

Abstract
Chemokine receptor CCR3 is highly expressed by eosinophils and signals in response to binding of the eotaxin family of chemokines, which are upregulated in allergic disorders. Consequently, CCR3 blockade is of interest as a possible therapeutic approach for the treatment of allergic disease. We have described previously a bi-specific antagonist of CCR1 and CCR3 named UCB35625, which was proposed to interact with the transmembrane residues Y41, Y113 and E287 of CCR1, all of which are conserved in CCR3. Here, we show that cells expressing the CCR3 constructs Y113A and E287Q are insensitive to antagonism by UCB35625 and also exhibit impaired chemotaxis in response to CCL11/Eotaxin suggesting that these residues are important for antagonist binding and also receptor activation. Furthermore, mutation of the residue Y113 to alanine was found to turn the antagonist UCB35625 into a CCR3 agonist. Screens of small molecule libraries identified a novel specific agonist of CCR3 named CH0076989. This was able to activate eosinophils and transfectants expressing both wild-type CCR3 and a CCR1:CCR3 chimaeric receptor lacking the CCR3 amino-terminus, indicating that this region of CCR3 is not required for CH0076989 binding. A direct interaction with the transmembrane helices of CCR3 was supported by mutation of the residues Y41, Y113 and E287 which resulted in complete loss of CH0076989 activity, suggesting that the compound mimics activation by CCL11. We conclude that both agonists and antagonists of CCR3 appear to occupy overlapping sites within the transmembrane helical bundle, suggesting a fine line between agonism and antagonism of chemokine receptors.
Asthma is characterised by the accumulation within the bronchial wall of leukocytes, of which the eosinophil predominates (1). Once recruited, eosinophils release a variety of mediators implicated in asthma pathology. Some of these, such as major basic protein, eosinophil cationic protein, and eosiniphil peroxidase, can directly induce tissue damage (2). Release of other mediators such as LTC4 and TGF-β can induce other responses such as bronchoconstriction, mucus hypersecretion (3) and airway remodelling (4).
Chemokines are a family of low molecular weight proteins which are key to the regulation of leukocyte migration, exerting their activity via G protein-coupled receptors expressed on the cell-surface (5). Whilst chemokines are associated with homeostatic cell migration and host defence, inadvertent or excessive production of chemokines has been implicated in the inflammatory components of many clinically important diseases including asthma (6). A chemokine central to the pathology of asthma is the CC chemokine CCL11/eotaxin (7). This chemokine interacts with the receptor CCR3 (8-10) the principal chemokine receptor expressed by the eosinophil (8-10) but also by other cells involved in the allergic response such as Th2 cells (11), the basophil (12) and mast cells (13). Activation of CCR3 by CCL11 occurs via a two step model (14) involving tethering of the ligand by the receptor amino-terminus and subsequent delivery to the extracellular loops (ECLs) (15,16). This is postulated to lead to conformational changes in the receptor, resulting in G protein recruitment and activation of intracellular signalling. Data from studies of activation of the related receptor, CCR5, suggest that the amino-terminus of the chemokine disrupts interactions between the side chains of neighbouring transmembrane helices, leading to the induction of the active receptor conformation (17,18). In contrast, the mechanism of CCR3 activation by CCL11 remains poorly understood, despite effort from several groups (19-24).
We have previously described the effects of a bi-specific CCR1/CCR3 antagonist UCB35625 (25), which interacts with the residues Y41, Y113, and E287 of CCR1 (26). A homology model of the structure of CCR1 transmembrane region, calculated using the structure of the related receptor rhodopsin as template (27), suggests these residues make up an intrahelical bundle in which the antagonist sits. Here we show that within CCR3, the conserved residues Y113 and E287 are important for the antagonist activity of UCB35625 and also for the agonist activity of CCL11. We also describe a novel small molecule agonist of CCR3, CH0076989, which binds to a site within the receptor overlapping that of the antagonist.
EXPERIMENTAL PROCEDURES
Materials
Reagents were purchased from Sigma-Aldrich (Poole, UK) and Invitrogen (Paisley, UK), unless stated otherwise. Recombinant human CCL11, was purchased from PeproTech EC Ltd. (London, UK). CH0076989 was provided by UCB-Celltech (Slough, UK). The monoclonal mouse anti-haemagglutinin (HA) antibody was purchased from Covance Research Products (Berkley, CA), and its corresponding IgG1 isotype control antibody was from Sigma-Aldrich. The rat monoclonal anti-CCR3 was from R & D Systems and its corresponding IgG2A isotype from BD Biosciences, Oxford, UK. Fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibody and the RPE conjugated goat anti-rat IgG were purchased from DAKO Cytomation (Ely, UK) and AbD Serotec (Kidlington, UK) respectively.
Generation of HA-CCR3 Point Mutants
A CCR3 construct tagged at the N-terminus with the HA-epitope was used as a template for mutagenesis. Point mutants of human HA-CCR3 were generated by PCR according to the manufacturer’s instructions, using the QuickChange™ site-directed mutagenesis kit (Stratagene, Amsterdam, Netherlands). Verification of mutation was performed by DNA sequencing on both strands (MWG Biotech, Ebersberg, Germany).
Cell Culture and transfection
The murine pre-B lymphoma cell line L1.2 was maintained in suspension in HEPES modified RPMI 1640 medium containing 10% foetal bovine serum (FBS), 100 units/ml penicillin, 0.1 mg/ml streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate, 50 μM β-mercaptoethanol and 1 × MEM non-essential amino acids at 37°C with 5% CO2 at a density of no more than 1 × 106 cells/ml. The previously described 4DE4 cell line stably transfected with CCR3 (15), was cultured in the aforementioned medium supplemented with 1 mg/ml G-418. L1.2 cells were transiently transfected by electroporation with 1 μg DNA/1 × 106 cells, as described previously (23) and were incubated overnight in the presence of 10 mM sodium butyrate to increase responsiveness.
Flow Cytometry
Approximately 5 × 105 cells were harvested, washed once with fluorescence-activated cell sorting (FACS) buffer (0.25% bovine serum albumin and 0.01% NaN3 in HEPES modified phosphate-buffered saline (PBS)) and incubated with 50 μg/ml anti-HA/anti-CCR3 antibody, or the appropriate isotype control, in 100 μl ice cold FACS buffer for 20 minutes. Cells were then washed with FACS buffer and incubated with 50μg/ml of the corresponding secondary antibody for 20 minutes in 50 μl of ice-cold FACS buffer. After this, cells were washed twice and resuspended in 400 μl FACS buffer before being analysed by flow cytometry, as described previously (26).
