

Abstract
Purpose
Two clinical trials were conducted to evaluate the clinical efficacy and immunologic impact of vaccination against the tyrosinase protein plus systemic interleukin 2 (IL-2) administration in patients with advanced metastatic melanoma.
Experimental Design
Full-length tyrosinase was employed as an immunogen to induce diverse immunologic responses against a commonly expressed melanoma antigen. Heterologous prime/boost vaccination with recombinant vaccinia and fowlpox vectors encoding tyrosinase was first explored in a randomized three-arm phase II trial, in which vaccines were administered alone or concurrently with low-dose or high-dose IL-2. In a subsequent single cohort phase II trial, all patients received the same vaccines and high-dose IL-2 sequentially rather than concurrently.
Results
Among a total of 64 patients treated on these trials, 8 objective partial responses (12.5%) were observed, all in patients receiving high-dose IL-2. Additional patients showed evidence of lesional regression (mixed tumor response) or overall regression that did not achieve partial response status (minor response). In vitro evidence of enhanced immunity against tyrosinase following protocol treatments was documented in 3 of 49 (6%) patients tested serologically, 3 of 23 (13%) patients tested for T-cell recognition of individual tyrosinase peptides, and 4 of 16 (25%) patients tested for T-cell recognition of full-length tyrosinase protein with real-time reverse transcription-PCR techniques.
Conclusions
Whereas prime/boost immunization with recombinant vaccinia and fowlpox viruses enhanced antityrosinase immunity in some patients with metastatic melanoma, it was ineffective alone in mediating clinical benefit, and in combination with IL-2 did not mediate clinical benefit significantly different from that expected from treatment with IL-2 alone.
Tyrosinase, an enzyme essential for melanin synthesis, is a commonly expressed melanoma/melanocyte lineage–specific protein capable of eliciting cellular and humoral immune responses in melanoma patients (1, 2). Immunogenic peptides spanning the entire length of this molecule include MHC class I–restricted (HLA-A1, -A2, -A24, -B35, and -B44) and class II–restricted (HLA-DR4, -DR8, and -DR15) epitopes recognized by CD8+ and CD4+ T cells, respectively (3–13). Immunization of melanoma patients with tyrosinase peptide vaccines restricted by HLA-A1, -A2, -A24, or -B44 has induced CTL responses occasionally associated with tumor regressions (14–18), showing the potential for tyrosinase to elicit immunologically diverse and clinically significant reactivities. Tyrosinase protein is more commonly and homogeneously expressed in metastatic melanoma lesions in situ than are other nonmutated melanoma antigens such as gp100, MART-1, or gp75 (19, 20), suggesting that tyrosinase might provide a better target for immunotherapy. Thus, we sought to develop a tyrosinase vaccine for clinical evaluation that would incorporate immunologic complexity, including the glycosylation that is required for generating certain tyrosinase epitopes and is available only under conditions of eukaryotic expression (6).
The poxviruses vaccinia and fowlpox have been used effectively as recombinant vaccines to immunize animals against a variety of pathogens (reviewed in refs. 21, 22), and when engineered to express tumor-associated antigens, they have mediated potent immunization and tumor regression in preclinical models (23, 24). The Surgery Branch of National Cancer Institute has previously conducted phase I/II clinical trials in which recombinant vaccinia or fowlpox vectors encoding gp100 or MART-1 were used to treat patients with advanced metastatic melanoma. From these trials, in which the same vaccine was administered repeatedly to raise immunity against the recombinant gene product (homologous prime/boost), in vitro or clinical evidence of effective immunization was infrequent (17, 25). The failure of vaccinia-based vectors to elicit robust antimelanoma responses was attributed to high preexisting serum titers of antivaccinia antibodies resulting from remote smallpox vaccination, and fowlpox-based vectors rapidly induced antiviral antibodies after the first inoculation (25). Importantly, antibodies generated against vaccinia or fowlpox in these patients did not seem to be cross-reactive, suggesting that heterologous prime/boost regimens might be efficacious. Indeed, immunizations with recombinant poxvirus vectors in murine tumor models showed the superiority of diversified versus homologous prime/boost regimens in generating CTL and mediating the regression of established micrometastatic disease (26–28).
Two investigational trials were designed to assess the clinical and immunologic effect of heterologous prime/boost vaccination with recombinant fowlpox/tyrosinase (rF-TYR) and vaccinia/tyrosinase (rV-TYR) in patients with advanced metastatic melanoma, based on the hypotheses that immunization against a complex full-length tumor antigen would generate HLA-diverse reactivities and that heterologous prime/boost vaccination would enhance efficacy. These were among the first immunotherapy trials to employ a diversified vaccine strategy. First, a randomized phase II study was conducted in which patients were treated with vaccines administered alone or concomitantly with low-dose interleukin 2 (IL-2) or high-dose IL-2 to augment antitumor immunity. This trial yielded in vitro evidence of successful immunization in some patients as well as an unexpectedly high clinical response rate in a nonrandomized subgroup of patients receiving high-dose IL-2 after failing to respond to vaccines alone. To further explore this finding, a single cohort phase II trial was conducted in which all patients received sequential rather than concomitant administration of vaccines and IL-2. We report here the clinical and immunologic results from these trials.
Patients and Methods
Patients
Patients eligible for protocol treatment had metastatic melanoma of cutaneous origin refractory to conventional therapy and measurable by standard imaging techniques or physical examination. Eligible patients were ≥16 years old, had an Eastern Cooperative Oncology Group performance status of 0 or 1 and a life expectancy of >3 months, had received no systemic cancer therapy within 3 weeks of protocol entry, and had documented disease progression since their most recent therapy. Patients with active infections, autoimmune diseases, immunosuppressive disorders, requirement for steroids, coagulation disorders, uncontrolled brain metastases, or cardiovascular, respiratory, hepatic, or renal dysfunction were excluded from study. Forty-seven patients were enrolled on the randomized phase II trial (July 1999–January 2002) and 19 patients were subsequently enrolled on the single cohort phase II trial (March 2003–May 2004). Patients previously treated with high-dose IL-2 were eligible for the randomized trial but not for the single cohort trial. Because the rV-TYR vaccine used in these studies is a replication-competent virus capable of causing clinically significant infections in susceptible individuals and because the vaccinia immune globulin indicated for managing such complications was unavailable from 1999 to 2001, patients treated on the randomized protocol during that time period were required to have been previously vaccinated against smallpox to reduce the potential for infectious complications. Eligible patients had no evidence of skin disorders such as eczema that might increase their susceptibility to vaccinia-related cutaneous infections. These clinical protocols were approved by the Investigational Review Board of the National Cancer Institute and the U.S. Food and Drug Administration. All patients signed an informed consent document before protocol entry.
Clinical trial design
In a randomized phase II protocol, patients were immunized every 4 weeks with rF-TYR or rV-TYR administered on an alternating schedule (Fig. 1A), starting with rF-TYR. Each inoculation was considered one treatment cycle, and four cycles constituted a treatment course. To minimize bias in assignment of treatments, patients were randomized to evaluate three treatment arms according to the Simon optimal two-stage design (29, 30): arm 1, vaccines alone; arm 2, vaccines followed immediately with s.c. low-dose IL-2; and arm 3, vaccines followed immediately by i.v. high-dose IL-2. Both arms 1 and 2 were designed to detect an objective response rate (complete response + partial response) of 25% as opposed to 5%, with 13 patients initially enrolled and the intent to accrue a total of 20 patients if at least one objective clinical response was observed. If at least three clinical responses were noted in 20 patients, then the treatment strategy employed in arm 1 and/or arm 2 would be considered worthy of further development. The design of arm 3 was based on a documented 15% objective response rate in patients with metastatic melanoma treated with a standard regimen of two cycles of high-dose IL-2 (31). To determine whether vaccines plus high-dose IL-2 could result in an improved response rate of 35% versus 15%, an initial group of 19 patients was treated with the intent to accrue 33 patients if at least four clinical responses were observed. If eight or more clinical responses were noted in 33 patients, then arm 3 would be considered worthy of further development. For patients with rapidly progressive disease at midcourse, treatment was discontinued. Otherwise, patients were evaluated for clinical response after one course of therapy. Patients with stable disease or any evidence of tumor regression were offered additional courses of treatment. According to the protocol design, patients with progressive disease in arm 1 or arm 2 were offered standard therapy with high-dose IL-2 if they had not previously received this and were still medically eligible (hereafter referred to as a nonrandomized “arm 4”).
Fig. 1.
