

Abstract
Motor-powered movement along microtubule tracks is important for membrane organization and trafficking. However, the molecular basis for membrane transport is poorly understood, in part because of the difficulty in reconstituting this process from purified components. Using video microscopic observation of organelle transport in vitro as an assay, we have purified two polypeptides (245 and 170 kD) from Dictyostelium extracts that independently reconstitute plus-end–directed membrane movement at in vivo velocities. Both polypeptides were found to be kinesin motors, and the 245-kD protein (DdUnc104) is a close relative of Caenorhabditis elegans Unc104 and mouse KIF1A, neuron-specific motors that deliver synaptic vesicle precursors to nerve terminals. A knockout of the DdUnc104 gene produces a pronounced defect in organelle transport in vivo and in the reconstituted assay. Interestingly, DdUnc104 functions as a dimeric motor, in contrast to other members of this kinesin subfamily, which are monomeric.
Keywords: kinesin, microtubule, organelle transport, Dictyostelium, dynein
Movement of membrane organelles along microtubules plays an important role in the organization of the cytoplasm and the delivery of macromolecules in most eukaryotes (reviewed in Goodson et al. 1997; Lane and Allan 1998). Distributions of large intracellular membrane compartments, such as the perinuclear location of the Golgi and lysosomes and the extended reticular network of the endoplasmic reticulum, are achieved through microtubule-based movement. Transport between compartments in the secretory or endocytic pathways also relies on active transport along a polarized microtubule network. In most cells, the microtubules extend from the centrosome, where their minus-ends are located, towards the plasma membrane. This organization provides a navigational system for membrane trafficking between the cell center and the periphery.
Membrane transport along microtubules is driven by several types of ATP-hydrolyzing molecular motors. Cytoplasmic dynein (minus-end directed) (Harada et al. 1998; Schnapp and Reese 1989; Schroer et al. 1989) and three classes of plus-end–directed kinesin proteins (the conventional kinesins) (Schroer et al. 1988; Hollenbeck and Swanson 1990; Tanaka et al. 1998), the heteromeric kinesins (e.g., KRP85/95, KIF3A/B) (Morris and Scholey 1997; Nonaka et al. 1998; Tuma et al. 1998), and the “monomeric” kinesins (e.g., Unc104, KIF1A, KIF1B) (Hall and Hedgecock 1991; Nangaku et al. 1994; Yonekawa et al. 1998) have all been shown to serve as motors for organelle transport. Rabkinesin-6 (Echard et al. 1998) and KIFC2 (Hanlon and Goldstein 1997; Saito et al. 1997) are also candidate organelle transport motors. None of the above motors, however, was discovered based on an ability to stimulate organelle movement. Rather, they were initially identified through in vitro microtubule gliding assays [kinesin (Vale et al. 1985), cytoplasmic dynein (Paschal et al. 1987)], antibody (e.g., KRP85/95; Cole et al. 1992), and PCR (KIFs; Aizawa et al. 1992) screens designed to uncover new kinesin superfamily members, or genetic screens in Caenorhabditis elegans for muscular defects (Unc104; Hall and Hedgecock 1991). The involvement of these motors in organelle transport was later established using approaches such as gene knockouts (Saxton et al. 1991; Seiler et al. 1997; Gindhart et al. 1998; Harada et al. 1998; Nonaka et al. 1998; Tanaka et al. 1998), in vivo antibody microinjections (Rodionov et al. 1991; Lippincott-Schwartz et al. 1995; Morris and Scholey 1997; Tuma et al. 1998), colocalization with membranes (Pfister et al. 1989; Okada et al. 1995), and inhibition of organelle transport in vitro by antibodies, pharmacologic inhibitors, or immunodepletion (Schnapp and Reese 1989; Schroer et al. 1988, Schroer et al. 1989; Blocker et al. 1997).
The above studies have provided valuable insight into both the biological roles and types of subcellular membranes transported by these motors. Nevertheless, the mechanisms by which motors interact with and move their cargoes are poorly understood. In vitro organelle transport studies, however, have shown that cytoplasmic dynein is incapable of moving organelles by itself and requires dynactin as a cytosolic activator (Gill et al. 1991; Schroer and Sheetz 1991). Dynactin's importance for dynein-based organelle transport in vivo was subsequently confirmed by genetic and dominant negative studies (McGrail et al. 1995; Tinsley et al. 1996; Burkhardt et al. 1997). Although dynactin's precise role is unclear, recent studies have shown that the Arp1 subunit of dynactin interacts with a spectrin isoform, which may provide a mechanism for docking dynein onto intracellular membranes (Holleran et al. 1996). Another study, however, has reported that rhodopsin acts as a direct dynein receptor in the absence of dynactin (Tai et al. 1999).
Less is known about the mechanism of organelle motility driven by plus-end–directed motors. Whether soluble activators are required for kinesin-based in vitro organelle motility has been controversial (Schroer et al. 1988; Schroer and Sheetz 1991; Urrutia et al. 1991; Schnapp et al. 1992). A potential receptor for conventional kinesin (kinectin) has been identified through affinity chromatography (Toyoshima et al. 1992; Kumar et al. 1995), but its role in vivo has not been established. Furthermore, unlike the in vitro assays for cytoplasmic dynein described above, plus-end–directed organelle transport has not been faithfully reconstituted using a biochemically defined system. Highly purified or recombinant conventional kinesin has been shown to bind to membranes (Skoufias et al. 1994), but not to reproducibly elicit their movement on microtubules (Schroer et al. 1988).
To make progress on understanding the molecular basis of plus-end–directed organelle transport, a system amenable to both in vitro biochemical reconstitution and genetic analysis is needed. Here, we have developed such a system using the cellular slime mold Dictyostelium discoideum. Using video microscopic observation of in vitro organelle movement as a biochemical fractionation assay, we have purified two polypeptides that independently reconstitute transport. When combined with salt-washed vesicles that do not move on their own, the purified proteins faithfully reconstitute plus-end–directed movement with the same properties observed in crude extracts and in vivo, suggesting that they are likely to be physiologically important for this process. One of the purified factors is a novel dimeric kinesin that is closely related to Unc104 and KIF1A, monomeric kinesin motors that transport synaptic vesicle precursors in higher eukaryotes. Disruption of this kinesin gene reveals that it is the dominant plus-end–directed organelle transport motor in Dictyostelium.
Materials and Methods
Purification of Plus-End–directed Transport Activity
Dictyostelium high speed supernatant (HSS)1 was prepared as previously described (Pollock et al. 1998). Wild-type cells [5 liters at a density of 4–8 × 106 cells/ml in HL-5 media (Sussman 1987) containing 100 μg/ml streptomycin and 100 U/ml penicillin] were collected by centrifugation at 4,000 g for 15 min at 4°C and washed in 1 liter ice-cold Sorenson's phosphate buffer, pH 6.0 (Malchow et al. 1972). Cells were resuspended in 1:1 wt/vol lysis buffer (LB: 30 mM Tris-HCl, pH 8.0, 4 mM EGTA, 3 mM DTT, 5 mM benzamidine, 10 μg/ml soybean trypsin inhibitor, 5 μg/ml TPCK/TAME, 10 μg/ml leupeptin, pepstatin A, and chymostatin, and 5 mM PMSF) containing 30% (wt/vol) sucrose. The suspension was split into thirds and each third was lysed by one passage through a nucleopore polycarbonate filter (5-μm pore size, 47-mm diameter; Costar Corp.) using a 10-ml syringe. The lysate was centrifuged at 2,000 g for 5 min at 4°C. The resulting post-nuclear supernatant was layered over a 1-ml cushion of LB/25% sucrose and centrifuged in a rotor (TLA 100.4; Beckman Instruments, Inc.) at 180,000 g for 15 min at 4°C to obtain an HSS. 5 liters of cells generated ∼20 ml of HSS.
A microtubule–affinity-purified fraction (ATP releasate) was then prepared from the HSS as previously described (Pollock et al. 1998). In brief, HSS was incubated with 15 U/ml hexokinase, 3 mM glucose, 4 mM AMP-PNP/MgCl2, 20 μM taxol, and 0.5 mg/ml taxol-stabilized microtubules for 20 min on ice. Microtubules and associated proteins were centrifuged through a 1-ml cushion of LB/25% sucrose containing 20 μM taxol at 85,000 g for 15 min (4°C). The microtubule pellet was resuspended in LB/5% sucrose containing 0.3 M KCl (1:12 vol of the original HSS volume) and immediately recentrifuged at 85,000 g for 15 min (4°C). The resulting supernatant (“salt wash”), containing predominantly minus-end–directed transport activity, was removed. The microtubule pellet was resuspended in phosphate buffer (10 mM potassium phosphate, pH 6.8, 1 mM EGTA, 3 mM DTT, 5 mM benzamidine, 10 μg/ml soybean trypsin inhibitor, 5 μg/ml TPCK/TAME, 10 μg/ml leupeptin, pepstatin A, and chymostatin, and 5 mM PMSF)/5% sucrose containing 5 mM ATP/MgCl2 (1:20 vol of the original HSS volume) to elute the plus-end–directed transport activity. After incubation for 15 min on ice, microtubules were separated from the released proteins by centrifuging at 90,000 g for 15 min (4°C). The supernatant (ATP releasate) was collected and either assayed directly (see below) or frozen in liquid nitrogen.
1.5–2 ml of ATP releasate in phosphate buffer/5% sucrose was diluted to 2.26 ml in phosphate buffer/5% sucrose and CaCl2 was added to 1.2 mM. 2.2 ml of the releasate was then loaded onto a 1.8-ml hydroxyapatite column (20 μm ceramic hydroxyapatite; American International Chemical) equilibrated in phosphate buffer/5% sucrose (for hydroxyapatite and Mono Q chromatography buffers, protease inhibitors included only 5 mM benzamidine, 5 μg/ml TPCK/TAME, and 1 mM PMSF). The column was run on the SMART chromatography system (Amersham Pharmacia Biotechnology). Sample was loaded at a flow rate of 250 μl/min and 600-μl fractions were eluted at 500 μl/min in two steps of 200 and 300 mM potassium phosphate (3.6 ml each). For motility assays, 45-μl aliquots of all fractions were dialyzed for 2 h against LB pH 8.0/5% sucrose using a microdialyzer system 100 (Pierce Chemical Co.), exchanging the buffer in the chamber once after 30 min of dialysis. For further chromatography, the 200-mM potassium phosphate peak fractions were pooled and dialyzed against LB pH 9.0/5% sucrose using a microdialyzer system 500 (Pierce Chemical Co.).