Chemotaxis Assay
The chemotactic responsiveness of cells was ascertained using ChemoTx™ plates (NeuroProbe, Gaithersburg, MD), as described previously (26). Using an assay buffer of RPMI supplemented with 0.1% BSA, duplicate concentrations of agonist in the presence or absence of UCB35625 were applied in a final volume of 31 μl to the lower wells of the chemotaxis chamber. The filter was put in place and 2 × 105 cells in the same assay buffer were applied to the upper surface. Following a 5 hour incubation in a humidified chamber at 37°C in the presence of 5% CO2, the number of migrating cells traversing a 5 μm pore filter were counted using a hemocytometer. Data are shown as the percentage of migrating cells.
Internalisation Assay
Chemokine receptor internalisation was assessed by the loss of cell surface receptor expression, as determined by flow cytometry using an antibody directed against CCR3 (28). 1 × 106 cells were resuspended in duplicate in 100 μl of ice cold culture medium with an additional tube serving as a buffer control. One set of cells was incubated at 37°C for 20 minutes prior to the addition of appropriate stimuli whilst the duplicates remained on ice. Either 100 nM CCL11, 10 μM CH0076989 or 10 μM UCB35625 were added to tubes incubating at 37°C or their controls on ice. All samples were then incubated for 20 minutes at either 37°C or on ice, before washing, staining and receptor expression analysis by flow cytometry. The remaining receptor expression was calculated as the mean fluorescence of antibody stained chemokine cells - mean fluorescence of isotype stained chemokine treated cells. This was expressed as a percentage of mean fluorescence of buffer treated cells.
Gated autofluorescence forward scatter (GAFS) assay
GAFS assays were performed as described previously (29). Blood was taken from healthy donors according to a protocol approved by the St Mary’s Hospital Ethics Committee protocol and granulocytes (comprising neutrophils and eosinophils) were isolated over discontinuous platelet-depleted plasma/Percoll gradients (30). Granulocytes were pre-incubated at a concentration of 5×106 cells/ml for 30 min at room temperature in filtered GAFS buffer (PBS containing 0.9 mM CaCl2, 0.5 mM MgCl2, 10 mM HEPES, 10 mM glucose and 0.1% BSA). The cells were washed and then incubated in GAFS buffer with varying concentrations of CH0076989 for 4 minutes at 37°C in a shaking water bath. Cells were then placed on ice and 250 μl of ice-cold fixative added. Finally the cells were analysed on a FACScaliber (BD Biosciences, Oxford, UK) as described previously (29)
Homology modelling
Generation of a CCR3 model structure and docking of antagonists and agonists was carried out as previously described (26), using our earlier model of CCR1 as a template.
Data and statistical analysis
Data are expressed as the mean ± SEM of at least three separate experiments, and were analysed with a relevant statistical test, where stated, using PRISM v4.03 software (GraphPad, San Diego, CA, USA).
RESULTS
Identification of the binding pocket within CCR3 for UCB35625
We have previously described the bispecific CCR1/CCR3 antagonist UCB35625 (Figure 1A) which antagonises responses mediated by either CCR1 and CCR3, with the compound an order of magnitude more potent at CCR1 (25). The mode of action of this compound at either receptor is intriguing. Whilst the compound is unable to effectively displace chemokines from either receptor (as deduced by radiolabelled binding studies), in assays of eosinophil shape change, the compound induces profiles of insurmountable antagonism (reduced maximal response) from CCR1 expressing cells and profiles of surmountable antagonism (right shifted response) from CCR3 expressing cells (25). Using L1.2 CCR3 transfectants, chemotactic responses to increasing concentrations of CCL11 were assessed in the presence or absence of 500 nM UCB35625 (Figure 1B). In keeping with earlier reported GAFS data (25) antagonism was surmountable, with high concentrations of CCL11 able to induce migration albeit with reduced efficacy. This is indicative of a competitive element to the antagonist activity of UCB35625 at CCR3.
Figure 1. The CCR1/3 antagonist UCB35625 displays surmountable antagonism at CCR3 in assays of chemotaxis.

Panel A shows the structure of the small molecule antagonist of CCR1/3 UCB35625. Panel B shows the chemotactic responses of L1.2 transfectants transiently expressing CCR3 to CCL11, in the presence or absence of 500nM UCB35625. Data is represented as the mean ± SEM from three separate experiments.
A previous study from our group determined the binding site of UCB35625 within CCR1, with mutation of the transmembrane (TM) amino acids Y41 (TM1), Y113 (TM3) and E287 (TM7) leading to resistance to UCB35625 (26). Since CCR3 and CCR1 exhibit a high level of homology at the amino acid level and all three residues are conserved between both receptors, we postulated that the compound might interact with the same residues within CCR3. HA-tagged Y41A, Y113A and E287Q mutant CCR3 constructs were generated via site-directed mutagenesis. These were transiently transfected into L1.2 cells along with wild type (WT) HA-CCR3 and surface receptor expression measured by flow cytometry using an anti-HA antibody (Figure 2A). All three mutants were expressed at the cell surface, although the Y41A mutant appeared to be expressed at slightly reduced levels compared to WT CCR3. The same transfectants were subsequently examined in a chemotaxis assay in response to increasing concentrations of CCL11 in the presence or absence of 500 nM UCB35625 (Figure 2B-E). In contrast to our previous findings with CCR1, mutation of Y41 to alanine in CCR3 did not lead to resistance to blockade by UCB35625, with cells expressing this construct responding to CCL11 with an identical profile to cells expressing the WT receptor (Figures 2C and B respectively), albeit with a reduced maximum. This correlates with the apparent reduced expression of the Y41A construct. However, mutagenesis of both Y113 and E287 yielded CCR3 constructs resistant to antagonism of the CCL11 response by UCB35625 (Figure 2D and E). Also of interest was the reduced potency of CCL11 for cells expressing the Y113A and E287Q constructs, with little activity observed at the 1nM concentration of CCL11 compared to cells expressing WT CCR3. This suggests that Y113 and E287 are involved in both antagonist binding and receptor activation by the agonist CCL11. On closer inspection of the response of Y113A transfectants (Figure 2D), we identified an apparent increase in basal chemotaxis in the absence of CCL11 but in the presence of 500 nM UCB35625, suggesting that the compound might possess agonist activity at this mutated receptor. This was subsequently verified by assessment of the chemotaxis of Y113A and WT CCR3 transfectants to increasing concentrations of the antagonist (Figure 2F). Whilst UCB35625 was not chemotactic for the WT CCR3 transfectants, Y113A transfectants responded with a typical bell-shaped profile, although with significantly lower efficacy compared to the natural ligand CCL11. Collectively, these data suggest that Y113 and E287 are involved in both antagonist binding and receptor activation by agonist and that mutation of Y113 to alanine converts UCB35625 from an antagonist of CCR3 to a partial agonist of the receptor.