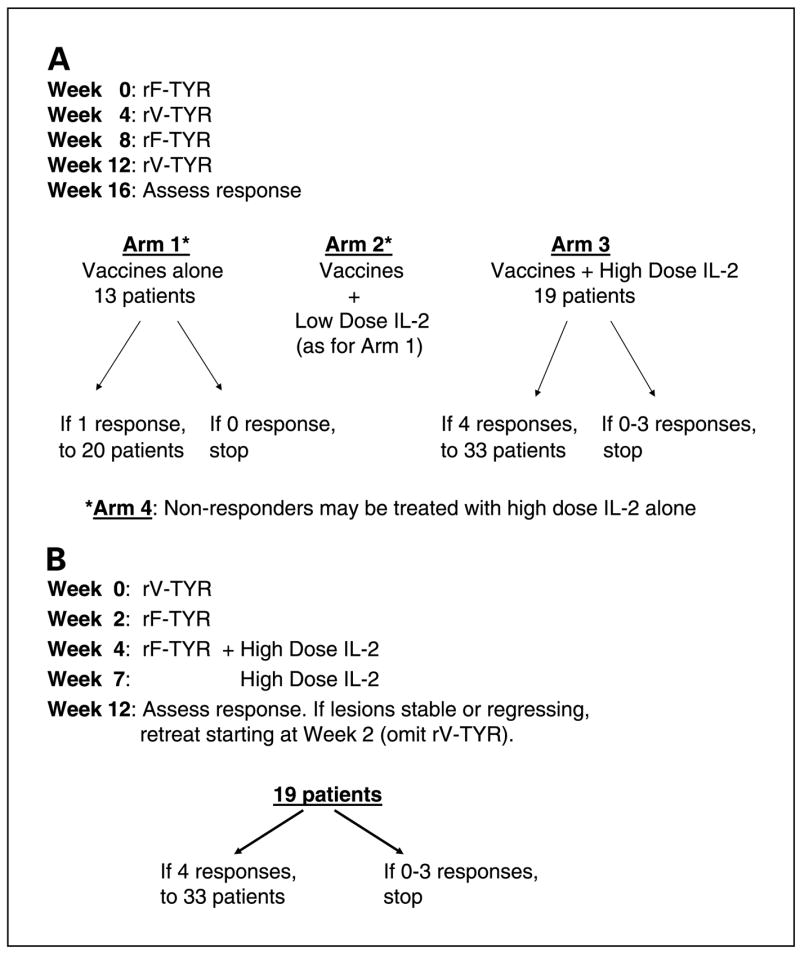
Protocol schemas for the randomized phase II trial that accrued patients from 1999 to 2002 (A) and the single cohort phase II trial that accrued patients from 2003 to 2004 (B). Vaccines were administered i.m. at 2 × 109 plaque-forming units per dose.”Response” refers to complete or partial clinical response.
In a subsequent single cohort phase II trial, patients received three immunizations at 2-week intervals (rV-TYR, rF-TYR, and rF-TYR) followed immediately by two cycles of high-dose IL-2 (Fig. 1B). The strategy of administering vaccines and IL-2 sequentially rather than concurrently was based on an observation from the randomized trial that three of eight patients who received vaccines alone (arm 1) followed by IL-2 alone (arm 4) showed partial responses (37.5% objective response rate). Although this observation was derived from a small group of patients, the lower bound of a one-sided 90% confidence interval about 3/8 is 14.7%; thus, there was >90% probability that the response rate associated with this treatment was at least as good, but possibly better, than the historical response rate of 15% to treatment with IL-2 alone. The rationale for modifying the vaccine sequence and treatment schedule is discussed in Results. Statistical considerations for this follow-up trial were identical to those for arm 3 of the first trial, evaluating the efficacy of the combination treatment relative to that of standard treatment with high-dose IL-2 alone.
The primary end point for both protocols was objective tumor response (complete response + partial response). The secondary end point was to characterize the immunologic responses generated by the protocol treatments, including serologic responses against the poxvirus vectors and tyrosinase protein and cell-mediated immune responses against tyrosinase. To this end, serum was collected from whole blood and lymphocytes from apheresis specimens at intervals and cryopreserved for future analysis.
Clinical response assessment
Bidimensional measurements of all metastatic lesions were recorded from computed tomography scans, magnetic resonance imaging scans, and/or physical examinations done within 4 weeks before protocol treatment and ~ 4 weeks after completing one treatment course (Fig. 1). A complete response was defined as the disappearance of all clinical evidence of disease for at least 4 weeks following the completion of therapy. A partial response was defined as ≥50% decrease in the sum of the products of the longest perpendicular diameters of all measurable lesions, and a minor response was defined as a 25% to 49% reduction with no new lesions appearing and no lesion increasing by >25%. A mixed response was defined as the regression of some lesions but simultaneous progression of others. Patients with any evidence of tumor regression or stable disease after one treatment course were eligible to receive another course of the same treatment; otherwise treatment was discontinued. Following a partial response or complete response, the appearance of new lesions or a >25% increase in size of known lesions was considered a relapse. Response duration was measured from the date of achieving the response, and stable disease duration from the initiation of therapy.
Clinical reagents
Recombinant poxviruses encoding the full-length human tyrosinase gene product were constructed and manufactured by Therion Biologics Corporation (Cambridge, MA). The rF-TYR vaccine was constructed from a live attenuated plaque-purified isolate from the POXVAC-TC strain of fowlpox virus, with the tyrosinase gene inserted through homologous recombination into the BamHI J region of the fowlpox genome under control of the vaccinia 40K promoter. The rV-TYR vaccine was constructed from a live plaque-purified isolate of the Wyeth (New York City Board of Health) strain of vaccinia virus. A tyrosinase insert identical to that contained in rF-TYR was inserted through homologous recombination into the thymidine kinase gene, located in the HindIII J region of the vaccinia virus genome under transcriptional control of the vaccinia 40K promoter. Both rF-TYR and rV-TYR were manufactured under sterile conditions by infection of primary chicken embryo dermal cells. Purified viruses were stored at −70°C until use. Each dose was administered i.m. at 2 × 109 plaque-forming units. Appropriate biohazard precautions for vaccine administration and care of the injection site were approved by the NIH Institutional Biosafety Committee and the NIH Office of Biotechnology Activities. Fowlpox virus can infect mammalian cells but cannot replicate in nonavian species; thus, systemic infections in humans are unlikely. However, because vaccinia virus is replication competent in mammalian cells, patients were instructed in proper disposal of vaccine site dressings and to avoid close contact with individuals who might be at increased risk for developing clinically significant vaccinia infections, including young children, pregnant women, and immunosuppressed individuals, for 2 weeks following each rV-TYR inoculation.
Recombinant human IL-2 was supplied by Chiron Corporation (Emeryville, CA). Patients in arm 2 of the randomized clinical trial self-administered IL-2 at a dose of 125,000 IU/kg s.c. daily for 12 days, commencing within 24 hours of each vaccine inoculation. Grade 3 or 4 toxicities according to Common Toxicity Criteria5 were managed by suspending and then resuming IL-2 administration at a 30% reduced dose. Patients on arm 3 of the randomized clinical trial received IL-2 as hospital inpatients at 720,000 IU/kg administered by i.v. bolus every 8 hours, beginning on the day after each vaccination and continuing for up to 4 days depending on patient tolerance. Doses were delayed or skipped as necessary to manage grade 3 or 4 toxicities. Patients on arm 4 of the first protocol or in the subsequent single-cohort trial received i.v. bolus IL-2 as two treatment cycles separated by a 7- to 10-day hiatus, a regimen considered to be standard-of-care for metastatic melanoma. Expected toxicities from IL-2 administration and their clinical management have been described elsewhere (31, 32).
Antiviral antibody titers
Titers of immunoglobulin G (IgG) antibodies against the vaccinia and fowlpox viruses were determined from blinded serum samples by ELISA. Vaccinia or fowlpox antigens were prepared as homogenized lysates of infected RK-13 cells or chicken embryonic dermal cells, respectively, and coated onto microtiter plates (Nunc Maxisorb) at 5 μg total protein/well. Serum samples were serially diluted from 50-fold to 3 million-fold and combined with plate-bound antigen. Plates were then treated with antihuman IgG antibody conjugated to horseradish peroxidase and developed with TMB peroxidase substrate, and the absorbance at A450 was determined. The antiviral IgG titer was defined as the reciprocal of the serum dilution generating an A450 of 0.500, which was approximately half maximal.
Neutralizing titers of antivaccinia antibodies were determined by the ability of patient sera to reduce infection of the susceptible cell line BSC-40 by a given number of viral plaque forming units. Serial dilutions of blinded patient serum samples were preincubated with live vaccinia virus at 37°C for 3 hours, and then the serum-virus mixtures were incubated with adherent BSC-40 cells in six-well tissue culture plates for 30 minutes followed by culturing for 2 days. Viral plaques in triplicate wells were visualized by crystal violet staining and counted. The neutralizing serum antibody titer was defined as the reciprocal of the serum dilution required to neutralize 50% of input plaques.
Antityrosinase antibodies detected by Western blotting
To assess the potential for protocol treatments to induce or enhance serologic responses against tyrosinase protein in melanoma patients, sera collected at intervals were used to probe Western blots containing tyrosinase protein. Blots containing poxvirus proteins were used as positive controls. Detergent lysates of COS-7 cells transfected with the plasmid pcDNA3.1/tyrosinase, or infected with the nonrecombinant vector TBC-Wy (vaccinia) or TBC-FPV (fowlpox), were electrophoresed in 4% to 20% Tris-glycine gels and then transferred to nitrocellulose membranes. Membranes were incubated with patient sera diluted 1:50 in buffer, then washed and incubated with peroxidase-conjugated rabbit anti-human IgG, IgA, IgM, or goat anti-human IgG (Sigma, St. Louis, MO) at 1:1,000 dilution. Blots were developed with 3,3′-diaminobenzidine peroxidase substrate (Sigma Fast DAB). As a positive control for the expression of tyrosinase protein, Western blots were probed with the tyrosinase-specific murine monoclonal antibody T311 (1 μg/mL; Vector Labs, Burlingame, CA) and counterstained with peroxidase-conjugated sheep anti-mouse IgG F(ab′)2 (Amersham Pharmacia Biotech, Piscataway, NJ).
