Mutant Branched-Chain α-Keto Acid Dehydrogenase E1s
BCKDH subcomplex reconstituted with wild-type E1 and native E2 displays enzyme activity and kinetic parameters similar to those of native BCKDH complex (Table I).5 Mutations of the two known phosphorylation sites on the E1α subunit (S293 and S303) produce differing effects on activity and kinetic parameters. S303A and S303E mutant E1s display a specific activity and kinetic parameters unchanged from that of wild-type E1 (Table I). S293A E1 displays a 12-fold increase in Km for KIV without a significant change in Vmax and S293E E1 is enzymatically inactive.5 These data suggest a role for this residue in α-keto acid substrate binding, and it has been shown that this residue is completely conserved in the amino acid sequences of all known BCKDH E1α proteins as well as those of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase.8 Alanine substitutions of three other residues surrounding S293 also abolish the enzymatic activity of E1.8 These include R288A, H292A, and D296A. These three mutant E1s are devoid of enzyme activity at all pH values tested and all substrate and cofactor concentrations tested; however, they appear to bind to the E2 subunit as efficiently as the wild-type E1 (Fig. 3). Evidence has been obtained for the involvement of His-292 in thiamine pyrophosphate binding.8
TABLE I.
Kinetic Parameters of Wild-Type and Mutant E1 Proteinsa
| Recombinant E1 | Km (α-ketisovalerate) (μM) | Vmax (U/mg protein)b |
|---|---|---|
| Wild-type | 45 ± 13c | 5.4 ± 0.7 |
| S293E | — | — |
| S293A | 532 ± 98 | 5.5 ± 0.6 |
| S303E | 56 ± 9 | 5.2 ± 0.2 |
| S303A | 58 ± 15 | 5.8 ± 0.7 |
Reproduced in part with permission from Y. Zhao, J. W. Hawes, K. M. Popov, J. Jaskiewicz, Y. Shimomura, D. W. Crabb, and R. A. Harris, J. Biol. Chem. 269, 18583 (1994).
Refers to units (μmoles of NADH produced per minute) per milligram of recombinant E1 protein.
Mean ± SE; data obtained with four concentrations of α-ketoisovalerate spanning the Km value.
Acknowledgments
This work was supported by grants from the U.S. Public Health Services (NIH DK19259 to R.A.H.), the Diabetes Research and Training Center of Indian University School of Medicine (AM 20542), the Grace M. Showalter Trust (to R.A.H.), the Uehara Memorial Foundation (to Y.S), and the University of Tsukuba Project Research Fund (to Y.S.).
References
- 1.Harris RA, Popov KM, Zhao Y, Shimomura Y. J Nutr. 1994;124:1499S. doi: 10.1093/jn/124.suppl_8.1499S. [DOI] [PubMed] [Google Scholar]
- 2.Paxton R, Kuntz MJ, Harris RA. Arch Biochem Biophys. 1986;244:187. doi: 10.1016/0003-9861(86)90108-6. [DOI] [PubMed] [Google Scholar]
- 3.Cook KG, Bradford AP, Yeaman SJ, Aitken A, Fearnley IM, Walker JE. Eur J Biochem. 1984;145:587. doi: 10.1111/j.1432-1033.1984.tb08597.x. [DOI] [PubMed] [Google Scholar]
- 4.Harris RA, Hawes JW, Popov KM, Zhao Y, Shimomura Y, Sato J, Jaskiewicz J, Hurley TD. Adv Enzyme Regul. 1997;37:271. doi: 10.1016/s0065-2571(96)00009-x. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Y, Hawes JW, Popov KM, Jaskiewicz J, Shimomura Y, Crabb DW, Harris RA. J Biol Chem. 1994;269:18583. [PubMed] [Google Scholar]
- 6.Cook KG, Bradford AP, Yeaman SJ. Biochem J. 1985;225:731. doi: 10.1042/bj2250731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimomura Y, Paxton R, Ozawa T, Harris RA. Anal Biochem. 1987;163:74. doi: 10.1016/0003-2697(87)90094-7. [DOI] [PubMed] [Google Scholar]
- 8.Hawes JW, Schnepf RJ, Jenkins AE, Shimnomura Y, Popov KM, Harris RA. J Biol Chem. 1995;270:31071. doi: 10.1074/jbc.270.52.31071. [DOI] [PubMed] [Google Scholar]
- 9.Goodwin GW, Kuntz MJ, Paxton R, Harris RA. Anal Biochem. 1987;162:536. doi: 10.1016/0003-2697(87)90430-1. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Popov KM, Shimomura Y, Kedishvili NY, Jaskiewicz J, Kuntz MJ, Kain J, Zhang B, Harris RA. Arch Biochem Biophys. 1994;308:446. doi: 10.1006/abbi.1994.1063. [DOI] [PubMed] [Google Scholar]