Figure 2. The CCR3 binding pocket of the small molecule antagonist UCB35625 involves the helical residues Y113 and E287Q.

Panel A shows the relative expression levels of CCR3 and the mutants Y41A, Y113A and E287Q following transient transfection of L1.2 cells. Panels B through E show the chemotactic responses of the same transfectants to CCL11 in the presence or absence of 500nM UCB35625. Panel F shows the chemotactic response of the of WT CCR3 and Y113A-CCR3 transfectants in response to increasing concentrations of UCB35625 or 10nM CCL11. Data is represented as the mean ± SEM from three separate experiments.
The small molecule CH0076989 is an agonist of CCR3
A subsequent screen of a small molecule library identified a compound CH0076989 (Figure 3A) which to our surprise raised the baseline of CCL11 responses in the GAFS assay (data not shown). We subsequently investigated whether CH0076989 had agonist activity independent of CCL11. Eosinophils isolated from 4 different donors were found to respond in a dose-dependent manner to the compound (Figure 3B), with optimal activity at 1 μM CH0076989, suggesting that the compound was indeed an agonist at an unidentified receptor. Since eosinophils express CCR3 but can also express CCR1 (29,31) we hypothesized that one of these receptors might be mediating the response to CH0076989. Stable transfectants expressing CCR3 or CCR1 were used in chemotaxis assays and the responses to increasing amounts of CH0076989 determined (Figure 4A). Whilst CCR1 transfectants were unresponsive to the compound, CCR3 transfectants migrated in a concentration-dependent fashion to CH0076989 with optimal chemotaxis observed at 1 μM, identical to the concentration required for optimal activity in the GAFS assay. Pre-incubation of CCR3 transfectants with anti-CCR3 antibodies reduced chemotactic responses to both 1 μM CH0076989 and 10 nM CCL11 (Figure 4B), thus confirming that CH0076989 is a specific agonist of CCR3.
Figure 3. CH0076789 induces eosinophil shape change in a dose dependent manner.

Panel A shows the structure of CH0076789. Panel B shows eosinophil shape change to increasing concentrations of CH0076789 from four different donors.
Figure 4. The small molecule agonist CH0076789 induces chemotaxis via CCR3 but not CCR1.
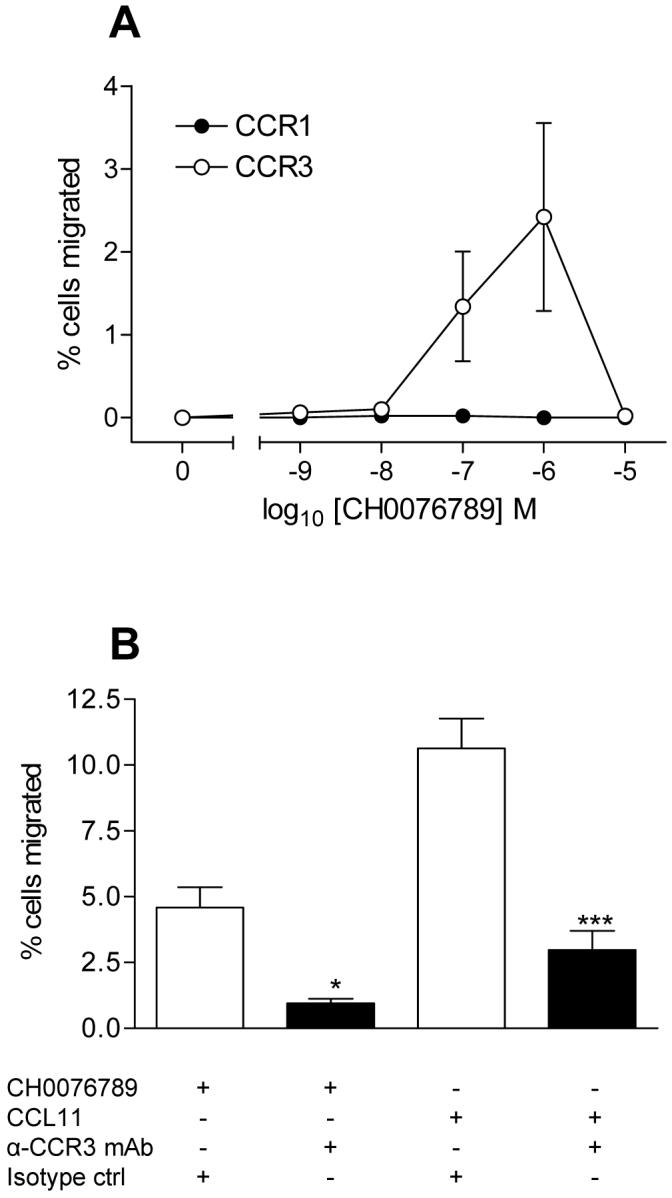
Panel A shows the chemotactic responses of L1.2 cells stably expressing either CCR1 or CCR3 to increasing concentrations of CH0076789. Panel B shows the response of L1.2 cells stably expressing CCR3 to either 1 μM CH0076789 or 10 nM CCL11 in the presence of a CCR3-specific blocking antibody or an isotype control. Basal responses to buffer alone were subtracted. Data is from four separate experiments. ***denotes p value of less than 0.001 and * denotes p value of less than 0.05 using one-way Anova with Bonferroni’s multiple comparisons test.