Assessing T-cell reactivity to tyrosinase peptides and protein
Patient HLA types were determined by the NIH W.G. Magnuson Clinical Center HLA Laboratory using sequence-specific PCR techniques. Peripheral blood lymphocytes (PBL) from patients whose HLA types were compatible with previously defined tyrosinase epitopes were tested for specific peptide recognition by two different methods. In conventional in vitro sensitization, PBL were stimulated repetitively with peptides (see Table 1) and IL-2 over 2 to 4 weeks, followed by ELISAs to measure the secretion of IFNγ protein from activated T cells incubated overnight with peptide-pulsed autologous peripheral blood mononuclear cells. Alternatively, fresh peripheral blood mononuclear cells were incubated with peptides 10 μg/mL for only 2 to 4 hours, and IFNγ mRNA copy numbers in activated T cells were quantified with real-time reverse transcription-PCR (RT-PCR). Detailed methods for these procedures have previously been published in a study of T-cell reactivities in 10 patients from the randomized protocol (33). Commonly recognized epitopes from EBV (nuclear antigens 4 and 6) and influenza (hemagglutinin, matrix protein, nucleoprotein, and basic polymerase 1) were used as positive controls for specific T-cell activation in these experiments. T-cell reactivity against tyrosinase peptides was considered significant if it exceeded backgrounds from no peptide or irrelevant peptide stimulation by at least 2-fold.
Table 1.
Peptides used in this study
| Antigen* | HLA allele | Designation | Sequence |
|---|---|---|---|
| Tyrosinase | A1 | Ty 146-156 | SSDYVIPIGTY |
| A1 | Ty 243-251 | KCDICTDEY | |
| A1 | Ty 243-251, C244S | KSDICTDEY | |
| A*0201 | Ty 1-9 | MLLAVLYCL | |
| A*0201 | Ty 8-17 | CLLWSFQTSA | |
| A*0201 | Ty 369-377, N371D | YMDGTMSQV | |
| A*2402 | Ty 206-214 | AFLPWHRLF | |
| B*3501/3503 | Ty 312-320 | LPSSADVEF | |
| B*4403 | Ty 192-200 | SEIWRDIDF | |
| B*4403 | Ty 192-200, S192Y† | YEIWRDIDF | |
| DRB1*1501 | Ty 386-406 | FLLHHAFVDSIFEQWLRRHRP | |
| DRB1*1501 | Ty 386-406, R402Q† | FLLHHAFVDSIFEQWLQRHRP | |
| Influenza HA | DRB1*0101 | HA 307-319 | PKYVKQNTLKLAT |
| Influenza PB1 | A1 | PB1591-599 | VSDGGPNLY |
| Influenza M1 | A*0201 | M158-66 | GILGFVFTL |
| Influenza NP | A3 | NP 265-273 | ILRGSVAHK |
| EBNA 4 | A11 | EBNA4 416-424 | IVTDFSVIK |
| EBNA 6 | B*4403 | EBNA6 130-139 | EENLLDFVRF |
NOTE: Peptides were synthesized with Fmoc chemistry and their purity confirmed with mass spectrometry.
Tyrosinase peptides are described in refs. 3–13. Viral peptides were used as controls in all experiments evaluating T-cell function to assess general immune competence.
Polymorphic variant sequences.
To investigate T-cell recognition of the full-length tyrosinase protein, a recombinant adenovirus vector encoding tyrosinase (Ad2/Ty, Genzyme Corporation, Framingham, MA) was used to infect autologous dendritic cells. Dendritic cells expressing tyrosinase protein were then used to stimulate patient PBL in 12-day in vitro sensitization cultures followed by IFNγ ELISAs, or in 2- to 4-hour in vitro sensitizations followed by IFNγ real-time RT-PCR, according to methods previously described (33). Values exceeding backgrounds from PBL stimulated with dendritic cells infected with Ad2/green fluorescent protein by at least 2-fold were considered significant.
Tumor biopsies
Excisional biopsy specimens embedded in paraffin or frozen in optimum cutting temperature compound were sectioned and stained with antibodies against the following molecules using reagents and methods previously described (19): tyrosinase, MART-1/Melan-A, gp100, S-100, MHC class I, and HLA-DR. Lesions were scored in a blinded manner for percent of tumor cells expressing each antigen. Specimens were also stained with anti-CD3, CD4, and CD8, and intratumoral lymphocyte infiltration was scored on a 0 to 3+ scale. Cells obtained from fine-needle aspirates of s.c. or lymph node metastases were processed as paraffin-embedded cell blocks, fixed in formalin, and stained with H&E or with antibodies as above (34).
Results
Clinical responses
From 1999 to 2002, 47 patients with advanced metastatic melanoma were randomized to receive treatment with poxvirus vaccines encoding tyrosinase, alone or in combination with low-dose or high-dose IL-2 (Fig. 1A). Characteristics of patients in arms 1 to 3 were similar with respect to sex (overall, 58% male), age (average, 47 years), performance status (92% Eastern Cooperative Oncology Group 0), and prior treatments (surgery, 100%; chemotherapy, 38%; radiotherapy, 19%; immunotherapy, 81%; data not shown). Forty-five of 47 patients were evaluated for response: patients enrolled but not treated included one with a concomitant breast cancer and another with a metastatic carcinoma of unknown origin. By American Joint Committee on Cancer 2002 staging criteria, 11% of evaluable patients presented at stage III (9% stage IIIB and 2% stage IIIC) and 89% presented at stage IV (16% M1A, 9% M1B, and 64% M1C), distributed evenly among the three treatment arms. Twenty-six patients received at least one full course of therapy (four cycles over 4 months) whereas treatment was discontinued midcourse in 19 patients due to rapid disease progression (five patients from arm 1, eight from arm 2, and six from arm 3). No patient receiving vaccines alone showed an objective response. Two patients in arm 3 showed an objective clinical response (Table 2), one of whom has remained disease-free for over 3 years following resection of a residual periportal lymph node metastasis. Nine patients had biologically interesting “mixed” tumor responses. Although the mixed responses did not meet partial response criteria, some involved a significant overall reduction in tumor burden. For example, patient 42 in arm 3 experienced a 95% overall regression of disseminated multiorgan metastases following one treatment course, despite concomitant progression in s.c. and gallbladder sites (Table 3). Following a second course of treatment, a regressing s.c. metastasis and a progressing gall bladder metastasis were resected and analyzed for antigen expression and lymphoid infiltration (Fig. 2). Intense CD4+ and CD8+ lymphoid infiltrates were observed only in the regressing s.c. lesion, suggesting that tumor regression was immune mediated. Lymphocytic infiltration in an atypical nevus resected simultaneously suggested an immune response against a shared melanoma/melanocyte lineage antigen. All specimens expressed tyrosinase.
Table 2.
Clinical response evaluation of patients on the randomized trial
| No. patients (response duration in months)* |
||||
|---|---|---|---|---|
| Arm 1 | Arm 2 | Arm 3 | Arm 4† | |
| Randomized | 13 | 14 | 20 | NA |
| Evaluable | 13 | 13 | 19 | 17 |
| CR | 0 | 0 | 0 | 0 |
| PR | 0 | 0 | 2 (4, 8) | 5 (3, 4, 9,18, 40+) |
| MR | 0 | 0 | 0 | 0 |
| MXR | 1 | 2 | 6 | 3 |
| SD | 1 (6) | 1 (4) | 1 (4) | 1 (5) |
| PD | 11 | 10 | 10 | 8 |
Abbreviations: NA, not applicable; CR, complete response; PR, partial response; MR, minor response; MXR, mixed response; SD, stable disease; PD, progressive disease.
Duration of stable disease was measured from initiation of treatment. Duration of response was measured from date of achieving response.
In nonrandomized Arm 4, select patients from Arms 1 and 2 with progressive disease subsequently received high-dose IL-2 as single-agent therapy.
Table 3.
Mixed clinical response in patient 42
| Tumor site | Tumor area (cm2)* |
||
|---|---|---|---|
| Pretreatment (7/20/01) | After course 1 (11/27/01) | After course 2 (3/22/02) | |
| Thyroid gland | 3.3 | 0 | 0 |
| Adrenal gland | 4.0 | 0 | 0 |
| Kidney | 6.8 | 0 | 0 |
| Lymph node 1 | 14.4 | 0 | 0 |
| Lymph node 2 | 10.3 | 0 | 0 |
| S.c.1 | 5.0 | 0 | 0 |
| S.c. 2 | 4.0 | 0 | 0 |
| S.c. 3 | 0 | 1.4 | 0 |
| Gall bladder | 0.3 | 1.1 | 1.5 |
| Total | 48.1 | 2.5 | 1.5 |
NOTE: Patient 42 was treated with vaccines and high-dose IL-2 on Arm 3 of the randomized trial.
Bidimensional measurements obtained from computed tomography scans. Numerous additional s.c. lesions assessable only by physical exam regressed partially or completely during treatment.