The dialysate of the pooled hydroxyapatite fractions was diluted to 2.26 ml in LB pH 9.0/5% sucrose and 2.2 ml was loaded onto a 100-μl SMART system Mono Q column (Mono Q PC 1.6/5; Amersham Pharmacia Biotechnology) equilibrated in LB pH 9.0/5% sucrose. The column was loaded at 50 μl/min, and 100-μl fractions were eluted at 100 μl/min with a 115-mM NaCl step elution (500 μl), followed by a 170–300 mM NaCl gradient (1 ml). 45-μl aliquots of fractions were dialyzed against LB pH 8.0/5% sucrose as above for use in motility assays.
In Vitro Organelle Transport Assays
Sea urchin sperm axoneme-nucleated microtubule structures were prepared in flow cells (10 μl) as described previously (Pollock et al. 1998). KI vesicles were also prepared as described previously (Pollock et al. 1998); these vesicles, which are a mixed population isolated from the crude extract, were isolated by sedimentation through a sucrose cushion, treated with 0.3 M KI to dissociate weakly bound proteins, and then resedimented through a sucrose cushion. The flow cells were first washed with 10 μl of LB pH 8.0 (without PMSF)/15% sucrose to remove any free microtubules, followed by the introduction of 10 μl of assay mix consisting of, in the case of HSS, 5 μl of HSS, 3.5 μl LB/15% sucrose, 1 μl KI-washed vesicles, and 0.5 μl ATP-regenerating mix (Pollock et al. 1998). Assay mixes for the ATP releasate consisted of 5 μl of ATP releasate, 3.0 μl LB/15% sucrose, 1 μl KI-washed vesicles, 0.5 μl of a 20-mg/ml casein stock, and 0.5 μl ATP-regenerating mix. Assay mixes for fractions from the hydroxyapatite and Mono Q columns (including the hydroxyapatite load) consisted of 8 μl of the fraction, 1 μl KI vesicles, 0.5 μl casein, and 0.5 μl ATP regenerating mix. The movement of organelles was observed using a microscope (Axioplan; Carl Zeiss, Inc.) equipped with differential interference contrast (DIC) optics, a 50- or 100-W mercury arc lamp, and a 63×, 1.4 NA Plan-Neofluor objective. Images were detected using a camera (Newvicon; Hamamatsu Photonics); contrast enhancement and background subtraction were performed with an image processor (Argus10; Hamamatsu Photonics), and recordings were made with a super VHS video tape recorder (AG-5700; Panasonic).
Organelle motility was quantified by counting the number of movements in each direction on a single axoneme/microtubule structure. Only axonemes with clearly defined polarity were used. Recordings were performed on axonemes with between 6 and 12 microtubules (8–14 μm each in length) polymerized from the plus ends. If an organelle paused briefly and then continued in the same direction, it was scored as a single movement. Velocities of movements were measured using an NIH-IMAGE–based measuring program developed by J. Hartman; only vesicles that moved smoothly over a distance of at least 1.5–2 μm were scored.
In Vivo Transport Assays
To observe linear organelle movements in live cells, 20 μl of null or control cells (grown to 3–7 × 106 cells/ml in HL-5 media containing 100 μg/ml streptomycin, 100 U/ml penicillin, and 5 μg/ml blasticidin (ICN Biomedicals Inc.) were introduced into flow cells (20 μl) made with coverslips that had been incubated overnight in 1 M HCl, and then washed extensively with water. The flow cells were inverted for 2 min, and then viewed by DIC optics as described above. Each cell was observed for 4 min, and the slide was discarded after 10 min of observation. Only linear and continuous movements >1 μm in length were scored.
To observe the movement of mitochondria in live cells, 108 cells were collected by centrifugation at 800 g for 3 min at room temperature (25°C). Cells were resuspended in 10 ml HL5 containing P/S/blasticidin and 50 nM Mitotracker (Molecular Probes, Inc.). Cells were incubated for 15 min at 22°C while shaking at 180 rpm. The cells were repelleted and washed once in 10 ml Sorenson's phosphate buffer at room temperature (25°C) and resuspended in 5 ml HL5 plus P/S/blasticidin. Cells were incubated for 30 min at 22°C while shaking at 180 rpm. Cells were then diluted 1:5 in phosphate buffer and flowed into flow cells (20 μl) prepared with acid-washed coverslips as described above. The flow cell was inverted for 2 min, and then viewed by fluorescent optics using a 63×, 1.4 NA objective and a SIT camera (Hamamatsu Photonics). Each cell was viewed for 1 min and the slide was discarded after a maximum of 15 min of observation. Only linear and continuous movements >1 μm in length were scored.
Hydrodynamic Analysis
Sucrose Density Gradients.
200 μl ATP releasate in LB pH 8.0/5% sucrose/0.15 M NaCl was loaded onto a 2.2 ml 10–25% continuous sucrose gradient (in LB/0.15 M NaCl) and spun for 5 h at 200,000 g in a rotor (TLS-55; Beckman) (4°C). In parallel, a 200-μl mix of calibration standards (BSA, 4.3S; aldolase, 7.4S; catalase, 11.3S; ferritin, 17.6S; and thyroglobulin, 19.4S) at 1 mg/ml each in LB/5% sucrose/0.15 M NaCl was run on a separate gradient. 200 μl fractions were collected from each gradient. The sedimentation profiles of the calibration proteins were monitored by Coomassie staining, while the profile of DdUnc104 was monitored by immunoblotting with an affinity-purified DdUnc104 peptide antibody (see below). The S value of DdUnc104 was determined by comparing its position in the gradient to the positions of the standards plotted against their S values.
Gel Filtration.
50 μl of the DdUnc104 peak from the sucrose gradient was loaded onto a 2.4-ml gel filtration column (SMART system: Superose 6 PC 3.2/30; Amersham Pharmacia Biotechnology) equilibrated in 30 mM Tris-HCl, pH 8.0, 4 mM EGTA, 3 mM DTT, 5 mM benzamidine, 5 μg/ml TPCK/TAME, and 0.15 M NaCl. The column was run at 30 μl/min and 50-μl fractions were collected. The elution profile of DdUnc104 was again monitored by immunoblotting. The elution profiles of calibration standards with known Stokes radii (3.1 nm ovalbumin, 4.8 nm aldolase, 5.2 nm catalase, 6.1 nm ferritin, and 8.5 nm thyroglobulin) prepared in the same buffer were assessed by Coomassie stain. Linear plots of the (−logK av)1/2 versus Stokes radius for each standard were used to determine the Stokes radius of DdUnc104 from its K av (K av = Ve − Vo/Vt − Vo, where Ve = elution volume, Vo = void volume as determined by the elution volume of blue dextran, and Vt = total column volume or the total accessible volume as assessed by the elution profile of 1 M NaCl).
Partial specific volume and axial ratio calculations were made using the Sednterp program developed by John Philo. Coiled-coil predictions were made using the program COILS (Lupas et al. 1991).
Peptide Sequencing
Mono Q fractions containing the 245- and 170-kD polypeptides were pooled, TCA precipitated, and separated on 4–12% gradient polyacrylamide gels (Novex) under denaturing and reducing conditions followed by staining with Coomassie blue. After cutting out the protein bands, in-gel digestion with Endoproteinase Lys-C (Boehringer Mannheim Biochemicals) was carried out as described (Hellman et al. 1995). Gel-extracted peptides were then fractionated using a Vydac microbore C8 column (The Separations Group) and individual peptides were subjected to Edman degradation with a protein sequencer (#492; Perkin-Elmer, Applied Biosystems Division).
Peptide Antibody Production and Immunoblotting
A peptide based on DdUnc104 amino acids 359–373 was synthesized with a COOH-terminal cysteine and used for immunization of a rabbit (QCB, Inc.). Peak bleeds were pooled, and the antibody was purified against the synthetic peptide coupled to a thiol-coupling resin using the QCB standard antibody purification protocol. The purified antibody was dialyzed into 80 mM Tris-HCl, pH 8.0, 4 mM EGTA for storage.
For immunoblotting, samples were separated on 4–12% gradient polyacrylamide gels (Novex) and electroblotted to nitrocellulose membranes at 100 mA for 75 min. The blots were incubated with the DdUnc104 peptide antibody (1:500) overnight at 4°C, and then incubated in HRP-conjugated secondary antibody (1:2,000; Amersham Life Sciences) for 1 h at room temperature. Blots were developed using a chemiluminescence kit (NEN Life Sciences) and exposed to Hyperfilm (Amersham Life Sciences).