CH0076989 is a partial agonist of CCR3 and induces receptor internalisation
We subsequently compared chemotactic responses to both CCL11 and CH0076989 in parallel, using CCR3 transfectants. CH0076989 was notably less potent and efficacious than CCL11, with maximal chemotactic responses to CCL11 observed at 3nM compared to an optimal concentration of 1μM CH0076989 (Figure 5A). Upon incubation with a concentration of ligand in excess of that required for optimal chemotaxis, chemokine receptors typically undergo endocytosis or internalization. Since the small molecule showed activity at CCR3 in GAFS and chemotaxis assays we assessed whether or not the compound could modulate receptor internalization as assayed by flow cytometry using CCR3 specific antibodies. Cell surface expression of CCR3 was seen to be reduced following treatment of L1.2 CCR3 transfectants cells with 100 nM CCL11 for 20 minutes at 37°C (Figure 5B), in agreement with previous findings (28). A 10 μM concentration of CH0076989 was also seen to induce a loss of CCR3 at the cell surface. In contrast to the reduced efficacy of the compound in chemotaxis assays, CH0076989 reduced surface receptor expression levels to those observed with CCL11 (59.4% and 59.6% of original expression, respectively). Surface expression was not significantly reduced following incubation with 10 μM of the previously characterised CCR1/CCR3 bi-specific antagonist UCB 35625 (25) suggesting that agonist activity at the receptor, and not simply the ability to bind to CCR3, is needed to induce receptor endocytosis.
Figure 5. CH0076789 is a partial agonist of CCR3 and induces receptor downregulation.
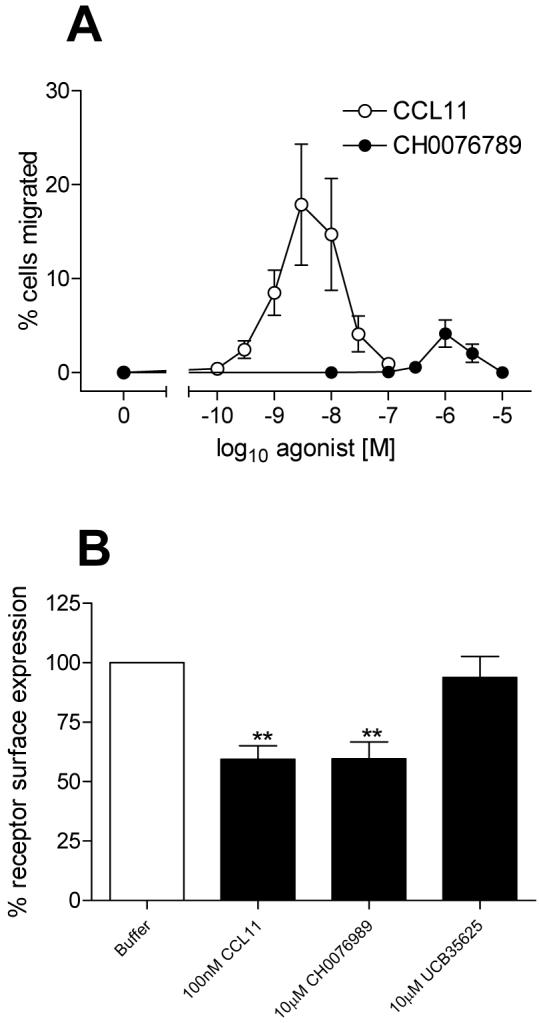
Panel A shows the comparative chemotactic responses of CCR3 transfectants to increasing concentrations of CCL11 and CH0076789. Panel B shows the ability of fixed concentrations of CCL11, CH0076789 or the CCR1/3 antagonist UCB35625 to induce CCR3 down regulation, as observed by flow cytometry. Data is represented as the mean ± SEM from three separate experiments. ** denotes p value of less than 0.01 using one-way Anova with Bonferroni’s multiple comparisons test.
We have previously shown that in order to activate CCR3, CCL11 has to bind to the amino-terminus and extracellular regions of the receptor (15,24). Indeed CCR1/3 chimaeric constructs in which the amino-termini of both receptors are exchanged are unable to efficiently activate G proteins, despite being able to bind ligand (15). Since CH0076989 is several orders of magnitude smaller than CCL11, we hypothesized that it may activate CCR3 by binding to different receptor determinants than the natural ligand CCL11. To test this hypothesis, a chimaeric receptor (Chi-1) containing amino acids 1-32 from CCR1 fused to amino acids 33-355 of CCR3 (15) was employed in chemotaxis assays (Figure 6A). Cells transiently expressing either the chimaeric construct Chi-1 or wild-type CCR3 were assessed for their chemotactic responses to increasing concentrations of CH0076989 or a fixed concentration of 10nM CCL11 (Figures 6B and C). CCR3 transfectants responded robustly to CCL11 and CH0076989 as seen before (Figure 5B). Cells expressing Chi-1 transfectants were able to respond to CH0076989 in a manner identical to CCR3 transfectants but showed markedly reduced responsiveness to CCL11 as previously described (15) suggesting that whilst the amino-terminus of CCR3 is required for effective CCL11 activity, it is dispensable for functional responses to CH0076989.
Figure 6. The small molecule agonist CH0076789 activates CCR3 by binding to a site distinct from that of CCL11.