Fig. 2.
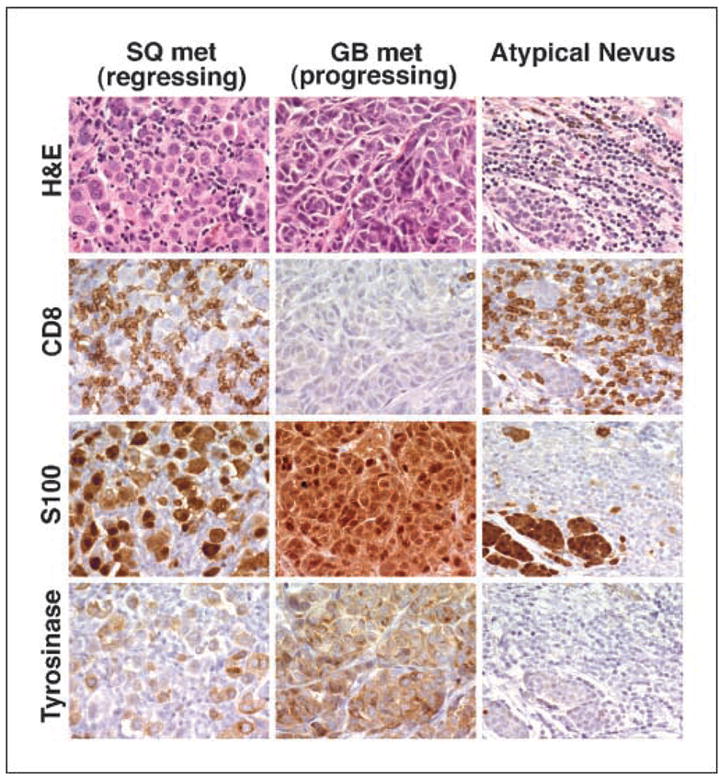
Tumor regression associated with intratumoral lymphoid infiltrates in patient 42, who manifested a mixed response following treatment with poxvirus/tyrosinase vaccines plus high-dose IL-2 (arm 3 on the randomized trial). S100 and tyrosinase are expressed in normal/atypical melanocytes as well as melanoma cells. Whereas tyrosinase protein is expressed in both a regressing s.c. (SQ) lesion and a progressing gall bladder (GB) metastasis, CD8+ lymphoid infiltrates are seen only in the regressing lesion. Staining with anti-CD4 gave similar results (not shown). Both metastatic lesions expressed MHC I uniformly and MHC II poorly (not shown). Concomitant infiltration of immune cells into an atypical nevus correlated clinically with the development of cutaneous depigmentation surrounding benign or atypical nevi and suggested the development of immunity against a shared melanoma-melanocyte lineage antigen.
According to the Simon optimal two-stage protocol design (29), the number of complete responses + partial responses observed in each of the three randomized treatment arms did not suggest a therapeutic effect significant enough to warrant additional patient accrual beyond the first stage in each arm. However, an apparently high response rate was observed in the nonrandomized treatment arm 4, including select patients from arms 1 and 2 with progressive disease, who then received standard treatment with two cycles of high-dose IL-2 (Table 2). Specifically, three of eight patients with progressive disease following treatment with vaccines alone for 2 to 6 months (arm 1) experienced partial responses after receiving IL-2 (37.5% response rate). Hypothetically, repeated immunizations to induce high-affinity antigen-specific T cells, followed by systemic IL-2 to further activate T cells, might provide an advantage over the concomitant administration of both agents which could cause activation-induced T-cell death (16) and/or the expansion of regulatory T cells (35). To explore this possibility in a larger group of patients, a second clinical trial was conducted in which all patients received vaccines and high-dose IL-2 sequentially rather than concomitantly (Fig. 1B). Additional treatment modifications were made based on findings from the previous trial. Because vaccines alone had no significant clinical effect in the first trial, vaccines in the second trial were administered at 2-week, instead of 4-week, intervals to shorten this part of the treatment. Furthermore, based on neutralizing antivaccinia antibody titer data (discussed below), rV-TYR was administered only once to each patient. The clinical characteristics of the 19 patients enrolled on this trial were similar to those on the previous randomized trial [average age, 47 years; male predominance, 68%; Eastern Cooperative Oncology Group 0 performance status, 89%; prior treatments including surgery (100%), chemotherapy (21%), radiotherapy (16%), and immunotherapy (68%)] as was the American Joint Committee on Cancer staging [stage III, 5%; stage IV, 95%, including M1A (11%), M1B (5%), and M1C (79%)]. Among these patients, one partial response, two minor responses, and four mixed responses were observed; thus, 7 of 19 patients showed some evidence of tumor regression following this protocol treatment. However, the objective response rate of 5% did not indicate an advantage compared with our historical experience with IL-2 alone. Of note, treatment-related toxicities observed in patients on both trials were similar to those expected from IL-2 therapy (31, 32).
Sera, lymphocytes, and tumor specimens archived from patients on both protocols were studied to assess the immunologic effect of these treatments as described below.
Serologic immunity against poxvirus vectors
Heterologous prime/boost vaccination regimens were used in these clinical trials to minimize immune responses against the viral vectors and maximize immunity against the tyrosinase transgene. To assess the validity of this approach from the standpoint of antiviral immunity, sera collected from patients on the randomized trial were tested for the presence of IgG antibodies against vaccinia and fowlpox viruses and for neutralizing antibodies against vaccinia. As shown in Fig. 3A, there was no evidence of serologic cross-reactivity between the two poxviruses: antivaccinia IgG titers did not increase significantly after the first fowlpox inoculation, nor did anti-fowlpox titers increase significantly after the first vaccinia inoculation. We also tested sera from patients who had received at least two inoculations with the same recombinant virus to evaluate antiviral immunity following repeated inoculations. As shown in Fig. 3B, pretreatment antivaccinia titers were already positive secondary to childhood smallpox vaccination compared with a vaccinia-naïve control titer of <50. Titers rose sharply after the first rV-TYR inoculation and then stabilized with repeated inoculations. In contrast, in a patient population that was initially fowlpox naïve, anti-fowlpox titers continued to increase after the second rF-TYR inoculation and then stabilized with repeated inoculations. Of note, antivaccinia titers were generally 1 log-fold higher than anti-fowlpox titers, consistent with the enhanced immunogenicity of replication-competent pathogenic viruses. Although small numbers of patients from each of the protocol treatment arms were analyzed, there did not seem to be major differences in antiviral titers, depending on whether or not IL-2 was administered.
Fig. 3.
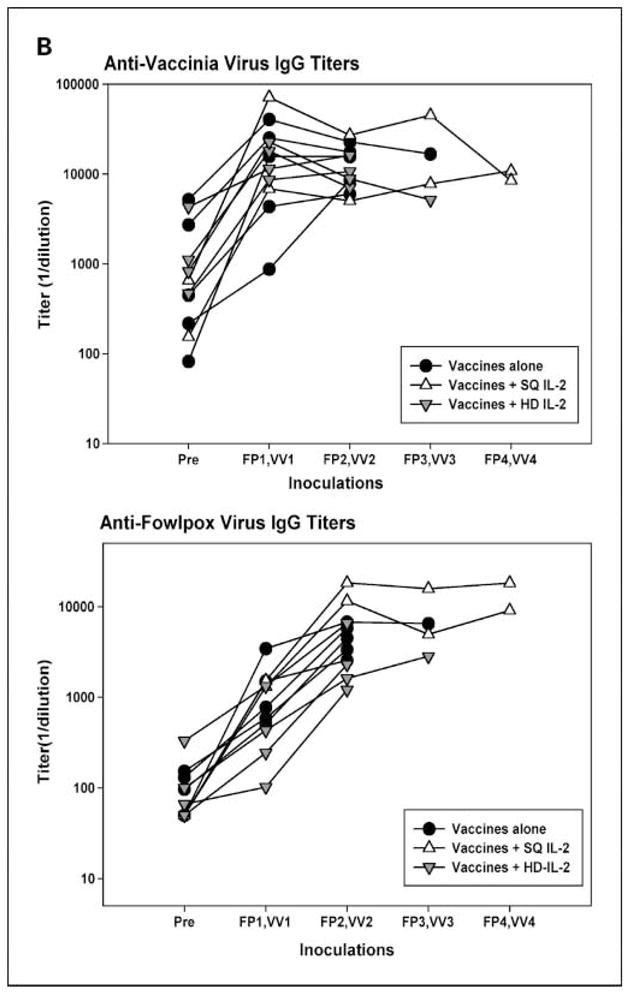
Antiviral IgG titers induced by vaccination. Patients on the randomized clinical trial received rF-TYR (FP) and rV-TYR (VV) on an alternating schedule every 4 weeks, starting with rF-TYR. Sera collected before treatment or 4 weeks following each inoculation were tested by ELISA for antiviral IgG. A, following one round of heterologous prime/boost inoculation in 12 patients, there was no evidence for serologic cross-reactivity between the two poxviruses. Naïve serum titers in the same assays were <50 for vaccinia virus and <50 to 229 for fowlpox virus. B, repeat immunizations with the same recombinant viruses in 11 patients showed maximal antivaccinia titers after the first rV-TYR and maximal anti-fowlpox titers after the second rF-TYR.