Cloning of the DdUnc104 Gene
One of the peptide sequences of the 245-kD protein was class-conserved among members of the Unc104/KIF1A subfamily. Therefore, fully degenerate oligonucleotides corresponding to the VVNEDAQ peptide (amino acids 360–366) obtained from peptide sequencing and to a sequence highly conserved in the Unc104/KIF1A family, KSYTMMG, were used to prime a PCR reaction using Dictyostelium oligo (dT)-primed first-strand cDNA as a template (Superscript Preamplification System; GIBCO BRL). The expected ∼780-bp PCR product was cloned and sequenced. Primers made to this PCR product were used to generate a 470-bp probe that was used to screen a Dictyostelium vegetative cDNA library (λZAP; Stratagene Inc.; kindly provided by Dr. Rick Firtel, University of California, San Diego, San Diego, CA). An ∼2-kb DdUnc104 cDNA clone containing the starting methionine and an upstream stop codon was obtained from the screen; however, this cDNA terminated prematurely. To obtain further downstream sequence, total RNA was extracted from vegetative cells (Trizol; GIBCO BRL), and poly A+ RNA was further purified using an mRNA separator kit (Clontech). The poly A+ RNA was used as a template to generate cDNA that was then used for a 3′ rapid amplification of cDNA ends (RACE) reaction (Marathon cDNA Amplification Kit; Clontech) using a primer based on the 3′ end of the λZAP fragment. This method yielded a 1.8-kb DdUnc104 fragment that was continuous with the λZAP fragment, but which still terminated prematurely. Primers based on the 3′ end of this new fragment were used in a new 3′ RACE reaction, using the same cDNA pool, to obtain a 3-kb fragment containing the remainder of the DdUnc104 gene. (Note: the DdUnc104 gene is also known as ksnA in Dictyostelium nomenclature.) To confirm the sequences obtained from the RACE products, two additional independent PCR reactions were performed for each segment and the resulting clones were sequenced. Percent identity between protein sequences was calculated using the worldwide web version of the program Blast available at http://www.ncbi.nlm.nih.gov/BLAST. Multiple sequence alignment was done with the program Pileup (Genetics Computer Group) and the output was shaded using MACBOXSHADE.
Disruption of the DdUnc104 Gene
A 1.4-kb cassette conferring blasticidin resistance (Adachi et al. 1994) was cloned between the adjacent BglII sites (nucleotides 2115–2134) in the DdUnc104 sequence. The 3.2-kb construct was released from the plasmid by digestion with EcoRV (nucleotide 1484) and XbaI (nucleotide 3306), and transformed into cells by electroporation as described (de Hostos et al. 1993). Transformants were selected on petri plates containing liquid DD-broth20 media (Manstein et al. 1995) supplemented with 5 μg/ml blasticidin (ICN Biomedicals Inc.). Isolated colonies were picked and transferred to 24-well plates. After further growth, the cells were collected for screening by PCR. Approximately 106 cells were resuspended in 50 μl lysis buffer (10 mM Tris, pH 8.0, 1 mM EDTA, 0.3% Tween-20, 60 μg/ml proteinase K) and incubated for 45 min at 56°C and 10 min at 94°C. 1 μl of the crude lysate was used as a template for 25-μl PCR reactions set up using standard protocols. Pairs of a primer specific to the blasticidin resistance cassette and a DdUnc104-specific primer flanking the disruption site were used for the PCR. Two independent null cell lines were obtained and were shown to behave identically. Genomic DNA was prepared from selected transformant strains (QIAGEN Inc.) and analyzed by Southern blotting as described previously (de Hostos et al. 1998) using the EcoRV-XbaI fragment of the DdUnc104 cDNA as a probe. For Northern analysis, RNA was prepared using Trizol (GIBCO BRL) and the EcoRV-XbaI fragment was used as a probe. For Western analysis, 5 μl of ATP releasates prepared from null and control cells (see below) were immunoblotted using the affinity-purified DdUnc104 antibody.
Preparation of ATP Releasates from DdUnc104 Null and Control Cells
For analysis of in vitro motility, ATP releasates were prepared from null and control cells (500–800 ml at a density of 0.6–1 × 107 cells/ml in HL5 containing P/S/blasticidin) as described above, with two modifications. First, the 0.3-M KCl wash before ATP elution of the microtubule pellet was omitted to preserve minus-end motility in the ATP releasate. Second, the buffer for the ATP release was LB pH 8.0/5% sucrose containing 5 mM ATP/MgCl2.
Results
Purification of Proteins that Stimulate Plus-End–directed Organelle Transport from Dictyostelium Extracts
Previously, we developed a method of preparing extracts from D. discoideum that supported abundant bidirectional microtubule-based vesicle transport; the velocities and travel distances of moving organelles were similar to those displayed in vivo (Pollock et al. 1998; Roos et al. 1987). We also showed that motion of KI-washed organelles, which did not move on their own, could be reconstituted by the addition of an HSS (Pollock et al. 1998). Here, we wished to purify the factor(s) responsible for plus-end–directed vesicle transport using a biochemical fractionation scheme. Microtubules grown in a polarized manner from salt-extracted axonemes (Allen and Borisy 1974) were used to determine the direction of vesicle movement in these experiments. Microtubule affinity was used as a first purification step since both plus- and minus-end vesicle transport activities cosediment with exogenously added microtubules in the absence of ATP and the presence of AMP-PNP and can be subsequently released from the microtubules by ATP (Pollock et al. 1998). Such procedures are known to enrich for microtubule motors and associated proteins. Furthermore, the plus-end–directed organelle transport activity could be partially separated from the more abundant minus-end–directed activity by incubating the microtubules with 0.3 M KCl (to extract minus-end–directed activity selectively) before the ATP release step. This microtubule affinity procedure resulted in an 80-fold increase in specific activity for plus-end–directed organelle transport relative to the HSS (Table ).
Table 1.
Purification of Organelle Transport Activity
| Protein concentration | Specific activity | Fold enrichment | ||||
|---|---|---|---|---|---|---|
| Minus end | Plus end | Minus end | Plus end | |||
| mg/ml | U/mg protein | Relative to HSS | ||||
| A | HSS | 23.8 | 0.13 | 0.03 | 1 | 1 |
| B | Salt wash | 3.1 | 0.41 | 0.06 | 3 | 2 |
| C | ATP releasate | 0.6 | 0.56 | 2.30 | 4 | 77 |
| D | Hydroxyapatite peak | 0.16 | — | 3.75 | — | 125 |
| E | Mono Q peak | 0.03 | — | 11.67 | — | 389 |
Protein concentrations were measured by Bradford assay for the HSS, salt wash, and ATP releasate fractions (A–C). Measurements were obtained for two to three separate preparations and the results were averaged. Because the hydroxyapatite and Mono Q peaks (D and E) were too dilute to obtain protein concentrations by Bradford assay, concentrations were measured by comparative densitometry of Coomassie-stained fractions relative to BSA standards. Values reported for D and E correspond to hydroxyapatite fraction 17 and Mono Q fraction 13 in Fig. 1. For specific activity calculations, one unit of activity in the organelle transport assay was defined as one movement per minute on a single axoneme/microtubule structure of defined polarity. Multiple assays were performed for each fraction and the results were averaged. The average number of movements per minute supported by the fraction over a 4-min assay was divided by the number of microliters of the fraction used in the total 10-μl assay volume to obtain the activity concentration in movements per minute per microliter. This value was then divided by the protein concentration of the fraction to calculate specific activity in units per microgram. Little or no minus-end–directed movement was observed in the last two columns (—).
To rule out the possibility that the vesicle transport observed was due to nonspecific interactions with membrane lipids, we tested whether treatment of KI vesicles with trypsin (100 μg/ml for 30 min on ice followed by soybean trypsin inhibitor, SBTI, at 4 mg/ml for 10 min) reduced their movement in the presence of the ATP release fraction. Plus-end–directed vesicle motility was decreased threefold by this treatment compared with a control in which trypsin was preblocked with SBTI before addition to the vesicles. Thus, proteins on the vesicle surface play an important role in motility.
Since the ATP release fraction contained numerous polypeptides (Fig. 1 A, LOAD), we further purified the plus-end–directed vesicle transport activity using hydroxyapatite chromatography (Fig. 1 A). The activity bound to the column and could be eluted by potassium phosphate. Three prominent polypeptides appeared in the activity-containing fractions: the ∼540-kD dynein heavy chain and two polypeptides of 245 and 170 kD. Even though dynein was prominent in the hydroxyapatite eluate, very little (less than two movements/axoneme) or no minus-end–directed transport was observed in several independent purifications.
Figure 1.

Purification of two factors supporting plus-end–directed vesicle transport activity. (A) A microtubule affinity-purified fraction was prepared from HSS (see Materials and Methods) and loaded onto a hydroxyapatite column (LOAD). Three sets of fractions (flow-through, and 200- and 300-mM potassium phosphate step elutions) were collected, and each fraction was independently assayed for its ability to transport KI-washed organelles in the plus-end direction. Activity in each fraction was quantified as plus-end–directed movements per axoneme-microtubule complex over a 4-min assay. Two assays were performed for each fraction, and the results were averaged. Activity was spread over three fractions (16, 17, and 18) in the 200-mM potassium phosphate eluate and consistently copeaked with the 170- and 245-kD polypeptides. Although there was a significant amount of dynein heavy chain (DHC) that was eluted by 200 mM potassium phosphate, virtually no minus-end–directed transport activity was observed. Myosin, which is an abundant contaminant in the ATP releasate and also migrates at 245 kD, eluted at higher potassium phosphate (300 mM; lane 22) and could therefore be separated from the other 245-kD polypeptide; the myosin fraction displayed no transport activity. The fractions with peak activity from the hydroxyapatite column were pooled, dialyzed, and applied to a Mono Q column (B). The column was eluted with a step of 115 mM NaCl, followed by a 170–275-mM NaCl gradient; fractions were collected and assayed as above. The activity again coeluted with the 245- and 170-kD polypeptides (fractions 13 and 14). Although in this column polypeptides in the 60–80-kD range copeaked with activity, they were not observed to copeak in other purifications.
The peak fractions of the hydroxyapatite column were pooled, applied to a Mono Q column, and eluted with a shallow NaCl gradient, which proved necessary to separate the 245- and 170-kD polypeptides from one another (Fig. 1 B). The organelle transport activity peaked consistently with the 245-kD polypeptide in six purification efforts, but activity was also detected in the later-eluting fractions that contained both the 170- and 245-kD polypeptides. In the Mono Q column, cytoplasmic dynein eluted earlier than the peak of plus-end–directed vesicle transport (Fig. 1 B). Again, little or no minus-end transport activity was observed in the peak dynein fractions.