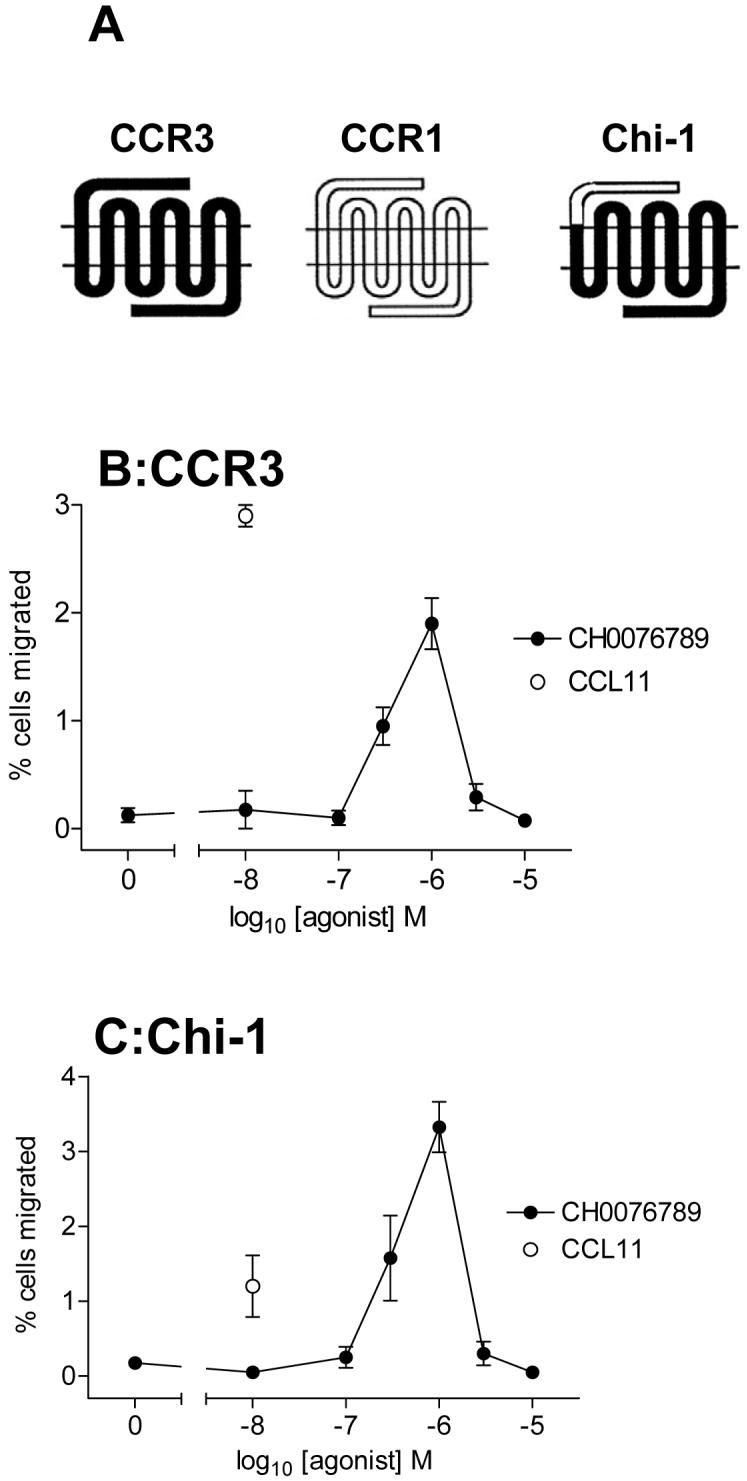
Panel A shows the pedigree of the chimeric construct Chi-1 which has the backbone of CCR3 and the amino terminus of CCR1. Panels B and C show the chemotactic responses to CCL11 or CH0076789 of L1.2 cells transiently transfected with cDNA encoding either CCR3 or Chi-1. Data is represented as the mean ± SEM from three separate experiments.
CH0076989 interacts with a binding pocket within CCR3 which overlaps with that of UCB35625
Since CH0076989 is predominantly hydrophobic and does not appear to interact with the amino-terminus of CCR3 we hypothesised that, like UCB35625, CH0076989 might bind to CCR3 within a pocket located between the TM helices. We therefore examined the three mutants of CCR3 that we had generated in our analysis of UCB35625 function, namely Y41A, Y113A and E287Q. Whilst transfectants expressing WT CCR3 were able to migrate in response to CH0076989, to our surprise, chemotaxis to CH0076989 was ablated in cells expressing either of the three point mutants (Figure 7A). As a control, the same transfectants were simultaneously assessed for their ability to respond to a fixed concentration of 10 nM CCL11 (Figure 7B). Responses of the same transfectants to CCL11 remained intact as seen previously (Figure 2B-D), suggesting that the lack of efficacy of CH0076989 at the three mutant constructs was specifically related to the inability of the small molecule to activate the receptor and not simply due to an impaired ability of the transfectants to undergo chemotaxis. Since the CCR3 residues Y113 and E287 also line the binding pocket for the antagonist UCB35625 we hypothesized that the sites for both antagonist and agonist binding might overlap. If this is the case then UCB35625 should be an antagonist of CH0076989 induced responses. We therefore assessed chemotactic responses of L1.2 CCR3 transfectants towards a fixed 1 μM concentration of CH0076989 in the presence of increasing amounts of UCB35625 (Figure 7C). In agreement with our hypothesis, UCB35625 was observed to readily inhibit the chemotactic activity of CH0076989 with equimolar concentrations of the antagonist greatly reducing cell migration. Thus, despite having diametrically opposed activities at CCR3, the compounds UCB35625 and CH0076989 appear to utilize an overlapping binding site within the transmembrane intrahelical bundle of CCR3.
Figure 7. The CCR3 binding pocket of the small molecule agonist CH0076789 overlaps that of the antagonist UCB35625.
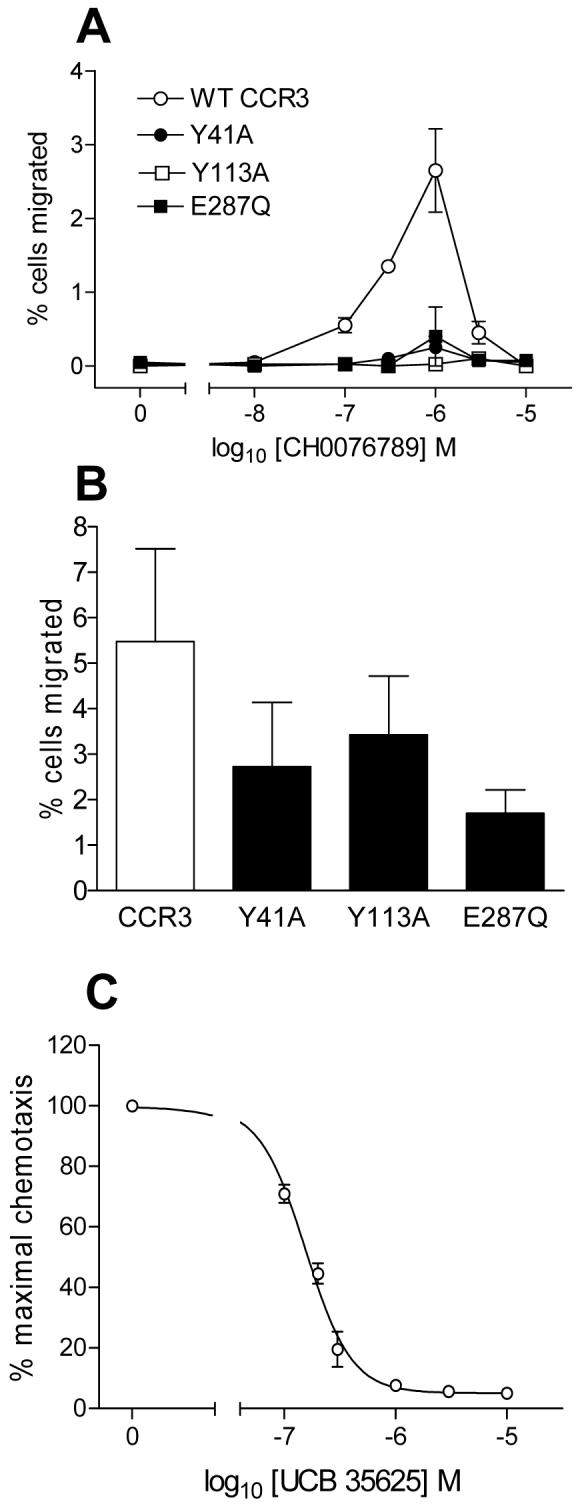
Panel A shows the chemotactic responses to CH0076789 of L1.2 cells transiently expressing CCR3 and the mutants Y41A, Y113A and E287Q. Panel B shows the chemotactic responses of the same transfectants to a fixed concentration of 10nM CCL11. Panel C shows dose-dependent inhibition of the chemotactic response of CCR3 transfectants to 1 μM CH0076789 by increasing concentrations of UCB35625. Basal migration to buffer alone was subtracted. Data is represented as the mean ± SEM from three separate experiments.