Antiviral IgG antibodies detected by ELISA do not necessarily equate with neutralizing activity that would affect the ability to repeatedly immunize patients. Thus, neutralizing antivaccinia antibody titers were measured in 24 patients from the randomized trial before and after the first rV-TYR inoculation. Neutralizing antibody titers were undetectable (<50) in 63% of patients before vaccination although all patients tested positive by ELISA. However, neutralizing titers rose sharply in all 24 patients after a single rV-TYR inoculation (data not shown). Again, administration of IL-2 did not seem to influence neutralizing antibody titers. These results, combined with the ELISA results described above, suggested that repeated inoculations with vaccinia vectors in patients with a remote history of smallpox vaccination were unlikely to further augment antityrosinase immunity. Based on these findings, patients on the subsequent single cohort trial (90% of whom had been vaccinated against smallpox) received only one rV-TYR vaccine, followed by multiple rF-TYR inoculations.
Serologic immunity against tyrosinase
Serologic responses against the full-length tyrosinase protein were assessed by probing Western blots containing tyrosinase with diluted patient sera. Western blotting is a relatively insensitive technique for this purpose compared with ELISA. However, multiple attempts to develop a tyrosinase ELISA proved unsuccessful because of the toxic effects of this large hydrophobic molecule in prokaryotic or eukaryotic protein expression systems, limiting protein availability.
Figure 4 shows a representative Western blot probed with sera from a patient treated with vaccines plus s.c. IL-2, who developed antityrosinase IgG antibodies following the second treatment cycle. Also shown is a control blot containing vaccinia proteins, which was simultaneously probed with the same sera, showing baseline reactivity that intensified dramatically after the first rV-TYR inoculation, consistent with our ELISA data (Fig. 3). Among 39 patients from the randomized trial and 10 patients from the single cohort trial who were assessed in this manner, none showed baseline antityrosinase serology but three patients developed reactivity following protocol treatment (6%). There was no correlation of serologic reactivity with clinical response.
Fig. 4.
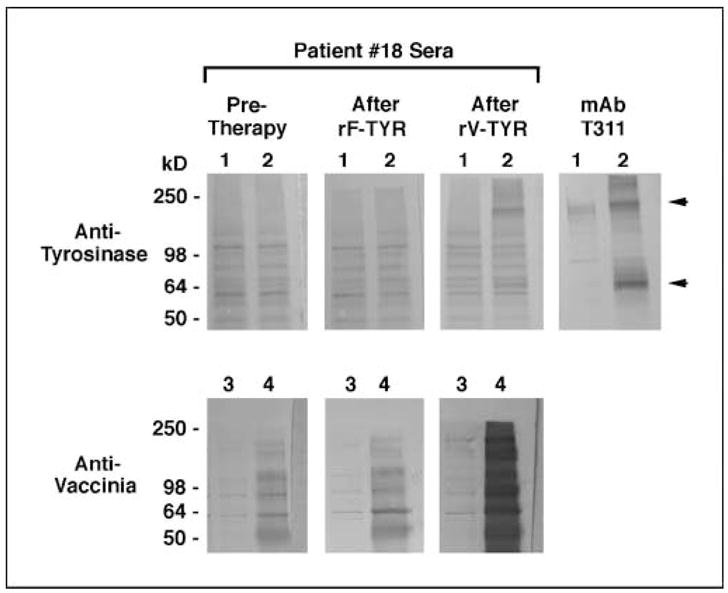
Antityrosinase IgG response developing in patient 18 after vaccination plus low-dose IL-2 (arm 2 on the randomized trial). Western blots containing lysates of COS-7 cells transfected with pcDNA3.1/tyrosinase (1.2 × 105 cell equivalents/lane) or infected with the nonrecombinant vaccinia vector TBC-Wy (0.7 × 105 cell equivalents/lane) were probed simultaneously with sera (1:50) collected before treatment, after the first treatment cycle (rF-TYR + IL-2), and after the second cycle (rV-TYR + IL-2). Lane 1, untransfected COS-7 cells; lane 2, COS-7 transfected with pcDNA3.1/tyrosinase; lane 3, uninfected COS-7; lane 4, COS-7 infected with TBC-Wy. The murine antityrosinase monoclonal antibody T311 provides a positive control for tyrosinase protein expression; arrows, monomeric and multimeric tyrosinase bands. Tyrosinase protein was visualized only with serum collected after the second treatment cycle. Antivaccinia IgG was evident pretreatment but increased significantly after the rV-TYR inoculation.
The demonstration of antityrosinase IgG antibodies in select patients from both clinical trials suggested the involvement of tyrosinase-specific CD4+ T cells in mediating immunoglobulin isotype switching. Because of the relative insensitivity of Western blotting, these data may underestimate actual antityrosinase serologic responses.
Cellular immunity against tyrosinase: peptide-based monitoring
MHC class I–restricted peptide–based in vitro immunomonitoring has become a routine adjunct for cancer immunotherapy protocols, even for patients immunized against full-length gene products such as carcinoembryonic antigen (36) or prostate-specific antigen (37). In our patients who were treated regardless of HLA type, we sought to determine if culturing PBL with tyrosinase peptides according to conventional repetitive in vitro sensitization techniques could reveal antityrosinase immune responses in patients whose HLA types were compatible with known epitopes (Table 1). Fourteen patients from the randomized clinical trial with amenable HLA types were assessed for recognition of tyrosinase epitopes restricted by five different HLA alleles, including HLA-A1 (seven patients), HLA-A2 (two patients), HLA-A24 (five patients), HLA-B35 (three patients), and HLA-B44 (two patients). For each HLA type, all relevant peptides listed in Table 1 were tested. PBL cultured for 2 to 4 weeks were assessed for recognition of peptide-pulsed autologous peripheral blood mononuclear cells after an overnight coculture, followed by an IFNγ ELISA to measure specific cytokine secretion from activated T cells. The results of these assays are summarized in Table 4. Two of 14 patients developed reactivity against a tyrosinase peptide over the course of treatment: PBL from patient 34 recognized Ty 192-200 (HLA-B44-restricted) and PBL from patient 35 recognized Ty 243-251 C244S (hereafter referred to as Ty 244S; HLA-A1-restricted; Fig. 5A). In addition, two patients were noted to have pretreatment reactivity against Ty 206-214 and Ty 244S (patients 16 and 28, respectively), which did not increase following protocol treatment. General immunocompetence in these patients was evidenced by T-cell recognition of HLA compatible viral epitopes (Table 1).
Table 4.
Summary of antityrosinase T-cell reactivities in 22 patients
| Arm (patient no.) | Response* | HLA† | Protein‡ |
Peptide§ |
||
|---|---|---|---|---|---|---|
| IVS | RT-PCR | IVS | RT-PCR | |||
| 1 (1) | PD | A2 | □ | ND | □ | ND |
| 1 (2) | PD | A24, B35 | ND | ND | □ | ND |
| 1, 4 (3)|| | PR | A1, A24 | □ | ■ | □ | □ |
| 1 (4)|| | MXR | A2, B35 | □ | ◀ | □ | □ |
| 1 (5) | PD | A24 | ND | ND | □ | ND |
| 1 (6) | PD | A1 | ND | ND | □ | ND |
| 1 and 4 (7)|| | PR | B35 | □ | ◀ | □ | ◀ Ty312 |
| 1 and 4 (13) | PR | B35 | ND | □ | ND | □ |
| 2 and 4 (16)|| | PR | A1, A24 | □ | □ | ◀ Ty206 | □ |
| 2 (18) | PD | A1 | ND | ND | □ | ND |
| 2 and 4 (20) | MXR | A1 | ND | □ | ND | □ |
| 2 and 4 (22)|| | PR | A24 | ▶ | □ | ND | □ |
| 3 (28)|| | MXR | A1 | ◀ | ◀ | ◀ Ty244S | ■ Ty244S |
| 3 (29) | PD | A1 | ND | ND | □ | ND |
| 3 (34)|| | SD | B44 | ▶ | ◀ | ■ Ty192 | ◀ Ty192Y |
| 3 (35)|| | MXR | A1 | ■ | ■ | ■ Ty244S | ■ Ty244S |
| 3 (37)|| | MXR | A1, B44 | □ | □ | ND | □ |
| 3 (38)|| | PR | A24, B35 | □ | □ | ND | □ |
| 3 (41) | MXR | A24, B44 | ND | □ | □ | □ |
| 3 (42) | MXR | None | ND | □ | ND | ND |
| 3 (43) | PR | A1, B35 | ND | ■ | ND | □ |
| 3 (44) | MXR | A24, B35 | ND | ■ | ND | □ |
NOTE: ■, new or increased reactivity following treatment; □, negative; ◀, positive before but not enhanced after treatment; ▶, positive after treatment but less than gp100 response.
Abbreviation: ND, not done.
Clinical response, defined in Patients and Methods.
Patient HLA types, for which the tested peptides, are listed in Table 1.
Response to full-length tyrosinase protein was assessed by repetitive in vitro stimulation (IVS) or real-time RT-PCR.
Peptides evoking specific T-cell reactivity are listed.
Results of select assays on PBL from these patients have previously been reported (33).
Fig. 5.