The cofractionation of activity with the 245-kD polypeptide in the Mono Q column strongly implicated it as a factor that can stimulate plus-end motility. However, in the hydroxyapatite column, an early-eluting fraction containing the 170- but not the 245-kD polypeptide (fraction 16 in Fig. 1 A) also stimulated transport. This finding raised the possibility that the 170-kD polypeptide was also capable of promoting plus-end–directed organelle movement. To address whether both polypeptides were truly independent transport-stimulating factors, we performed two additional experiments. First, we diluted a peak 245-kD Mono Q fraction (containing nominal 170-kD polypeptide) to a level of activity that was 25% of that in the 170-kD peak fraction. By silver staining, the quantity of the 245-kD polypeptide in this diluted fraction was greater than that found in an equal volume of the fourfold more active 170-kD peak fraction. This result indicates that organelle transport in the 170-kD peak fraction could not be stimulated solely by the 245-kD polypeptide contaminating this fraction.
Next, we measured the velocities of plus-end–directed movements from column fractions that had minimal overlap in their content of 170- and 245-kD polypeptides (Fig. 2). Movement velocities from the 245- or 170-kD–enriched fractions were 2.62 ± 0.50 (n = 27) and 1.86 ± 0.74 (n = 23) μm/s, respectively (mean ± SD; P < 0.001). The velocity histogram of movements generated by the 170-kD fraction appeared bimodal. The majority of movements (65%) was between 1 and 2 μm/s; by contrast, this velocity range represented only 11% of the movements stimulated by the 245-kD fraction. The 170-kD fraction also contained a subset of fast movements between 2 and 3.5 μm/s, which corresponded to the average velocity produced by the 245-kD fraction. Since the 170-kD fraction contained some contaminating 245-kD even in our purest fractions (Fig. 2), it seems likely that these few fast movements were due to the 245-kD polypeptide. Thus, we conclude that the 245- and 170-kD polypeptides both stimulate plus-end–directed organelle transport, but at different velocities.
Figure 2.
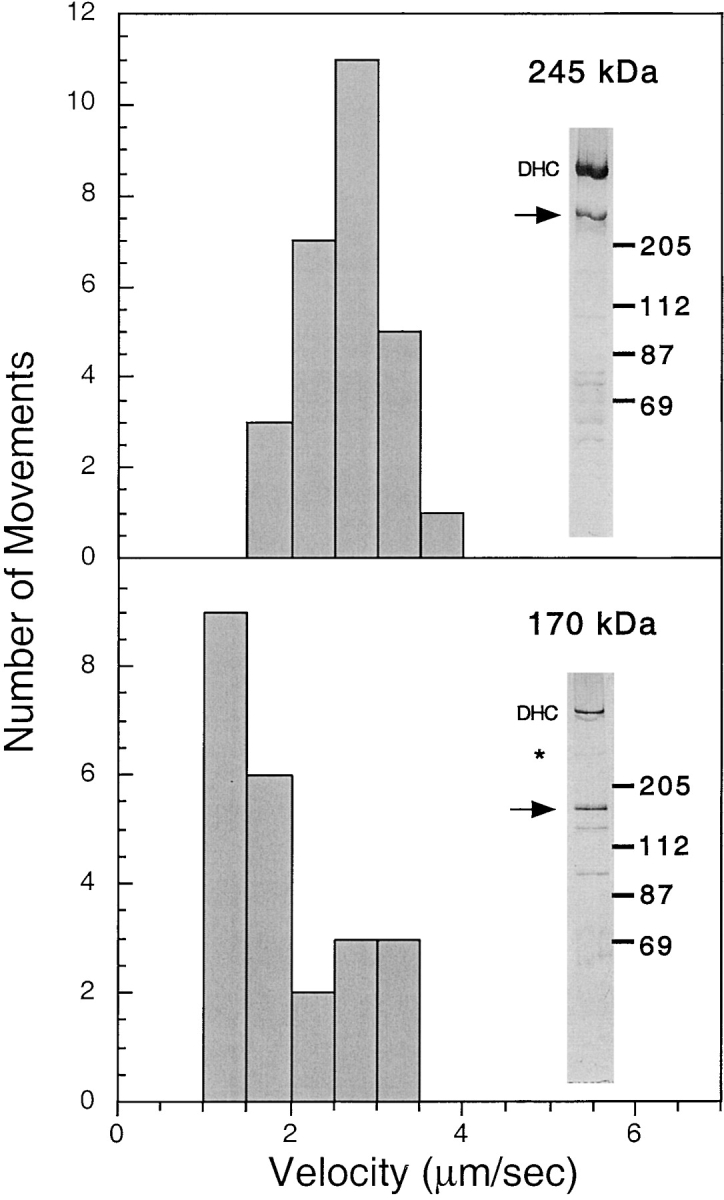
Velocities of plus-end–directed vesicle movements stimulated by the 245- and 170-kD–containing fractions. Velocities of plus-end–directed vesicle movements were measured in Mono Q fractions that had minimal overlap between the 245- and 170-kD polypeptides by silver stain (see inset for each histogram). The 245-kD (top) and the 170-kD (bottom) polypeptides are indicated by the arrows. *A small amount of contaminating 245-kD polypeptide in the 170-kD fraction (bottom) is present. Dynein heavy chain (DHC) was present in both fractions, but minus-end–directed motion was not observed. Histograms demonstrate that the average velocity of vesicle movements generated by the 245-kD polypeptide is significantly higher than that of the 170-kD polypeptide. The small number of fast movements generating the apparent bimodal distribution of the 170-kD histogram is likely due to the small amount of 245-kD polypeptide present in the fraction.
Our hydroxyapatite and Mono Q fractionation experiments showed that the amount of organelle transport in our assay was always in quantitative agreement with the amount of the 245- and 170-kD polypeptides in the fraction. From examination of silver-stained gels from many columns, we could not identify any other polypeptide that clearly and consistently copurified with either the 245- or 170-kD polypeptides (though our gel system would not have clearly resolved polypeptides <25 kD). From this data, we believe that the 245- and 170-kD polypeptides can stimulate transport in the absence of associated polypeptides, although a low molecular weight factor or a catalytic activity present in low stoichiometry could have been present and not detected in this analysis.
Identification of the 245- and 170-kD Polypeptides as Kinesin-related Proteins
To establish the molecular identity of the 245- and 170-kD polypeptides, we obtained peptide sequence from tryptic fragments. Both polypeptides contained peptide sequences that implicated them as members of the kinesin superfamily. A peptide from the 170-kD protein contained the sequence LYLVDLAGSEK, which includes the highly conserved DLAGSE motif of the switch II region of the kinesin nucleotide binding site (Sablin et al. 1996). The 245-kD polypeptide yielded a tryptic fragment containing the conserved VIxAL motif found in the α4 helix of kinesin motors. Since the 245-kD polypeptide was the more robust motor in our in vitro organelle transport assay, we chose this kinesin for further cloning and characterization.
The 245-kD polypeptide was cloned using a combination of degenerate PCR, library screening, and RACE (see Materials and Methods). The complete open reading frame predicts a protein of 2,205 amino acids of 248 kD that contains all five peptides identified by direct sequencing (Fig. 3 A). We are therefore confident that we cloned the correct polypeptide identified in our biochemical purification. Sequence alignments revealed a clear homology of the 245-kD polypeptide to the Unc104/KIF1A class of kinesin motors (Fig. 3 A). Two founding members of this class, mouse (Mm) KIF1A (190 kD) and the C. elegans (Ce) ortholog Unc104 (174 kD), are monomeric kinesin motors that have been implicated in the transport of synaptic vesicle precursors in neurons (Hall and Hedgecock 1991; Okada et al. 1995; Yonekawa et al. 1998). The Unc104/KIF1A class also includes motors implicated in other functions such as mitochondrial transport (KIF1B; Nangaku et al. 1994), Golgi-ER transport (KIF1C; Dorner et al. 1998), and cytokinesis (Klp38B; Ohkura et al. 1997).
Figure 3.