DISCUSSION
We describe here a small molecule named CH0076989 which is an agonist of the chemokine receptor CCR3 and appears to activate the receptor by binding to a site also used by a well characterized receptor antagonist, UCB35625 (25). Chemokines are thought to activate their receptors via a two-step model (14) involving an initial tethering of the chemokine by the amino-terminus of the chemokine receptor followed by interactions of the chemokine with the receptor extracellular loops. In the case of the related receptor CCR5, this has been postulated to facilitate the interaction of the chemokine amino-terminus with side chains of residues located within the transmembrane helices, notably residues within helices 2 and 3 (17,18). An earlier study from our group using chimaeric CCR1/3 constructs supports the notion that both CCR1 and CCR3 follow the two-step model of receptor activation as exchange of their respective amino-termini resulted in constructs with a considerably reduced affinity for ligand and ability to induce receptor activation (15). This model is also supported by NMR data describing interactions in solution between a amino-terminal peptide of CCR3 and CCL11 (19). Our present results indicate that the molecule CH0076989 bypasses such restraints and utilizes the transmembrane residues Y41, Y113 and E287 of CCR3 to interact with the receptor, leading to activation. Figure 8A shows a cartoon representation of CCR3 based on our previous model of the highly homologous CCR1 (26), with Y41 of TM1, Y113 of TM3 and E287 of TM7 highlighted. A possible docking of CH0076989 to these residues is shown in Figure 8B. Here the acidic side chain of E287 is postulated to form a salt bridge with the positively charged quaternary nitrogen of the agonist, whilst the hydroxyl groups of Y41 and Y113 hydrogen bond to oxygen moieties within the ring structure (Figure 8C).
Figure 8. Interpretation of molecular interactions of CCR3 with artificial and natural agonists.

Panel A depicts a homology model of the seven helical regions of CCR3 (cyan) with the side chains Y41, Y113 and E287 highlighted in yellow. Panels B and C show the same model with the small molecule agonist CH0076789 (magenta) docked. Putative hydrogen bonds are denoted by dashed lines. Panels D and E show the interaction of the amino-terminus of CCL11 (magenta) with the side chains Y113 and E287 of CCR3. Potential hydrogen bonds between the sidechains Y113 and E287 of CCR3 with the sidechain of S4 and the backbone oxygen of P2 of CCL11 are denoted by dashed lines.
In keeping with the importance of Y113 and E287 in the activation of CCR3, Y113A and E287Q transfectants exhibited reduced potency in chemotactic responses to CCL11, when compared to those of wild type expressing cells. Relating this data to the two-step model for CCR3 activation (15), we therefore postulate that CH0076989 mimics the interactions of the CCL11 amino-terminus with Y113 and E287 and induces receptor activation. Such a model would also be in keeping with other class A GPCRs such as rhosposin and the β2 adrenergic receptor, where movement of both TM3 and TM7 has been reported to occur during the activation process (32,33). The solution structure of CCL11 has been previously solved by NMR (34) and backbone dynamics calculations have shown the amino-terminal region of CCL11 to be highly flexible upstream of the constraining CC disulfide bridge with little interaction with the core of the chemokine (35,36). Using this NMR structure (PDB accession number 1EOT), we subsequently docked the amino-terminus of CCL11 within our model of CCR3 (Figure 8D and E) In this scenario, the hydroxyl group of Y113 is postulated to hydrogen bond with the hydroxyl group of the S4 residue of CCL11 and E287 hydrogen bond with the backbone oxygen of the P2 residue of CCL11. Whilst these interactions are at present speculative, such a model fits previous experimental data generated by other groups. Firstly, that removal of the first two amino acids of CCL11 by CD26/DPPIV severely reduces its biological activity (37) and, secondly, mutation of S4 of CCL11 to alanine results in a loss of both binding affinity and biological activity (20).
Y113 and E287 of CCR3 are also contact points for the small molecule antagonist UCB35625, which has activity at both CCR3 and its close relative CCR1 (26). We have previously shown that Y113 and E287, together with Y41, are critical for the activity of UCB35625 at CCR1 (26). Whilst it is not surprising that the antagonist interacts with a similar pocket in both receptors, it is curious that the same residues should be implicated in receptor activation by both the natural ligand CCL11 and the small molecule CH0076989. This may help explain the surmountable (right-shifted) profiles of antagonism obtained with UCB35625 following activation of CCR3 by CCL11, both here and in a previous study (25).
Homology modelling of CCR3 produced a helical bundle of very similar structure to that of CCR1, which is unsurprising as the receptors share considerable identity in these regions and the homology model of CCR3 was obtained using the previous presented model of CCR1 as template (25,38). While allowing the docking of agonists and antagonists to CCR3, such a model does not help to explain the different activities we observed at each receptor. For example, Y113 and E287 are conserved in both CCR1 and CCR3, yet CH0076989 only had activity at CCR3. High resolution structural information will be required to definitively answer these points. In its absence, recent developments in the ab initio modelling of chemokine receptors show much promise (39) and may help the refinement of our current models, highlighting structural differences between such closely related proteins.