Antityrosinase T-cell responses generated in patients undergoing treatment. A, PBL were collected from patient 35 before treatment, after four cycles (course 1), and after six cycles (mid-course 2) of vaccines plus high-dose IL-2 (arm 3 on the randomized trial). After repetitive in vitro sensitization for 4 weeks with the HLA-A1-restricted Ty 244S peptide, PBL obtained posttreatment, but not before treatment, could specifically recognize Ty 244S. Results of an IFNγ ELISA following an overnight stimulation of cultured PBL with Ty 244S or other HLA-A1-restricted peptides are shown. Simultaneous in vitro sensitization with Ty 146-156 or Ty 243-251 failed to raise reactive T cells (not shown). Clinically, this patient had a mixed tumor regression. B, in real-time RT-PCR assays quantifying IFNγ mRNA, PBL from patient 3 (HLA-A1 and -A24) failed to recognize known tyrosinase epitopes but showed specific reactivity against tyrosinase protein expressed in autologous dendritic cells. Reactivity increased following sequential treatments with recombinant vaccines and high-dose IL-2 (arms 1 and 4 on the randomized trial). Clinically, this patient had a partial response after IL-2 therapy. Stimulation index for peptide reactivity, copies IFNγ mRNA/copies CD8 mRNA in PBL stimulated with peptide, compared with no peptide; for protein reactivity, PBL response to dendritic cells infected with Ad2/Ty or Ad2/gp100, compared with Ad2/green fluorescent protein control. Indices ≥2 are considered positive. Modified from ref. 33.
Because of the labor-intensive nature of repetitive in vitro sensitization and the possibility of introducing experimental artifacts through prolonged cell culture, we sought to develop sensitive and efficient methods of immunomonitoring that would more closely reflect the functional state of T cells in vivo. As an alternative to conventional in vitro sensitization, real-time RT-PCR techniques afforded an opportunity to query fresh uncultured T cells for immune reactivity against tyrosinase peptides in a rapid assay with an exquisitely sensitive read-out (33, 38). Fresh peripheral blood mononuclear cells were stimulated with tyrosinase peptides in vitro for 2 to 4 hours; after which, real-time RT-PCR was used to quantify IFNγ mRNA copies in activated T cells. PBL from 15 patients from the randomized clinical trial were assessed in this manner, and 4 showed specific recognition of a tyrosinase peptide. Patients 28 and 35 (HLA-A1+) developed specific responses against Ty 244S over the course of treatment whereas patients 7 and 34 showed baseline recognition of Ty 312-320 and Ty 192-200 192Y, respectively, which did not increase following treatment. Eight patients assessed with real-time RT-PCR had been previously tested against the same peptides with conventional in vitro sensitization (patients 3, 4, 7, 16, 28, 34, 35, and 41) and similar results were observed in six of these eight patients (Table 4).
Based on these findings as well as the advantages of assaying fresh PBL from vaccinated patients, real-time RT-PCR was adopted as the method of choice for detecting peptide-specific T-cell reactivity. Among the 19 patients treated in the single cohort trial, 8 were assessed for T-cell recognition of HLA-compatible tyrosinase peptides using real-time RT-PCR, including 6 patients expressing HLA-A1, 2 HLA-A24, 1 HLA-B35, 1 HLA-B44, and 3 HLA-DR15 (some patients expressed multiple alleles). Three of eight patients showed tyrosinase-specific reactivity as follows. Two HLA-A1+ patients recognized Ty 244S but this reactivity was present pretreatment and decreased during treatment. Two HLA-DR15+ patients recognized Ty 386-406 R402Q: in one case, reactivity increased following treatment, and in another case, reactivity existed pretreatment but did not increase subsequently. Overall, among 23 patients from both trials assessed for tyrosinase peptide-specific T-cell activity with real-time RT-PCR, 3 (13%) manifested reactivity that increased following protocol treatments.
Cellular immunity against tyrosinase: protein-based monitoring
The observation that most patients failed to show antityrosinase cellular immunity in peptide-based assays provided an impetus to develop new methods for immunomonitoring against full-length proteins. This approach would be most appropriate to protocols such as ours in which full-length tumor antigens were used as immunogens and patients were treated regardless of HLA type, broadening the scope of analysis beyond the library of known immunogenic epitopes and individual HLA alleles. To this end, autologous dendritic cells infected with a recombinant adenovirus/tyrosinase vector were used to stimulate patient PBL in vitro. Patients were monitored for immune responses against tyrosinase protein by measuring IFNγ secretion from PBL cultured in a 14-day in vitro sensitization with tyrosinase-expressing dendritic cells or by quantifying IFNγ mRNA with real-time RT-PCR in fresh PBL stimulated for 2 to 4 hours with the same dendritic cells (33). Responses to autologous dendritic cells expressing gp100, a shared melanoma antigen irrelevant to the vaccines used in our trial, were monitored simultaneously as a specificity control. Among 11 patients tested with in vitro sensitization, 4 recognized tyrosinase protein, although only patient 35 manifested tyrosinase-specific reactivity that increased following immunization (Table 4). In comparison, 8 of 16 patients recognized tyrosinase protein when assessed with real-time RT-PCR, including 4 (25%) patients with tyrosinase-specific T-cell reactivity that increased following protocol treatment. In 10 patients for whom both in vitro sensitization and RT-PCR testing were done simultaneously, RT-PCR seemed to have enhanced sensitivity for antigen-specific reactions as well as the advantage of investigating fresh instead of cultured PBL.
In the context of clinical trials using full-length tyrosinase as an immunogen, the importance of protein-based versus peptide-based immunomonitoring was illustrated by real-time RT-PCR analyses of PBL reactivity in 15 patients (Table 4). PBL from every patient recognizing a tyrosinase peptide by RT-PCR analysis (patients 7, 28, 34, and 35) also recognized tyrosinase protein. Conversely, PBL from four patients (patients 3, 4, 43, and 44) reacting against tyrosinase protein did not recognize HLA-compatible tyrosinase peptides, implying the existence of as yet undescribed tyrosinase epitopes. This phenomenon is illustrated in studies of patient 3, who expressed HLA-A1 and -A24 but whose PBL failed to react against the known tyrosinase epitopes restricted by these alleles in conventional peptide in vitro sensitization or real-time RT-PCR experiments. This patient’s PBL nevertheless reacted specifically against tyrosinase protein expressed in autologous dendritic cells as measured by real-time RT-PCR (Fig. 5B).
Of note, only 6 among the 22 patients selected for the in vitro studies summarized in Table 4 had progressive disease. Because limited numbers of patients with progressive disease were assessed, it is not possible to correlate in vitro findings with clinical outcome.
Tumor biopsies
The emergence of antigen loss tumor variants has been documented in the context of tumor antigen-specific immunotherapies (39), at once suggesting a therapeutic effect and providing a possible explanation for treatment failures. Because previous work from our laboratory and others’ has shown that the vast majority of metastatic melanoma lesions express tyrosinase (20), documentation of expression was not an eligibility criterion for protocol entry. However, consistent with previous findings, pretreatment fine-needle aspiration biopsies of tumors from patients on the randomized vaccine trial revealed that 22 of 23 (96%) lesions in s.c., lymph nodal, intramuscular, and glandular sites expressed tyrosinase protein detectable by immunohistochemical staining. Twenty lesions in 13 patients were selected prospectively for serial fine-needle aspiration biopsies before and after treatment. Whereas the expression of tyrosinase, gp100, and MART-1 proteins fluctuated in individual lesions during protocol treatment, no correlation was found between the degree of tyrosinase expression (percent positive tumor cells) and tumor growth characteristics (progressing, stable, or regressing; data not shown). To examine the issue of immunoselection from a different perspective, melanoma antigen expression in 12 new metastatic lesions appearing in eight patients during treatment was assessed (Table 5). In 4 of 12 lesions, <25% of tumor cells expressed tyrosinase, suggesting the possibility that deficiencies in tyrosinase antigen expression might have been causally related to tumor recurrence in these cases. However, tyrosinase antigen loss was not associated with the majority of treatment failures observed.
Table 5.
Melanoma-associated antigen expression in 12 new metastatic lesions appearing during therapy in eight patients on the randomized trial
| Arm (patient) | Biopsy site | Biopsy process* | % Positive tumor cells
|
||
|---|---|---|---|---|---|
| Tyrosinase | gp100 | MART-1 | |||
| 1 (3) | Brain | FS | 0 | <25 | <25 |
| 1 (6) | Bone | FS | >75 | >75 | >75 |
| 1 (9) | Brain | FS | >75 | ND | ND |
| 1 (10) | S.c. | CB | >75 | 0 | 50–75 |
| S.c. | CB | 50–75 | <25 | <25 | |
| 2 (15) | Lymph node | CB | >75 | 50–75 | >75 |
| 2 (20) | Lymph node | FS | <25 | 25–50 | 50–75 |
| S.c. | FS | <25 | 50–75 | 50–75 | |
| S.c. | FS | <25 | 0 | 0 | |
| 3 (29) | S.c. | CB | >75 | >75 | <25 |
| S.c. | CB | >75 | >75 | 0 | |
| 3 (43) | S.c. | CB | >75 | >75 | >75 |
FS, frozen section of excised specimen; CB, paraffin-embedded cell block of fine-needle aspiration specimen.