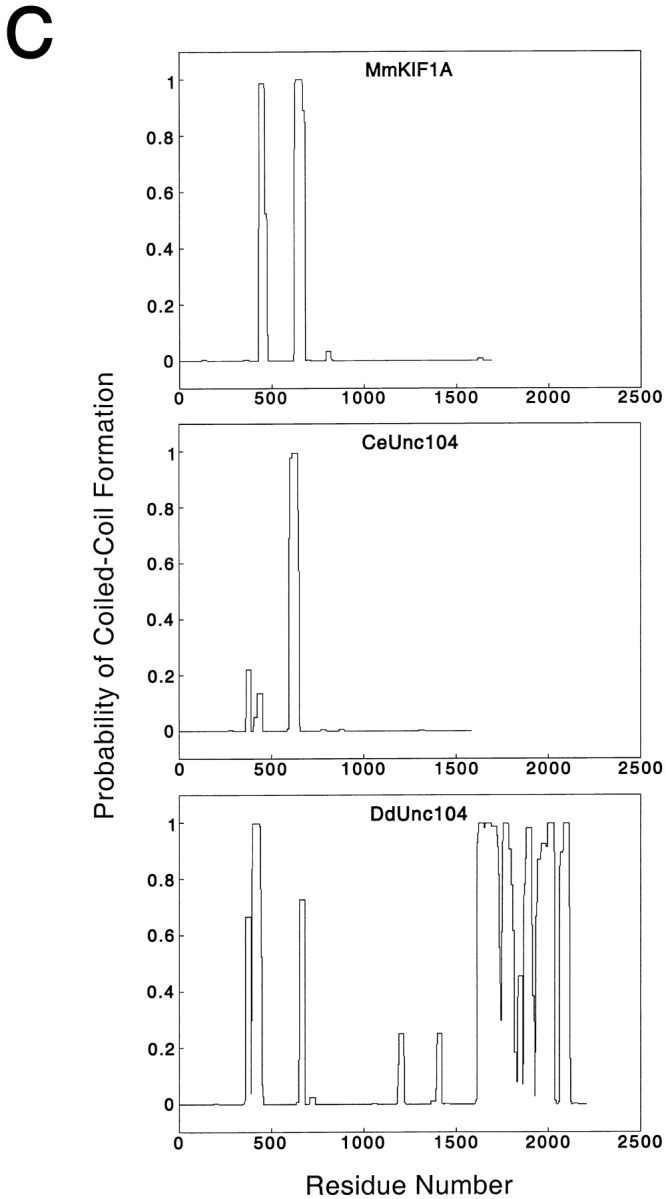
The 245-kD protein is a member of the Unc104/KIF1A family. The complete 2,205 amino acid sequence of the 245-kD protein, which we hereafter refer to as DdUnc104, is indicated in A. The amino acid sequences of homologous regions of mouse KIF1A and C. elegans Unc104 are displayed; regions of MmKIF1A and CeUnc104 that are not homologous are omitted. Amino acid identities between all three motors are indicated in black shading, while similar amino acids shared by all three are shaded gray. Peptide sequences obtained from tryptic fragments (see Materials and Methods) are underscored with black lines. The predicted positions of loop 3 (L3: DdUnc104, amino acids 63–73) and loop 12 (L12: DdUnc104, amino acids 296–309) are also indicated (see text for discussion of these loops). Amino acids 1523–1616 of DdUnc104 comprise a pleckstrin homology domain. This region (bounded by asterisks) is homologous to a similar region in MmKIF1A and CeUnc104. Both MmKIF1A and CeUnc104 end just after the PH domain, while DdUnc104 continues for an additional 589 amino acids. (B) Dot matrix plot comparisons of DdUnc104 with CeUnc104 (top) and MmKIF1B (bottom). The locations of the catalytic core, AF-6/cno, and PH domains are indicated (arrows for the core and AF-6/cno domains mark the COOH-terminal boundaries of these domains). The homology of DdUnc104 to CeUnc104 and MmKIF1A (not shown) extends well past the AF-6/cno domain, and additional homology (including the PH domain) is evident in the COOH-terminal portions of the two motors. In contrast, the homology of DdUnc104 to MmKIF1B ends shortly after the AF-6/cno domain, and no COOH-terminal homology is apparent. Dot matrix plots comparing DdUnc104 to human (Hs) KIF1C and Drosophila (Dm) Klp38B (not shown) looked similar to the plot for MmKIF1B. (C) The probability of coiled-coil formation along the lengths of the MmKIF1A, CeUnc104, and DdUnc104 proteins. MmKIF1A and DdUnc104 share two regions of high probability of coiled-coil formation in the NH2-terminal half of the molecule, one of which is also present in CeUnc104. Uniquely, the large COOH-terminal region of DdUnc104 also has a high probability of coiled-coil formation.


The sequence of the 245-kD kinesin reveals the following domain structure. The first 353 residues show high amino acid identity to the conserved catalytic core that defines the kinesin superfamily (Vale and Fletterick 1997). Within the kinesin superfamily, the catalytic core of the 245-kD kinesin is most similar to the Unc104/KIF1A class (40–60% identical residues). All members of the Unc104/KIF1A class also contain a highly conserved extension in loop 3 (L3), which is unique among kinesins (R. Case and R. Vale, unpublished observations). This L3 extension is also found in the 245-kD kinesin (Fig. 3 A). Several, but not all, members of the Unc104/KIF1A class contain several lysine residues in the microtubule-binding loop (L12) (Okada and Hirokawa 1999; Fig. 3 A). In the 245-kD kinesin, this loop contains only two lysines, and thus is much less charged.
For ∼250 amino acids after the catalytic core, the 245-kD kinesin shows significant identity to all kinesins in the Unc104/KIF1A class (Fig. 3a and Fig. b). The latter half of this class-conserved region is homologous to a domain found in nonmotor proteins such as human AF-6 and Drosophila cno (Ponting 1995), and whose function is poorly understood. Beyond the AF-6/cno domain, the 245-kD kinesin continues to exhibit strong homology to CeUnc104 and MmKIF1A for an additional ∼370 amino acids; however, sequence identity to other Unc104/KIF1A class kinesins (e.g., KIF1B) does not continue far past the AF-6/cno domain (Fig. 3 B). The sequence in the middle portion of the 245-kD kinesin (residues 973–1522) is novel and contains repeats of glutamine or serine that are often found in Dictyostelium proteins (Shaw et al. 1989; de Hostos et al. 1998). Residues 1523–1616 are predicted to form a pleckstrin homology (PH) domain whose greatest similarity in blast searches is to a COOH-terminal PH domain in MmKIF1A and CeUnc104 (Fig. 3 A). This PH domain is not found in other motors belonging to this kinesin class (Fig. 3 B). The sequences of MmKIF1A and CeUnc104 end shortly after the PH domain, but the 245-kD kinesin extends for another 589 amino acids. This region does not exhibit significant sequence identity to any other protein in the data base. In conclusion, the 245-kD kinesin shows more identity to the two ortholog motors CeUnc104 and MmKIF1A than to other members of the Unc104/KIF1A class. Since there is somewhat more nonmotor domain homology to CeUnc104 than to MmKIF1A (Fig. 3 A), we term the 245-kD kinesin Dictyostelium (Dd) Unc104.
We also examined DdUnc104 for possible coiled-coil domains using a sequence prediction program (Lupas et al. 1991). This analysis revealed that both MmKIF1A and DdUnc104 contain two regions in the NH2-terminal half of the molecule (approximately amino acids 359–448 and 652–679 in DdUnc104; the second is also present in CeUnc104), which have a propensity towards coiled-coil formation (Fig. 3 C). However, hydrodynamic analyses of MmKIF1A (Okada et al. 1995) and CeUnc104 (Pierce et al. 1999) indicated that these regions do not cause dimerization. Interestingly, the majority of the unique ∼600–amino acid extension of DdUnc104 was also predicted to have a high probability of coiled-coil formation (Fig. 3 C), raising the possibility that DdUnc104, in contrast to MmKIF1A and CeUnc104, is dimeric.
DdUnc104 Is a Homodimer
To determine the quaternary structure of DdUnc104, we analyzed its hydrodynamic behavior using velocity sedimentation through sucrose density gradients and gel filtration (see Materials and Methods). In two separate experiments, the S value was 10.9 and 10.2 S (average 10.6). The sucrose gradient peak was then subjected to gel filtration, and a Stokes radius of 10.5 nm was measured in both experiments. Together with a partial specific volume of 0.73 cm3/g estimated from the DdUnc104 amino acid sequence, the native molecular weight was determined to be 480 kD. Given the polypeptide's molecular weight of 248 kD, this result indicates that DdUnc104 is a dimer and also argues against the presence of a stoichiometrically associated polypeptide of significant mass. These hydrodynamic data also suggest that DdUnc104 has an extended shape with an axial ratio of 15.1. Thus, DdUnc104 is the first dimeric motor described for the Unc104/KIF1A class of kinesin motors.
A Knockout of DdUnc104 Causes In Vitro and In Vivo Defects in Organelle Transport
To learn more about the function of DdUnc104 in vivo, the DdUnc104 gene was disrupted by homologous recombination. A gene-disruption construct was made by inserting a blasticidin resistance cassette into a DdUnc104 cDNA fragment. The linearized construct was transformed into wild-type cells by electroporation, and transformants were screened by PCR (see Materials and Methods). A cell line was identified that gave the PCR products expected from a gene disruption event. Disruption of the DdUnc104 gene was established by Southern analysis, and absence of the DdUnc104 RNA and protein was confirmed by Northern and Western blotting (Fig. 4). A blasticidin-resistant cell line in which the construct had integrated elsewhere in the genome was used as a control (Fig. 4 A).
Figure 4.

Disruption of the DdUnc104 gene (ksnA in Dictyostelium nomenclature) by homologous recombination. A blasticidin resistance cassette was inserted into a DdUnc104 cDNA fragment (see Materials and Methods) and transformed into wild-type Dictyostelium. Blasticidin-resistant transformants were screened by PCR (see Materials and Methods). Disruption of the gene was confirmed by Southern blot (A), using the DdUnc104 cDNA fragment as a probe. Wild-type cells (WT) display the 1.8-kb fragment expected for the undisrupted DdUnc104 gene. In contrast, the null cells (KO, knockout) have the 3.2-kb fragment expected if the disruption construct integrated uniquely and properly into the DdUnc104 gene. A blasticidin-resistant control cell line (Control) displays both the wild-type 1.8-kb fragment, indicating that the DdUnc104 gene is intact, and the gene-disruption construct fragment of 3.2 kb, indicating that the construct integrated elsewhere in the genome. The fainter high molecular weight band (Control) is most likely due to a small amount of partial digestion. The absence of production of DdUnc104 RNA in the null cells was confirmed by Northern blot (B) using the DdUnc104 cDNA fragment as a probe. A single ≥6-kb DdUnc104 transcript (arrow) is present in control cells (Control) but not null cells (KO). The absence of the DdUnc104 protein in the null cells was confirmed by immunoblot (C). ATP release fractions were prepared from control and null cells and immunoblotted using an affinity-purified peptide antibody (see Materials and Methods). The 245-kD DdUnc104 protein (arrow) is present in fractions from control cells (Control) but not null cells (KO).
The DdUnc104 null cells grew normally and showed no gross morphological defects. In addition, after starvation, the null cells aggregated and differentiated normally and at a rate like that of wild-type cells. Thus, this motor is not essential for viability, cell division, or differentiation.
The DdUnc104 knockout provided an opportunity to assess whether DdUnc104 was a bona fide organelle transport motor in vivo. To address this question, linear (>1 μm) movements of organelles in control and DdUnc104 null cells were observed by video DIC microscopy; earlier studies have shown that these movements are predominately microtubule based (Roos et al. 1987). Quantitative measurements revealed that the mutant cells exhibited a 62% reduction in overall movements (Fig. 5 A). In Dictyostelium, organelle movement is not perfectly radial from the center to periphery, and hence it is very difficult to score the polarity of movement in vivo. Nevertheless, this result indicates that DdUnc104 drives organelle transport in vivo. Interestingly, when we examined the movement of mitochondria by labeling cells with a mitochondrial dye (Mitotracker; Poot et al. 1996), no difference in the frequency of mitochondrial movements in null and control cells was observed (Fig. 5 B). This result indicates that DdUnc104 is not required for the movement of mitochondria. Thus, the transport defect in the DdUnc104 null cells is not due to a generalized reduction in the movement of all organelles, but rather reflects a specific defect in the transport of an as yet unidentified organelle population(s).