Our finding that antagonists and agonists of CCR3 have overlapping binding sites and that a single point mutation in the receptor can turn an antagonist into an agonist, suggests that there is a fine line between agonist and antagonist activity at CCR3. Supportive of this, other CCR3 antagonist programmes have also reported the identification of small molecule agonists of the receptor following optimization of the structure-activity relationships of lead compounds (40,41). This may also be the case for other chemokine receptors, with the recent reports of small molecule agonists of CCR5, 8 and CXCR3 (42-44). Since genetic variations such as SNPs are relatively abundant within the genes encoding GPCRs (45) it is also plausible that certain individuals within a population harbour mutations which unexpectedly turn receptor antagonists into agonists or render the antagonists ineffective. It may therefore be beneficial screening individuals prior to their entry into clinical trials of such compounds.
In summary, we describe here the characterisation of a small molecule CCR3 agonist, CH0076989, which we postulate activates the receptor by mimicking the amino-terminus of the natural ligand CCL11. The elucidation of the binding site of CH0076989 within CCR3 along with that of the proteotypic antagonist UCB35625 gives insight into the mechanisms of chemokine receptor activation and may aid the development of more potent, selective, CCR3 antagonists. These have potential as therapeutics for the future treatment of allergic diseases such as asthma.
Acknowledgments
This work was supported by the Wellcome Trust, Project Grant 076036/Z/04/Z.
The abbreviations used are
- CCR
CC chemokine receptor
- ECLs
extracellular loops
- GAFS
gated autofluorescence/forward scatter
- HA
hemagglutinin
- LTC4
leukotriene C4
- TGF-β
transforming growth factor-β
- TM
transmembrane
Contributor Information
Emma L. Wise, Leukocyte Biology Section, NHLI Division, Faculty of Medicine, Sir Alexander Fleming Building, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
Cécile Duchesnes, Leukocyte Biology Section, NHLI Division, Faculty of Medicine, Sir Alexander Fleming Building, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK.
Paula C.A. da Fonseca, Structural Electron Microscopy Team, Section of Structural Biology, The Institute of Cancer Research, Chester Beatty Laboratories, London, SW3 6JB, UK .
Rodger A. Allen, UCB, Slough, SL1 4EN, UK. .
Timothy J. Williams, Leukocyte Biology Section, NHLI Division, Faculty of Medicine, Sir Alexander Fleming Building, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
James E. Pease, Leukocyte Biology Section, NHLI Division, Faculty of Medicine, Sir Alexander Fleming Building, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
REFERENCES
- 1.Gleich GJ. J Allergy Clin Immunol. 2000;105:651–663. doi: 10.1067/mai.2000.105712. [DOI] [PubMed] [Google Scholar]
- 2.Venge P, Dahl R, Fredens K, Peterson CG. Am.Rev.Respir.Dis. 1988;138:S54–57. doi: 10.1164/ajrccm/138.6_Pt_2.S54. [DOI] [PubMed] [Google Scholar]
- 3.Flavahan NA, Slifman NR, Gleich GJ, Vanhoutte PM. Am.Rev.Respir.Dis. 1988;138:685–688. doi: 10.1164/ajrccm/138.3.685. [DOI] [PubMed] [Google Scholar]
- 4.Kay AB, Phipps S, Robinson DS. Trends Immunol. 2004;25:477–482. doi: 10.1016/j.it.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 5.Rot A, Von Andrian UH. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- 6.Pease JE, Williams TJ. J Allergy Clin Immunol. 2006;118:305–318. doi: 10.1016/j.jaci.2006.06.010. quiz 319-320. [DOI] [PubMed] [Google Scholar]
- 7.Jose PJ, Griffiths-Johnson DA, Collins PD, Walsh DT, Moqbel R, Totty NF, Truong O, Hsuan JJ, Williams TJ. J.Exp.Med. 1994;179:881–887. doi: 10.1084/jem.179.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponath PD, Qin S, Post TW, Wang J, Wu L, Gerard NP, Newman W, Gerard C, Mackay CR. J Exp Med. 1996;183:2437–2448. doi: 10.1084/jem.183.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daugherty BL, Siciliano SJ, DeMartino J, Malkowitz L, Sirontino A, Springer MS. J Exp Med. 1996;183:2349–2354. doi: 10.1084/jem.183.5.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitaura M, Nakajima T, Imai T, Harada S, Combadiere C, Tiffany HL, Murphy PM, Yoshie O. J Biol Chem. 1996;271:7725–7730. doi: 10.1074/jbc.271.13.7725. [DOI] [PubMed] [Google Scholar]
- 11.Sallusto F, Mackay CR, Lanzavecchia A. Science. 1997;277:2005–2007. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 12.Uguccioni M, Mackay CR, Ochensberger B, Loetscher P, Rhis S, LaRosa GJ, Rao P, Ponath PD, Baggiolini M, Dahinden CA. J Clin Invest. 1997;100:1137–1143. doi: 10.1172/JCI119624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romagnani P, De Paulis A, Beltrame C, Annunziato F, Dente V, Maggi E, Romagnani S, Marone G. Am J Pathol. 1999;155:1195–1204. doi: 10.1016/S0002-9440(10)65222-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monteclaro FS, Charo IF. J Biol Chem. 1996;271:19084–19092. doi: 10.1074/jbc.271.32.19084. [DOI] [PubMed] [Google Scholar]
- 15.Pease JE, Wang J, Ponath PD, Murphy PM. J Biol Chem. 1998;273:19972–19976. doi: 10.1074/jbc.273.32.19972. [DOI] [PubMed] [Google Scholar]