Five of 19 patients on the single cohort trial underwent resections of a total of eight metastatic lesions following immunotherapy, either to remove residual tumors following minor or mixed regressions or to obtain a source of tumor-infiltrating lymphocytes for possible treatment in other protocols. Tissue sections were analyzed by immunohistochemical staining for the expression of melanoma-associated antigens, MHC molecules, and T-cell infiltrates (Table 6). Among four separate lesions resected from patient 5, two lymph nodal metastases that were stable or regressing at the time of resection expressed tyrosinase as well as MHC antigens and contained significant lymphoid infiltrates, whereas progressing lesions in the lung and bowel had lost expression of tyrosinase as well as MART-1 and gp100. Among a total of six progressive lesions in five patients, five lesions were devoid of either tyrosinase or MHC protein expression. It is unknown how these expression profiles might have been influenced by protocol therapy because the same lesions were not biopsied before treatment. In addition, this analysis is limited to tumor antigens and MHC molecules and does not address the potential loss of other molecules essential for immune recognition, such as the components of the intracellular antigen processing machinery, nor does it address tumor-associated molecules, such as B7-H1 or transforming growth factor β, which might negatively influence immune recognition. However, these results suggest that antigen loss or MHC loss is a possible mechanism for lesional tumor progression in some patients following protocol therapy.
Table 6.
Tumor antigen expression following treatment with tyrosinase vaccines and IL-2 on the single cohort trial
| Patient no. (response) | Site | Lesion growth | % Positive tumor cells* |
|||||
|---|---|---|---|---|---|---|---|---|
| Tyrosinase | gp100 | MART-1 | MHC I | HLA-DR | CD3† | |||
| 5 (MXR) | LN-1 | Regr‡ | 50 | 20 | 5 | >50 | 20 | 2+ |
| LN-2 | Stable | 5–50 | >50 | 0 | ND | 5–50 | 3+ | |
| Lung | Progr | 0 | 0 | 0 | 10–50 | 0 | 1+ | |
| Bowel | New | 0 | 0 | 0 | ND | ND | ND | |
| 7 (MR) | LN | Progr | >50 | >50 | >50 | 0 | 0 | 1+ |
| 8 (MXR) | LN | Progr | 0 | <5 | 0 | 5–50 | 5–50 | 3+ |
| 13 (MXR) | S.c. | New | >50 | >50 | >50 | 5–50 | <5 | 1+ |
| 19 (NR) | S.c. | Progr | 0 | 0 | 0 | >50 | >50 | 3+ |
Immunostaining of paraffin-embedded tissue sections.
Presence of CD3+ cells was scored on a 0 to 3+ scale.
Regr, regressing; progr, progressing.
Discussion
Clinically meaningful treatment-associated tumor regressions (i.e., regressions with the potential for significantly improving patient survival) are considered to include only complete and partial responses. According to these strict criteria, none of our phase II treatments based on heterologous prime/boost antityrosinase immunization, alone or with IL-2 administration, were effective enough to warrant further clinical evaluation. Vaccines alone had no clinical effect and vaccines plus IL-2 induced objective responses at a rate similar to that expected from high-dose IL-2 alone. However, 25 of 64 patients with advanced melanoma treated on these clinical trials showed, in addition to “classic” partial responses, some biologically interesting evidence of tumor response including lesional regression (mixed responses) or overall regression that did not achieve partial response status (minor responses). At the same time, evidence of enhanced immunity against tyrosinase following protocol treatments was documented in 3 of 49 (6%) patients tested serologically, 3 of 23 (13%) patients tested for T-cell recognition of individual tyrosinase peptides with real-time RT-PCR, and 4 of 16 (25%) patients tested for T-cell recognition of full-length tyrosinase protein with real-time RT-PCR. These results, combined with biopsy data showing that some progressing lesions failed to express tyrosinase or MHC proteins, suggest that our treatments induced antityrosinase immunity in some patients but nevertheless did not exceed a threshold necessary to result in long-term clinical benefit significantly different than that expected from treatment with IL-2 alone.
Many cancer vaccine trials to date have involved peptide immunizations designed to activate CD8+ T cells. Whereas this approach affords the opportunity for precisely focused immunomonitoring and the advantages of low cost and minimal toxicity, it restricts therapy to patients with particular HLA types and sets the stage for immunoselection of treatment-refractory tumor variants. Furthermore, it lacks the immunologic diversity normally associated with effective immune responses. Our choice of full-length tyrosinase as an immunogen was intended to induce an immunologically complex response which could potentially be more effective than responses mediated solely by one subset of immune cells reacting against a single peptide (40), and indeed we showed examples of both serologic and cellular immune responses in treated patients. Monitoring such complex immune responses was challenging and required the development of new methodologies described in this report. Immunomonitoring validated our vaccination approach in selected patients, causing us to consider whether the low incidence of clinical responses was related to a suboptimal choice of target antigen. Abundance of tyrosinase expression in melanoma lesions has led some investigators to study its use as a single marker in evaluating sentinel lymph node biopsies (41) and serum samples (42) for molecular evidence of tumor metastasis. However, despite its abundance, tyrosinase is a normal self antigen that is subject to immunologic tolerance. In this regard, therapies designed to bypass tolerance such as lymphodepleting chemotherapy plus adoptive T-cell transfer (43), or to break tolerance such as systemic anti-CTLA4 administration (44), have proved effective. Furthermore, tyrosinase is not essential in maintaining the malignant phenotype of melanoma cells. In this vein, additional diversification of the immunization strategy by inclusion of other shared nonmutated melanoma antigens might not afford a therapeutic advantage. Indeed, examples of progressive melanoma lesions that failed to express gp100 and MART-1, as well as tyrosinase, are shown in Tables 5 and 6.
Based on the findings from these two trials, we have begun to examine the immunologic relevance of proteins essential to maintaining the malignant characteristics of melanoma cells, which might serve as better therapeutic targets than tyrosinase. One example is BRAF, a signaling molecule in the mitogen-activated protein kinase cascade, which in ~ 60% of melanoma patients contains a constitutively activating somatic V599E mutation (45). We have shown in vitro that melanoma-specific CD4+ T cells from patients harboring this mutation can specifically recognize mutant BRAF (46), opening possibilities for raising tumor-specific immunologic responses in vivo that would be directed against a neoantigen of which the essential function dictates persistent expression for cancer cell survival.
Acknowledgments
We thank James C. Yang, Douglas Schwartzentruber, Patrick Hwu, Francesco Marincola, and Surgery Branch Clinical Associates for contributions in patient care; Claudia Seipp and Kathleen Morton-Cooper for nursing research; Armando Filie, Paul Duray, David Berman, and David Kleiner for pathologic analysis; Melissa Corbitt, Glenda O’Neill, and Melonise Battle for data management; Franck Housseau, Gregory Lizée, Marisa Shiina, Sima Patel, Kelledy Manson, Niem Nguyen, Mai Nguyen, and Michelle Mail for technical contributions; and Gail Mazzara for intellectual support.