Figure 5.
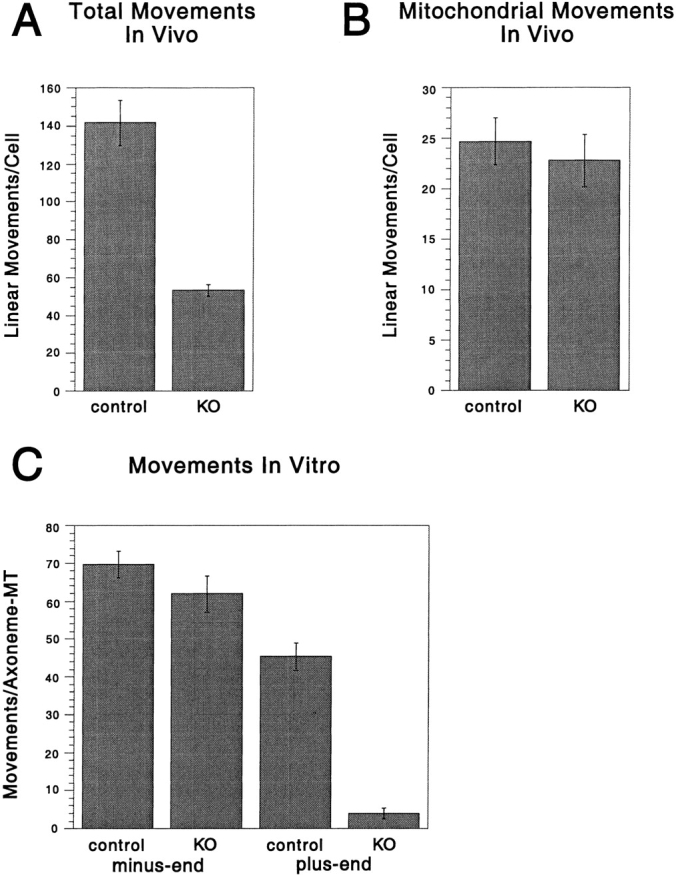
The DdUnc104 null cell line has in vivo and in vitro defects in organelle transport. (A) In vivo movements in control and null cells (from Fig. 4) were observed by differential interference contrast microscopy. Only linear and continuous movements >1 μm in length were scored, and each cell was observed for 4 min. The total number of movements per cell was averaged over all cells observed (values shown are the mean ± SEM; n = 10 cells for each cell type). The number of movements was significantly reduced in the null cells (KO, knockout) relative to the control cells (t test; P < 0.001). (B) In vivo movements of mitochondria labeled with a mitochondrial dye (Mitotracker) were observed by fluorescence microscopy. Only linear and continuous movements of >1 μm were scored, and each cell was observed for 1 min. The total number of movements per cell was averaged over all cells observed (values shown are the mean ± SEM; control, n = 27 cells; null, n = 22 cells). There was no statistically significant difference in the control and null means by t test. (C) ATP releasates were prepared from the control and null cells and combined with KI vesicles in our in vitro assay (see Materials and Methods), and minus- and plus-end–directed vesicle movements were scored. Multiple 4-min assays were performed on each of two separate ATP release preparations for each cell line, and the results for all assays were averaged (values shown are the mean ± SEM; control, n = 6 assays; null, n = 8 assays). The number of plus-end–directed movements was significantly reduced in the null cells relative to the control (t test; P < 0.001), while the levels of minus-end–directed movement were statistically equivalent by t test.
Since the polarity of organelle movement could not be determined in vivo, we analyzed whether the organelle motility defect in the DdUnc104 null cells was specific for the plus-end direction using our in vitro assay. An ATP release fraction prepared from either the control or null cells was incubated with KI-washed vesicles and scored for motility on our polarity-defined microtubule substrate. Minus-end–directed motility driven by dynein (Pollock et al. 1998) was nearly identical for the fractions obtained from control and null cells. However, dramatically, plus-end motility was decreased by 90% in the null cells (Fig. 5 C). This result confirms that the DdUnc104 polypeptide is responsible for the majority of the plus-end–directed motor activity seen in our biochemical assay.
Discussion
Purification of Motors Involved in Organelle Transport
Microtubule-based motor proteins have been purified using in vitro microtubule gliding activity (Vale et al. 1985; Paschal et al. 1987) or reactivity with pan-kinesin antibodies (Cole et al. 1992). However, these highly purified motors have not efficiently transported organelles. Here, we have approached the problem differently by directly purifying soluble factors needed for plus-end–directed movement of organelles. This strategy led to the discovery of two kinesin motors that drive organelle movement.
Several findings argue that the purified motors are the ones that power plus-end–directed organelle transport in Dictyostelium. First, the specific activity of plus-end–directed organelle transport increased throughout the purification, indicating that the relevant activities from the starting extract were being enriched at each step. Second, the speed and quality of movement (continuous motion for several microns) produced by the two purified motors was virtually identical to that observed in extracts (Pollock et al. 1998) and in living cells (Roos et al. 1987). The role of the 245-kD DdUnc104 motor in organelle transport was further confirmed by a gene knockout. Although several other kinesin genes have been identified in Dictyostelium (de Hostos et al. 1998), the 245-kD DdUnc104 and 170-kD kinesin are likely to represent the major organelle transport motors, since the DdUnc104 knockout results in the loss of the vast majority of plus-end–directed organelle movement in vitro. It is likely that the remainder of the movement is produced by the 170-kD motor, and this can be established in the future by analyzing cells that have knockouts in both the DdUnc104 and 170-kD kinesin genes.
In principle, our approach could have purified a soluble activator of an organelle-bound motor or a combination of an activator and a motor. Although these possibilities cannot be ruled out and further studies are required, our data does not favor these scenarios. First, there is no polypeptide that copurifies stoichiometrically with DdUnc104. This data, along with the hydrodynamic analysis, argues that this motor does not have an associated light chain, although it is possible that a small subunit (<25 kD) eluded detection in our SDS-PAGE analysis. Furthermore, based on many purification trials, we have not detected any substoichiometric polypeptide that consistently cofractionates with DdUnc104. We also believe that DdUnc104 is not activated by a protein that overlaps but does not copurify with the DdUnc104 fractions, since the relative amounts of organelle transport activity mirror the concentrations of DdUnc104. With regard to the membranes, all components required for transport are vesicle-associated after 0.3-M KI treatment, but are partially destroyed by trypsin. From these studies, we hypothesize that plus-end–directed organelle transport is driven by a direct interaction of DdUnc104 with a tightly associated, and possibly integral, membrane protein(s).
Our results differ somewhat from in vitro studies of plus-end–directed organelle transport using other systems. A recombinant KIF1B motor was reported to transport mitochondria in vitro (Nangaku et al. 1994), although the average frequency and velocity of movements were not reported, and several examples of moving mitochondria shown in this paper reveal transport rates (<0.1 μm/s) slower than those seen in vivo. In contrast, vesicle movements produced by the two motors purified in our study occur over long distances and match in vivo velocities. For conventional kinesin, fractionation experiments by Schroer and Sheetz 1991 indicated that kinesin requires an activator for transporting organelles, although its identity has not been established through further biochemical purification. Interestingly, many of the plus-end–directed organelle movements in this activator fraction were faster than kinesin (>2 μm/s) and comparable in speed with the movements observed in our study. It is possible that the movement observed in this fraction was due not to a kinesin activator, but to an Unc104/KIF1A-type motor.
DdUnc104: A Relative of the Synaptic Vesicle Precursor Transporter Unc104/KIF1A
The sequence of the 245-kD kinesin reveals that it belongs to the Unc104/KIF1A class of motors, since its amino acid identity extends well past the superfamily-conserved catalytic core. Based on phylogenetic trees of kinesin sequences from the database, the Unc104/KIF1A class encompasses ∼16 members from various organisms. Of these various members, DdUnc104 appears to be most similar to C. elegans Unc104 and mouse KIF1A. Most notably, its homology to CeUnc104/MmKIF1A uniquely extends well past the AF-6/cno domain found in many family members, and these three motors are also the only ones that contain a conserved PH domain at or near their COOH termini. However, DdUnc104 and CeUnc104/MmKIF1A must carry different types of cargo. MmKIF1A and CeUnc104 are expressed only in the nervous system, where they specifically transport synaptic vesicle precursors (Hall and Hedgecock 1991; Yonekawa et al. 1998). Thus, a DdUnc104-like motor from unicellular organisms most likely evolved to acquire a more specialized function in the nervous system of higher eukaryotes. However, the conservation of nonmotor domains in DdUnc104 and CeUnc104/MmKIF1A suggests that the mechanisms by which they bind their cargo may be closely related.
DdUnc104 is the first member of the Unc104/KIF1A kinesin subfamily identified and fully sequenced from a unicellular organism. A partial clone of a KIF1B homologue, TLKIF1, has recently been cloned from the thermophilic fungus Thermomyces lanuginosus (Sakowicz et al. 1999), suggesting that members of this kinesin class may be common in lower eukaryotes. Since DdUnc104 is an evolutionarily distant member of the Unc104/KIF1A kinesin class, sequences that are uniquely conserved between it and other family members are likely to define regions that serve important functions for this class of motors. One such conserved motif is a unique extension in loop 3. Based on the location of this loop in the conventional kinesin and Ncd crystal structures, these extra amino acids would be expected to extend towards the nucleotide and hence may be involved in modulating enzyme kinetics. The AF-6/cno homology domain is also highly conserved between DdUnc104 and its higher eukaryotic relatives; its position adjacent to the catalytic core suggests that it may be involved in motor mechanics or regulation. In the COOH-terminal tail domain, the PH domains of DdUnc104, CeUnc104, and MmKIF1A are more similar to one another than to the PH domains from other proteins. PH domains are thought to be involved in membrane interactions (Saraste and Hyvonen 1995; Rebecchi and Scarlata 1998), and the similar PH domains of these motors may reflect a conserved function in cargo binding.