- 16.Xanthou G, Duchesnes CE, Williams TJ, Pease JE. Eur J Immunol. 2003;33:2241–2250. doi: 10.1002/eji.200323787. [DOI] [PubMed] [Google Scholar]
- 17.Blanpain C, Doranz BJ, Bondue A, Govaerts C, De Leener A, Vassart G, Doms RW, Proudfoot A, Parmentier M. J Biol Chem. 2003;278:5179–5187. doi: 10.1074/jbc.M205684200. [DOI] [PubMed] [Google Scholar]
- 18.Govaerts C, Bondue A, Springael JY, Olivella M, Deupi X, Le Poul E, Wodak SJ, Parmentier M, Pardo L, Blanpain C. J Biol Chem. 2003;278:1892–1903. doi: 10.1074/jbc.M205685200. [DOI] [PubMed] [Google Scholar]
- 19.Ye J, Kohli LL, Stone MJ. J Biol Chem. 2000;275:27250–27257. doi: 10.1074/jbc.M003925200. [DOI] [PubMed] [Google Scholar]
- 20.Mayer MR, Stone MJ. J Biol Chem. 2001;276:13911–13916. doi: 10.1074/jbc.M011202200. [DOI] [PubMed] [Google Scholar]
- 21.Ye J, Mayer KL, Mayer MR, Stone MJ. Biochemistry. 2001;40:7820–7831. doi: 10.1021/bi010252s. [DOI] [PubMed] [Google Scholar]
- 22.Martinelli R, Sabroe I, LaRosa G, Williams TJ, Pease JE. J Biol Chem. 2001;276:42957–42964. doi: 10.1074/jbc.M103933200. [DOI] [PubMed] [Google Scholar]
- 23.Auger GA, Pease JE, Shen X, Xanthou G, Barker MD. Eur J Immunol. 2002;32:1052–1058. doi: 10.1002/1521-4141(200204)32:4<1052::AID-IMMU1052>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 24.Duchesnes CE, Murphy PM, Williams TJ, Pease JE. Mol Immunol. 2006;43:1221–1231. doi: 10.1016/j.molimm.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 25.Sabroe I, Peck MJ, Jan Van Keulen B, Jorritsma A, Simmons G, Clapham PR, Williams TJ, Pease JE. J Biol Chem. 2000;275:25985–25992. doi: 10.1074/jbc.M908864199. [DOI] [PubMed] [Google Scholar]
- 26.de Mendonca FL, da Fonseca PC, Phillips RM, Saldanha JW, Williams TJ, Pease JE. J Biol Chem. 2005;280:4808–4816. doi: 10.1074/jbc.M412267200. [DOI] [PubMed] [Google Scholar]
- 27.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le T I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 28.Sabroe I, Jorritsma A, Stubbs VE, Xanthou G, Jopling LA, Ponath PD, Williams TJ, Murphy PM, Pease JE. Eur J Immunol. 2005;35:1301–1310. doi: 10.1002/eji.200425171. [DOI] [PubMed] [Google Scholar]
- 29.Sabroe I, Hartnell A, Jopling LA, Bel S, Ponath PD, Pease JE, Collins PD, Williams TJ. J Immunol. 1999;162:2946–2955. [PubMed] [Google Scholar]
- 30.Haslett C, Guthrie LA, Kopaniak MM, Johnston RB, Henson PM. Am.J.Pathol. 1985;119:101. [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips R, Stubbs VELS, Henson MR, Williams TJ, Pease JE, Sabroe I. J Immunol. 2003;170:6190–6201. doi: 10.4049/jimmunol.170.12.6190. [DOI] [PubMed] [Google Scholar]
- 32.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 33.Gether U, Lin S, Ghanouni P, Ballesteros JA, Weinstein H, Kobilka BK. Embo J. 1997;16:6737–6747. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crump MP, Rajarathnam K, Kim KS, Clark-Lewis I, Sykes BD. J Biol Chem. 1998;273:22471–22479. doi: 10.1074/jbc.273.35.22471. [DOI] [PubMed] [Google Scholar]
- 35.Crump MP, Spyracopoulos L, Lavigne P, Kim KS, Clark-lewis I, Sykes BD. Protein Sci. 1999;8:2041–2054. doi: 10.1110/ps.8.10.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye J, Mayer KL, Stone MJ. J Biomol NMR. 1999;15:115–124. doi: 10.1023/a:1008376728947. [DOI] [PubMed] [Google Scholar]
- 37.Struyf S, Proost P, Schols D, De Clercq E, Opdenakker G, Lenaerts JP, Detheux M, Parmentier M, De Meester I, Scharpe S, Van Damme J. J Immunol. 1999;162:4903–4909. [PubMed] [Google Scholar]
- 38.Alkhatib G, Berger EA, Murphy PM, Pease JE. J Biol Chem. 1997;272:20420–20426. doi: 10.1074/jbc.272.33.20420. [DOI] [PubMed] [Google Scholar]
- 39.Vaidehi N, Schlyer S, Trabanino RJ, Floriano WB, Abrol R, Sharma S, Kochanny M, Koovakat S, Dunning L, Liang M, Fox JM, de Mendonca FL, Pease JE, Goddard WA, 3rd, Horuk R. J Biol Chem. 2006;281:27613–27620. doi: 10.1074/jbc.M601389200. [DOI] [PubMed] [Google Scholar]
- 40.Ting PC, Lee JF, Wu J, Umland SP, Aslanian R, Cao J, Dong Y, Garlisi CG, Gilbert EJ, Huang Y, Jakway J, Kelly J, Liu Z, McCombie S, Shah H, Tian F, Wan Y, Shih NY. Bioorg Med Chem Lett. 2005;15:1375–1378. doi: 10.1016/j.bmcl.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 41.Anderskewitz R, Bauer R, Bodenbach G, Gester D, Gramlich B, Morschhauser G, Birke FW. Bioorg Med Chem Lett. 2005;15:669–673. doi: 10.1016/j.bmcl.2004.11.039. [DOI] [PubMed] [Google Scholar]
- 42.Saita Y, Kodama E, Orita M, Kondo M, Miyazaki T, Sudo K, Kajiwara K, Matsuoka M, Shimizu Y. J Immunol. 2006;177:3116–3122. doi: 10.4049/jimmunol.177.5.3116. [DOI] [PubMed] [Google Scholar]
- 43.Haskell CA, Horuk R, Liang M, Rosser M, Dunning L, Islam I, Kremer L, Gutierrez J, Marquez G, Martinez AC, Biscone MJ, Doms RW, Ribeiro S. Mol Pharmacol. 2006;69:309–316. doi: 10.1124/mol.105.014779. [DOI] [PubMed] [Google Scholar]
- 44.Stroke IL, Cole AG, Simhadri S, Brescia MR, Desai M, Zhang JJ, Merritt JR, Appell KC, Henderson I, Webb ML. Biochem Biophys Res Commun. 2006;349:221–228. doi: 10.1016/j.bbrc.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 45.Tang CM, Insel PA. Expert Opin Ther Targets. 2005;9:1247–1265. doi: 10.1517/14728222.9.6.1247. [DOI] [PubMed] [Google Scholar]