Grant support: Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
References
- 1.Robbins PF, Kawakami Y. Human tumor antigens recognized by T cells. Curr Opin Immunol. 1996;8:628–36. doi: 10.1016/s0952-7915(96)80078-1. [DOI] [PubMed] [Google Scholar]
- 2.Sahin U, Tureci O, Schmitt H, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci U S A. 1995;92:11810–3. doi: 10.1073/pnas.92.25.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawakami Y, Robbins PF, Wang X, et al. Identification of new melanoma epitopes on melanosomal proteins recognized by tumor infiltrating T lymphocytes restricted by HLA-A1, -A2, and -A3 alleles. J Immunol. 1998;161:6985–92. [PubMed] [Google Scholar]
- 4.Robbins PF, El-Gamil M, Kawakami Y, et al. Recognition of tyrosinase by tumor-infiltrating lymphocytes from a patient responding to immunotherapy. Cancer Res. 1994;54:3124–6. [PubMed] [Google Scholar]
- 5.Topalian SL, Gonzales MI, Parkhurst M, et al. Melanoma-specific CD4+ T cells recognize nonmutated HLA-DR-restricted tyrosinase epitopes. J Exp Med. 1996;183:1965–71. doi: 10.1084/jem.183.5.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Housseau F, Moorthy A, Lange DA, et al. N-linked carbohydrates in tyrosinase are required for its recognition by human MHC class II-restricted CD4+ T cells. Eur J Immunol. 2001;31:2690–701. doi: 10.1002/1521-4141(200109)31:9<2690::aid-immu2690>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 7.Kittlesen DJ, Thompson LW, Gulden PH, et al. Human melanoma patients recognize an HLA-A1-restricted CTL epitope from tyrosinase containing two cysteine residues: implications for tumor vaccine development. J Immunol. 1998;160:2099–106. [PubMed] [Google Scholar]
- 8.Wölfel T, Van Pel A, Brichard V, et al. Two tyrosinase nonapeptides recognized on HLA-A2 melanomas by autologous cytolytic T lymphocytes. Eur J Immunol. 1994;24:759–64. doi: 10.1002/eji.1830240340. [DOI] [PubMed] [Google Scholar]
- 9.Riley JP, Rosenberg SA, Parkhurst MR. Identification of a new shared HLA-A2. 1 restricted epitope from the melanoma antigen tyrosinase. J Immunother. 2001;24:212–20. [PubMed] [Google Scholar]
- 10.Kang X, Kawakami Y, El-Gamil M, et al. Identification of a tyrosinase epitope recognized by HLA-A24-restricted, tumor-infiltrating lymphocytes. J Immunol. 1995;155:1343–8. [PubMed] [Google Scholar]
- 11.Morel S, Ooms A, van Pel A, et al. A tyrosinase peptide presented by HLA-B35 is recognized on a human melanoma by autologous cytotoxic T lymphocytes. Int J Cancer. 1999;83:755–9. doi: 10.1002/(sici)1097-0215(19991210)83:6<755::aid-ijc10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 12.Brichard VG, Herman J, Van Pel A, et al. A tyrosinase nonapeptide presented by HLA-B44 is recognized on a human melanoma by autologous cytolytic T lymphocytes. Eur J Immunol. 1996;26:224–30. doi: 10.1002/eji.1830260135. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi H, Kokubo T, Sato K, et al. CD4+ T cells from peripheral blood of a melanoma patient recognize peptides derived from nonmutated tyrosinase. Cancer Res. 1998;58:296–301. [PubMed] [Google Scholar]
- 14.Jäeger E, Ringhoffer M, Dienes HP, et al. Granulocyte-macrophage-colony-stimulating factor enhances immune responses to melanoma-associated peptides in vivo. Int J Cancer. 1996;67:54–62. doi: 10.1002/(SICI)1097-0215(19960703)67:1<54::AID-IJC11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 15.Schaed SG, Klimek VM, Panageas KS, et al. T-cell responses against tyrosinase 368–376(370D) peptide in HLA*A0201+ melanoma patients: randomized trial comparing incomplete Freund’s adjuvant, granulocyte macrophage colony-stimulating factor, and QS-21 as immunological adjuvants. Clin Cancer Res. 2002;8:967–72. [PubMed] [Google Scholar]
- 16.Slingluff CL, Petroni GR, Yamshchikov GV, et al. Immunologic and clinical outcomes of vaccination with a multiepitope melanoma peptide vaccine plus low-dose interleukin-2 administered either concurrently or on a delayed schedule. J Clin Oncol. 2004;22:4474–85. doi: 10.1200/JCO.2004.10.212. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheibenbogen C, Schadendorf D, Bechrakis NE, et al. Effects of granulocyte-macrophage colony-stimulating factor and foreign helper protein as immunologic adjuvants on the T-cell response to vaccination with tyrosinase peptides. Int J Cancer. 2003;104:188–94. doi: 10.1002/ijc.10961. [DOI] [PubMed] [Google Scholar]
- 19.Cormier JN, Abati A, Fetsch P, et al. Comparative analysis of the in vivo expression of tyrosinase MART-1/Melan-A, and gp100 in metastatic melanoma lesions: implications for immunotherapy. J Immunother. 1998;21:27–31. doi: 10.1097/00002371-199801000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y-T, Stockert E, Tsang S, et al. Immunophenotyping of melanomas for tyrosinase: implications for vaccine development. Proc Natl Acad Sci U S A. 1995;92:8125–9. doi: 10.1073/pnas.92.18.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paoletti E. Applications of poxvirus vectors to vaccination: an update. Proc Natl Acad Sci U S A. 1996;93:11349–53. doi: 10.1073/pnas.93.21.11349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moss B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc Natl Acad Sci U S A. 1996;93:11341–8. doi: 10.1073/pnas.93.21.11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Bronte V, Chen PW, et al. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol. 1995;154:4685–92. [PMC free article] [PubMed] [Google Scholar]
- 24.Irvine KR, McCabe BJ, Rosenberg SA, et al. Synthetic oligonucleotide expressed by a recombinant vaccinia virus elicits therapeutic CTL. J Immunol. 1995;154:4651 –7. [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenberg SA, Yang JC, Schwartzentruber DJ, et al. Recombinant fowlpox viruses encoding the anchor-modified gp100 melanoma antigen can generate anti-tumor immune responses in patients with metastatic melanoma. Clin Cancer Res. 2003;9:2973–80. [PMC free article] [PubMed] [Google Scholar]
- 26.Irvine KR, Chamberlain RS, Shulman EP, et al. Enhancing efficacy of recombinant anticancer vaccines with prime/boost regimens that use two different vectors. J Natl Cancer Inst. 1997;89:1595–601. doi: 10.1093/jnci/89.21.1595. [DOI] [PubMed] [Google Scholar]
- 27.Hodge JW, McLaughlin JP, Kantor JA, et al. Diversified prime and boost protocols using recombinant vaccinia virus and recombinant non-replicating avian pox virus to enhance T-cell immunity and antitumor responses. Vaccine. 1997;15:759–68. doi: 10.1016/s0264-410x(96)00238-1. [DOI] [PubMed] [Google Scholar]
- 28.Chen C-H, Wang T-L, Hung C-F, et al. Boosting with recombinant vaccinia increases HPV-16 E7-specific T cell precursor frequencies of HPV-16 E7-expressing DNA vaccines. Vaccine. 2000;18:2015 –22. doi: 10.1016/s0264-410x(99)00528-9. [DOI] [PubMed] [Google Scholar]
- 29.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 30.Simon RM, Steinberg SM, Hamilton M, et al. Clinical trial designs for the early clinical development of therapeutic cancer vaccines. J Clin Oncol. 2001;19:1848–54. doi: 10.1200/JCO.2001.19.6.1848. [DOI] [PubMed] [Google Scholar]
- 31.Rosenberg SA, Lotze MT, Yang JC, et al. Experience with the use of high-dose interleukin-2 in the treatment of 652 cancer patients. Ann Surg. 1989;210:474–85. doi: 10.1097/00000658-198910000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mavroukakis SA, Muehlbauer PM, White RL, et al. Clinical pathways for managing patients receiving interleukin 2. Clin J Oncol Nurs. 2001;5:207–17. [PubMed] [Google Scholar]
- 33.Housseau F, Lindsey KR, Oberholtzer SD, et al. Quantitative real-time RT-PCR as a method for monitoring T lymphocyte reactivity to full-length tyrosinase protein in vaccinated melanoma patients. J Immunol Methods. 2002;266:87–103. doi: 10.1016/s0022-1759(02)00104-7. [DOI] [PubMed] [Google Scholar]
- 34.Fetsch PA, Riker AI, Marincola FM, et al. Tyrosinase immunoreactivity in fine-needle aspiration samples of metastatic malignant melanoma. Cancer. 2000;90:252–7. doi: 10.1002/1097-0142(20000825)90:4<252::aid-cncr9>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 35.Zhang H, Chua KS, Guimond M, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+ CD25+ regulatory T cells. Nat Med. 2005;11:1238–43. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 36.Marshall JL, Hoyer RJ, Toomey MA, et al. Phase I study in advanced cancer patients of a diversified prime-and-boost vaccination protocol using recombinant vaccinia virus and recombinant nonreplicating avipox virus to elicit anti-carcinoembryonic antigen immune responses. J Clin Oncol. 2000;18:3964–73. doi: 10.1200/JCO.2000.18.23.3964. [DOI] [PubMed] [Google Scholar]
- 37.Kaufman HL, Wang W, Manola HJ, et al. Phase II randomized study of vaccine treatment of advanced prostate cancer (E7897): a trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2004;22:2122–32. doi: 10.1200/JCO.2004.08.083. [DOI] [PubMed] [Google Scholar]
- 38.Kammula U, Marincola FM, Rosenberg SA. Real-time quantitative polymerase chain reaction assessment of immune reactivity in melanoma patients after tumor peptide vaccination. J Natl Cancer Inst. 2000;92:1336–44. doi: 10.1093/jnci/92.16.1336. [DOI] [PubMed] [Google Scholar]
- 39.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99:16168–73. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chapman PB. T-cell chauvinists versus antibody advocates-Can’t we all just get along? J Clin Oncol. 2004;22:4446–8. doi: 10.1200/JCO.2004.06.939. [DOI] [PubMed] [Google Scholar]
- 41.Kammula US, Ghossein R, Bhattacharya S, Coit DG. Serial follow-up and the prognostic significance of reverse transcriptase-polymerase chain reaction-staged sentinel lymph nodes from melanoma patients. J Clin Oncol. 2004;22:3989–96. doi: 10.1200/JCO.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 42.Voit C, Kron M, Rademaker J, et al. Molecular staging in stage II and III melanoma patients and its effect on long-term survival. J Clin Oncol. 2005;23:1218–27. doi: 10.1200/JCO.2005.04.098. [DOI] [PubMed] [Google Scholar]
- 43.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Attia P, Phan GQ, Maker AV, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol. 2005;23:6043–53. doi: 10.1200/JCO.2005.06.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davies H, Bignell GR, Cox C, et al. Mutation of the BRAF gene in human cancer. Nature. 2002;471:949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 46.Sharkey MS, Lizée G, Gonzales MI, Patel S, Topalian SL. CD4+ T-cell recognition of mutated B-RAF in melanoma patients harboring the V599E mutation. Cancer Res. 2004;64:1595–9. doi: 10.1158/0008-5472.can-03-3231. [DOI] [PubMed] [Google Scholar]