A surprising difference between DdUnc104 and other members of this class concerns their oligomeric state. KIF1A (Okada et al. 1995), KIF1B (Nangaku et al. 1994), and CeUnc104 (the NH2-terminal half) (Pierce et al. 1999) are monomeric, and sequence analysis of other members suggests that they also function using a single motor domain. Hence, this class has been referred to as the “monomeric kinesins” (Vale and Fletterick 1997; Hirokawa 1998). However, the dimeric nature of DdUnc104 raises the question of whether other motors in this class can function as dimers under some circumstances. For DdUnc104, dimerization is constitutive due to the long coiled-coil COOH terminal to its PH domain. However, KIF1A and its higher eukaryotic relatives also contain sequences that are predicted to form coiled-coils (Fig. 3 B), and perhaps binding of the motor tail to receptors on the membrane can trigger dimerization in a functionally equivalent manner.
An alternative model is that higher eukaryotic Unc104/KIF1A family members have evolved to have efficient motor function as monomers. Consistent with this idea, some of the Unc104/KIF1A family members from higher eukaryotes have an insertion of several lysines in the microtubule binding loop. For MmKIF1A, these lysines have been shown to be important in enabling this monomeric motor to move processively along microtubules in vitro (Okada and Hirokawa 1999). In contrast, the equivalent loop in DdUnc104 has fewer lysines, perhaps because they are not needed for motility by this dimeric motor.
Remarkably, although the knockout of the DdUnc104 motor dramatically decreases organelle transport, it has little effect on the morphology, division, and differentiation of these cells. This result suggests either that membrane trafficking can be achieved by vesicle diffusion, that DdUnc104 activity is redundant with another activity (perhaps with the 170-kD kinesin), or that other mechanisms act to compensate for the loss of this motor. However, with regard to this latter point, it is interesting that minus-end–directed motility is identical in extracts from the null and wild-type cells, indicating that the decrease in plus-end–directed motility does not elicit any compensatory change in the level of dynein-based movement.
An Assay for Identifying Additional Organelle Transport Factors
Our reconstituted assay system provides a starting point for identifying additional factors involved in organelle transport and for understanding their function. As documented in earlier work (Schroer and Sheetz 1991), we find that Dictyostelium cytoplasmic dynein loses its ability to transport organelles during purification, suggesting that it becomes separated from a required cytosolic factor. It will be interesting to determine if this factor is dynactin (Gill et al. 1991) or whether another type of activator is used by Dictyostelium. In addition, this assay provides an opportunity for identifying the membrane receptors that interact with the DdUnc104 and 170-kD kinesin motors so that movement can eventually be reconstituted in a completely defined system. The molecular genetics offered by Dictyostelium also provides a testing ground for determining whether membrane proteins that bind kinesins in vitro indeed serve as bona fide receptors in vivo.
Acknowledgments
We thank the members of the Vale lab for their support and their helpful discussions. Nora Hom-Booher provided valuable assistance with the gene cloning. Kimberley Lucas provided technical assistance.
N. Pollock is a student in the Medical Scientist Training Program. E.L. de Hostos is supported by a National Science Foundation grant (MCB-9996186) and the Tropical Disease Research Unit, Department of Pathology, UCSF. R.D. Vale is supported in part by the National Institutes of Health (grant 38499).
Footnotes
1.used in this paper: DIC, differential interference contrast; HSS, high speed supernatant; PH, pleckstrin homology; RACE, rapid amplification of cDNA ends
References
- Adachi H., Hasebe T., Yoshinaga K., Ohta T., Sutoh K. Isolation of Dictyostelium discoideum cytokinesis mutants by restriction enzyme–mediated integration of the blasticidin S resistance marker. Biochem. Biophys. Res. Commun. 1994;205:1808–1814. doi: 10.1006/bbrc.1994.2880. [DOI] [PubMed] [Google Scholar]
- Aizawa H., Sekine Y., Takemura R., Zhang Z., Nangaku M., Hirokawa N. Kinesin family in murine central nervous system. J. Cell Biol. 1992;119:1287–1296. doi: 10.1083/jcb.119.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen C., Borisy G.G. Structural polarity and directional growth of microtubules of Chlamydomonas flagella. J. Mol. Biol. 1974;90:381–402. doi: 10.1016/0022-2836(74)90381-7. [DOI] [PubMed] [Google Scholar]
- Blocker A., Severin F.F., Burkhardt J.K., Bingham J.B., Yu H., Olivo J.C., Schroer T.A., Hyman A.A., Griffiths G. Molecular requirements for bi-directional movement of phagosomes along microtubules. J. Cell Biol. 1997;137:113–129. doi: 10.1083/jcb.137.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt J.K., Echeverri C.J., Nilsson T., Vallee R.B. Overexpression of the dynamitin (p50) subunit of dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J. Cell Biol. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole D.G., Cande W.Z., Baskin R.J., Skoufias D.A., Hogan C.J., Scholey J.M. Isolation of a sea urchin egg kinesin-related protein using peptide antibodies. J. Cell Sci. 1992;101:291–301. doi: 10.1242/jcs.101.2.291. [DOI] [PubMed] [Google Scholar]
- de Hostos E.L., McCaffrey G., Sucgang R., Pierce D.W., Vale R.D. A developmentally regulated kinesin-related motor protein from Dictyostelium discoideum . Mol. Biol. Cell. 1998;9:2093–2106. doi: 10.1091/mbc.9.8.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hostos E.L., Rehfuess C., Bradtke B., Waddell D.R., Albrecht R., Murphy J., Gerisch G. Dictyostelium mutants lacking the cytoskeletal protein coronin are defective in cytokinesis and cell motility. J. Cell Biol. 1993;120:163–173. doi: 10.1083/jcb.120.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner C., Ciossek T., Muller S., Moller N.P.H., Ullrich A., Lammers R. Characterization of KIF1C, a new kinesin-like protein involved in vesicle transport from the Golgi apparatus to the endoplasmic reticulum. J. Biol. Chem. 1998;273:20267–20275. doi: 10.1074/jbc.273.32.20267. [DOI] [PubMed] [Google Scholar]
- Echard A., Jollivet F., Martinez O., Lacapere J.J., Rousselet A., Janoueix-Lerosey I., Goud B. Interaction of a Golgi-associated kinesin-like protein with Rab6. Science. 1998;279:580–585. doi: 10.1126/science.279.5350.580. [DOI] [PubMed] [Google Scholar]
- Gill S.R., Schroer T.A., Szilak I., Steuer E.R., Sheetz M.P., Cleveland D.W. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J. Cell Biol. 1991;115:1639–1650. doi: 10.1083/jcb.115.6.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindhart J.G., Desai C.J., Beushausen S., Zinn K., Goldstein L.S. Kinesin light chains are essential for axonal transport in Drosophila . J. Cell Biol. 1998;141:443–454. doi: 10.1083/jcb.141.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodson H.V., Valetti C., Kreis T. Motors and membrane traffic. Curr. Opin. Cell Biol. 1997;9:18–28. doi: 10.1016/s0955-0674(97)80147-0. [DOI] [PubMed] [Google Scholar]
- Hall D.H., Hedgecock E.M. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans . Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- Hanlon D.W., Goldstein L.S.B. Characterization of KIFC2, a neuronal kinesin superfamily member in mouse. Neuron. 1997;18:439–451. doi: 10.1016/s0896-6273(00)81244-1. [DOI] [PubMed] [Google Scholar]
- Harada A., Takei Y., Kanai Y., Tanaka Y., Nonaka S., Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J. Cell Biol. 1998;141:51–59. doi: 10.1083/jcb.141.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman U., Wernstedt C., Gonez J., Heldin C.H. Improvement of an in-gel digestion procedure for the micropreparation of internal protein fragments for amino acid sequencing. Anal. Biochem. 1995;224:451–455. doi: 10.1006/abio.1995.1070. [DOI] [PubMed] [Google Scholar]
- Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- Hollenbeck P.J., Swanson J.A. Radial extension of macrophage tubular lysosomes supported by kinesin. Nature. 1990;346:864–866. doi: 10.1038/346864a0. [DOI] [PubMed] [Google Scholar]
- Holleran E.A., Tokito M.K., Karki S., Holzbaur E.L.F. Centractin (ARP1) associates with spectrin revealing a potential mechanism to link dynactin to intracellular organelles. J. Cell Biol. 1996;135:1815–1830. doi: 10.1083/jcb.135.6.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J., Yu H., Sheetz M.P. Kinectin, an essential anchor for kinesin-driven vesicle motility. Science. 1995;267:1834–1837. doi: 10.1126/science.7892610. [DOI] [PubMed] [Google Scholar]
- Lane J., Allan V. Microtubule-based membrane movement. Biochim. Biophys. Acta. 1998;1376:27–55. doi: 10.1016/s0304-4157(97)00010-5. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Cole N.B., Marotta A., Conrad P.A., Bloom G.S. Kinesin is the motor for microtubule-mediated Golgi-to-ER membrane traffic. J. Cell Biol. 1995;128:293–306. doi: 10.1083/jcb.128.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A., Van Dyke M., Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Malchow D., Naegele B., Schwarz H., Gerisch G. Membrane-bound cyclic AMP phosphodiesterase in chemotactically responding cells of Dictyostelium discoideum . Eur. J. Biochem. 1972;28:136–142. doi: 10.1111/j.1432-1033.1972.tb01894.x. [DOI] [PubMed] [Google Scholar]
- Manstein D.J., Schuster H.P., Morandini P., Hunt D.M. Cloning vectors for the production of proteins in Dictyostelium discoideum . Gene. 1995;162:129–134. doi: 10.1016/0378-1119(95)00351-6. [DOI] [PubMed] [Google Scholar]
- McGrail M., Gepner J., Silvanovich A., Ludmann S., Serr M., Hays T.S. Regulation of cytoplasmic dynein function in vivo by the Drosophila Glued complex. J. Cell Biol. 1995;131:411–425. doi: 10.1083/jcb.131.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R.L., Scholey J.M. Heterotrimeric kinesin-II is required for the assembly of motile 9+2 ciliary axonemes on sea urchin embryos. J. Cell Biol. 1997;138:1009–1022. doi: 10.1083/jcb.138.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku M., Sato-Yoshitake R., Okada Y., Noda Y., Takemura R., Yamazaki H., Hirokawa N. KIF1B, a novel microtubule plus end–directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Nonaka S., Tanaka Y., Okada Y., Takeda S., Harada A., Kanai Y., Kido M., Hirokawa N. Randomization of left–right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Ohkura H., Torok T., Tick G., Hoheisel J., Kiss I., Glover D.M. Mutation of a gene for a Drosophila kinesin-like protein, Klp38B, leads to failure of cytokinesis. J. Cell Sci. 1997;110:945–954. doi: 10.1242/jcs.110.8.945. [DOI] [PubMed] [Google Scholar]
- Okada Y., Hirokawa N. A processive single-headed motorkinesin superfamily protein KIF1A. Science. 1999;283:1152–1157. doi: 10.1126/science.283.5405.1152. [DOI] [PubMed] [Google Scholar]
- Okada Y., Yamazaki H., Sekine-Aizawa Y., Hirokawa N. The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell. 1995;81:769–780. doi: 10.1016/0092-8674(95)90538-3. [DOI] [PubMed] [Google Scholar]
- Paschal B.M., Shpetner H.S., Vallee R.B. MAP 1C is a microtubule-activated ATPase which translocates microtubules in vitro and has dynein-like properties. J. Cell Biol. 1987;105:1273–1282. doi: 10.1083/jcb.105.3.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister K.K., Wagner M.C., Stenoien D.L., Brady S.T., Bloom G.S. Monoclonal antibodies to kinesin heavy and light chains stain vesicle-like structures, but not microtubules, in cultured cells. J. Cell. Biol. 1989;108:1453–1463. doi: 10.1083/jcb.108.4.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce D.W., Hom-Booher N., Otsuka A.J., Vale R.D. Single-molecule behavior of monomeric and heteromeric kinesins. Biochemistry. 1999;38:5412–5421. doi: 10.1021/bi9830009. [DOI] [PubMed] [Google Scholar]
- Pollock N., Koonce M.P., de Hostos E.L., Vale R.D. In vitro microtubule-based organelle transport in wild-type Dictyostelium and cells overexpressing a truncated dynein heavy chain. Cell Motil. Cytoskelet. 1998;40:304–314. doi: 10.1002/(SICI)1097-0169(1998)40:3<304::AID-CM8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Ponting C.P. AF-6/cnoneither a kinesin nor a myosin, but a bit of both. TIBS (Trends Biochem. Sci.) 1995;20:265–266. doi: 10.1016/s0968-0004(00)89040-4. [DOI] [PubMed] [Google Scholar]
- Poot M., Zhang Y.Z., Kramer J.A., Wells K.S., Jones L.J., Hanzel D.K., Lugade A.G., Singer V.L., Haugland R.P. Analysis of mitochondrial morphology and function with novel fixable fluorescent stains. J. Histochem. Cytochem. 1996;44:1363–1372. doi: 10.1177/44.12.8985128. [DOI] [PubMed] [Google Scholar]
- Rebecchi M.J., Scarlata S. Pleckstrin homology domainsa common fold with diverse functions. Annu. Rev. Biophys. Biomol. Struct. 1998;27:503–528. doi: 10.1146/annurev.biophys.27.1.503. [DOI] [PubMed] [Google Scholar]
- Rodionov V.I., Gyoeva F.K., Gelfand V.I. Kinesin is responsible for centrifugal movement of pigment granules in melanophores. Proc. Natl. Acad. Sci. USA. 1991;88:4956–4960. doi: 10.1073/pnas.88.11.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos U.-P., De Brabander M., Nuydens R. Movements of intracellular particles in undifferentiated amebae of Dictyostelium discoideum . Cell Motil. Cytoskelet. 1987;7:258–271. [Google Scholar]
- Sablin E.P., Kull F.J., Cooke R., Vale R.D., Fletterick R.J. Crystal structure of the motor domain of the kinesin-related motor ncd. Nature. 1996;380:555–559. doi: 10.1038/380555a0. [DOI] [PubMed] [Google Scholar]
- Saito N., Okada Y., Noda Y., Kinoshita Y., Kondo S., Hirokawa N. KIFC2 is a novel neuron-specific C-terminal type kinesin superfamily motor for dendritic transport of multivesicular body-like organelles. Neuron. 1997;18:425–438. doi: 10.1016/s0896-6273(00)81243-x. [DOI] [PubMed] [Google Scholar]
- Sakowicz R., Farlow S., Goldstein L.S.B. Cloning and expression of kinesins from the thermophilic fungus Thermomyces lanuginosus . Prot. Sci. 1999;In press doi: 10.1110/ps.8.12.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M., Hyvonen M. Pleckstrin homology domainsa fact file. Curr. Opin. Struct. Biol. 1995;5:403–408. doi: 10.1016/0959-440x(95)80104-9. [DOI] [PubMed] [Google Scholar]
- Saxton W.M., Hicks J., Goldstein L.S., Raff E.C. Kinesin heavy chain is essential for viability and neuromuscular functions in Drosophila, but mutants show no defects in mitosis. Cell. 1991;64:1093–1102. doi: 10.1016/0092-8674(91)90264-y. [DOI] [PubMed] [Google Scholar]
- Schnapp B.J., Reese T.S. Dynein is the motor for retrograde axonal transport of organelles. Proc. Natl. Acad. Sci. USA. 1989;86:1548–1552. doi: 10.1073/pnas.86.5.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnapp B.J., Reese T.S., Bechtold R. Kinesin is bound with high affinity to squid axon organelles that move to the plus-end of microtubules. J. Cell Biol. 1992;119:389–399. doi: 10.1083/jcb.119.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer T.A., Schnapp B.J., Reese T.S., Sheetz M.P. The role of kinesin and other soluble factors in organelle movement along microtubules. J. Cell Biol. 1988;107:1785–1792. doi: 10.1083/jcb.107.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer T.A., Sheetz M.P. Two activators of microtubule-based vesicle transport. J. Cell Biol. 1991;115:1309–1318. doi: 10.1083/jcb.115.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer T.A., Steuer E.R., Sheetz M.P. Cytoplasmic dynein is a minus-end-directed motor for membranous organelles. Cell. 1989;56:937–946. doi: 10.1016/0092-8674(89)90627-2. [DOI] [PubMed] [Google Scholar]
- Seiler S., Nargang F.E., Steinberg G., Schliwa M. Kinesin is essential for cell morphogenesis and polarized secretion in Neurospora crassa . EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:3025–3034. doi: 10.1093/emboj/16.11.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D.R., Richter H., Giorda R., Ohmachi T., Ennis H.L. Nucleotide sequences of Dictyostelium discoideum developmentally regulated cDNAs rich in (AAC) imply proteins that contain clusters of asparagine, glutamine, or threonine. Mol. Gen. Genet. 1989;218:453–459. doi: 10.1007/BF00332409. [DOI] [PubMed] [Google Scholar]
- Skoufias D., Cole D.G., Wedaman K.P., Scholey J.M. The carboxyl-terminal domain of kinesin heavy chain is important for membrane binding. J. Biol. Chem. 1994;269:1477–1485. [PubMed] [Google Scholar]
- Sussman M. Cultivation and synchronous morphogenesis of Dictyostelium under controlled experimental conditions. Methods Cell Biol. 1987;28:9–29. doi: 10.1016/s0091-679x(08)61635-0. [DOI] [PubMed] [Google Scholar]
- Tai A.W., Chuang J.-Z., Bode C., Wolfrum U., Sung C.-H. Rhodopsin's carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain Tctex-1. Cell. 1999;97:877–887. doi: 10.1016/s0092-8674(00)80800-4. [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Kanai Y., Okada Y., Nonaka S., Takeda S., Harada A., Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Tinsley J.H., Minke P.F., Bruno K.S., Plamann M. P150Glued, the largest subunit of the dynactin complex, is nonessential in Neurospora but required for nuclear distribution. Mol. Biol. Cell. 1996;7:731–742. doi: 10.1091/mbc.7.5.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima I., Yu H., Steuer E.R., Sheetz M.P. Kinectin, a major kinesin-binding protein on ER. J. Cell Biol. 1992;118:1121–1131. doi: 10.1083/jcb.118.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuma M.C., Zill A., Le Bot N., Vernos I., Gelfand V. Heterotrimeric kinesin II is the microtubule motor protein responsible for pigment dispersion in Xenopus melanophores. J. Cell Biol. 1998;143:1547–1558. doi: 10.1083/jcb.143.6.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrutia R., McNiven M.A., Albanesi J.P., Murphy D.B., Kachar B. Purified kinesin promotes vesicle motility and induces active sliding between microtubules in vitro. Proc. Natl. Acad. Sci. USA. 1991;88:6701–6705. doi: 10.1073/pnas.88.15.6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale R.D., Fletterick R.J. The design plan of kinesin motors. Annu. Rev. Cell Dev. Biol. 1997;13:745–777. doi: 10.1146/annurev.cellbio.13.1.745. [DOI] [PubMed] [Google Scholar]
- Vale R.D., Reese T.S., Sheetz M.P. Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell. 1985;42:39–50. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonekawa Y., Harada A., Okada Y., Funakoshi T., Kanai Y., Takei Y., Terada S., Noda T., Hirokawa N. Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J. Cell Biol. 1998;141:431–441. doi: 10.1083/jcb.141.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]